

Abstract
Inorganic nitrate (NO3 −), nitrite (NO2 −) and NO are nitrogenous species with a diverse and interconnected chemical biology. The formation of NO from nitrate and nitrite via a reductive ‘nitrate–nitrite–NO’ pathway and resulting in vasodilation is now an established complementary route to traditional NOS‐derived vasodilation. Nitrate, found in our diet and abundant in mammalian tissues and circulation, is activated via reduction to nitrite predominantly by our commensal oral microbiome. The subsequent in vivo reduction of nitrite, a stable vascular reserve of NO, is facilitated by a number of haem‐containing and molybdenum‐cofactor proteins. NO generation from nitrite is enhanced during physiological and pathological hypoxia and in disease states involving ischaemia–reperfusion injury. As such, modulation of these NO vascular repositories via exogenously supplied nitrite and nitrate has been evaluated as a therapeutic approach in a number of diseases. Ultimately, the chemical biology of nitrate and nitrite is governed by local concentrations, reaction equilibrium constants, and the generation of transient intermediates, with kinetic rate constants modulated at differing physiological pH values and oxygen tensions.
Linked Articles
This article is part of a themed section on Nitric Oxide 20 Years from the 1998 Nobel Prize. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.2/issuetoc
Abbreviations
- CBS
cystathionine β‐synthase
- cPTIO
2‐(4‐carboxyphenyl)‐4,4,5,5‐tetramethylimidazoline‐1‐oxyl‐3‐oxide
- Cygb
cytoglobin
- eNOS
endothelial NOS
- Hb
haemoglobin
- HfPEF
heart failure with preserved ejection fraction
- I/R
ischaemia–reperfusion
- mARC‐1 and mARC‐2
mitochondrial amidoxime‐reducing component–1 and 2
- Mb
myoglobin
- N2O3
dinitrogen trioxide
- Ngb
neuroglobin
- NiR
nitrite reductase
- RSNO
S‐nitrosothiol
- RBCs
red blood cells
- sGC
soluble GC
- XO
xanthine oxidase
Introduction
We are celebrating the 20th anniversary of the Nobel Prize awarded for the discovery of NO as a physiological signalling molecule with roles in neurobiology, immune defence and the regulation of cardiovascular homeostasis. Canonical de novo NO synthesis occurs via the oxidation of L‐arginine by oxygen catalysed by NOS. In the vasculature, NO is generated by the endothelial NOS (eNOS) and then diffuses to smooth muscle where high picomolar to low nanomolar concentrations activate soluble GC (sGC), ultimately triggering a signalling cascade leading to vasodilation, described elsewhere in this special issue.
Here, we focus on the chemical biology of a secondary complementary pathway leading to NO generation, the oxygen‐independent reduction of nitrate to nitrite to NO. This pathway to NO formation is reductive and NOS‐independent, with enhanced activity during conditions of physiological and pathological hypoxia or ischaemia, such as occurs during the arterial‐to‐venous deoxygenation of blood or during pathological episodes of ischaemia and reperfusion (I/R). While nitrite is readily reduced to form NO by proton and electron transfer reactions in the mammalian vasculature by haem and molybdopterin‐containing enzymes, efficient nitrate reduction requires more specialized nitrate reductase enzymes typically present in bacteria. Thus, nitrate reduction in mammals largely requires enteral symbiotic bacteria, generating nitrite, which is then further reduced to NO in the stomach or absorbed into the circulation where it is reduced to NO by haemoglobin or members of the molybdopterin family of enzymes. In the context of this review, nitrate (NO3 −) and nitrite (NO2 −) both refer to their inorganic forms: anions of various salts with alkali cations (e.g. sodium or potassium).
