

Abstract
Background and Purpose
Abdominal aortic aneurysm (AAA) is a degenerative disease affecting human health, but there are no safe and effective medications for AAA therapy. Cycloastragenol (CAG), derived from Astragali Radix, has various pharmacological effects. However, whether CAG can protect against AAA remains elusive. In this study, we investigated whether CAG has an inhibitory effect on AAA and its related mechanism.
Experimental Approach
The AAA mouse model was induced by incubating the abdominal aorta with elastase. CAG was administered by gavage at different doses beginning on the same day or 14 days after inducing AAA to explore its preventive or therapeutic effects respectively. The preventive effects of CAG on AAA were verified in another AAA mouse model induced by angiotensin II in ApoE−/− mouse. In vitro experiments were implemented on rat vascular smooth muscle cells (VSMCs) stimulated by TNF‐α.
Key Results
Compared to the control AAA model group, CAG (125 mg·kg−1 body weight day−1) reduced the incidence of AAA, the dilatation of aorta and elastin degradation in media in both mouse models of AAA. CAG suppressed the inflammation, oxidation, phenotype switch and apoptosis in TNF‐α‐stimulated VSMCs, ameliorated the expression and activity of MMPs and decreased the activation of the ERK/JNK signalling pathway. CAG also inhibited the degradation of elastin in TNF‐α‐stimulated VSMCs.
Conclusion and Implications
CAG presents protective effects against AAA through down‐regulation of the MAPK signalling pathways and thus attenuates inflammation, oxidation, VSMC phenotype switch and apoptosis and the expression of MMPs as well as increasing elastin biosynthesis.
Abbreviations
- AAA
abdominal aortic aneurysm
- ECM
extracellular matrix
- VSMC
vascular smooth muscle cell
- AST
astragaloside IV
- CAG
cycloastragenol
- SD
Sprague Dawley
- MCP‐1
monocyte chemo‐attractant protein‐1
- HO‐1
haem oxygenase‐1
- Nrf‐2
nuclear factor‐erythroid‐2‐related factor 2
Introduction
Abdominal aortic aneurysm (AAA) is the localized dilatation of the abdominal aorta, which is manifested as abnormal dilatation of the abdominal aorta to more than 50% of its diameter (Kumar et al., 2017). AAA occurs in up to 8% of men aged >65 years yet usually remain asymptomatic until rupture (Nordon et al., 2011). Rupture of the AAA results in an overall mortality rate of more than 80% in patients (Jeanmonod and Jeanmonod, 2017). The risk factors for AAA include smoking, aging, male, hypertension and pre‐existing vascular diseases (Cornuz et al., 2004; Diehm et al., 2007). Currently, the treatment of confirmed aortic aneurysms is to perform open or endovascular surgical repair when the aortic diameter is over 5.5 cm, while only surveillance, but no medication, is applied to the patients who have a small AAA with an aortic diameter of 3.0–5.4 cm (Erbel et al., 2014), because, at present, there is no effective drug in clinical use. Therefore, there is an urgent need to develop a medical therapy to limit the progressive expansion of small AAAs.
AAA is characterized by the degradation of the extracellular matrix (ECM), chronic inflammatory infiltration, oxidative stress and apoptosis of vascular smooth muscle cells (VSMCs) (Ryer et al., 2015). Chronic inflammation is thought to be the leading cause of AAA (Ryer et al., 2015). In the incipient stage of AAA, macrophages infiltrate into the aortic wall and secrete inflammatory mediators (Egan et al., 1990), which then recruit monocyte‐macrophages and lymphocytes and trigger an inflammatory response. Furthermore, inflammatory cells secrete MMPs, which assist in the degradation of the ECM (Malik et al., 1996). In response to local inflammatory cytokines, VSMCs undergo phenotypic switching early on in aneurysm formation and secrete MMPs, which damage the aortic wall matrix, and inflammatory cytokines that also induce the apoptosis of VSMCs (Owens et al., 2004; Ailawadi et al., 2009). In addition, oxidative injury of blood vessels is among the risk factors during the initial phases of vascular diseases (Emeto et al., 2016). An increase in oxidative stress in local aorta is one of the known causes and consequences of aortic inflammation. Moreover, inflammatory cytokines and ROS activate ERK1/2 (Hao et al., 2014) and JNK1/2 (Zhang et al., 2015) and other MAPKs in VSMCs, which is an important signalling pathway in the pathogenesis of AAA.
Astragali Radix has been used as a herbal medicine to treat cardiovascular disorders in China for thousands of years, and astragaloside IV (AST) has been identified as the main active compound in Astragali Radix (Zhao et al., 2015). Since most of the AST is metabolized to cycloastragenol (CAG) in vivo, CAG is also defined as a natural active compound in Astragali Radix and an active form of AST (Zhou et al., 2012). There are no related reports about the anti‐AAA effects of AST and CAG so far. However, the pharmacological properties of CAG, which include anti‐oxidant, anti‐inflammatory and cardiovascular protective effects (Zhao et al., 2015; Sun et al., 2017), indicate that it may have potential as an inhibitor of AAA. Therefore, we explored the effect of CAG on AAA in mice and its related mechanisms.
Methods
Ethics of animal experiments
Healthy 8‐ to 10‐week‐old male C57BL/6 mice weighing 20 to 25 g, used for the Elastase model of AAA, and healthy 4‐month‐old male ApoE−/− mice weighing about 30 g, used for the angiotensin (Ang) II model of AAA, were obtained from the Department of Laboratory Animal Science, Peking University Health Science Centre (Beijing, China). Mice were housed in open‐top conventional cages with usual bedding material along with appropriate environmental enrichment, and were kept in pathogen‐free conditions. A maximum of five mice were housed in a single cage. All mice were exposed to a 12 h light/dark cycle under defined environmental conditions at 25 ± 2°C with a relative humidity of 50% and were given free access to food and water. All animal care and experimental procedures complied with the Animals (Scientific procedures) Act 1986 and all procedures involving animals conformed to the Regulations for the Administration of Affairs Concerning Experimental Animals published by the State Science and Technology Commission of China and were approved by the Biomedical Ethics Committee of Peking University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010).
Pharmacokinetic studies using UPLC‐MS
Healthy 8 to 10‐week‐old male C57BL/6 mice were administered CAG by p.o. gavage at a dose of 125 mg·kg−1. And 0.1 mL blood samples were drawn from the orbital vein of mice using blood collection tube at 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 6.0, 9.0, 12.0 and 24.0 h after the p.o. administration of CAG. All blood samples were stored at 80°C until further analysis. The peak plasma concentration (Cmax) and time to reach Cmax (Tmax) values, reaction half‐time (t1/2), the total area under the plasma concentration–time curve from time zero to infinity (AUC0~∞) and mean residence time (MRT0~∞) were determined by applying Kinetica software version 4.4 (more details about instrument conditions, sample extraction and methods used are shown in the Supporting Information).
Elastase‐induced AAA in mice
AAA was induced by local application of 1.5 U pancreatic elastase on the abdominal aortas of C57BL/6 mice as described previously (Wang et al., 2017a). Briefly, the mice were anaesthetized with an i.p. injection of sodium pentobarbital (50 mg·kg−1) and placed in a supine position on an animal operating table. After a 1.5 cm midline incision had been made in the abdominal wall, the abdominal aorta of the mice from infra‐renal aorta to bifurcation of the aorta was isolated by blunt dissection. The separated abdominal aorta was then wrapped circumferentially with blotting (bibulous) paper soaked with 1.5 U pancreatic elastase (mice in Sham group were treated with saline) for 50 min, after which the blotting paper was removed and the abdomen was closed up with sutures.
Ang II‐induced AAA in mice
ApoE−/− mice were anaesthetized and Ang II (1000 mg·kg−1·min−1; Sigma, St. Louis, MO), or saline was administered via s.c. implanted osmotic mini‐pumps (Model 2004, Durect, Cupertino, CA), as described previously (Lu et al., 2015). Briefly, the mice were anaesthetized with an i.p. injection of sodium pentobarbital (50 mg·kg−1) and placed in a prostrate position on the animal operating table. The area to be incised was shaved, swabbed with betadine followed by three wipes with 70% ethanol. A surgical scalpel was used to make a 1 cm incision behind the ear over the shoulder blade of the front leg and a haemostat used to make a s.c. tunnel under the skin. Then the haemostat tip was advanced towards the tail to create a pocket for the pump. The pump was then inserted into the incision with the moderator head positioned to the rear of the mouse and sutured in place.
