

Abstract
Endothelial NOS (eNOS), and its product NO, are vital components of the control of vasomotor function and cardiovascular homeostasis. In the present review, we will take a deep dive into eNOS enzymology, function and mechanisms regulating endothelial NO. The mechanisms regulating eNOS and NO synthesis discussed here include alterations to transcriptional, post‐translational modifications and protein–protein regulations. Also, we will discuss the phenotypes associated with various eNOS mutants and the consequences of a disrupted eNOS/NO cascade, highlighting the importance of eNOS function and vascular homeostasis.
Linked Articles
This article is part of a themed section on Nitric Oxide 20 Years from the 1998 Nobel Prize. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.2/issuetoc
Abbreviations
- BH4
tetrahydrobiopterin
- CaM
calmodulin
- Cav‐1
caveolin‐1
- CSD
Cav‐1 scaffold domain
- EC
endothelial cells
- eNOS/NOS3
endothelial NOS
- Hbα
haemoglobin α
- LEENE
lncRNA that enhances eNOS expression
- lncRNAs
long non‐coding RNAs
- ox‐LDL
oxidized LDL
- STEEL
spliced‐transcript endothelial‐enriched lncRNA
Introduction
The discovery of endothelial‐derived relaxing factor, identified as NO, revolutionized our understanding of local control of vasomotor function and cardiovascular homeostasis. The identification of endothelial NOS (eNOS, NOS3) as the enzyme responsible for generating endothelial‐derived NO has led to the exploration of how this enzyme is regulated in health and disease. The central focus of this review is to highlight recent advances in the field and perspectives for the future.
Overview of eNOS enzymology and function
Akin to all NOS isoforms, eNOS is a homodimeric, haem‐containing oxidoreductase that couples the metabolism of L‐arginine in the oxygenase domain with NADPH‐dependent flux of electrons from the reductase domain. Upon changes in local calcium, calcium‐activated calmodulin (CaM) facilitates interdomain electron transfer and accelerates NO synthesis. The rates of electron transfer and haem iron reduction differ amongst NOS isoforms and requires the critical co‐factor tetrahydrobiopterin (BH4) for optimal NO synthesis. Unlike the other NOS isoforms, eNOS is predominately membrane bound due to amino‐terminal fatty acylation by myristic and palmitic acid, and dual acylation brings eNOS into the proximity of biological membranes such as the cytoplasmic face of the Golgi or plasmalemmal caveolae. The proper subcellular localization of eNOS ensures optimal regulation by mechanical forces (shear stress or pressure gradients), calcium ions and kinases. Therefore, any agonist that mobilizes intracellular calcium (VEGF, bradykinin, histamine, etc.) or alterations in intracellular signalling pathways leading to enhanced CaM binding or reduced CaM dissociation has the potential to promote eNOS activity and NO release. Under conditions of BH4 depletion or L‐arginine deficiency, eNOS can become uncoupled and generate oxygen‐derived free radicals (Forstermann and Sessa, 2012).
Activation of eNOS promotes endothelium‐dependent vasodilation and regulates systemic blood pressure. Accordingly, mice lacking eNOS also lack endothelium‐dependent relaxation of large blood vessels, have markedly elevated blood pressure, impaired vascular remodelling and are more susceptible to ischaemic injury, myocardial infarction and atherosclerosis, consistent with the known cardioprotective actions of the endothelial‐derived NO. Although the endothelium can produce other gasotransmitters such as carbon monoxide and hydrogen sulfide, vasodilatory eicosanoids and other vasoactive lipids, these substances do not adequately compensate for the loss of eNOS in mice. Interestingly, recent data suggest that eNOS is required for the actions of hydrogen sulfide (King et al., 2014) and that there are multiple levels of crosstalk between hydrogen sulfide and the NO‐cGMP pathway (Bucci et al., 2010; Bucci et al., 2012). Despite the importance of eNOS in whole body cardiovascular homeostasis, the role of eNOS in controlling microvascular flow is less prominent. Mechanistically, eNOS‐derived NO can activate soluble guanylate cyclase, thereby increasing the formation of the second messenger, cGMP and activation of protein kinase G. Protein kinase G phosphorylates several downstream substrates that are responsible for many of the biological actions of NO. Additionally, eNOS can provide NO equivalents used in additional chemical reactions throughout the body. The actions and targets of eNOS‐derived NO will be reviewed in other articles in this series, and the remainder of this review will focus on regulation of eNOS gene expression and post‐translational control of eNOS activity.