In vivo metabolism of nitrate and nitrite and regulation of NO‐signalling in the vasculature
Inorganic nitrite was long regarded as an inert oxidation product of NO and a biomarker of NO formation, despite some indications to the contrary (Haldane et al., 1897; Brooks, 1937). This thinking was based on the relatively low potency of nitrite as a vasodilator of preconstricted aortic rings; high concentrations of nitrite, in the micromolar to millimolar range, were required to observe significant vasodilation (Furchgott and Bhadrakom, 1953). Murad and Ignarro demonstrated that nitrite can activate GC and vasodilate aortic rings but also at relatively high concentrations or in the presence of thiols (Mittall et al., 1978; Ignarro and Gruetter, 1980; Ignarro et al., 1981). Notably, all of these experiments were conducted at physiological pH values, with high oxygen tensions and in buffered systems without red blood cells (RBCs). More recently, Modin and colleagues showed that vasodilation could occur at approximately 10 μM nitrite concentrations during acidification (Modin et al., 2001). Our group observed arterial‐to‐venous consumption of nitrite in the human circulation at rest, with exercise, and during NO gas inhalation, the latter associated with vasodilation of the human forearm suggesting that bioactivation of nitrite to a vasodilator (vide infra) might be possible in vivo. Two studies directly tested this hypothesis with one negative trial showing no vasodilation (Lauer et al., 2001) and a second positive study showing significant forearm circulation vasodilation at both pharmacological and slightly supraphysiological concentrations (Cosby et al., 2003). The latter was later confirmed by other investigators in animal and human studies (Hunter et al., 2004; Webb et al., 2004; Kozlov et al., 2005; Dejam et al., 2007; Ingram et al., 2009; Patel et al., 2011). Further studies established that NO‐dependent vasodilation by nitrite can occur in the physiological range (100–200 nM) (Dejam et al., 2005) and afforded insights into the mechanism of bioactivation including the key role of low oxygen (hypoxic) tensions, reactions with deoxyhaemoglobin and accentuation by lower pH (Cosby et al., 2003; Crawford et al., 2006; Dalsgaard et al., 2007; Maher et al., 2008; Pinder et al., 2009). The oxygen and pH effects are elegantly illustrated in exercise, in which plasma nitrite consumption is higher compared with resting values and linked to increases in local blood flow (Bailey et al., 2017).
The half‐life of NO in whole blood is calculated to be less than 2 ms (Crawford et al., 2006), whereas that of nitrite is approximately 42–52 min (Dejam et al., 2007; Pluta et al., 2011; Rix et al., 2015). As reductive mechanisms discussed here lead to NO generation, nitrite is considered a ‘stable’ reserve of NO. Nitrite thus signals via a number of mechanisms (Figure 1), including the canonical NO–sGC pathway, by activating sGC via iron nitrosylation and stimulating vasodilation (Jeffers et al., 2005). NO/nitrite are also involved in generation of nitrosating agents such as dinitrogen trioxide (N2O3), leading to nitrosation of thiols (vide infra) such as those in complex I of the mitochondrial electron transport chain. This action is cytoprotective, as nitrosation of these critical thiols occurs during I/R injury resulting in inhibition of complex I activity, preventing generation of damaging ROS as well as cytochrome c export following reoxygenation (Shiva et al., 2007; Chouchani et al., 2013). Additionally, nitrite oxidation (vide infra) to the radical nitrogen dioxide (NO2 •), an oxidant, results in nitration of several species, including unsaturated fatty acids, generating signalling nitro‐fatty acids (Villacorta et al., 2016). All of these topics (the canonical NO pathway, NO in the mitochondria and nitro‐fatty acids) are described at length in separate reviews within this special issue.
Figure 1.
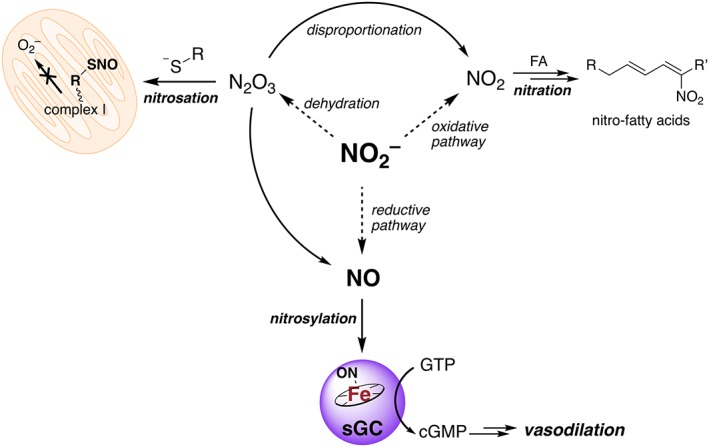
Myriad chemistries of nitrite (NO2 −) leading to protective and signalling effects. Nitrite can be reduced to NO (mechanisms discussed below), which nitrosylates a ferrous haem in sGC, triggering the production of cGMP from GTP. cGMP triggers a signal cascade resulting in vasodilation. Alternatively, nitrate can be oxidized (conditions described below) to yield the oxidant NO2 •. NO2 • reacts with a number of targets, but under conditions of excess NO2 •, can participate in nitration chemistry of fatty acids (FAs). The generated nitro‐fatty acids are electrophilic and modify critical redox active thiols to mediate signalling, for example, by activating transcription factors like Nrf‐Keap1. Nitrite can also be dehydrated catalytically or in the presence of acid (vide infra) to generate the strong nitrosating agent N2O3. N2O3 can react with nucleophiles such as thiols resulting in S‐nitrosothiol (RSNO) formation. Here, this activity is depicted on a critical thiol of complex I of the mitochondrial electron transport chain, preventing formation of damaging superoxide (especially under I/R injury). N2O3 can also disproportionate into NO2 • and NO. Dotted arrows represent reactions that are also catalysed.
