

Abstract
This review briefly summarizes what was known about NOS enzymology at the time of the Nobel Prize award in 1998 and then discusses from the author's perspective some of the advances in NOS enzymology over the subsequent 20 years, focused on five aspects: the maturation process of NOS enzymes and its regulation; the mechanism of NO synthesis; the redox roles played by the 6R‐tetrahydrobiopterin cofactor; the role of protein conformational behaviour in enabling NOS electron transfer and its regulation by NOS structural elements and calmodulin, and the catalytic cycling pathways of NOS enzymes and their influence on NOS activity.
Linked Articles
This article is part of a themed section on Nitric Oxide 20 Years from the 1998 Nobel Prize. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.2/issuetoc
Abbreviations
- AI
auto‐inhibitory structural element that is present in the FMN domain of NOSs
- Arg
L‐arginine
- CaM
calmodulin
- CT
C‐terminal tail structural element that is present in NOS enzymes
- BH4
6R‐tetrahydro‐L‐biopterin
- Haem
iron protoporphyrin IX
- Hsp90
heat shock protein 90
- NOHA
N‐hydroxy‐L‐arginine
- NOSoxy
oxygenase domain of NOS
- SNO
S‐nitroso modified
What did we know by 1998?
The three principal mammalian NOS enzymes (EC 1.14.13.39; NOS 1, 2, 3; neuronal, inducible and endothelial NOS respectively) had already been cloned and successfully expressed in E. coli or in insect cells, so their characterization was underway by 1998. Regarding the chemical mechanism of NO synthesis, we knew that NOS catalysed a two‐step oxidation of L‐arginine (Arg) with N‐hydroxy‐L‐Arg (NOHA) forming as an enzyme‐bound intermediate, and we knew basic information such as the moles of NADPH and O2 consumed per NO formed and the source of the oxygen atoms incorporated into the NO and citrulline products (Knowles and Moncada, 1994; Masters et al., 1996; Stuehr, 1997). We knew that active NOS enzymes are homodimeric with subunits consisting of an N‐terminal oxygenase domain (NOSoxy) and a C‐terminal reductase domain that are linked together by a calmodulin (CaM) binding sequence and that two NOSoxy domains must interact with one another to form the active homodimer. We knew that NOS subunits utilize a unique combination of four redox cofactors, two of which bind in the NOSoxy domain [iron protoporpyrin IX (haem) and 6R‐tetrahydrobiopterin (BH4)] and two in the reductase domain (FAD and FMN) and that during catalysis, the flow of NADPH‐derived electrons is first into FAD and then into FMN. We knew that the Ca2+‐promoted CaM binding event allows cross‐subunit electron transfer from the FMN to the haem and is the essential electron transfer step because it enables O2 to bind to the NOS haem and thus start the process of NO biosynthesis (Knowles and Moncada, 1994; Masters et al., 1996; Stuehr, 1997). We knew that BH4 has important structural and thermodynamic effects on NOS (Marletta, 1994; Presta et al., 1998a; Presta et al., 1998b) but had not yet uncovered or appreciated its essential redox role. Stopped‐flow and rapid‐quench experimental approaches had just begun to be deployed by 1998 and were being used to study the steps taking place during each of the two reactions of NO synthesis (Arg to NOHA and NOHA to citrulline + NO) in single or multiple catalytic turnover settings (Abu‐Soud et al., 1997a; Abu‐Soud et al., 1997b). The stopped‐flow studies characterized the NOS ferrous haem‐dioxy complex regarding its UV‐visible spectrum and the kinetics of its formation and disappearance and showed that bound BH4 hastened its disappearance but for reasons that were not immediately clear. The crystal structures of the NOSoxy domain dimer with bound Arg and BH4 were first reported in 1998 (Crane et al., 1998; Raman et al., 1998), and this provided solid foundation for subsequent NOS structure–function studies. Finally, mechanisms that regulate the intracellular localization and activity of NOS enzymes were coming into focus, including roles for caveolin and post‐translational protein phosphorylation, palmitoylation and myristoylation, particularly for the neuronal and endothelial NOS enzymes (Nakane et al., 1991; Garcia‐Cardena et al., 1996; Dudzinski et al., 2006). Spectroscopic studies revealed an additional important aspect of the NOS reaction mechanism, namely, that the NO generated by NOS first bound to the enzyme ferric haem before it escaped into solution (Abu‐Soud et al., 1997b) and showed that the build‐up of NOS haem‐NO species could be significant during NO synthesis and could affect the enzyme catalytic activity and oxygen concentration‐dependence (apparent KmO2), both in vitro (Abu‐Soud et al., 1996) and in the human lung (Dweik et al., 1998). Thus, many of the key experimental approaches and concepts were either in place or being established by the end of 1998, which guided and enabled studies of NOS enzymology over the past 20 years.
What's new?
NOS maturation and modes of its regulation
The fact that NOS proteins acquire four cofactors (FAD, FMN, haem and BH4) and then form a homodimer to become active provides opportunities to regulate their activity through control of NOS maturation and assembly. Since 1998, we have improved our understanding of NOS maturation, particularly the haem insertion process, which is a key step in creating the active enzyme and have uncovered several ways that NO can negatively regulate NOS maturation and the stability of the NOS homodimer.
Haem insertion into at least two NOS enzymes was discovered to involve heat shock protein 90 (hsp90) (Ghosh et al., 2011; Peng et al., 2012). Hsp90 associates with the haem‐free, immature form of NOS in cells, drives haem insertion in an ATP‐dependent process and then dissociates from the haem‐containing NOS. Based on the known functions of hsp90, it is possible that its mechanism of action may be to alter the protein conformation of the NOS oxygenase domain to facilitate haem cofactor insertion into the binding pocket. Such facilitation of haem insertion might also explain how hsp90 ‘stimulates’ the activity of NOS enzymes. But much about this process, including the molecular mechanism of action and the possible involvement of co‐chaperones, remains to be defined. Of note, hsp90 also drives haem insertion into other haem proteins including soluble GCs and haemoglobin (Ghosh and Stuehr, 2017; Ghosh et al., 2018) and thus may function more generally in haem protein maturation in biology.
Work done prior to the Nobel Prize showed that NO can affect the level of mature active NOS in at least three ways: NO can increase NOS protein expression through its effect on iron‐responsive transcription factors (Weiss et al., 1994). NO can restrict NOS maturation by lowering haem production in cells, through NO inactivating ferrochelatase (Furukawa et al., 1995), which is the mitochondrial enzyme that catalyses iron insertion into the protoporphyrin ring during haem biosynthesis. NO can also directly inhibit haem insertion into iNOS (Albakri and Stuehr, 1996). After 1998, it was discovered that exposing cells to NO (i.e. at levels equivalent to the NO generated by activated macrophages or Kupffer cells) broadly inhibited haem insertion into all three NOS enzymes and several other haem proteins, including haemoglobin, cytochrome P450 and catalase (Waheed et al., 2010). For iNOS, NO inhibition of its haem insertion was shown to be dependent on the S‐nitrosation (SNO) of a Cys residue in GAPDH (Cys152 in human GAPDH) (Chakravarti et al., 2010). Indeed, GAPDH was recently shown to bind and allocate the bioavailable haem to iNOS during its maturation in cells (Sweeny et al., 2018).
NO can also diminish the level of active NOS that builds up in cells by acting at points downstream from the haem insertion process. For example, NO can react with the haem‐containing NOS monomers during their maturation in a way that prevents them from forming a homodimer (Chen et al., 2002). NO can also destabilize and break up mature NOS homodimers in cells (Mitchell et al., 2005; Li et al., 2006). In these circumstances, the evidence suggests that NO may first react with the NOS ferric haem to enable nitrosonium‐based covalent protein modifications, specifically SNO modification or oxidation of Cys residues in NOS that are located at the dimer interface and involved in Zn2+ binding and dimer stabilization.