Transcriptional and post‐transcriptional regulation of eNOS
The human eNOS gene is located on Chromosome 7 and contains a promoter region rich in several transcription factor binding sites for a variety of transcription factors including KLF2, Sp1, Sp3, Ets1,Ets2, Smad2, GATA1, GATA2, GATA4, AP‐1, cJun/Fos, NFkB, CHOP10 and p53. Although eNOS was initially considered to be a constitutively expressed gene [relative to inducible NOS (iNOS) or NOS2], there are several circumstances where it can be induced several‐fold. Most importantly, eNOS gene expression is up‐regulated by fluid shear stress (Nishida et al., 1992) and cyclic stretch (Awolesi et al., 1995) in cultured endothelial cells (EC), and this has been observed also in exercised animals (Sessa et al., 1994; Fukai et al., 2000). Short‐term changes in shear stress rapidly activate eNOS (see below section on phosphorylation), and this is critical for flow‐mediated regulation of vascular tone while sustained shear promotes eNOS gene expression and chronic remodelling of blood vessels. Some additional stimuli that induce eNOS include VEGF‐A (Papapetropoulos et al., 1997; Bouloumie et al., 1999), TGFβ (Saura et al., 2002), lysophosphotidycholine (Zembowicz et al., 1995), statins (Laufs et al., 1998), cyclosporine A (Navarro‐Antolin et al., 2000), H202 (Searles, 2006), and oestrogen (Tan et al., 1999). Conversely, pro‐inflammatory stimuli such as oxidized LDL (ox‐LDL) (Liao et al., 1995), TNFα (Nishida et al., 1992; Neumann et al., 2004), LPS (Lu et al., 1996) and hypoxia (McQuillan et al., 1994; Fish et al., 2010) can reduce eNOS mRNA levels. The stability of eNOS mRNA has been reported to range between 24 and 48 h (Yoshizumi et al., 1993; McQuillan et al., 1994), and a significant decrease in eNOS mRNA stability has been observed under TNFα, ox‐LDL and hypoxic conditions. Mechanistically, DNA methylation of the eNOS promoter can dramatically reduce Sp1, Sp3 and Ets1 transcription factor binding and eNOS mRNA levels (Chan et al., 2004). Hypoxia promotes dramatic changes to eNOS expression through a variety of mechanisms, including changes to transcription factor dynamics, decreases in mRNA stability and most recently changes to histones associated with the eNOS promoter (Fish et al., 2010). Additional work has described a dynamic ‘histone code’ where changes to histones at the proximal promoter of eNOS regulate eNOS mRNA levels (Fish et al., 2005). This code includes the acetylation of histones H3 and H4 and the dimethylation and trimethylation of histone H3. In fact, the decrease in transcription observed in hypoxia is in part attribute to a reduction in acetylation and methylation of eNOS proximal promoter histones (Fish et al., 2010).
Recently, work exploring the role of long noncoding RNAs (lncRNAs) in EC uncovered two distinct lncRNAs: spliced‐transcript endothelial‐enriched lncRNA (STEEL) and lncRNA that enhances eNOS expression (LEENE) that are able to influence eNOS mRNA levels. STEEL can up‐regulate the important EC transcription factor KLF2 and thereby increases eNOS mRNA levels (Man et al., 2018). Using a combination of transcriptome and chromatin conformation profiling, LEENE was discovered from an enhancer that has proximal association with the eNOS genomic locus (Miao et al., 2018). Both STEEL and LEENE have strong ties to KLF2, an established driver of eNOS transcription (Lin et al., 2005), with STEEL and LEENE not only influencing the KLF2 promoter but also relying on KLF2 at the transcriptional level. In addition to lncRNAs, a role of micro RNAs (miRNAs) on eNOS levels has been described. The loss of Dicer in EC, which blocks the synthesis of all mature miRNAs, increases eNOS mRNA and protein levels, effects rescued by miR‐221/222. This effect is likelyto be indirect as there are no miR221/222 seed sequences in the eNOS 3′ untranslated region (Suarez et al., 2007). mIR‐92a is the best studied miRNA regulating eNOS mRNA levels via targeting the transcription factor KLF2. Indeed, antagonism of miR‐92 increases KLF2 levels and eNOS activity and promotes beneficial effects on cardiac function, arteriogenesis and angiogenesis (Bonauer et al., 2009).
eNOS regulation by post‐translational modifications
Initially, eNOS was believed to be regulated only by the essential allosteric regulator of all NOS isoforms, calcium‐activated CaM. However, detailed work on the subcellular localization od eNOS and mapping of its post‐translational modifications has provided novel insights into how mechanical forces, growth factors, bioactive peptides and lipid mediators regulate NO synthesis in EC. The purification of eNOS as a membrane protein and the subsequent cloning and identification of the mechanisms of membrane targeting via acylation have led to a series of studies identifying post‐translational control mechanisms such as phosphorylation and protein–protein interactions that regulate eNOS activity beyond CaM, thereby fine tuning eNOS activation. In addition to these modifications, eNOS can be S‐nitrosylated (at C94 and C98) (Erwin et al., 2005) reducing its activity, acetylated (K609, S765 and S771) increasing its activity (Jung et al., 2010) or glutathionylated in the C‐terminal reductase domain (C689 and C908) uncoupling eNOS to generated superoxide anion (Chen et al., 2010).