Administration regimens
The administration regimens are as follows: (I) To explore the preventive effects of CAG on elastase‐induced AAA, the mice were randomly divided into a sham group (Sham), elastase‐induced model group (Model), high‐dose CAG group (CAG‐H) and low‐dose CAG group (CAG‐L). Mice in the Sham and Model group were administered 200 μL 0.01 M PBS (pH = 7.4) p.o. once per day; the CAG‐H group and the CAG‐L group received CAG treatment at a dose of 125 and 62.5 mg·kg−1 of body weight, respectively, p.o. once per day beginning on the same day the AAA was induced and continuing for two weeks. (II) To explore the therapeutic effects of CAG on elastase‐induced AAA, the mice were randomly assigned to a sham group (Sham), elastase‐induced model group (Model) and CAG‐H. Mice in the Sham and Model groups were administered with 200 μL 0.01 M PBS p.o. once per day, and the CAG‐H group received CAG treatment at a dose of 125 mg·kg−1 of body weight p.o. once per day, 14 days after the induction of AAA and continuing for four weeks. (III) In order to explore whether CAG treatment simply delays the growth of AAA in mice or prevents its occurrence, we then examine the mice at a later stage. The mice were randomly divided into a sham group (Sham), elastase‐induced model group (Model), CAG‐H. High‐dose CAG was given to the mice from the day of elastase‐induced AAA and lasted for 14 days, then CAG was displaced by 0.01 M PBS, which was also continued for 14 days, while 0.01 M PBS was given in the other two groups for 4 weeks. (IV) To explore the preventive effects of CAG on Ang II‐induced AAA, the mice were randomly assigned to Vehicle group (Vehicle), Ang II‐induced model group (Ang II) and CAG‐H (Ang II + CAG‐H). Mice in the Vehicle and the Ang II groups were administered 200 μL 0.01 M PBS (pH = 7.4) p.o. once per day; the Ang II + CAG‐H group received CAG at a dose of 125 mg·kg−1 of body weight p.o. once per day beginning on the same day that the AAA was induced and continuing for four weeks. The administration regimens (I–IV) are shown in Figure 1A. All animals were fed a standard chow diet.
Figure 1.

(A) Induction of AAA and administration regimens. (B) Molecular structure of CAG. (C) Mean plasma concentration–time curve of CAG in mice after a single p.o. administration. Each data point represents six mice.
At the end of the experiments, all mice were killed with an overdose of sodium pentobarbital. The aortas were excised under a dissection microscope and photographed; the aortic diameters were then measured by two different investigators with Image J software. AAA was defined when the dilatation of the mice aortas exceeded that of normal mice aortas by 50%. The tissues obtained were kept at −80°C to extract RNA and protein for PCR or Western blot analysis or fixed in 4% paraformaldehyde in PBS (pH 7.4) and embedded in Tissue‐Tek O.C.T. compound for histological analysis.
Histological analysis
Specimens from the dilated aortas in the infra‐renal region of all mice from Sham, Model or CAG‐treated group were embedded in Tissue‐Tek O.C.T. compound in liquid nitrogen and then cut into 5 μm serial sections. Haematoxylin–eosin (H&E) staining and aldehyde fuchsin staining were used to analyse the morphology and evaluate elastin degradation of the mice aortas respectively.
Immunohistochemistry staining of CD68, MMP‐2 and MMP‐9 was used to observe the macrophage infiltration and inflammation as well as expression of MMP‐2 and MMP‐9 in the mice aorta. Briefly, the aortic slices were subjected to peroxidase quenching with 3% hydrogen peroxide then incubated overnight with 100‐fold diluted polyclonal primary antibodies at 4°C. Subsequently, the slices were washed with PBS and then incubated with 500‐fold diluted goat anti‐rabbit or mice secondary antibodies conjugated with peroxidase at 37°C for 60 min. After bing counterstained with haematoxylin, the slices were incubated with diaminobenzidine peroxidase substrate to visualize peroxidase activity by light microscopy.
Cell culture
Male Sprague Dawley (SD) rats weighing about 100 g were provided by the Department of Laboratory Animal Science, Peking University Health Science Centre (Beijing, China).
Primary VSMCs were isolated from male SD rats according to the protocol described previously (Wang et al., 2017b). Briefly, SD rats were killed by administration of 50 mg·kg−1 sodium pentobarbital i.p., and the whole aorta was excised and washed with 0.01 M PBS (containing 100 U·mL−1 benzylpenicillin sodium and 100 mg·mL−1 streptomycin sulfate). Following removal of the intima and adventitia, arterial tissues were cut into 1 mm2 segments and then transferred to a culture flask. After being incubated in the bottom of a flask for 6 h with DMEM (containing 20% FBS and 100 U·mL−1 benzylpenicillin sodium and 100 mg·mL−1 streptomycin sulfate) at 37 °C (5% CO2), the flask was turned over and incubated to obtain a static culture for a week. After being washed with 0.01 M PBS (pH = 7.4), the cells were trypsinized at 37°C for 5 min. The cells from passages five to seven were used in the following experiments.
MTT assay for cell viability
The VSMCs were cultured in 96‐well plates (1 × 104 cells per well) for 24 h. The cells were then treated with different concentrations of CAG for 24 h. After treatment, MTT was added to each well to a final concentration of 0.5 mg·mL−1 and incubated for 4 h at 37°C in a humidified incubator containing 5% CO2. Then 200 μL DMSO was used to dissolve formazan, and absorbance at 490 nm was measured with an elisa reader (No. 550, Hercules, California, USA).
Induction of AAA micro‐environment in VSMCs and drug treatment
To establish an aneurysm micro‐environment in vitro in VSMCs, 100 ng·mL−1 TNF‐α was applied to stimulate the VSMCs for 24 h. These cells were then treated with 31.25 μg·mL−1 CAG (TNF‐α + CAG). After incubation for 24 h, the cells were collected and RNA or proteins were extracted for PCR or Western blot analysis. Parallel experiments were done in TNF‐α‐treated‐only (TNF‐α) or non TNF‐α‐treated normal cells (Untreated).
Inhibition of ERK1/2 activation
The MAPK kinase (MEK) inhibitor, U0126 (purchased from Promega), was used to examine the effects of CAG on MAPK signalling pathway. VSMCs were incubated with 10 μM U0126 plus TNF‐α or CAG for 24 h according to the manufacturer's instruction. And total RNA was extracted for further study.
RT‐PCR analysis
Total RNA was extracted from tissues or cells using Trizol Reagent (Molecular Research Center, USA), and cDNA synthesis was performed using the TransScript First‐Strand cDNA Synthesis Super Mix (TransGen Biotech). EvaGreen qPCR MasterMix (abm, Canada) was used to evaluate mRNA expression levels according to the manufacturer's instructions. The primers used for real‐time PCR are shown in Table 1. And transcript levels were normalized to an internal control of β‐actin in tissues or GAPDH in cells.
Table 1.