NOS maturation and homodimer stability have also been considered as potential drug targets: this was first indicated by steric‐bulky imidazoles preventing haem insertion into iNOS in cells, which prevented formation of the active homodimer (Sennequier et al., 1999). Several drug prototypes have been developed that are NOS isoform‐specific and work by blocking dimerization of haem‐containing NOS monomers in cells (Blasko et al., 2002; Gahman et al., 2011), but they have not been developed for clinical use. Figure 1 illustrates how hsp90, NO and related factors act at different points in the process to influence the expression, maturation and stability of active NOS homodimers.
Figure 1.
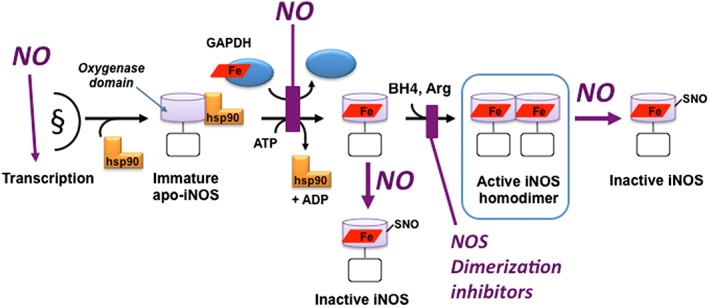
The NOS maturation pathway and points of regulation by NO or by dimerization inhibitors. The model shows results obtained primarily for iNOS but may be generally applicable for the three mammalian NOS. After transcription and translation, the immature NOS monomer exists in cells in complex with chaperone hsp90. Haem, likely to be provided by GAPDH, is inserted into the NOS monomer in an ATP‐dependent, hsp90‐driven process, to form a haem‐containing NOS monomer. The haem‐containing monomeric species dissociates hsp90 and then can form a mature NOS homodimer in the presence of BH4 and Arg. NO can influence these processes at the points shown by increasing transcription of NOS, inhibiting GAPDH‐dependent haem insertion and causing SNO of NOS either prior to or after it forms a homodimer, which blocks or reverses homodimer formation respectively. Dimerization inhibitors block homodimer assembly. Light purple cylinder is the NOSoxy domain; open rectangle is the NOS reductase domain. Haem is the red rhombus.
NOS mechanism of NO synthesis
Rapid‐mixing stopped‐flow and rapid‐quench approaches were just beginning to be deployed around the time of the Nobel award to study the mechanism of NO synthesis. These experiments typically held the NOS haem in its ferrous state under an inert atmosphere and then studied its stepwise reaction with O2 and the subsequent conversion of Arg to NOHA or the conversion of NOHA into NO + citrulline, within a single catalytic turnover (Figure 2). Coupling these approaches with the information obtained from recent crystal structures of the NOSoxy domains (Crane et al., 1998; Raman et al., 1998) fuelled numerous investigations into the reaction mechanism and NOS structure–function relationships.
Figure 2.
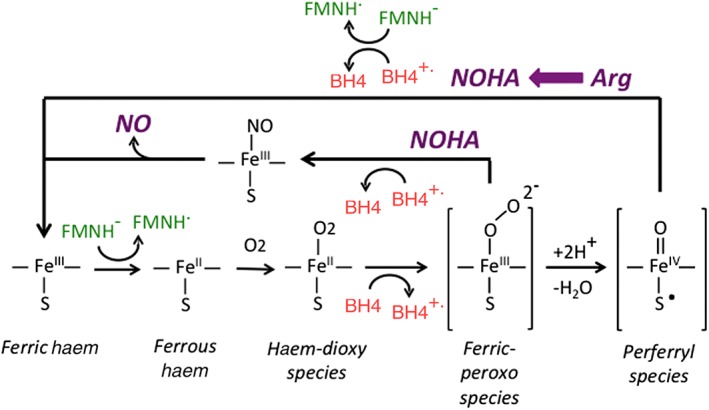
NOS O2 activation steps and the accompanying BH4 redox transitions that occur during the Arg and NOHA oxidation reactions. The catalysis starts with the ferric enzyme being reduced by an electron from the reductase domain (FMNH−). This allows the haem to bind O2 and form the haem‐dioxy species, which is then reduced by BH4 to form the reactive haem‐oxy species that react either with Arg or with NOHA as indicated. After Arg is oxidized to NOHA, the reductase domain provides an electron to reduce the NOS‐bound BH4 radical back to BH4 and, in doing so, resets the ferric enzyme for catalysis of NOHA oxidation. During the NOHA reaction, the BH4 radical that forms receives an electron from a NOS reaction/product species, and this allows generation of the enzyme ferric haem‐NO species that releases NO.
The rapid‐mixing approaches allowed measures of the kinetics of various individual steps in NOS that take place during Arg hydroxylation or NOHA conversion to NO, primarily focusing on the changes that occur at the haem, BH4 or flavin centres. Generally, the NOS enzymes were found to activate oxygen in a stepwise process and form intermediate haem‐oxy species like those that form in the cytochrome P450, which are haem‐thiolate enzymes similar to NOS (Krest et al., 2013). Discussions about the NOS haem‐oxy species that may form downstream from the initial haem‐O2 species in the Arg and NOHA reactions (Woodward et al., 2009; Davydov et al., 2014; Shamovsky et al., 2018) and theoretical studies of the NOS reaction mechanism (de Visser, 2009; Shamovsky et al., 2018) have been published elsewhere and so are not discussed here in detail.
In NOS, one can directly observe the build‐up and disappearance of the initial haem‐O2 species, and this has allowed a fairly comprehensive understanding of its electronics, stability and reactivity (Abu‐Soud et al., 1997a; Li et al., 2007; Davydov et al., 2014). The same holds for the immediate product species that builds up after the NOHA oxidation reaction, namely, the NOS ferric haem‐NO complex (Pant and Crane, 2006; Wang et al., 2010). We have far less data on the various reactive haem‐oxy species that are expected to form in between these two points in the reaction trajectory (Figure 2), because the relative kinetics are such that these species do not build‐up for observation under normal experimental conditions. However, based on different approaches including low temperature annealing experiments (Woodward et al., 2009; Davydov et al., 2014), a general consensus has emerged that the Arg hydroxylation and NOHA oxidation steps are likely to involve two distinct haem‐oxy species that form within NOS during oxygen activation. Specifically, Arg is thought to react with a NOS haem iron‐perferryl species, while NOHA is thought to react with a haem iron‐peroxo species that typically forms prior to the perferryl species during stepwise oxygen activation (Figure 2).
Along with these findings, the investigations since 1998 have uncovered aspects of the reaction mechanism unique to NOS enzymes. These include how NOS manages odd‐electron chemistry through the use of its BH4 cofactor, how it regulates electron transfer into the NOSoxy domain to enable the reduction of the haem and of the BH4 radical that forms during catalysis and how NOS catalysis goes on despite its generating NO, which is typically a poison for haem‐dependent enzymes. What we've learned about these aspects is discussed below.