Multisite phosphorylation of eNOS regulates activity
Work in the 1990s documented that eNOS is a phosphoprotein. In 1999, the most abundant phosphoserine site (S1176, S1177 or S1179 in murine, human or bovine eNOS, respectively) was discovered using mass spectrometry (Dimmeler et al., 1999; Fulton et al., 1999). The residue S1177 in human eNOS is a canonical phospho‐acceptor site for the protein kinase Akt; however, additional AGC kinase family members (protein kinase A, protein kinase G, protein kinase C and AMP‐activated kinase; AMPK) can phosphorylate this site in vitro and perhaps in vivo. Since the discovery of S1177, six additional phosphorylation sites have been mapped including Y81, S114, T495, S615, S633 and Y657. Investigations into these various phosphorylation sites have shown eNOS activity to be activated or inhibited depending on which particular site is phosphorylated. Moreover, there are stimulus‐dependent kinetic changes in most of these sites depending on the method of stimulation and the duration of the response (Bauer et al., 2003). Y81, S615, S633 and S1177 have been identified as stimulatory sites for eNOS activity while S114, T495 and Y657 are recognized as inhibitory sites (Figure 1). Src kinase is the dominant kinase phosphorylating Y81, thus increasing eNOS activity and NO production (Fulton et al., 2005). The kinases PKA, AMPK and Akt have been implicated in the phosphorylation of the S615 and S633 enhancing enzyme activity (Fulton, 2016). The inhibitory site S114 is increased under conditions of shear stress and influenced by ERK while the Y495 site has been shown to be phosphorylated by AMPK, ROCK and PKC (Navarro‐Antolin et al., 2000). Most recently, proline‐rich proline‐rich tyrosine kinase 2 (PYK2) phosphorylates Y657 and impairs eNOS enzyme activity (Loot et al., 2009).
Figure 1.
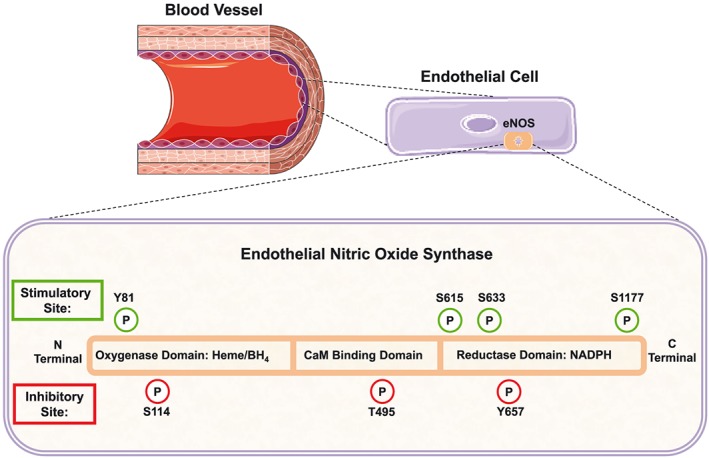
Diagram illustrating the various phosphorylation sites (stimulatory and inhibitory) located in human eNOS.
Despite the presence of many sites for the phosphorylation of eNOS, there has been little mechanistic work to characterize how each site directly regulates enzymic activity, using purified eNOS. Early work demonstrated that eNOS contained two autoinhibition sites, near the CaM binding domain and at the extreme carboxy terminus of the protein and that putative phosphorylation sites were in the vicinity of these inhibitory sites (Salerno et al., 1997). As shown for the S1179 site (bovine eNOS) within the carboxy‐terminal inhibitory domain, mutation of S1179 to aspartate (eNOS S1179D) renders eNOS constitutively active and coupled suggesting that the negative charge of phosphate or aspartate dampens autoinhibition. Indeed, purified eNOS S1179D is more active than non‐phosphorylated eNOS and has enhanced electron flux from the reductase to the oxygenase domain and reduced CaM dissociation at lower concentrations of calcium (McCabe et al., 2000).
Although there is ample in vitro evidence for each of the phosphorylation sites influencing eNOS activity, the only definitive in vivo evidence supporting a physiological role of eNOS phosphorylation is for S1176 (murine site), because the relevant mutant mice are available. Using a gene‐targeting strategy, mice were generated expressing a serine to alanine mutation (eNOS S1176A) or serine to aspartate mutation (eNOS S1176D; Figure 2). Biochemically, eNOS S1176A cannot be phosphorylated and eNOS S1176D is a ‘gain of function’ mutant. Interestingly, eNOS S1176A mice exhibit reduced endothelial‐dependent responses, enhanced blood pressure, insulin resistance and increased weight. These effects are diminished in eNOS S1176D mice that demonstrate enhanced endothelial‐dependent responses and are protected from insulin resistance and stroke (Atochin et al., 2007; Kashiwagi et al., 2013; Li et al., 2013). Interestingly, atherosclerotic mice lacking the ApoE gene have larger lesions when bred to eNOS S1176A mice (Park et al., 2016), similar to eNOS‐deficient mice bred to the same atherosclerosis‐prone strain (Kuhlencordt et al., 2001). Phosphorylation of S1176 in eNOS is also critical for vascular permeability changes induced by VEGF, histamine and non‐immune irritants, as eNOS S1176A mice show diminished permeability while eNOS S1176D mice show normal or enhanced changes in vascular leakage (Di Lorenzo et al., 2013).