Primer sequences used in amplification PCR and semi‐quantitative RT‐PCR
| Genes | Sequences | |
|---|---|---|
| β‐actin (mouse) | Forward | GGCTGTATTCCCCTCCATCG |
| Reverse | CCAGTTGGTAACAATGCCATGT | |
| IL‐6 (mouse) | Forward | CCAAGAGGTGAGTGCTTCCC |
| Reverse | CTGTTGTTCAGACTCTCTCCCT | |
| MCP‐1 (mouse) | Forward | TTAAAAACCTGGATCGGAACCAA |
| Reverse | GCATTAGCTTCAGATTTACGGGT | |
| IL‐1β (mouse) | Forward | TGAATTGGTCATAGCCCGCA |
| Reverse | GTTGCCCTCAGCAGTAAGGA | |
| GAPDH (mouse) | Forward | TGATGACATCAAGAAGGTGGTGAAG |
| Reverse | TCCTTGGAGGCCATGTAGGCCAT | |
| IL‐6 (rat) | Forward | CCTTCTTGGGACTGATGT |
| Reverse | CTCTGGCTTTGTCTTTCT | |
| MCP‐1 (rat) | Forward | AATGAGTCGGCTGGAGAA |
| Reverse | GTGCTTGAGGTGGTTGTG | |
| IL‐1β (rat) | Forward | GACCCCAAAAGATTAAGGA |
| Reverse | CACAATGAGTGACACTGCC | |
| HO‐1 (rat) | Forward | ACAGAAGAGGCTAAGACCG |
| Reverse | CAGGCATCTCCTTCCATT | |
| Nrf‐2 (rat) | Forward | TTTGTAGATGACCATGAGTCGC |
| Reverse | GCCAAACTTGCTCCATGTCC | |
| Fibulin‐5 (rat) | Forward | AGACTTGCTGAACTCGTGGG |
| Reverse | CTGGGTCGCATACCCTTTGT | |
| Fibrillin‐1 (rat) | Forward | TGCTCTGAAAGGACCCAATGT |
| Reverse | CGGGACAACAGTATGCGTTATAAC | |
Western blot analysis
Tissues or cells were collected and protein concentrations were quantified with a biscinchonic acid kit (Pierce Biotechnology, Rockford, IL, USA). For Western blot, 30 μg of protein was loaded in each well and resolved by 10% SDS‐PAGE, and the protein bands were then electro‐transferred onto a PVDF membrane using the Bio‐Rad MiniProtean II apparatus (Bio‐Rad Laboratories, Carlsbad, CA, USA). The blots were subsequently incubated with anti‐rabbit elastin (1:1000), anti‐rabbit MMP‐2 (1:1000), anti‐rabbit MMP‐9 (1:100), anti‐rabbit ERK1/2 (1:2000), anti‐rabbit phosphorylated ERK1/2 (1:2000), anti‐rabbit JNK1/2 (1:2000), anti‐rabbit phosphorylated JNK1/2 (1:2000), anti‐rabbit SM22α (1:1000) and anti‐rabbit caspase‐3 (1:1000) or anti‐mouse GAPDH (1:5000) at 4°C overnight followed by HRP‐conjugated secondary antibodies (1:5000) for 1 h and visualized with an enhanced chemiluminescence system (Pierce Biotechnology, Rockford, IL, USA). GAPDH was used as an internal control for data normalization. All bands were quantified by Image J software.
Gelatin zymography analysis
To determine the enzyme activity of MMP‐2 and MMP‐9, gelatin zymography was performed. Protein extracts (10 μg) were mixed with SDS buffer, and electrophoresis was conducted (10% SDS‐PAGE with 0.1% gelatin as substrate). Then the gels were washed with 2.5% Triton X‐100 and incubated at 37°C for 48 h with renaturing buffer followed by staining with coomassie brilliant blue and destained with destaining solution containing 10% acetic acid and 40% methanol. Gels were scanned using Image‐analyser LAS‐4000 (Fujifilm, Tokyo, Japan), and images were assessed by Image J software.
Statistical analysis
All data are presented as mean ± SEM. P < 0.05 was considered as a statistically significant difference. Statistical significance of differences among the groups was analysed by one‐way ANOVA for multiple comparisons with Tukey's test. Tukey's tests were run only when F achieved P < 0.05 and there was no significant variance inhomogeneity. And elastin degradation scores between multiple groups were analysed by one‐tailed Wilcoxon test. All statistical analyses were performed using GraphPad Prism for Windows (Version 4, San Diego, CA, USA). The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Chemicals
CAG was purchased from Chengdu Kingtiger pharmaceutical chemical Co., Ltd. (Sichuan, China, molecular structure of CAG was shown in Figure 1B). TNF‐α was purchased from Peprotech (Rocky Hill, USA). FBS, DMEM, trypsin and EDTA were purchased from GIBCO (Grand Island, USA). Tissue‐Tek O.C.T. compound was purchased from Sakura Finetek Japan Co., Ltd. (Tokyo, Japan). Antibody of CD68 (cat# BA3638) was purchased from Boster Biological Technology Co., Ltd. (Hubei, China). Mouse anti‐GAPDH (cat# sc‐365062) primary antibody and HRP‐conjugated secondary antibodies were all purchased from Santa Cruz (Dallas, USA). Rabbit anti‐MMP‐2 (cat# ab37150), rabbit anti‐MMP‐9 (cat# ab38898), rabbit anti‐elastin (cat# ab217356), rabbit anti‐ERK1/2 (cat# ab17942) and phosphorylated ERK1/2, rabbit anti‐JNK1/2 and phosphorylated JNK1/2 (cat# ab131499) primary antibodies were purchased from Abcam (Massachusetts, USA); rabbit anti‐SM22α (cat# ab14106) was purchased from Abcam, and rabbit anti‐caspase‐3 (cat# 9662S) was purchased from CST (Samoa, USA). Rat TNF‐α was purchased from PeproTech (USA). Elastase, penicillin, streptomycin, methylthiazolyl tetrazolium (MTT), gelatin and all other reagents were purchased from Sigma‐Aldrich (Beijing, China).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18(Alexander et al., 2017a, 2017b).
Results
Pharmacokinetic studies of CAG
The structure of CAG is shown in Figure 1B. The mean plasma concentration versus time curves of CAG after its p.o. administration to mice at a dose of 125 mg·kg−1 body weight is shown in Figure 1C, and the pharmacokinetic parameters were calculated and summarized in Table 2. The mean Tmax and t1/2 were 2.10 ± 0.22 and 2.33 ± 0.61 h respectively. The maximum concentrations of CAG was obtained with Cmax (787.49 ± 95.81 ng·mL−1), the total area under the plasma concentration–time curve from time zero to infinity was 2962.06 ± 625.06 h·ng·mL−1 and mean residence time was 4.36 ± 1.18 h.
Table 2.
The main pharmacokinetic parameters of CAG in mice after p.o. administration
| Parameters | CAG p.o. 125 mg·kg−1 |
|---|---|
| Tmax (h) | 2.10 ± 0.22 |
| Cmax (ng·mL−1) | 787.49 ± 95.81 |
| t1/2 (h) | 2.33 ± 0.61 |
| AUC0~∞ (h·ng·mL−1) | 2962.06 ± 625.06 |
| MRT0~∞ (h) | 4.36 ± 1.18 |
n = 6 in each group.
Protective effects of CAG on AAA formation
After administration of elastase for 14 days, mice in the Model group developed severe AAA, but CAG‐H inhibited the development of AAA (Figure 2A). For quantitative analysis, we counted the incidence and the largest external diameters of arteries in all mice groups and found that the CAG‐H group had a substantially reduced incidence of AAA (16.7% vs. 100%) and dilation of infra‐renal aortic lumen (3.29 ± 0.83 vs. 1.28 ± 0.29), compared to the Model group (Figure 2B, C). However, the CAG‐L had no significant effects on AAA. We also confirmed that CAG treatment prevented the occurrence rather than simply delaying the growth of AAA in mice (Supporting Information Figure S1). And CAG also inhibited Ang II‐induced AAA in mice (Figure 2D–F).
Figure 2.