Multiple new roles for BH4
One of the most exciting stories in NOS enzymology is how they utilize their BH4 cofactor. Around the time of the Nobel Prize, it was known that bound BH4 played important ‘non‐redox’ roles in NOS, such as influencing the midpoint potential of the haem iron and stabilizing NOS protein structural elements that in turn stabilize the dimer interface or restrict access of small molecules to the haem distal pocket (Abu‐Soud et al., 1998; Presta et al., 1998a; Presta et al., 1998b). Initially, a mechanism was proposed that had BH4 participating in the chemistry of NO synthesis by forming a reactive pterin‐hydroperoxy species, similar to the process in aromatic amino acid hydroxylase enzymes, but the NOSoxy crystal structures showed that the bound BH4 is located to the side of the haem and not in a position that would allow a pterin‐hydroperoxy species to react with the bound Arg or NOHA substrates. Rather, the relative positions of BH4 and haem in NOS were reminiscent of how ascorbate is held next to the haem in ascorbate peroxidase (Sharp et al., 2003), which allows ascorbate to act as a one‐electron donor to the haem. This structural similarity was prescient, because rapid‐mixing and rapid‐freezing approaches soon showed that the bound BH4 in NOS donates an electron to the haem‐dioxy species that forms during oxygen activation and so becomes a bound, BH4 cation radical (Hurshman and Marletta, 2002; Stuehr et al., 2005). This presented a novel use for BH4 cofactor in biology as a one‐electron donor. Moreover, BH4 only donates the ‘second electron’ in the NOS oxygen activation process, which is critical, because it allows NOS to generate haem‐oxy species that react with Arg or NOHA (Figure 2). Importantly, the rate of electron transfer from BH4 to the haem‐dioxy species was found to be about 10 times faster than the rate of electron transfer from the NOS flavoprotein domain to haem (Stuehr et al., 2005). The kinetic advantage of BH4 helped to explain why BH4 was needed to ‘couple’ NOS haem reduction to NO synthesis. The quicker BH4 reduction of the initial haem‐O2 species prevents it from breaking down into superoxide and ferric haem. But despite its speed, the BH4 electron transfer was still slower than the subsequent downstream reaction steps of NO synthesis (Stuehr et al., 2005) (Figure 2). This precludes the build‐up and observation of any downstream haem‐oxy based reaction intermediates, except when the reaction is studied under special cryogenic conditions, as noted above. In the subsequent work (see Hurshman and Marletta, 2002; Stuehr et al., 2005), the rate and extent of BH4 electron transfer to the NOS haem‐dioxy species were found to depend on the electronic properties of the pterin and the haem, the NOS amino acids that surround the bound BH4 and the identity of the NOS isoform itself, because eNOS, iNOS and nNOS showed surprisingly large differences in their rates of BH4 radical formation and the radical lifetimes (Wei et al., 2005).
By 1998, we knew that the bound BH4 is not easily lost from NOS and appeared capable of supporting multiple rounds of NO synthesis (Witteveen et al., 1996; Gorren and Mayer, 2002; Stuehr et al., 2005). Indeed, the stoichiometry of NADPH usage by NOS, along with other considerations, implied that the BH4 radical that forms during Arg hydroxylation is reduced back to BH4 within NOS, in order for NOS to continue the second round of oxygen activation that is needed for NOHA oxidation (Figure 2). Ultimately, the NOS reductase domain was shown to be the source of this electron, and the BH4 radical reduction step was found to be CaM‐dependent (Wei et al., 2008). A subsequent study showed that the rate and extent of BH4 radical reduction in various NOS mutants matches their rates and extents of haem reduction (Ramasamy et al., 2016). This result implied that the mechanism of BH4 radical reduction involves the NOS flavoprotein domain passing an electron through the NOS haem in order to reach the bound BH4 radical. Importantly, this solves a physical problem that exists in NOS enzymes concerning BH4 radical reduction, due to their BH4 cofactor being located too far from the surface of the NOS oxygenase domain to allow a direct electron transfer to take place from the FMN in the reductase domain, even when the FMN domain docks onto the oxygenase domain. Thus, the NOS haem can handle the electron that it receives from the NOS reductase domain in two ways: the electron can be used to enable oxygen binding at the haem iron, or it can be utilized to reduce the bound BH4 radical, depending on need and where the NOS enzyme is in the catalytic cycle (Figure 2).
There is also a BH4 radical formed in NOS during the NOHA oxidation step, but in this case, it is quickly reduced back to BH4, independent of the NOS reductase domain, according to a time course that matches the formation rate of the NOS ferric haem‐NO product species (Stuehr et al., 2005). This consecutive one‐electron oxidation and reduction of BH4 is unprecedented in biology but makes mechanistic sense, because it solves the fundamental challenge that all NOS enzymes must overcome in generating their odd‐electron product (NO). Specifically, in the NOHA oxidation step, the BH4 first functions as a one‐electron donor to the haem‐O2 species to enable oxygen activation, just as it functions in the Arg hydroxylation step (Figure 2). But subsequently, the BH4 radical acts as a one‐electron oxidant to receive an electron from a NOS product species that forms during the NOHA oxidation process, such as the ferrous haem‐NO species or possibly even from nitroxyl that may be released within the active site (Wei et al., 2003; Woodward et al., 2010). Significantly, it is this electron‐accepting role of the BH4 radical that allows the ferric haem‐NO product species to form at the end of the reaction (Figure 2). Because such a precisely timed redox chemistry cannot be performed by the attached NOS reductase domain, BH4‐free NOS enzymes generate haem‐bound nitroxyl instead of NO (Rusche et al., 1998; Adak et al., 2000). Thus, of all the structural and electronic roles that BH4 performs to enable NO synthesis, acting as a sequential one‐electron reductant and oxidant during the NOHA oxidation reaction step may be its key role, because it is this function that allows NOS to generate the free radical NO as a product.
Electron transfer and its regulation in NOS
Conformational equilibrium and motions of the NOS reductase domain
By 1998, reductase domain function was already known to involve an electron transfer between its FAD and FMN cofactors and that catalysis depended on electrons transferring out of the FMN domain. In 2004, the crystal structure of the NOS reductase domain (Garcin et al., 2004) was illuminating but also presented a quandary because, while it showed the FMN cofactor in position to accept electrons directly from FAD, it also showed that the FMN in this position was buried in the structure and inaccessible to solvent and therefore not in a position to transfer electrons out of the reductase domain to external acceptors or to the NOSoxy domain. This led to proposals that the FMN domain must undergo relatively large movements to twist and swing away from its position in the ‘closed’ conformation that is depicted in the crystal structure, in order to interact with and transfer electrons to external acceptors or to the NOSoxy domain. Thus, the reductase domain was envisioned to swing back and forth between closed and open conformations during NO synthesis, which would allow its FMN domain to both accept electrons and deliver them to NOSoxy. We have since learned that this model is generally accurate and understand a bit more about what controls the motions and the interactions of the FMN domain to enable its electron receiving and donating functions. Biophysical approaches (electron paramagnetic and NMR, ensemble and single molecule fluorescence) clearly show that the NOS reductase domain populates a number of more open conformations along with the closed conformation that is identical or similar to the conformation depicted in the crystal structure. Accordingly, the biophysical data place the FMN residing at various distances from the FAD and the haem cofactors in NOS, including some where the FMN resides close enough to the haem (within 17 Å) to engage in electron transfer between them (Astashkin et al., 2010; Hedison et al., 2017). However, the data suggest that the lifetime of such species is relatively short. Ensemble values for the open versus closed conformational equilibrium (Keq) have been determined for the reduced eNOS and nNOS reductase domains, based on their reactivity towards the electron acceptor protein cytochrome c, along with estimates of their average conformational closing and opening rates to switch between the reactive (open) and non‐reactive (closed) states (Haque et al., 2014). Overall, the studies reveal that the two NOS enzymes differ in the degree to which their reductase domains favour closed versus open conformations (the conformational Keq open/closed order is eNOS < nNOS) and also differ greatly in their average rates of conformational opening and closing (rates in nNOS are approximately 10× faster than in eNOS) (Haque et al., 2014). There is also evidence that the reduction state of the NOS flavins and the occupancy of the NADPH binding site may influence the conformational equilibrium of NOS reductase domains, particularly between the zero and one or two electron‐reduced states (Dunford et al., 2007; Hedison et al., 2017). Whether reduction beyond the one‐electron level has any further significant influence on the conformational equilibrium remains a matter of debate.