Figure 2.
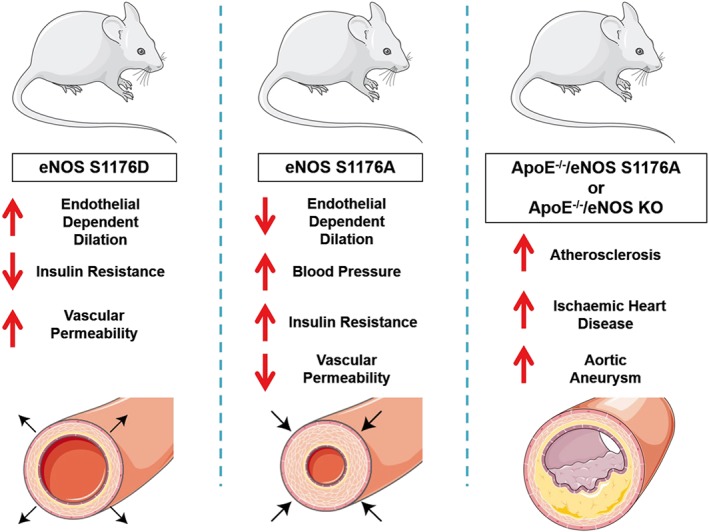
Diagram illustrating the phenotypes observed in eNOS mutant mice including eNOS S1176D, eNOS S1176A and the double mutant ApoE−/−/eNOS S1176A and ApoE−/−/eNOS KO mice.
As mentioned before, many kinases can phosphorylate eNOS at S1176, and the importance of each kinase in vivo is virtually unknown. In order to test the physiological importance of Akt as an eNOS kinase, global Akt1‐deficient mice were bred to eNOS mutant mice (Schleicher et al., 2009). Notably, eNOS S1176D mice rescued impaired wound healing and ischaemic angiogenesis typically observed in Akt1‐deficient mice; effects attenuated in Akt1‐deficient/eNOS S1176A mice. Thus, this genetic epistasis experiment implies that eNOS is an important Akt1 substrate in vivo and that S1176 is a critical site of eNOS phosphorylation, independent of the additional sites of phosphorylation.
Flow or shear‐induced eNOS activation is one of the most important physiological stimuli regulating acute changes in blood flow and long‐term vascular remodelling. Increases in blood flow (and the attendant changes in shear stress) can activate the endothelial cation channel, PIEZO1, promoting ATP release and activation of the purinergic receptor, P2Y2, which is coupled to the G protein, G11. Activation of this pathway stimulates Akt phosphorylation and eNOS phosphorylation on S1176 (Wang et al., 2016). Consequently, mice lacking PIEZO1 in endothelium have reduced NO formation and eNOS activation and develop hypertension. Recently, endothelial‐specific Akt1‐deficient mice were generated, and these mice have reduced circulating NO levels, reduced eNOS S1176 phosporylation in blood vessels, increased blood pressure and impaired vessel function and ischaemic arteriogenesis (Lee et al., 2018). However, these deficits were not as severe as eNOS or global Akt1‐deficient mice suggesting additional levels of eNOS regulation by additional kinases. Similar genetic epistasis experiments are clearly needed for other eNOS kinases, and mutant strains lacking the other sites of eNOS phosphorylation are critical to delineate the complex role of eNOS phosphorylation in vivo.
Regulation of eNOS by protein–protein interactions
In addition to the dynamic, post‐translational regulation of eNOS, there is extensive evidence demonstrating the importance of additional proteins that stimulate or inhibit eNOS function. The most important protein regulator of eNOS is CaM. Calcium‐activated CaM regulates all NOS isoforms and is an essential allosteric regulator facilitating NADPH‐dependent electron flux from the reductase domain to the oxygenase domain. In endothelial‐lined vessels and in cultured EC, any agonist that triggers an influx of extracellular calcium or mobilizes intracellular calcium can promote endothelial‐dependent relaxations and NO release. Early evidence suggested that fluid shear stress can induce calcium‐independent NO release since neutralization of either extracellular or intracellular calcium did not affect shear‐induced NO release and sustained shear did not mobilize intracellular calcium, as measured with fluorometric dyes. However, it was subsequently shown that low levels of calcium‐activated CaM can bind eNOS in a manner similar to iNOS and that phosphorylation of eNOS may change the association and dissociation rates of CaM binding to eNOS (McCabe et al., 2000). In addition, there is evidence that the binding of heat shock protein 90 to eNOS regulates the sensitivity of CaM towards eNOS (Brouet et al., 2001; Fontana et al., 2002). As mentioned above, there are many eNOS interacting proteins and this topic has been extensively reviewed recently (Siragusa and Fleming, 2016). However, we will highlight work on caveolin‐1 (Cav‐1) and haemoglobin α (Hbα) as these interactions that have been best characterized in vitro and are physiologically relevant in vivo.