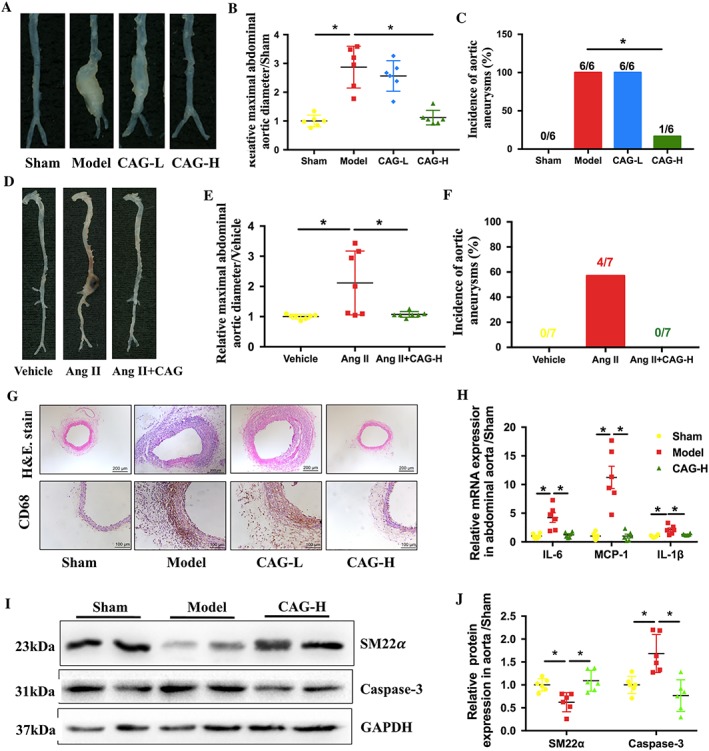
CAG prevented AAA formation and inhibited inflammation in aortic wall of the mice. (A) Morphology of aorta, (B) relative maximal external abdominal aortic diameter and (C) incidence of elastase‐induced AAA in mice, n = 6 in each group. (D) Morphology of aorta, (E) relative maximal external abdominal aortic diameter and (F) incidence of Ang II‐induced AAA, n = 7 in each group. The formation of AAA was defined as ≥50% dilation of normal abdominal aorta diameter. (G) Representative images of H&E staining of aortic cross sections, scale bar: 200 μm, and representative images of immunohistochemical staining of CD‐68 in aortic sections from the indicated groups of mice, scale bar: 100 μm, and (H) relative mRNA expression of inflammatory cytokines MCP‐1, IL‐6 and IL‐1β in the aorta respectively. (I) Protein expression of SM22α and caspase‐3 in the aorta. (J) Quantitative results of (F). n = 6 in each group. *Indicates that there is a statistically significant difference between the two groups.
Two weeks after AAA induction, mice arteries from the Model group presented with severe dilatation in the aortic lumen. Also, flattening, fragmentation and degeneration of the elastic laminae in the medial layer, as well as thickening and remodelling in the aortic adventitia, were observed from H&E staining (Figure 2G). Compared to the Model group, a high dose of CAG substantially reduced the elastin degradation in the arterial media, thus preserved the intact structure of the aortic wall, while the CAG‐L had little effect.
Local inflammatory responses in the Model group occurred afer the induction and development of AAA, which were characterized by severe macrophage infiltration and overexpression of inflammatory cytokines. In comparison with mice in the Model group, the mice treated with CAG at a high dose had a significantly decreased expression of CD68, the marker of macrophages, and a down‐regulated mRNA expression of monocyte chemo‐attractant protein (MCP)‐1 (CCL2), IL‐6 and IL‐1β (Figure 2G, H). Elastase also induced the phenotype switch and apoptosis of VSMCs. The expression of SM22α, a marker of contractile VSMCs, was decreased in the AAA while the expression of caspsase‐3 was raised. And CAG treatment up‐regulated the expression of SM22α and down‐regulated the expression of caspsase‐3 (Figure 2I, J).
Elastase induced elastin degradation in the Model group and administration of CAG at the high dose attenuated the degradation of elastin (Figure 3A). However, the CAG‐H decreased the expression of MMP‐2 and MMP‐9 in the aorta (Figure 3A–C).
Figure 3.
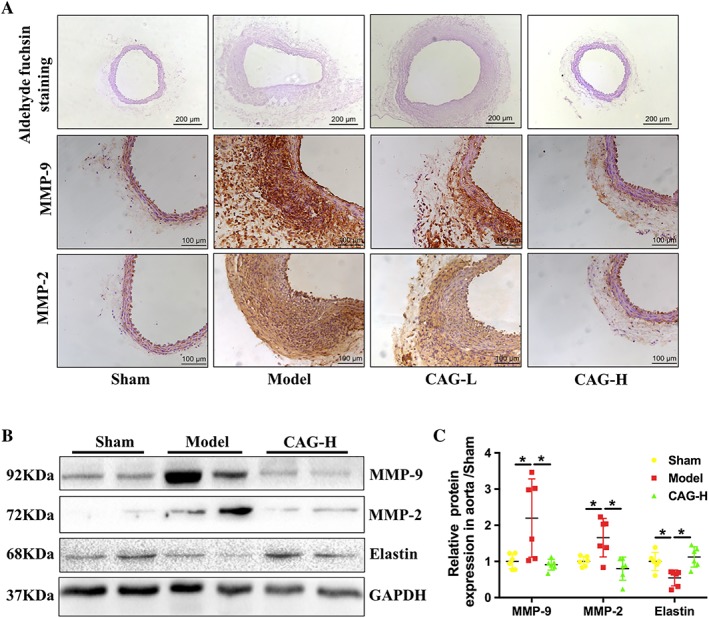
CAG inhibited elastase‐induced vascular remodelling and ECM degradation. (A) Representative images of aldehyde fuchsin staining of aortic sections 14 days after induction of AAA. Scale bar: 200 μm, and representative images of immunohistochemical staining of MMP‐2 and MMP‐9 in aortic sections from the indicated groups of mice. Scale bar: 100 μm. (B) Protein expression of MMP‐9, MMP‐2 and elastin in aortas from mice in the indicated groups. (C) Quantitative results of (B). n = 6 in each group. *Indicates that there is a statistically significant difference between the two groups.
Therapeutic effects of CAG on the established AAA
To explore the therapeutic effects of CAG on already‐formed AAA, the high dose of CAG was administered to the mice 14 days after the induction of AAA when it was already established. Compared to the Model group, the CAG‐H group had a substantially reduced dilatation of infra‐renal aortic lumen (1.444 ± 0.254 vs. 2.25 ± 0.256) and decreased incidence of AAA (50% vs. 100%) (Figure 4A–C).
Figure 4.
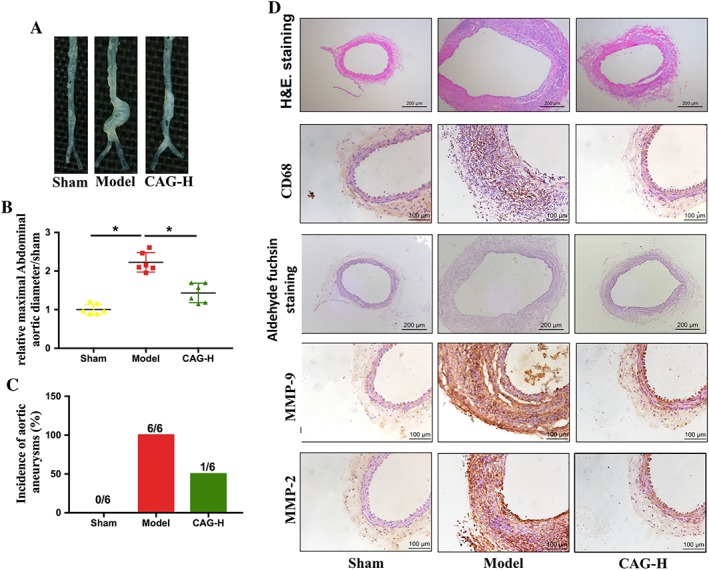
Therapeutic effects of CAG on the established AAA. (A) Morphology of aorta, (B) relative maximal external abdominal aortic diameter and (C) incidence of elastase‐induced AAA in mice. The formation of AAA was defined as ≥50% dilation of normal abdominal aorta diameter. n = 6 in each group. (D) Representative images of H&E staining of aortic cross sections, scale bar: 200 μm, representative images of immunohistochemical staining of CD‐68 in aortic sections from the indicated groups of mice, scale bar: 100 μm, representative images of aldehyde fuchsin staining of aortic sections. Scale bar: 200 μm, and representative images of immunohistochemical staining of MMP‐2 and MMP‐9 in aortic sections from the indicated groups. Scale bar: 100 μm. *Indicates that there is a statistically significant difference between the two groups.
In the established AAA, mice arteries presented with seatvere dilation in the aortic lumen. In addition, degeneration of the elastic laminae in the medial layer, as well as thickening and remodelling in the aortic adventitia, can be seen from H&E staining and aldehyde fuchsin staining (Figure 4D). Compared to the Model group, the CAG‐H group had substantially reduced elastin degradation in the media, thus the structure of the aortic wall was preserved. Simultaneously, the CAG‐H decreased the infiltration of CD68‐positive macrophages into the aortic wall, as well as the expression of MMP‐2 and MMP‐9 (Figure 4D).