Structural regulatory elements within NOS
By 1998, it was appreciated that NOS enzymes repress their electron transfer reactions relative to related dual‐flavin reductase enzymes (Knowles and Moncada, 1994; Marletta, 1994; Masters et al., 1996; Stuehr, 1997). Functionally, the most important repression point is the FMN to haem electron transfer that initiates NO synthesis and whose repression is relieved by CaM binding. We now know that NOS enzymes contain several protein structural elements that help to repress and govern electron transfer both within and out of the reductase domain. Some of the structural elements are common to and present in related dual‐flavin reductases, while others are unique to the NOS enzymes. The common structural elements studied thus far include the protein linker that connects the FAD and FMN domains in NOS, the surface‐charged residues on the FAD, FMN and NOSoxy domains and a loop near the bound FMN which helps to stabilize the FMN in its semiquinone oxidation state (Li et al., 2008; Haque et al., 2012). In general, electron flux through NOS enzymes is partly governed by each of these elements. For example, electron flux through eNOS and nNOS is sensitive to the length and amino acid composition of their FAD‐FMN domain linkers. The surface charge‐pairing interactions of the FMN domain have different affects, depending on the domain partner: charge pairings in the FAD‐FMN domain interface help to repress electron flux through the reductase domain, while those formed at the FMN‐NOSoxy domain interface aid NOS haem reduction. In the cases where it has been studied, the effects of the structural element on the NOS conformational equilibrium could explain how it changes the electron flux through NOS. For example, the surface charge interactions between the FAD and FMN domains were shown to stabilize the closed conformation of the reductase domain, consistent with their repressing electron flux (Haque et al., 2013).
Unique regulatory elements that are present in NOS enzymes include a C‐terminal tail (CT) that is present in all three mammalian NOS and an autoinhibitory insert (AI) that is located in the FMN domains of eNOS and nNOS but is absent in iNOS. Deletion studies clearly showed that both elements help to repress electron transfer through NOS in the absence of bound CaM and that the AI also helps to determine the Ca2+ concentration threshold that enables CaM binding (Roman and Masters, 2006). The crystal structure of nNOS reductase domain (Garcin et al., 2004) revealed that the CT acts to clasp and hold the FMN domain against the FAD domain, thereby stabilizing the closed conformation, and its ability to do so was confirmed by conformational measures recorded for NOS CT deletion and point mutants (Tiso et al., 2007). In contrast, the crystal structure did not shed light on how the AI may function, but it is thought to possibly antagonize the FMN domain interactions that would otherwise help haem reduction to occur in NOS and thus help to ensure that haem reduction does not occur in the absence of bound CaM. Indeed, combined deletion of the CT and AI elements allows CaM‐independent haem reduction to take place in eNOS (Chen and Wu, 2003). Phosphorylation can occur at residues within the CT and AI elements, and this typically diminishes their ability to repress electron transfer or to antagonize CaM binding in NOS and so provides another means to physiologically activate NO synthesis independent of increasing the intracellular Ca2+level (Dudzinski et al., 2006; Feng et al., 2008; Tran et al., 2008; Garcia and Sessa, 2018). In general, what's emerged is that the common and unique structural elements present in NOS enzymes affect NOS electron transfer reactions ultimately by controlling the conformational behaviours of the FMN domain.
Regulation by CaM
By 1998, we knew that Ca2+‐driven CaM binding plays a key role by relieving repression on three critical electron transfer steps in the NOS catalytic pathway, namely, the electron transfers between NADPH and FAD, FAD and FMN, and FMN and haem. In fact, NOS enzymes presented the first example where electron transfer within a redox enzyme is controlled by a Ca2+‐binding protein (Abu‐Soud et al., 1994). Over the last 20 years, an impressive number of approaches that include protein crystallization (Garcin et al., 2004; Xia et al., 2009), cryo‐electron microscopy (Volkmann et al., 2014; Yokom et al., 2014), hydrogen‐deuterium exchange MS (Smith et al., 2013), electron paramagnetic and NMR, ensemble and single molecule fluorescence and molecular dynamics (Salerno et al., 2013; Campbell et al., 2014; Arnett et al., 2015; He et al., 2015; Sheng et al., 2015; Hollingsworth et al., 2016; Hedison et al., 2017) have been employed to understand how CaM controls NOS catalysis. We now know that CaM binding to NOS is a stepwise process: two Ca2+ ions first bind in the two higher affinity lobes of CaM, and this enables CaM to bind to NOS and relieve the repression on the NOS electron transfer steps within the reductase domain (NADPH to FAD, FAD to FMN) and to cause intermediate changes in the conformational distribution. But critically, this binding is not sufficient to trigger the electron transfer from the FMN to the haem, which requires that two additional Ca2+ ions bind to fill all four lobes of CaM. Mechanistically, we know that CaM binding does not alter the relative thermodynamics of the electron transfer reactions between the two flavins or between the FMN and haem nor does it ‘unlock’ the NOS reductase conformation from the closed structure that is shown in the crystal structure, because data show that the reductase domain exists as a dynamic mix of open and closed conformational species in all of its physiologically relevant states (i.e. from one‐ to four‐electron reduced). Instead, we know that CaM binding enables the NOS electron transfer events and catalysis in at least four ways (Figure 3): (i) CaM shifts the conformational equilibrium of the reductase domain towards more open conformations (Haque et al., 2014; Yokom et al., 2014; Arnett et al., 2015; He et al., 2015; Hedison et al., 2017) that must become populated in order for the FMN domain to have a chance at interacting with the NOSoxy domain for electron transfer to the haem. (ii) CaM shortens the dwell time of any one conformation (Salerno et al., 2013; He et al., 2015) and thus speeds the transitions between various conformations. This in turn makes possible a faster electron flux through the reductase domain. (iii) CaM tightens the geometric and lifetime distributions of the conformations (He et al., 2015), possibly through the bound CaM forming a salt‐bridging interaction with the FMN domain (Xia et al., 2009). (iv) CaM helps to limit and guide the movements of the FMN domain through space. This is likely to involve CaM forming temporary ionic and hydrophobic interactions with the surface of the NOSoxy domain that ultimately help to direct FMN domain docking and perhaps also involves CaM relieving a physical block to FMN domain docking by the AI regulatory element (Smith et al., 2013; Campbell et al., 2014; Volkmann et al., 2014; Yokom et al., 2014; Sheng et al., 2015; Hollingsworth et al., 2016). Thus, CaM binding to NOS increases electron flux and triggers electron transfer to the haem through causing a combination of kinetic and entropic changes that help to free its FMN domain and then help to speed, restrict and direct FMN domain movements towards the docking site on the NOSoxy domain (Figure 3).
Figure 3.