eNOS regulation and interaction with caveolin‐1
Caveolae are flask‐shaped invaginations of the plasma membrane that are highly enriched in endothelia. Indeed, caveolae are the major plasmalemmal vesicle in EC and are prominent in all endothelia lining large and small blood vessels. Cav‐1 is the key protein required for the formation of caveolae since the genetic loss of Cav‐1 eliminates all measurable caveolae (Drab et al., 2001; Razani et al., 2001). In addition to Cav‐1, additional proteins called cavins were discovered which modify caveolae number and function (Chidlow and Sessa, 2010). Early insights into the eNOS–Cav‐1 connection was determined by co‐localization and co‐fractionation of eNOS with Cav‐1 (Garcia‐Cardena et al., 1996; Shaul et al., 1996)). Moreover, experiments with purified components showed that eNOS can interact with at least two domains of Cav‐1 and this interaction negatively regulated eNOS activity and NO release from cells. One juxtamembrane domain of Cav‐1, termed the Cav‐1 scaffolding domain (CSD), was shown to be sufficient for eNOS docking and inhibition of eNOS activity. One issue that is still unresolved in vivo is whether eNOS directly interacts with Cav‐1 in vivo, although in vitro experiments support this model (Garcia‐Cardena et al., 1997; Ju et al., 1997). Physiologically, the interaction of eNOS is functionally relevant since one of the main phenotypes in mice lacking Cav‐1 is enhanced endothelium‐dependent relaxations, cardiac and pulmonary vascular changes and cardiomyopathy (Drab et al., 2001; Razani et al., 2001; Murata et al., 2007). Paradoxically, although acetylcholine‐mediated relaxations are enhanced, flow‐induced dilation and proportional vascular remodelling are attenuated (Yu et al., 2006). This deficit in flow‐mediated mechanosignalling is now linked to the role of caveolae as source of residual plasma membrane that dynamically respond to alterations in membrane tension (Sinha et al., 2011).
Interestingly, generation of a cell permeable version of the CSD has been used as a Cav‐1 mimetic in experimental models in vivo. Early work demonstrated that treatment of mice with cell permeable CSD reduces acute changes in vascular permeability in response to non‐immune irritants (Bucci et al., 2000). Since that time, several papers have shown that the CSD can reduce tissue oedema, tumour growth, asthma, retinal inflammation angiogenesis and neuroinflammation (Kraehling and Sessa, 2017). Mechanistically, eNOS can contribute to these underlying processes, but it is likely that additional Cav‐1 targets are influenced by the CSD and additional work is needed to identify the molecular targets of the CSD in vivo.
Haemoglobin α
The relationship between the amount of NO generated from NOS and the biological actions of authentic NO gas can be less than stoichiometric. Recent evidence showing that EC express the α subunit of Hbα suggests that the biological activity of NO can be decreased through sequestration by Hb (Straub et al., 2012). Hbα is localized at the interface between EC and smooth muscle cells (called myoendothelial junctions) in resistance arteries, and Hbα can directly interact with eNOS and sequester NO when Hb is in its high affinity, reduced Fe2+‐form. This binding results in the oxidation of Hbα to the Fe3+ state and the generation of nitrate and the cycling of the Fe ions back to the reduced state is via the enzyme NADH‐cytochrome b5 reductase 3. The Fe3+ Hbα has a much lower affinity for NO, thereby permitting the limited diffusion of NO to the underlying vascular smooth muscle. Pharmacological manipulation of this pathway using a novel peptide to disrupt the interaction of eNOS with Hbα reduces blood pressure in a model of hypertension (Straub et al., 2014).
Concluding remarks and future perspectives
The discovery of a unique NOS isoform regulating endothelial‐dependent responses has led to an explosion of information on the molecular and cellular pathways regulating eNOS function. In experimental models and in humans, eNOS is critical for normal vascular homeostasis, vascular remodelling and adaptation to stress and exercise. In most cardiovascular diseases, impaired endothelium‐dependent responses is a hallmark of disease progression, but it is not clear if correction of this abnormality in humans would delay or prevent disease. Perhaps a deeper understanding of the multifaceted regulation of eNOS will permit the identification of new therapeutic approaches for the treatment of vascular dysfunction and disease.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b, 2017c).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by Grants R35 HL139945 from the National Institutes of Health, an AHA MERIT and the Leducq Fondation (MIRVAD network) to W.C.S., and the Vascular Research Training Grant 5T32HL007950‐15 to V.G.