CAG ameliorated inflammation and oxidative stress in VSMCs
To further explore the mechanism by which CAG inhibited the development and progression of AAA, VSMCs were stimulated with 100 ng·mL−1 TNF‐α to mimic the micro‐environment of AAA in vitro. The results of the MTT assay showed that CAG has no cytotoxic effects (cell viabilities higher than 80% of the normal cell control) on VSMCs at concentrations less than 62.5 μg·mL−1 (Figure 5A). Thus, according to preliminary experiments, CAG concentrations of 31.25 μg·mL−1 were chosen for the following in vitro experiments.
Figure 5.
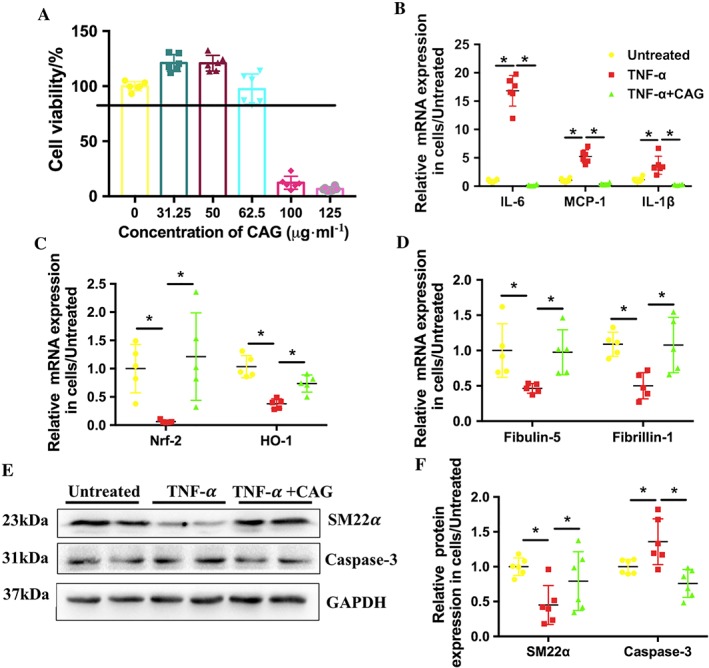
CAG attenuated inflammation and oxidative stress and facilitated elastin biosynthesis in VSMCs stimulated with 100 ng·mL−1 TNF‐α. (A) Cytotoxicity of CAG in rat VSMCs. (B) Effects of CAG on mRNA expression level of inflammatory cytokines of IL‐6, MCP‐1 and IL‐1β. (C) Effects of CAG on mRNA expression level of oxidation genes Nrf‐2 and HO‐1. (D) Effects of CAG on mRNA expression level of elastin biosynthesis related genes of fibrillin‐1 and fibulin‐5. (E) Protein expression of SM22α and Caspase‐3 in VSMCs. (F) Quantitative results of (E). n = 6 in each group. *Indicates that there is a statistically significant difference between the two groups.
After treatment with TNF‐α for 24 h, mRNA expression of pro‐inflammatory cytokines MCP‐1 and IL‐6 significantly increased compared to that of the untreated cells (Untreated). However, the mRNA expression level of the above two cytokines were significantly decreased by treating the cells with CAG (Figure 5B). After treatment with TNF‐α for 24 h, the mRNA expression of nuclear factor‐erythroid‐2‐related factor (Nrf)‐2 and haem oxygenase (HO)‐1, the most important anti‐oxidative genes, were dramatically suppressed, indicating oxidative stress in VSMCs. Nevertheless, CAG treatment ameliorated the decrease in Nrf‐2 and HO‐1 expression in VSMCs caused by TNF‐α (Figure 5C). TNF‐α also induce the phenotype switch and apoptosis of VSMCs in vitro. The expression of SM22α, a marker of contractile VSMCs, was decreased in TNF‐α‐stimulated VSMCs while the expression of caspsase‐3 was raised. However, CAG treatment up‐regulated the expression of SM22α and down‐regulated the expression of caspsase‐3 (Figure 5E, F), indicating that CAG could prevent the phenotype switch and apoptosis of VSMCs. These results are consistent with those observed in in vivo.
CAG inhibited the expression and activity of MMPs and facilitated elastin biosynthesis in VSMCs
The mRNA expression of fibulin‐5 and fibrillin‐1, two genes that facilitate elastin biosynthesis, were significantly reduced in VSMCs when the cells were treated with TNF‐α, while CAG treatment attenuated the reduction of these two genes caused by TNF‐α (Figure 5D).
The synthesis and secretion of MMP‐2 and MMP‐9 in VSMCs were increased after the cells were treated with TNF‐α, which further induced the degradation of elastin fibres. Results of Western blot demonstrate that CAG down‐regulated the expression of MMP‐2 and MMP‐9 and inhibited degradation of elastin, compared to the cells treated with TNF‐α alone (Figure 6A, B). Furthermore, the activity of MMP‐2 and MMP‐9 was also inhibited by CAG, as shown in the results of gelatin zymography (Figure 6C ,D).
Figure 6.

CAG treatment suppressed the activity and expression of MMPs and prevented the degradation of elastin in TNF‐α‐stimulated VSMCs. (A) Protein expression level of MMP‐2, MMP‐9 and elastin. (B) Quantifications of (A). (C) Enzymatic activity of MMP‐2 and MMP‐9 in the TNF‐α‐stimulated VSMCs and (D) quantification of data shown in (C). n = 6 in each group. *Indicates that there is a statistically significant difference between the two groups.
CAG inhibited ERK1/2 and JNK1/2 MAPK signalling pathway
To investigate the molecular mechanisms underlying the inhibitory effects of CAG on AAA development, the effects of CAG on the two classic subunits of MAPK signalling, p‐ERK and p‐JNK, were examined in mice aortas and VSMCs from the groups with different treatments. Elastase and TNF‐α treatment significantly activated both the ERK and JNK pathways in vivo and in vitro respectively. And CAG treatment down‐regulated the phosphorylation of ERK and JNK both in vivo and in vitro (Figure 7A–D). However, the anti‐inflammatory effect of CAG on TNF‐α‐stimulated VSMCs was inhibited when the cells were treated with the MEK inhibitor U0126 simultaneously (Figure 7E–G). These results suggest that suppression of the ERK and JNK signalling pathway may contribute to the inhibitory effects of CAG on AAA formation and development.
Figure 7.
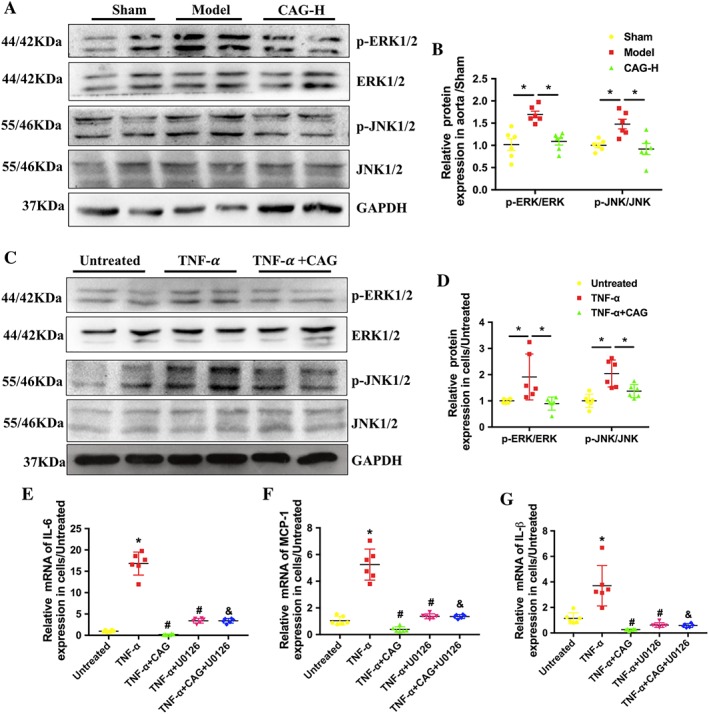
CAG inhibited ERK/JNK signalling pathways in elastase‐induced AAA and TNF‐α‐stimulated VSMCs. (A) Protein expression of p‐ERK, ERK, p‐JNK and JNK in aorta after 14 days of treatment. (B) Quantitative results of (A). (C) Protein expression of p‐ERK, ERK, p‐JNK and JNK in VSMCs at 24 h after TNF‐α/(TNF‐α + CAG) treatment. (D) Quantitative results of (C). (E–G) The mRNA expression of IL‐6, MCP‐1 and IL‐1β in cells treated with TNF‐α, TNF‐α plus CAG with or without U0126. *P<0.05 compared with Untreated group, #P <0.05 compared with TNF‐α group and &P<0.05 compared with TNF‐α + CAG group. n = 6 in each group.