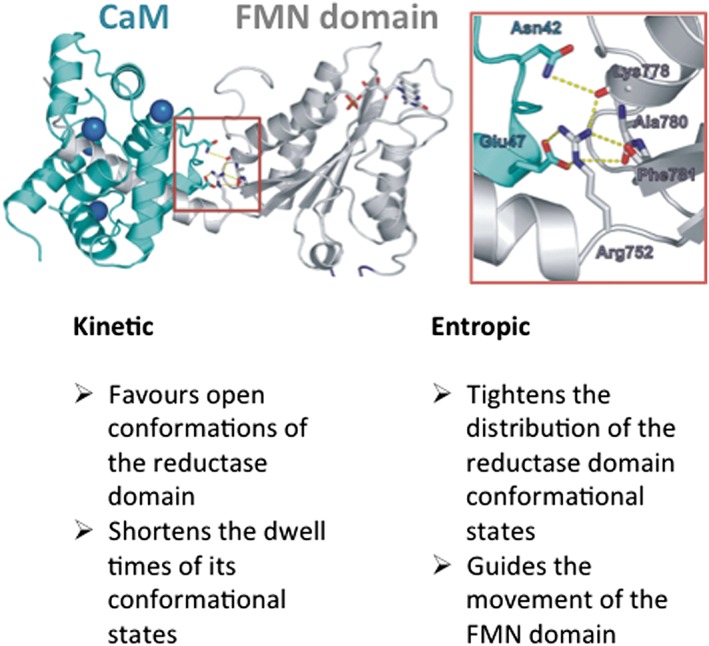
CaM binding and effect on the conformational behaviours of the NOS reductase domain. The picture shows CaM (blue‐green) bound to an FMN domain‐CaM site construct of iNOS (grey) (Xia et al., 2009), with bound calcium ions coloured blue. The boxed area, magnified to the right, illustrates a key stabilizing interaction that involves a conserved Arg residue of the FMN domain (Arg752 of rat nNOS shown), and other residues as indicated, with CaM residue Glu47, that is required for the effects of CaM on NOS. The lower portion of the figure lists some kinetic and entropic effects of CaM binding. See text for related details.
NOS catalytic cycling and the fundamental effects of NO
NOS enzymes produce a haem poison (NO) as their natural product. This unique circumstance prompted research to understand how NOS enzymes avoid being poisoned by their self‐generated NO. Over the last 20 years, research directed along these lines provided deep insights into NOS catalysis that can explain some of its more puzzling aspects, including the greater than 10‐fold range in NO synthesis activities observed between the three mammalian NOS enzymes and the 100‐fold range in their apparent KmO2 values (Stuehr et al., 2004). The following sections review the main features of NO‐based regulation.
All NOS enzymes bind their self‐generated NO before releasing it
The NOS haem is bound within a protected pocket that not only enables its chemistry but also restricts ingress and egress of low MW compounds from the haem. This causes newly generated NO to dwell within the haem pocket such that it binds and is released from the ferric haem many times before it escapes from the enzyme into solution. This behaviour is reflective of the approximate 90% probability that NO rebinds to the haem after the haem iron‐NO bond is broken by laser flash photolysis (Gautier et al., 2004), and importantly, it means that the NOS ferric haem‐NO species is de facto the immediate product of NOS catalysis rather than free NO released from the enzyme. This in turn makes it possible that NOS will become poisoned by its self‐generated NO, because the longer the ferric haem‐NO species persists within NOS, the more likely it will be reduced by an electron provided from the reductase domain to form the ferrous haem‐NO species, which releases NO very slowly and is in fact the NO‐poisoned form of NOS (Figure 4).
Figure 4.
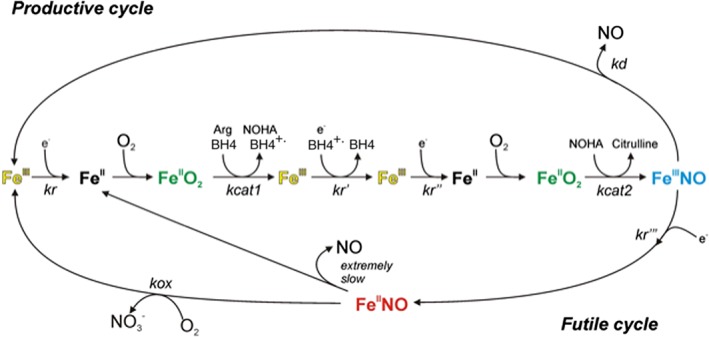
NOS enzyme productive and futile cycling during catalysis. The reduction of ferric enzyme (kr) is the rate‐limiting step for the NO biosynthetic reactions (central linear portion). This electron transfer is needed to reduce the ferric haem before each catalytic step (kr, kr′) and also to reduce the BH4 radical between the Arg and NOHA oxidation reactions (kr′). The kcat1 and kcat2 are the conversion rates of the NOS haem‐dioxy species (FeIIO2) to products in the L‐Arg and NOHA reactions respectively. The ferric haem‐NO product complex (FeIIINO) can either release NO according to rate kd as part of a productive cycle or be reduced by the reductase domain according to rate kr ′′′ to a ferrous haem–NO complex (FeIINO), which reacts with O2 according to rate kox in an NO dioxygenase reaction as part of a futile cycle, to generate nitrate and the ferric enzyme.
All NOS enzymes have inherent NOS and NO dioxygenase activities
Once the ferric haem‐NO species forms during NOS catalysis, it partitions between either being reduced or allowing NO release from the ferric enzyme. As Figure 4 shows, the partitioning ratio is determined by the rate of NO escape out of the enzyme haem pocket (designated kd) and the rate of haem reduction by the reductase domain (designated kr). When NO is released from the enzyme, it represents a ‘productive cycle’ because the enzyme returns to its ferric form and is ready to start another round of catalysis, and the released NO can go on to have its myriad biological effects. In contrast, if the ferric haem‐NO species becomes reduced, it forces the NOS enzyme molecule to participate in a ‘futile cycle’ where the haem‐bound NO becomes further oxidized to nitrate in order to regenerate the ferric enzyme and NO is not released (Figure 4). The futile cycle involves an NO dioxygenase reaction, in which a molecule of O2 reacts directly with the ferrous haem‐NO species according to the rate kox. Both the magnitude and O2 concentration‐dependence of the kox parameter differ broadly between the NOS isoforms (Tejero et al., 2009). Moreover, the O2 concentration‐dependence of the kox parameter differs markedly from the O2 concentration‐dependence for O2 binding to the ferrous NOS haem during NO biosynthesis (Stuehr et al., 2004). This means that the O2 concentration‐dependence of NOS activity (i.e. the apparent KmO2 for NO synthesis) reflects a blend of the two different O2 dependencies, and this gives NOS enzymes higher than expected apparent KmO2 values for their NO synthesis (Stuehr et al., 2004), such that in some cases (nNOS and iNOS), their observed activity varies with O2 concentration across the entire physiological range.
The extent to which a given NOS cycles through the productive versus futile pathways during its catalysis depends primarily on the settings of three kinetic parameters (kr, kox, and kd) (Figure 4). Interestingly, the set points of the three kinetic parameters differ among NOS enzymes, and this in turn causes marked differences regarding the observed steady state NO synthesis activities and the apparent KmO2 values for NO synthesis (Stuehr et al., 2004). It is tempting to speculate that these fundamental differences help to shape the biological functions of each NOS enzyme and to regulate their NO release in response to changes in tissue and cell oxygenation levels, but the biological consequences remain to be tested.