Garcia, V. , and Sessa, W. C. (2019) Endothelial NOS: perspective and recent developments. British Journal of Pharmacology, 176: 189–196. 10.1111/bph.14522.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Overview. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Other ion channels. Br J Pharmacol 174: S195–S207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atochin DN, Wang A, Liu VW, Critchlow JD, Dantas AP, Looft‐Wilson R et al (2007). The phosphorylation state of eNOS modulates vascular reactivity and outcome of cerebral ischemia in vivo . J Clin Invest 117: 1961–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awolesi MA, Sessa WC, Sumpio BE (1995). Cyclic strain upregulates nitric oxide synthase in cultured bovine aortic endothelial cells. J Clin Invest 96: 1449–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer PM, Fulton D, Boo YC, Sorescu GP, Kemp BE, Jo H et al (2003). Compensatory phosphorylation and protein‐protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric‐oxide synthase. J Biol Chem 278: 14841–14849. [DOI] [PubMed] [Google Scholar]
- Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A et al (2009). MicroRNA‐92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science 324: 1710–1713. [DOI] [PubMed] [Google Scholar]
- Bouloumie A, Schini‐Kerth VB, Busse R (1999). Vascular endothelial growth factor up‐regulates nitric oxide synthase expression in endothelial cells. Cardiovasc Res 41: 773–780. [DOI] [PubMed] [Google Scholar]
- Brouet A, Sonveaux P, Dessy C, Balligand JL, Feron O (2001). Hsp90 ensures the transition from the early Ca2+‐dependent to the late phosphorylation‐dependent activation of the endothelial nitric‐oxide synthase in vascular endothelial growth factor‐exposed endothelial cells. J Biol Chem 276: 32663–32669. [DOI] [PubMed] [Google Scholar]
- Bucci M, Gratton JP, Rudic RD, Acevedo L, Roviezzo F, Cirino G et al (2000). In vivo delivery of the caveolin‐1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat Med 6: 1362–1367. [DOI] [PubMed] [Google Scholar]
- Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Pyriochou A, Roussos C et al (2010). Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arterioscler Thromb Vasc Biol 30: 1998–2004. [DOI] [PubMed] [Google Scholar]
- Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Zaid A, Giannogonas P et al (2012). cGMP‐dependent protein kinase contributes to hydrogen sulfide‐stimulated vasorelaxation. PLoS One 7: e53319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan Y, Fish JE, D'Abreo C, Lin S, Robb GB, Teichert AM et al (2004). The cell‐specific expression of endothelial nitric‐oxide synthase: a role for DNA methylation. J Biol Chem 279: 35087–35100. [DOI] [PubMed] [Google Scholar]
- Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA et al (2010). S‐glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468: 1115–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidlow JH Jr, Sessa WC (2010). Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovasc Res 86: 219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lorenzo A, Lin MI, Murata T, Landskroner‐Eiger S, Schleicher M, Kothiya M et al (2013). eNOS‐derived nitric oxide regulates endothelial barrier function through VE‐cadherin and Rho GTPases. J Cell Sci 126 (Pt 24): 5541–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM (1999). Activation of nitric oxide synthase in endothelial cells by Akt‐dependent phosphorylation. Nature 399: 601–605. [DOI] [PubMed] [Google Scholar]
- Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B et al (2001). Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin‐1 gene‐disrupted mice. Science 293: 2449–2452. [DOI] [PubMed] [Google Scholar]
- Erwin PA, Lin AJ, Golan DE, Michel T (2005). Receptor‐regulated dynamic S‐nitrosylation of endothelial nitric‐oxide synthase in vascular endothelial cells. J Biol Chem 280: 19888–19894. [DOI] [PubMed] [Google Scholar]
- Fish JE, Matouk CC, Rachlis A, Lin S, Tai SC, D'Abreo C et al (2005). The expression of endothelial nitric‐oxide synthase is controlled by a cell‐specific histone code. J Biol Chem 280: 24824–24838. [DOI] [PubMed] [Google Scholar]
- Fish JE, Yan MS, Matouk CC, St Bernard R, Ho JJ, Gavryushova A et al (2010). Hypoxic repression of endothelial nitric‐oxide synthase transcription is coupled with eviction of promoter histones. J Biol Chem 285: 810–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana J, Fulton D, Chen Y, Fairchild TA, McCabe TJ, Fujita N et al (2002). Domain mapping studies reveal that the M domain of hsp90 serves as a molecular scaffold to regulate Akt‐dependent phosphorylation of endothelial nitric oxide synthase and NO release. Circ Res 90: 866–873. [DOI] [PubMed] [Google Scholar]