Discussion
AAA is a common and rapidly developing degenerative aortic disease that results in considerable mortality due to aneurysm rupture (Kadoglou and Liapis, 2004). In clinical research, β‐adrenoceptor antagonists, statins, renin‐angiotensin system inhibitors, oestrogen and doxycycline have been used to treat AAA (Davis et al., 2015; Fraga‐Silva et al., 2015). However, side effects and unknown mechanisms made these drugs undesirable for AAA treatment. Therefore, there is no effective drug in the clinic that can be used to limit the progressive developmentof AAA or reverse an established small AAA. To the best of our knowledge, this study provides the first evidence of the inhibitory effects of CAG on AAA formation and development. Besides, there is no report of the pharmacokinetics of CAG in mice until now. So the pharmacokinetic study was performed to provide more preclinical information on CAG.
In this study, AAA was mainly induced by incubating the infra‐renal aorta with pancreatic elastase. To investigate the preventive effects, CAG at two different doses was administered to the mice on the same day that AAA was induced and continued for 2 weeks. To explore the therapeutic effects of CAG on the already formed AAA, the high dose of CAG was administered to the mice fourteen days after the induction of AAA and continued for 4 weeks. Our results suggest that CAG can not only prevent the formation of AAA but also reverse the already‐formed small AAA, which are reflected by observing that CAG‐H inhibited the dilatation of aorta and reduced the incidence of AAA. After the CAG treatment, the flattening, fragmentation and degeneration of the elastic laminae in the medial layer, as well as thickening and remodelling in aortic adventitia, were significantly ameliorated. The inhibitory effect of CAG on Ang II‐induced AAA was similar to that in elastase‐induced AAA. All these outcomes indicate that CAG has a clear preventive effect on AAA and provide a basis for its clinical transformation and treatment for AAA.
The significant feature of AAA is chronic inflammation with infiltration of inflammatory cells into the aortic wall (Saraff et al., 2003). In the Model group, the infiltration of macrophages in aneurysmal tissue (Halpern et al., 1994), which was a leading cause of AAA, was found to be severe. Macrophages can secrete inflammatory mediators, such as TNF‐α, MCP‐1, IL‐6 and IL‐1β (Davis et al., 2015). The inflammatory cellular response is to a large extent mediated by these cytokines and constitutes a complex network that maintains the chronic inflammation in AAA. Genetic knockout or specific antagonism of these inflammatory factors can prevent the formation of AAA induced by Ang II or elastase in mice (Daugherty et al., 2010; Johnston et al., 2013).
During the development of AAA, macrophages recruit more inflammatory cells via the secretion of cytokines and proteases (mainly MMP‐2 and MMP‐9) (Longo et al., 2002; Nosoudi et al., 2015) and promote degradation of ECM proteins. These degradative products may exacerbate the inflammatory condition. VSMCs of the synthetic phenotype can secrete MMPs in response to inflammation and oxidative stress (McCormick et al., 2007; Ailawadi et al., 2009) and contribute greatly to the degradation and remodelling of ECM. This in turn causes the loss of vascular elasticity and tension accompanied by the degeneration of elastin and collagen.
In our study, CAG reduced the infiltration of CD68‐positive macrophages into the aortic wall and attenuated the expression of inflammatory mediators, such as MCP‐1, IL‐6 and IL‐1β in vivo and in vitro, which resulted in the suppression of local arterial inflammation, and suppressed the transformation of VSMCs and prevented the apoptosis of VSMCs. In the current study, the expression of MMP‐2 and MMP‐9 was increased in adventitia in both the Model mice group and TNF‐α‐stimulated VSMCs, but CAG administration down‐regulated the expression and activity of MMP‐9 and MMP‐2. Therefore, CAG inhibited the degradation of elastin and preserved the structural integrity of aorta wall, and thus reduced the occurrence and development of AAA.
Oxidative stress plays a critical role in the development of AAA (Usui et al., 2015). ROS levels are markedly increased within human AAA segments compared with adjacent non‐aneurysmal aortas and in the aortic walls of experimental animals (Emeto et al., 2016), implicating the critical role of ROS in AAA formation. ROS are thought to be a common link between inflammation, matrix degradation and apoptosis of VSMCs (Lum and Roebuck, 2001; Emeto et al., 2016). Nrf‐2 is a transcription factor and controls the basal and inducible expression of a battery of antioxidant genes. The overexpression of Nrf‐2 inhibits ROS production induced by hypertrophic factors (Kovac et al., 2015). HO‐1 is a stress response protein located downstream of Nrf‐2 and catalyses the oxidation of haem to biliverdin (Ryter et al., 2006). Its reactive products have potent anti‐inflammatory and anti‐oxidative functions. Shaw‐Fang Yet et al. demonstrated that a deficiency in HO‐1 exacerbates angiotensin II‐induced aortic aneurysm in mice (Ho et al., 2016).
In our study, TNF‐α, reduced the mRNA expression of HO‐1 and Nrf‐2 in VSMCs due to acute oxidative injury. However, CAG treatment elevated the mRNA expression of Nrf‐2 and HO‐1 in these TNF‐α‐treated VSMCs, thus it may reduce ROS and ROS‐linked inflammation, matrix degradation and apoptosis of VSMCs, which is beneficial in the prevention of AAA.
The main components of elastic fibres, elastin and fibrillin‐containing microfibrils play a structural and mechanical role in the arteries, and their essential function is to provide the tissues with elasticity and resilience (Lannoy et al., 2014). The formation of elastic fibre is a complex multi‐step process (Aya et al., 2015). The initial pericellular microassembly of tropoelastin generates elastin globules that are stabilized by lysyl oxidase (LOX). These globules are deposited on a fibrillin microfibril template, where they coalesce and undergo further cross‐linking to form the elastin core of mature fibres. Fibrillin‐1 is one of the foremost fibrillins (Lannoy et al., 2014), and mutations in fibrillin‐1 are mainly responsible for Marfan syndrome (Sengle et al., 2012). The secreted protein fibulin‐5 is necessary for elastic fibre assembly, and it interacts with both tropoelastin and LOX–like enzymes, facilitating elastin deposition onto microfibrils and the subsequent cross‐linking; knockdown of fibulin‐5 reduces elastin deposition on microfibrils (Choudhury et al., 2009).
A balance between the synthesis and degradation of elastin plays a vital role in the recovery of the injured aorta in aortic aneurysm. In our study, CAG treatment suppressed the expression of MMPs and attenuated elastin degradation, and thus preserved the intact structure of the aortic wall. Moreover, CAG treatment significantly increased mRNA expression of fibrillin‐1 and fibulin‐5, suggesting that elastin re‐synthesis is promoted by CAG. More importantly, even though elastase had already caused the degradation of elastin after 14 days of AAA induction, CAG treatment still reversed the pathological changes, indicating that the biosynthesis of elastin probably plays a major role in the therapeutic effects of CAG.
Several lines of evidence highlight the involvement of the MAPK signalling pathway in AAA formation and progression (Yang et al., 2014). JNK and ERK in MAPK pathways are highly activated in human and mouse AAA tissues. An ERK antagonist can prevent AAA formation by inhibiting MMP activation (Ghosh et al., 2012), and inhibition of JNK prevents the development of AAA and causes regression of the established AAA in vivo (Yoshimura et al., 2006). These pathways trigger extensive molecular patterns in several cell types that cooperatively promote the production of MMPs, activate pro‐inflammatory mediators and oxidative stress (Suh et al., 2009; Guo et al., 2015). And it is reported that oxidative stress mediates MAPK activation (Lum and Roebuck, 2001), which is associated with cell growth and differentiation and is extensively linked to inflammation, apoptosis and cell death in a positive‐feedback loop, which exacerbates the outcomes of AAA.