Computer simulations have been performed that incorporate the measured rate parameters into the catalytic cycling model, in order to model and understand NOS enzyme behaviours. These studies showed that the model depicted in Figure 4 is reasonably accurate in describing and predicting the behaviours of a given NOS enzyme regarding its relative NO synthesis and NO dioxygenase activities, its O2 concentration‐dependence and how the enzyme balances its kinetic parameters to avoid being poisoned by NO (Stuehr et al., 2004). These computer simulations have also improved our understanding by helping to interpret how site‐directed changes in NOS structural elements, and the consequent changes caused in the kinetic parameters, affect NOS catalytic cycling, which in turn has effects on NOS activities that are sometimes counter‐intuitive or hard to understand. For example, the computer simulations of NOS enzyme cycling have explained how changes in NOS structure that increase or decrease the haem reduction rate (kr) can either increase or diminish the observed NO synthesis activity of a NOS (Stuehr et al., 2004; Haque et al., 2009; Tejero et al., 2010; Haque et al., 2012).
Conclusions and perspectives
The last 20 years have improved our understanding on several aspects of NOS enzymology. Regarding structure, the significant advances in molecular imaging approaches, including cryo‐EM, MS and computational docking and dynamics have provided a decent understanding of NOS 3D structure and its domain interactions, despite our not having a full length NOS crystal structure in hand. Regarding catalysis, we better understand the electron transfer and redox chemistry of the haem and BH4 cofactors, how they are regulated and how their unique interplay allows NO synthesis. We also better appreciate the unique challenge that NO synthesis poses for NOS enzymes and how it creates a circumstance where NOS enzymes have evolved to balance intrinsic NO releasing and NO destroying (NO dioxygenase) activities. Going forward, a number of aspects remain less understood and could benefit from further research: what are the molecular mechanisms that enable and regulate NOS maturation and destruction in cells? Is NO production the sole function, or even the main function, of all the NOS and NOS‐like proteins that are found in nature (Santolini et al., 2017)? Why are the catalytic behaviours of the three mammalian NOS so different, and how are these differences related to individual biological functions? How do various cellular proteins (other than CaM) interact with NOS enzymes? How do such proteins and the many post‐translational modifications of NOS enzymes regulate NOS catalytic behaviour at the molecular level? The concepts, tools and approaches developed over the last 20 years should provide a solid foundation to address these and other outstanding questions in NOS enzymology.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors thank collaborators and laboratory members who have been involved in NOS research and thank the NIH for financial support primarily through NIH grants GM 51491 and CA 53914 to D.J.S.
Stuehr, D. J. , and Haque, M. M. (2019) Nitric oxide synthase enzymology in the 20 years after the Nobel Prize. British Journal of Pharmacology, 176: 177–188. 10.1111/bph.14533.
References
- Abu‐Soud HM, Gachhui R, Raushel FM, Stuehr DJ (1997a). The ferrous‐dioxy complex of neuronal nitric oxide synthase. Divergent effects of L‐arginine and tetrahydrobiopterin on its stability. J Biol Chem 272: 17349–17353. [DOI] [PubMed] [Google Scholar]
- Abu‐Soud HM, Presta A, Mayer B, Stuehr DJ (1997b). Analysis of neuronal NO synthase under single‐turnover conditions: conversion of N ω‐hydroxyarginine to nitric oxide and citrulline. Biochemistry 36: 10811–10816. [DOI] [PubMed] [Google Scholar]
- Abu‐Soud HM, Rousseau DL, Stuehr DJ (1996). Nitric oxide binding to the heme of neuronal nitric‐oxide synthase links its activity to changes in oxygen tension. J Biol Chem 271: 32515–32518. [DOI] [PubMed] [Google Scholar]
- Abu‐Soud HM, Wu C, Ghosh DK, Stuehr DJ (1998). Stopped‐flow analysis of CO and NO binding to inducible nitric oxide synthase. Biochemistry 37: 3777–3786. [DOI] [PubMed] [Google Scholar]
- Abu‐Soud HM, Yoho LL, Stuehr DJ (1994). Calmodulin controls neuronal nitric‐oxide synthase by a dual mechanism. Activation of intra‐ and interdomain electron transfer. J Biol Chem 269: 32047–32050. [PubMed] [Google Scholar]
- Adak S, Wang Q, Stuehr DJ (2000). Arginine conversion to nitroxide by tetrahydrobiopterin‐free neuronal nitric‐oxide synthase. Implications for mechanism. J Biol Chem 275: 33554–33561. [DOI] [PubMed] [Google Scholar]
- Albakri QA, Stuehr DJ (1996). Intracellular assembly of inducible NO synthase is limited by nitric oxide‐mediated changes in heme insertion and availability. J Biol Chem 271: 5414–5421. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Other proteins. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett DC, Persechini A, Tran QK, Black DJ, Johnson CK (2015). Fluorescence quenching studies of structure and dynamics in calmodulin‐eNOS complexes. FEBS Lett 589: 1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astashkin AV, Elmore BO, Fan W, Guillemette JG, Feng C (2010). Pulsed EPR determination of the distance between heme iron and FMN centers in a human inducible nitric oxide synthase. J Am Chem Soc 132: 12059–12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasko E, Glaser CB, Devlin JJ, Xia W, Feldman RI, Polokoff MA et al (2002). Mechanistic studies with potent and selective inducible nitric‐oxide synthase dimerization inhibitors. J Biol Chem 277: 295–302. [DOI] [PubMed] [Google Scholar]
- Campbell MG, Smith BC, Potter CS, Carragher B, Marletta MA (2014). Molecular architecture of mammalian nitric oxide synthases. Proc Natl Acad Sci U S A 111: E3614–E3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarti R, Aulak KS, Fox PL, Stuehr DJ (2010). GAPDH regulates cellular heme insertion into inducible nitric oxide synthase. Proc Natl Acad Sci U S A 107: 18004–18009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PF, Wu KK (2003). Structural elements contribute to the calcium/calmodulin dependence on enzyme activation in human endothelial nitric‐oxide synthase. J Biol Chem 278: 52392–52400. [DOI] [PubMed] [Google Scholar]
- Chen Y, Panda K, Stuehr DJ (2002). Control of nitric oxide synthase dimer assembly by a heme‐NO‐dependent mechanism. Biochemistry 41: 4618–4625. [DOI] [PubMed] [Google Scholar]
- Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ et al (1998). Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science 279: 2121–2126. [DOI] [PubMed] [Google Scholar]
- Davydov R, Labby KJ, Chobot SE, Lukoyanov DA, Crane BR, Silverman RB et al (2014). Enzymatic and cryoreduction EPR studies of the hydroxylation of methylated N (ω)‐hydroxy‐L‐arginine analogues by nitric oxide synthase from Geobacillus stearothermophilus . Biochemistry 53: 6511–6519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser SP (2009). Density functional theory (DFT) and combined quantum mechanical/molecular mechanics (QM/MM) studies on the oxygen activation step in nitric oxide synthase enzymes. Biochem Soc Trans 37: 373–377. [DOI] [PubMed] [Google Scholar]
- Dudzinski DM, Igarashi J, Greif D, Michel T (2006). The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol 46: 235–276. [DOI] [PubMed] [Google Scholar]