- Forstermann U, Sessa WC (2012). Nitric oxide synthases: regulation and function. Eur Heart J 33: 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukai T, Siegfried MR, Ushio‐Fukai M, Cheng Y, Kojda G, Harrison DG (2000). Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J Clin Invest 105: 1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton D, Church JE, Ruan L, Li C, Sood SG, Kemp BE et al (2005). Src kinase activates endothelial nitric‐oxide synthase by phosphorylating Tyr‐83. J Biol Chem 280: 35943–35952. [DOI] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K et al (1999). Regulation of endothelium‐derived nitric oxide production by the protein kinase Akt. Nature 399: 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton DJ (2016). Transcriptional and posttranslational regulation of eNOS in the endothelium. Adv Pharmacol 77: 29–64. [DOI] [PubMed] [Google Scholar]
- Garcia‐Cardena G, Martasek P, Masters BS, Skidd PM, Couet J, Li S et al (1997). Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo . J Biol Chem 272: 25437–25440. [DOI] [PubMed] [Google Scholar]
- Garcia‐Cardena G, Oh P, Liu J, Schnitzer JE, Sessa WC (1996). Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. Proc Natl Acad Sci U S A 93: 6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju H, Zou R, Venema VJ, Venema RC (1997). Direct interaction of endothelial nitric‐oxide synthase and caveolin‐1 inhibits synthase activity. J Biol Chem 272: 18522–18525. [DOI] [PubMed] [Google Scholar]
- Jung SB, Kim CS, Naqvi A, Yamamori T, Mattagajasingh I, Hoffman TA et al (2010). Histone deacetylase 3 antagonizes aspirin‐stimulated endothelial nitric oxide production by reversing aspirin‐induced lysine acetylation of endothelial nitric oxide synthase. Circ Res 107: 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi S, Atochin DN, Li Q, Schleicher M, Pong T, Sessa WC et al (2013). eNOS phosphorylation on serine 1176 affects insulin sensitivity and adiposity. Biochem Biophys Res Commun 431: 284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK et al (2014). Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase‐nitric oxide dependent. Proc Natl Acad Sci U S A 111: 3182–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraehling JR, Sessa WC (2017). Contemporary approaches to modulating the nitric oxide‐cGMP pathway in cardiovascular disease. Circ Res 120: 1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlencordt PJ, Gyurko R, Han F, Scherrer‐Crosbie M, Aretz TH, Hajjar R et al (2001). Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double‐knockout mice. Circulation 104: 448–454. [DOI] [PubMed] [Google Scholar]
- Laufs U, La Fata V, Plutzky J, Liao JK (1998). Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation 97: 1129–1135. [DOI] [PubMed] [Google Scholar]
- Lee MY, Gamez‐Mendez A, Zhang J, Zhuang Z, Vinyard DJ, Kraehling J et al (2018). Endothelial cell autonomous role of Akt1: Regulation of vascular tone and ischemia‐induced arteriogenesis. Arterioscler Thromb Vasc Biol 38: 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Atochin D, Kashiwagi S, Earle J, Wang A, Mandeville E et al (2013). Deficient eNOS phosphorylation is a mechanism for diabetic vascular dysfunction contributing to increased stroke size. Stroke 44: 3183–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao JK, Shin WS, Lee WY, Clark SL (1995). Oxidized low‐density lipoprotein decreases the expression of endothelial nitric oxide synthase. J Biol Chem 270: 319–324. [DOI] [PubMed] [Google Scholar]
- Lin Z, Kumar A, SenBanerjee S, Staniszewski K, Parmar K, Vaughan DE et al (2005). Kruppel‐like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res 96: e48–e57. [DOI] [PubMed] [Google Scholar]
- Loot AE, Schreiber JG, Fisslthaler B, Fleming I (2009). Angiotensin II impairs endothelial function via tyrosine phosphorylation of the endothelial nitric oxide synthase. J Exp Med 206: 2889–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JL, Schmiege LM 3rd, Kuo L, Liao JC (1996). Downregulation of endothelial constitutive nitric oxide synthase expression by lipopolysaccharide. Biochem Biophys Res Commun 225: 1–5. [DOI] [PubMed] [Google Scholar]
- Man HSJ, Sukumar AN, Lam GC, Turgeon PJ, Yan MS, Ku KH et al (2018). Angiogenic patterning by STEEL, an endothelial‐enriched long noncoding RNA. Proc Natl Acad Sci U S A 115: 2401–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe TJ, Fulton D, Roman LJ, Sessa WC (2000). Enhanced electron flux and reduced calmodulin dissociation may explain ‘calcium‐independent’ eNOS activation by phosphorylation. J Biol Chem 275: 6123–6128. [DOI] [PubMed] [Google Scholar]
- McQuillan LP, Leung GK, Marsden PA, Kostyk SK, Kourembanas S (1994). Hypoxia inhibits expression of eNOS via transcriptional and posttranscriptional mechanisms. Am J Physiol 267 (5 Pt 2): H1921–H1927. [DOI] [PubMed] [Google Scholar]