Elastase incubation and TNF‐α stimulation induced the phosphorylation of ERK and JNK, confirming that ERK and JNK of the MAPK pathways were activated in the AAA Model mice group and TNF‐α‐stimulated VSMCs, but such activation was largely blocked by CAG treatment. Also the anti‐inflammatory effect of CAG on TNF‐α‐stimulated VSMCs was inhibited by an MEK inhibitor U0126. There are many reports about the effect of AST on ERK and JNK MAPK signalling pathways (Li et al., 2015; Wang et al., 2017c). As an active form of AST, it was shown in this study that CAG can also affect ERK and JNK MAPK signalling pathways.
CAG suppressed the infiltration of macrophages into the adventitia and reduced the inflammation, oxidative stress and the expression and activity of MMP‐2 and MMP‐9. The mechanism by which CAG inhibited AAA was probably related to its inhibition of the ERK and JNK signalling pathway. On the one hand, inflammation and oxidative stress and the expression and activity of MMPs can be suppressed by blocking the MAPK signalling pathway. On the other hand, the inhibition of inflammation and oxidation facilitates inactivation of the MAPK signalling pathway.
In a previous study, we found that grape‐seed polyphenols (GSP) have ameliorative effects on AAA (Wang et al., 2017a). GSP are mixtures of flavonoids, epicatechin, flavanols and anthocyanins (Du et al., 2007). CAG is a kind of triterpenoid saponin extracted from Radix Astragali, which is completely different in molecular structure from the compounds in GSP. Therefore, they might modulate different signalling pathways, although the anti‐AAA effects of GSP and CAG are all related to their anti‐inflammatory and anti‐oxidative stress effects. The complicated components in GSP means it is difficult to clarify the active compounds in GSP and study their mechanisms of action. While the defined structure of CAG makes it possible to explore the mechanism behind its anti‐AAA effects, which was elucidated to be associated with the attenuation of ECM degradation, inhibition of chronic inflammatory responses and oxidative stress through the down‐regulation of the ERK/JNK signalling pathway. But beyond that, T‐lymphocytes have a vital role in AAA development (Meng et al., 2014), and in a previous study it was suggested that CAG also has an effect on lymphocytes (Sun et al., 2017), indicating that CAG might also inhibit the T cells involved the formation of AAA; this needs to be validated in future studies.
In this study, we found that CAG has not only preventive but also therapeutic effects on mice with AAA induced by either elastase or Ang II. The findings in the current study provide a basis for discovering new drugs for treating AAA and also support the potential application of CAG in the clinic for nonsurgical prevention and treatment of AAA in the future.
Author contributions
All authors contributed extensively to the work presented in this paper. Y.W. and R.Q. designed the experiments, wrote up the manuscript and prepared figures. Y.W. executed the in vivo and in vitro experiments. C.C., Q.W., Y.C. and L.X. provided help for the laboratory technique, experiments and data analysis. The manuscript has been reviewed and approved by all authors.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 CAG treatment prevented the occurrence rather than simply delay the growth of AAA in mice. (a) morphology of aorta (b) relative maximal external abdominal aortic diameter. In this segment, the high dose CAG (125 mg·kg‐1·d‐1) was given to the mice from the day of Elastase‐AAA induction and lasted for 14 days, then CAG were displaced by 0.01M PBS, which was also lasted for 14 days while 0.01M PBS was given in other two group for 4 weeks. And n=6 in each group.
Figure S2 Acute toxicity of CAG on mice. (a) Body weight change. (b) Weight of major organs/body weight. (c) Clinical chemistry parameters, glutamate pyruvate transaminase (ALT), aspartate aminotransferase (AST), total creatine phosphokinase (CK), creatine kinase isoenzyme (CK‐MB) and LDH. There is no obvious significance between CAG treated group and PBS treated group no matter male or female mice. n=5 in each group.
Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (81770268, 81360054) and the National Basic Research Program of China (2015CB932100).
Wang, Y. , Chen, C. , Wang, Q. , Cao, Y. , Xu, L. , and Qi, R. (2019) Inhibitory effects of cycloastragenol on abdominal aortic aneurysm and its related mechanisms. British Journal of Pharmacology, 176: 282–296. 10.1111/bph.14515.
References
- Ailawadi G, Moehle CW, Pei H, Walton SP, Yang Z, Kron IL et al (2009). Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J Thorac Cardiovasc Surg 138: 1392–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: enzymes. Br J Pharmacol 174 (Suppl. 1): S272–s359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: overview. Br J Pharmacol 174 (Suppl. 1): S1–s16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aya R, Ishiko T, Noda K, Yamawaki S, Sakamoto Y, Tomihata K et al (2015). Regeneration of elastic fibers by three‐dimensional culture on a collagen scaffold and the addition of latent TGF‐beta binding protein 4 to improve elastic matrix deposition. Biomaterials 72: 29–37. [DOI] [PubMed] [Google Scholar]
- Choudhury R, McGovern A, Ridley C, Cain SA, Baldwin A, Wang MC et al (2009). Differential regulation of elastic fiber formation by fibulin‐4 and ‐5. J Biol Chem 284: 24553–24567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornuz J, Sidoti Pinto C, Tevaearai H, Egger M (2004). Risk factors for asymptomatic abdominal aortic aneurysm: systematic review and meta‐analysis of population‐based screening studies. Eur J Public Health 14: 343–349. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty A, Rateri DL, Charo IF, Owens AP, Howatt DA, Cassis LA (2010). Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE−/− mice. Clin Sci (Lond) 118: 681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FM, Rateri DL, Daugherty A (2015). Abdominal aortic aneurysm: novel mechanisms and therapies. Curr Opin Cardiol 30: 566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehm N, Dick F, Schaffner T, Schmidli J, Kalka C, Di Santo S et al (2007). Novel insight into the pathobiology of abdominal aortic aneurysm and potential future treatment concepts. Prog Cardiovasc Dis 50: 209–217. [DOI] [PubMed] [Google Scholar]
- Du Y, Guo H, Lou H (2007). Grape seed polyphenols protect cardiac cells from apoptosis via induction of endogenous antioxidant enzymes. J Agric Food Chem 55: 1695–1701. [DOI] [PubMed] [Google Scholar]
- Egan TG, Monaghan SM, Muir RC, Gilmore RJ, Clarkson JE, Crooks TJ (1990). Prenatal screening of pregnant mothers for parenting difficulties: final results from the Queen Mary Child Care Unit. Soc Sci Med 30: 289–295. [DOI] [PubMed] [Google Scholar]
- Emeto TI, Moxon JV, Au M, Golledge J (2016). Oxidative stress and abdominal aortic aneurysm: potential treatment targets. Clin Sci (Lond) 130: 301–315. [DOI] [PubMed] [Google Scholar]
- Erbel R, Aboyans V, Boileau C, Bossone E, Bartolomeo RD, Eggebrecht H et al (2014). 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J 35: 2873–2926. [DOI] [PubMed] [Google Scholar]
- Fraga‐Silva RA, Trachet B, Stergiopulos N (2015). Emerging pharmacological treatments to prevent abdominal aortic aneurysm growth and rupture. Curr Pharm Des 21: 4000–4006. [PubMed] [Google Scholar]
- Ghosh A, DiMusto PD, Ehrlichman LK, Sadiq O, McEvoy B, Futchko JS et al (2012). The role of extracellular signal‐related kinase during abdominal aortic aneurysm formation. J Am Coll Surg 215: 668–680 e661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, He H, Fu ZC, Huang S, Chen T, Papasian CJ et al (2015). Adipocyte‐derived PAMM suppresses macrophage inflammation by inhibiting MAPK signalling. Biochem J 472: 309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpern VJ, Nackman GB, Gandhi RH, Irizarry E, Scholes JV, Ramey WG et al (1994). The elastase infusion model of experimental aortic aneurysms: synchrony of induction of endogenous proteinases with matrix destruction and inflammatory cell response. J Vasc Surg 20: 51–60. [DOI] [PubMed] [Google Scholar]
- Hao Q, Chen X, Wang X, Dong B, Yang C (2014). Curcumin attenuates angiotensin II‐induced abdominal aortic aneurysm by inhibition of inflammatory response and ERK signaling pathways. Evid Based Complement Alternat Med 2014: 270930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res 46: D1091–d1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y, Wu M, Gung P, Chen C, Kuo C, Yet S (2016). Heme oxygenase‐1 deficiency exacerbates angiotensin II‐induced aortic aneurysm in mice. Oncotarget 7: 67760–67776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanmonod D, & Jeanmonod R (2017). Aneurysm, Abdominal Aortic Rupture (AAA). In StatPearls. Treasure Island (FL).