- Dunford AJ, Rigby SE, Hay S, Munro AW, Scrutton NS (2007). Conformational and thermodynamic control of electron transfer in neuronal nitric oxide synthase. Biochemistry 46: 5018–5029. [DOI] [PubMed] [Google Scholar]
- Dweik RA, Laskowski D, bu‐Soud HM, Kaneko F, Hutte R, Stuehr DJ et al (1998). Nitric oxide synthesis in the lung. Regulation by oxygen through a kinetic mechanism. J Clin Invest 101: 660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng C, Roman LJ, Hazzard JT, Ghosh DK, Tollin G, Masters BS (2008). Deletion of the autoregulatory insert modulates intraprotein electron transfer in rat neuronal nitric oxide synthase. FEBS Lett 582: 2768–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Kohno H, Tokunaga R, Taketani S (1995). Nitric oxide‐mediated inactivation of mammalian ferrochelatase in vivo and in vitro: possible involvement of the iron‐sulphur cluster of the enzyme. Biochem J 310 (Pt 2): 533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahman TC, Herbert MR, Lang H, Thayer A, Symons KT, Nguyen PM et al (2011). Identification and SAR of selective inducible nitric oxide synthase (iNOS) dimerization inhibitors. Bioorg Med Chem Lett 21: 6888–6894. [DOI] [PubMed] [Google Scholar]
- Garcia‐Cardena G, Fan R, Stern DF, Liu J, Sessa WC (1996). Endothelial nitric oxide synthase is regulated by tyrosine phosphorylation and interacts with caveolin‐1. J Biol Chem 271: 27237–27240. [DOI] [PubMed] [Google Scholar]
- Garcia V, Sessa WC (2018). Endothelial nitric oxide synthase (eNOS): perspective and recent developments. Br J Pharmacol . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R et al (2004). Structural basis for isozyme‐specific regulation of electron transfer in nitric‐oxide synthase. J Biol Chem 279: 37918–37927. [DOI] [PubMed] [Google Scholar]
- Gautier C, Negrerie M, Wang ZQ, Lambry JC, Stuehr DJ, Collin F et al (2004). Dynamic regulation of the inducible nitric‐oxide synthase by NO: comparison with the endothelial isoform. J Biol Chem 279: 4358–4365. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Chawla‐Sarkar M, Stuehr DJ (2011). Hsp90 interacts with inducible NO synthase client protein in its heme‐free state and then drives heme insertion by an ATP‐dependent process. FASEB J 25: 2049–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Garee G, Sweeny EA, Nakamura Y, Stuehr DJ (2018). Hsp90 chaperones hemoglobin maturation in erythroid and nonerythroid cells. Proc Natl Acad Sci U S A 115: E1117–E1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Stuehr DJ (2017). Regulation of sGC via hsp90, cellular heme, sGC agonists, and NO: new pathways and clinical perspectives. Antioxid Redox Signal 26: 182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorren AC, Mayer B (2002). Tetrahydrobiopterin in nitric oxide synthesis: a novel biological role for pteridines. Curr Drug Metab 3: 133–157. [DOI] [PubMed] [Google Scholar]
- Haque MM, Bayachou M, Fadlalla MA, Durra D, Stuehr DJ (2013). Charge‐pairing interactions control the conformational setpoint and motions of the FMN domain in neuronal nitric oxide synthase. Biochem J 450: 607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque MM, Bayachou M, Tejero J, Kenney CT, Pearl NM, Im SC et al (2014). Distinct conformational behaviors of four mammalian dual‐flavin reductases (cytochrome P450 reductase, methionine synthase reductase, neuronal nitric oxide synthase, endothelial nitric oxide synthase) determine their unique catalytic profiles. FEBS J 281: 5325–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque MM, Fadlalla M, Wang ZQ, Ray SS, Panda K, Stuehr DJ (2009). Neutralizing a surface charge on the FMN subdomain increases the activity of neuronal nitric‐oxide synthase by enhancing the oxygen reactivity of the enzyme heme‐nitric oxide complex. J Biol Chem 284: 19237–19247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque MM, Fadlalla MA, Aulak KS, Ghosh A, Durra D, Stuehr DJ (2012). Control of electron transfer and catalysis in neuronal nitric‐oxide synthase (nNOS) by a hinge connecting its FMN and FAD‐NADPH domains. J Biol Chem 287: 30105–30116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS guide to pharmacology in 2018: updates and expansion to encompass the new guide to immunopharmacology. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Haque MM, Stuehr DJ, Lu HP (2015). Single‐molecule spectroscopy reveals how calmodulin activates NO synthase by controlling its conformational fluctuation dynamics. Proc Natl Acad Sci U S A 112: 11835–11840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedison TM, Hay S, Scrutton NS (2017). A perspective on conformational control of electron transfer in nitric oxide synthases. Nitric Oxide 63: 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth SA, Holden JK, Li H, Poulos TL (2016). Elucidating nitric oxide synthase domain interactions by molecular dynamics. Protein Sci 25: 374–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurshman AR, Marletta MA (2002). Reactions catalyzed by the heme domain of inducible nitric oxide synthase: evidence for the involvement of tetrahydrobiopterin in electron transfer. Biochemistry 41: 3439–3456. [DOI] [PubMed] [Google Scholar]
- Knowles RG, Moncada S (1994). Nitric oxide synthases in mammals. Biochem J 298 (Pt 2): 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krest CM, Onderko EL, Yosca TH, Calixto JC, Karp RF, Livada J et al (2013). Reactive intermediates in cytochrome p450 catalysis. J Biol Chem 288: 17074–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Hayden EY, Panda K, Stuehr DJ, Deng H, Rousseau DL et al (2006). Regulation of the monomer‐dimer equilibrium in inducible nitric‐oxide synthase by nitric oxide. J Biol Chem 281: 8197–8204. [DOI] [PubMed] [Google Scholar]
- Li D, Kabir M, Stuehr DJ, Rousseau DL, Yeh SR (2007). Substrate‐ and isoform‐specific dioxygen complexes of nitric oxide synthase. J Am Chem Soc 129: 6943–6951. [DOI] [PubMed] [Google Scholar]
- Li H, Das A, Sibhatu H, Jamal J, Sligar SG, Poulos TL (2008). Exploring the electron transfer properties of neuronal nitric‐oxide synthase by reversal of the FMN redox potential. J Biol Chem 283: 34762–34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marletta MA (1994). Nitric oxide synthase: aspects concerning structure and catalysis. Cell 78: 927–930. [DOI] [PubMed] [Google Scholar]
- Masters BS, McMillan K, Sheta EA, Nishimura JS, Roman LJ, Martasek P (1996). Neuronal nitric oxide synthase, a modular enzyme formed by convergent evolution: structure studies of a cysteine thiolate‐liganded heme protein that hydroxylates L‐arginine to produce NO. as a cellular signal. FASEB J 10: 552–558. [DOI] [PubMed] [Google Scholar]
- Mitchell DA, Erwin PA, Michel T, Marletta MA (2005). S‐Nitrosation and regulation of inducible nitric oxide synthase. Biochemistry 44: 4636–4647. [DOI] [PubMed] [Google Scholar]
- Nakane M, Mitchell J, Forstermann U, Murad F (1991). Phosphorylation by calcium calmodulin‐dependent protein kinase II and protein kinase C modulates the activity of nitric oxide synthase. Biochem Biophys Res Commun 180: 1396–1402. [DOI] [PubMed] [Google Scholar]
- Pant K, Crane BR (2006). Nitrosyl‐heme structures of Bacillus subtilis nitric oxide synthase have implications for understanding substrate oxidation. Biochemistry 45: 2537–2544. [DOI] [PubMed] [Google Scholar]