- Miao Y, Ajami NE, Huang TS, Lin FM, Lou CH, Wang YT et al (2018). Enhancer‐associated long non‐coding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nat Commun 9: 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T, Lin MI, Huang Y, Yu J, Bauer PM, Giordano FJ et al (2007). Reexpression of caveolin‐1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin‐1 knockout mice. J Exp Med 204: 2373–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro‐Antolin J, Rey‐Campos J, Lamas S (2000). Transcriptional induction of endothelial nitric oxide gene by cyclosporine A. A role for activator protein‐1. J Biol Chem 275: 3075–3080. [DOI] [PubMed] [Google Scholar]
- Neumann P, Gertzberg N, Johnson A (2004). TNF‐alpha induces a decrease in eNOS promoter activity. Am J Physiol Lung Cell Mol Physiol 286: L452–L459. [DOI] [PubMed] [Google Scholar]
- Nishida K, Harrison DG, Navas JP, Fisher AA, Dockery SP, Uematsu M et al (1992). Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J Clin Invest 90: 2092–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A, Garcia‐Cardena G, Madri JA, Sessa WC (1997). Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest 100: 3131–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Mima A, Li Q, Rask‐Madsen C, He P, Mizutani K et al (2016). Insulin decreases atherosclerosis by inducing endothelin receptor B expression. JCI Insight 1. pii: e86574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB et al (2001). Caveolin‐1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276: 38121–38138. [DOI] [PubMed] [Google Scholar]
- Salerno JC, Harris DE, Irizarry K, Patel B, Morales AJ, Smith SM et al (1997). An autoinhibitory control element defines calcium‐regulated isoforms of nitric oxide synthase. J Biol Chem 272: 29769–29777. [DOI] [PubMed] [Google Scholar]
- Saura M, Zaragoza C, Cao W, Bao C, Rodriguez‐Puyol M, Rodriguez‐Puyol D et al (2002). Smad2 mediates transforming growth factor‐beta induction of endothelial nitric oxide synthase expression. Circ Res 91: 806–813. [DOI] [PubMed] [Google Scholar]
- Schleicher M, Yu J, Murata T, Derakhshan B, Atochin D, Qian L et al (2009). The Akt1‐eNOS axis illustrates the specificity of kinase‐substrate relationships in vivo . Sci Signal 2: ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searles CD (2006). Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am J Physiol Cell Physiol 291: C803–C816. [DOI] [PubMed] [Google Scholar]
- Sessa WC, Pritchard K, Seyedi N, Wang J, Hintze TH (1994). Chronic exercise in dogs increases coronary vascular nitric oxide production and endothelial cell nitric oxide synthase gene expression. Circ Res 74: 349–353. [DOI] [PubMed] [Google Scholar]
- Shaul PW, Smart EJ, Robinson LJ, German Z, Yuhanna IS, Ying Y et al (1996). Acylation targets emdothelial nitric‐oxide synthase to plasmalemmal caveolae. J Biol Chem 271: 6518–6522. [DOI] [PubMed] [Google Scholar]
- Sinha B, Koster D, Ruez R, Gonnord P, Bastiani M, Abankwa D et al (2011). Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 144: 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siragusa M, Fleming I (2016). The eNOS signalosome and its link to endothelial dysfunction. Pflugers Arch 468: 1125–1137. [DOI] [PubMed] [Google Scholar]
- Straub AC, Butcher JT, Billaud M, Mutchler SM, Artamonov MV, Nguyen AT, et al (2014). Hemoglobin alpha/eNOS coupling at myoendothelial junctions is required for nitric oxide scavenging during vasoconstriction. Arterioscler Thromb Vasc Biol 34: 2594–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub AC, Lohman AW, Billaud M, Johnstone SR, Dwyer ST, Lee MY, et al (2012). Endothelial cell expression of haemoglobin alpha regulate nitric oxide signaling. Nature 491: 473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez Y, Fernandez‐Hernando C, Pober JS, Sessa WC (2007). Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res 100: 1164–1173. [DOI] [PubMed] [Google Scholar]
- Tan E, Gurjar MV, Sharma RV, Bhalla RC (1999). Estrogen receptor‐alpha gene transfer into bovine aortic endothelial cells induces eNOS gene expression and inhibits cell migration. Cardiovasc Res 43: 788–797. [DOI] [PubMed] [Google Scholar]
- Wang S, Chennupati R, Kaur H, Iring A, Wettschureck N, Offermanns S (2016). Endothelial cation channel PIEZO1 controls blood pressure by mediating flow‐induced ATP release. J Clin Invest 126: 4527–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizumi M, Perrella MA, Burnett JCJ, Lee ME (1993). Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half‐life. Circ Res 73: 205–209. [DOI] [PubMed] [Google Scholar]
- Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI et al (2006). Direct evidence for the role of caveolin‐1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest 116: 1284–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zembowicz A, Tang JL, Wu KK (1995). Transcriptional induction of endothelial nitric oxide synthase type III by lysophosphatidylcholine. J Biol Chem 270: 17006–17010. [DOI] [PubMed] [Google Scholar]