- Johnston WF, Salmon M, Su G, Lu G, Stone ML, Zhao Y et al (2013). Genetic and pharmacologic disruption of interleukin‐1beta signaling inhibits experimental aortic aneurysm formation. Arterioscler Thromb Vasc Biol 33: 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoglou NP, Liapis CD (2004). Matrix metalloproteinases: contribution to pathogenesis, diagnosis, surveillance and treatment of abdominal aortic aneurysms. Curr Med Res Opin 20: 419–432. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovac S, Angelova PR, Holmstrom KM, Zhang Y, Dinkova‐Kostova AT, Abramov AY (2015). Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim Biophys Acta 1850: 794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Y, Hooda K, Li S, Goyal P, Gupta N, Adeb M (2017). Abdominal aortic aneurysm: pictorial review of common appearances and complications. Ann Transl Med 5: 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannoy M, Slove S, Jacob MP (2014). The function of elastic fibers in the arteries: beyond elasticity. Pathol Biol (Paris) 62: 79–83. [DOI] [PubMed] [Google Scholar]
- Li M, Wang W, Geng L, Qin Y, Dong W, Zhang X et al (2015). Inhibition of RANKL‐induced osteoclastogenesis through the suppression of the ERK signaling pathway by astragaloside IV and attenuation of titanium‐particle‐induced osteolysis. Int J Mol Med 36: 1335–1344. [DOI] [PubMed] [Google Scholar]
- Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT (2002). Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest 110: 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Howatt DA, Balakrishnan A, Moorleghen JJ, Rateri DL, Cassis LA et al (2015). Subcutaneous angiotensin II infusion using osmotic pumps induces aortic aneurysms in mice. J Vis Exp. 10.3791/53191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum H, Roebuck KA (2001). Oxidant stress and endothelial cell dysfunction. Am J Physiol Cell Physiol 280: C719–C741. [DOI] [PubMed] [Google Scholar]
- Malik N, Greenfield BW, Wahl AF, Kiener PA (1996). Activation of human monocytes through CD40 induces matrix metalloproteinases. J Immunol 156: 3952–3960. [PubMed] [Google Scholar]
- McCormick ML, Gavrila D, Weintraub NL (2007). Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 27: 461–469. [DOI] [PubMed] [Google Scholar]
- Meng X, Yang J, Zhang K, An G, Kong J, Jiang F et al (2014). Regulatory T cells prevent angiotensin II‐induced abdominal aortic aneurysm in apolipoprotein E knockout mice. Hypertension 64: 875–882. [DOI] [PubMed] [Google Scholar]
- Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM (2011). Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol 8: 92–102. [DOI] [PubMed] [Google Scholar]
- Nosoudi N, Nahar‐Gohad P, Sinha A, Chowdhury A, Gerard P, Carsten CG et al (2015). Prevention of abdominal aortic aneurysm progression by targeted inhibition of matrix metalloproteinase activity with batimastat‐loaded nanoparticles. Circ Res 117: e80–e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR (2004). Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84: 767–801. [DOI] [PubMed] [Google Scholar]
- Ryer EJ, Garvin RP, Schworer CM, Bernard‐Eckroth KR, Tromp G, Franklin DP et al (2015). Proinflammatory role of stem cells in abdominal aortic aneurysms. J Vasc Surg 62: 1303–1311 e1304. [DOI] [PubMed] [Google Scholar]
- Ryter SW, Alam J, Choi AM (2006). Heme oxygenase‐1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86: 583–650. [DOI] [PubMed] [Google Scholar]
- Saraff K, Babamusta F, Cassis LA, Daugherty A (2003). Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II‐infused, apolipoprotein E‐deficient mice. Arterioscler Thromb Vasc Biol 23: 1621–1626. [DOI] [PubMed] [Google Scholar]
- Sengle G, Tsutsui K, Keene DR, Tufa SF, Carlson EJ, Charbonneau NL et al (2012). Microenvironmental regulation by fibrillin‐1. PLoS Genet 8: e1002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh S, Kim J, Jin U, Choi H, Chang Y, Lee Y et al (2009). Deoxypodophyllotoxin, flavolignan, from Anthriscus sylvestris Hoffm. inhibits migration and MMP‐9 via MAPK pathways in TNF‐alpha‐induced HASMC. Vascul Pharmacol 51: 13–20. [DOI] [PubMed] [Google Scholar]
- Sun C, Jiang M, Zhang L, Yang J, Zhang G, Du B et al (2017). Cycloastragenol mediates activation and proliferation suppression in concanavalin A‐induced mouse lymphocyte pan‐activation model. Immunopharmacol Immunotoxicol 39: 131–139. [DOI] [PubMed] [Google Scholar]
- Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T et al (2015). Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II‐induced aortic aneurysm. Arterioscler Thromb Vasc Biol 35: 127–136. [DOI] [PubMed] [Google Scholar]
- Wang C, Wang Y, Yu M, Chen C, Xu L, Cao Y et al (2017a). Grape‐seed polyphenols play a protective role in elastase‐induced abdominal aortic aneurysm in mice. Sci Rep 7: 9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhou LR, Qin Y, Cen Y, Hu L et al (2017b). IP‐10/CXCR3 axis promotes the proliferation of vascular smooth muscle cells through ERK1/2/CREB signaling pathway. Cell Biochem Biophys 75: 139–147. [DOI] [PubMed] [Google Scholar]
- Wang P, Luan J, Xu W, Wang L, Xu D, Yang C et al (2017c). Astragaloside IV downregulates the expression of MDR1 in Bel7402/FU human hepatic cancer cells by inhibiting the JNK/cJun/AP1 signaling pathway. Mol Med Rep 16: 2761–2766. [DOI] [PubMed] [Google Scholar]
- Yang C, Li W, Li S, Li J, Li Y, Kong S et al (2014). MCP‐1 stimulates MMP‐9 expression via ERK 1/2 and p38 MAPK signaling pathways in human aortic smooth muscle cells. Cell Physiol Biochem 34: 266–276. [DOI] [PubMed] [Google Scholar]
- Yoshimura K, Aoki H, Ikeda Y, Furutani A, Hamano K, Matsuzaki M (2006). Regression of abdominal aortic aneurysm by inhibition of c‐Jun N‐terminal kinase in mice. Ann N Y Acad Sci 1085: 74–81. [DOI] [PubMed] [Google Scholar]
- Zhang X, He C, Tian K, Li P, Su H, Wan J (2015). Ginsenoside Rb1 attenuates angiotensin II‐induced abdominal aortic aneurysm through inactivation of the JNK and p38 signaling pathways. Vascul Pharmacol 73: 86–95. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Li Q, Zhao W, Li J, Sun Y, Liu K et al (2015). Astragaloside IV and cycloastragenol are equally effective in inhibition of endoplasmic reticulum stress‐associated TXNIP/NLRP3 inflammasome activation in the endothelium. J Ethnopharmacol 169: 210–218. [DOI] [PubMed] [Google Scholar]
- Zhou R, Song Y, Ruan J, Wang Y, Yan R (2012). Pharmacokinetic evidence on the contribution of intestinal bacterial conversion to beneficial effects of astragaloside IV, a marker compound of astragali radix, in traditional oral use of the herb. Drug Metab Pharmacokinet 27: 586–597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 CAG treatment prevented the occurrence rather than simply delay the growth of AAA in mice. (a) morphology of aorta (b) relative maximal external abdominal aortic diameter. In this segment, the high dose CAG (125 mg·kg‐1·d‐1) was given to the mice from the day of Elastase‐AAA induction and lasted for 14 days, then CAG were displaced by 0.01M PBS, which was also lasted for 14 days while 0.01M PBS was given in other two group for 4 weeks. And n=6 in each group.
Figure S2 Acute toxicity of CAG on mice. (a) Body weight change. (b) Weight of major organs/body weight. (c) Clinical chemistry parameters, glutamate pyruvate transaminase (ALT), aspartate aminotransferase (AST), total creatine phosphokinase (CK), creatine kinase isoenzyme (CK‐MB) and LDH. There is no obvious significance between CAG treated group and PBS treated group no matter male or female mice. n=5 in each group.