- Peng HM, Morishima Y, Pratt WB, Osawa Y (2012). Modulation of heme/substrate binding cleft of neuronal nitric‐oxide synthase (nNOS) regulates binding of Hsp90 and Hsp70 proteins and nNOS ubiquitination. J Biol Chem 287: 1556–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presta A, Siddhanta U, Wu C, Sennequier N, Huang L, Abu‐Soud HM et al (1998a). Comparative functioning of dihydro‐ and tetrahydropterins in supporting electron transfer, catalysis, and subunit dimerization in inducible nitric oxide synthase. Biochemistry 37: 298–310. [DOI] [PubMed] [Google Scholar]
- Presta A, Weber‐Main AM, Stankovich MT, Stuehr D (1998b). Comparative effects of substrates and pterin cofactor on the heme midpoint potential in inducible and neuronal nitric oxide synthases. J Am Chem Soc 120: 9460–9465. [Google Scholar]
- Raman CS, Li H, Martasek P, Kral V, Masters BS, Poulos TL (1998). Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell 95: 939–950. [DOI] [PubMed] [Google Scholar]
- Ramasamy S, Haque MM, Gangoda M, Stuehr DJ (2016). Tetrahydrobiopterin redox cycling in nitric oxide synthase: evidence supports a through‐heme electron delivery. FEBS J 283: 4491–4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman LJ, Masters BS (2006). Electron transfer by neuronal nitric oxide synthase is regulated by concerted interaction of calmodulin and two intrinsic regulatory elements. J Biol Chem 281: 23111–23118. [DOI] [PubMed] [Google Scholar]
- Rusche KM, Spiering MM, Marletta MA (1998). Reactions catalyzed by tetrahydrobiopterin‐free nitric oxide synthase. Biochemistry 37: 15503–15512. [DOI] [PubMed] [Google Scholar]
- Salerno JC, Ray K, Poulos T, Li H, Ghosh DK (2013). Calmodulin activates neuronal nitric oxide synthase by enabling transitions between conformational states. FEBS Lett 587: 44–47. [DOI] [PubMed] [Google Scholar]
- Santolini J, Andre F, Jeandroz S, Wendehenne D (2017). Nitric oxide synthase in plants: where do we stand? Nitric Oxide 63: 30–38. [DOI] [PubMed] [Google Scholar]
- Sennequier N, Wolan D, Stuehr DJ (1999). Antifungal imidazoles block assembly of inducible NO synthase into an active dimer. J Biol Chem 274: 930–938. [DOI] [PubMed] [Google Scholar]
- Shamovsky I, Belfield G, Lewis R, Narjes F, Ripa L, Tyrchan C et al (2018). Theoretical studies of the second step of the nitric oxide synthase reaction: electron tunneling prevents uncoupling. J Inorg Biochem 181: 28–40. [DOI] [PubMed] [Google Scholar]
- Sharp KH, Mewies M, Moody PC, Raven EL (2003). Crystal structure of the ascorbate peroxidase‐ascorbate complex. Nat Struct Biol 10: 303–307. [DOI] [PubMed] [Google Scholar]
- Sheng Y, Zhong L, Guo D, Lau G, Feng C (2015). Insight into structural rearrangements and interdomain interactions related to electron transfer between flavin mononucleotide and heme in nitric oxide synthase: a molecular dynamics study. J Inorg Biochem 153: 186–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BC, Underbakke ES, Kulp DW, Schief WR, Marletta MA (2013). Nitric oxide synthase domain interfaces regulate electron transfer and calmodulin activation. Proc Natl Acad Sci U S A 110: E3577–E3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuehr DJ (1997). Structure‐function aspects in the nitric oxide synthases. Annu Rev Pharmacol Toxicol 37: 339–359. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S (2004). Update on mechanism and catalytic regulation in the NO synthases. J Biol Chem 279: 36167–36170. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ, Wei CC, Wang Z, Hille R (2005). Exploring the redox reactions between heme and tetrahydrobiopterin in the nitric oxide synthases. Dalton Trans : 3427–3435. [DOI] [PubMed] [Google Scholar]
- Sweeny SE, Singh AB, Chakravarti R, Martinez‐Guzman O, Saini A, Haque MM et al (2018). Glyceraldehyde‐3‐phosphate dehydrogenase is a chaperone that allocates labile heme in cells. J Biol Chem 293: 14557–14568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejero J, Haque MM, Durra D, Stuehr DJ (2010). A bridging interaction allows calmodulin to activate NO synthase through a bi‐modal mechanism. J Biol Chem 285: 25941–25949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejero J, Santolini J, Stuehr DJ (2009). Fast ferrous heme‐NO oxidation in nitric oxide synthases. FEBS J 276: 4505–4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiso M, Tejero J, Panda K, Aulak KS, Stuehr DJ (2007). Versatile regulation of neuronal nitric oxide synthase by specific regions of its C‐terminal tail. Biochemistry 46: 14418–14428. [DOI] [PubMed] [Google Scholar]
- Tran QK, Leonard J, Black DJ, Persechini A (2008). Phosphorylation within an autoinhibitory domain in endothelial nitric oxide synthase reduces the Ca(2+) concentrations required for calmodulin to bind and activate the enzyme. Biochemistry 47: 7557–7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann N, Martasek P, Roman LJ, Xu XP, Page C, Swift M et al (2014). Holoenzyme structures of endothelial nitric oxide synthase—an allosteric role for calmodulin in pivoting the FMN domain for electron transfer. J Struct Biol 188: 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed SM, Ghosh A, Chakravarti R, Biswas A, Haque MM, Panda K et al (2010). Nitric oxide blocks cellular heme insertion into a broad range of heme proteins. Free Radic Biol Med 48: 1548–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZQ, Wei CC, Stuehr DJ (2010). How does a valine residue that modulates heme‐NO binding kinetics in inducible NO synthase regulate enzyme catalysis? J Inorg Biochem 104: 349–356. [DOI] [PubMed] [Google Scholar]
- Wei CC, Wang ZQ, Durra D, Hemann C, Hille R, Garcin ED et al (2005). The three nitric‐oxide synthases differ in their kinetics of tetrahydrobiopterin radical formation, heme‐dioxy reduction, and arginine hydroxylation. J Biol Chem 280: 8929–8935. [DOI] [PubMed] [Google Scholar]
- Wei CC, Wang ZQ, Hemann C, Hille R, Stuehr DJ (2003). A tetrahydrobiopterin radical forms and then becomes reduced during Nω‐hydroxyarginine oxidation by nitric‐oxide synthase. J Biol Chem 278: 46668–46673. [DOI] [PubMed] [Google Scholar]
- Wei CC, Wang ZQ, Tejero J, Yang YP, Hemann C, Hille R et al (2008). Catalytic reduction of a tetrahydrobiopterin radical within nitric‐oxide synthase. J Biol Chem 283: 11734–11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss G, Werner‐Felmayer G, Werner ER, Grunewald K, Wachter H, Hentze MW (1994). Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J Exp Med 180: 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witteveen CF, Giovanelli J, Kaufman S (1996). Reduction of quinonoid dihydrobiopterin to tetrahydrobiopterin by nitric oxide synthase. J Biol Chem 271: 4143–4147. [DOI] [PubMed] [Google Scholar]
- Woodward JJ, Chang MM, Martin NI, Marletta MA (2009). The second step of the nitric oxide synthase reaction: evidence for ferric‐peroxo as the active oxidant. J Am Chem Soc 131: 297–305. [DOI] [PubMed] [Google Scholar]
- Woodward JJ, Nejatyjahromy Y, Britt RD, Marletta MA (2010). Pterin‐centered radical as a mechanistic probe of the second step of nitric oxide synthase. J Am Chem Soc 132: 5105–5113. [DOI] [PubMed] [Google Scholar]
- Xia C, Misra I, Iyanagi T, Kim JJ (2009). Regulation of interdomain interactions by calmodulin in inducible nitric‐oxide synthase. J Biol Chem 284: 30708–30717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokom AL, Morishima Y, Lau M, Su M, Glukhova A, Osawa Y et al (2014). Architecture of the nitric‐oxide synthase holoenzyme reveals large conformational changes and a calmodulin‐driven release of the FMN domain. J Biol Chem 289: 16855–16865. [DOI] [PMC free article] [PubMed] [Google Scholar]