Nitrate is ubiquitous in leafy green vegetables and beets, while nitrite is not typically found at significant levels in natural foods, although is added in the meat curing process as a preservative (Khatri et al., 2017). Therefore, humans can ingest high levels of nitrate, especially when consuming vegetable‐rich diets. The source of intravascular nitrite in vivo emanates from oxidation of eNOS‐derived NO and from reduction of the otherwise stable dietary nitrate, as it has a plasma half‐life of 5–6 h (Lundberg et al., 2008). Although human nitrate reductase activity has been posited as a secondary function of xanthine oxidase (XO, vide infra), especially for organic nitrates (R–ONO2) (Millar et al., 1998; Li et al., 2005; Khambata et al., 2015), inorganic nitrate reduction is largely attributed to our oral microbiome (Lundberg et al., 2008). When ingested, nitrate is reduced to nitrite by facultative anaerobes inhabiting the salivary glands via nitrate reductase enzymes akin to those found in soil bacteria. Further, nitrate is actively taken up from circulation by the salivary glands, resulting in nitrate concentrations 10‐fold higher in the oral cavity than in the plasma (Ahluwalia et al., 2016). Depletion of oral microbes attenuates nitrite formation from nitrate and abolishes the vasodilatory response of nitrate, implicating nitrite as the central active agent (Webb et al., 2008b). When swallowed, some nitrite is subsequently reduced to NO in the stomach by a non‐enzymic, low pH‐dependent mechanism where it modulates gastric fluid production and confers defence against infections (Lundberg et al., 2011). The rest of the nitrite is absorbed via the gastrointestinal (GI) tract and becomes part of the ‘nitrate‐nitrite‐nitric oxide pathway’ responsible for vasodilatory control under low oxygen conditions. It is important to note that other, yet‐to‐be elucidated mechanisms are likely to exist in the context of this pathway as the proton pump inhibitor esomeprazole, which lowers gastric acidity, blunts the effects of only orally ingested nitrite (Montenegro et al., 2017). Such findings imply a gastric activation step in swallowed nitrite, though probably not directly to short‐lived NO by a non‐enzymic path. Additionally, a recent clinical trial reported that the vasodilatory effects of oral nitrite were also blunted during co‐administration of conjugated linoleic acid (Hughan et al., 2017). As mentioned above, nitrite potently vasodilates during direct intra‐arterial infusion, so more work is required to understand the mechanisms for how gastric reactions can modulate its bioactivity.
A few investigators have reported that mammals reduce nitrate to nitrite through direct action of native XO (Li et al., 2003; Jansson et al., 2008; Piknova et al., 2015, 2016). In 2003, Li et al. characterized the nitrate reductase activity of XO using electron paramagnetic resonance (EPR), chemiluminescence and an NO electrode. The reduction was found to be acid‐catalysed. Jansson et al. (2008) observed nitrate reductase activity of rodent and human tissues that was dramatically blunted by the XO inhibitor, allopurinol. In addition, plasma nitrite was increased in vivo after nitrate infusions in rodents and, when allopurinol was also administered, these nitrite increases were blunted. Importantly, increases in plasma nitrite were observed in germ‐free mice as well, supporting the existence of a eukaryotic nitrate reductase (Jansson et al., 2008). Interestingly, the Schechter group has observed that nitrate is present in about threefold higher concentrations in skeletal muscle than in blood and is reduced to nitrite in the muscle (Piknova et al., 2015, 2016). Thus, there exists evidence of a mammalian nitrate reductase. Such mammalian nitrate reductase activity may be important in certain tissues and conditions such as ischaemia.
While this body of research suggests that the reduction of nitrate may occur by mammalian enzymes, placebo‐controlled crossover studies in normal volunteers, examining the effects of the mouthwash chlorhexidine to eliminate commensal oral bacteria, have reported that salivary, plasma and urinary nitrite levels were lowered and BP was increased (Kapil et al., 2013), supporting a major role for bacterial nitrate reduction occurring in the oral cavity. Several mouse and rat studies have also shown that effects of nitrate can be blocked in germ‐free mice or with oral antiseptic treatment (reviewed in Koch et al., 2017). Thus, the weight of evidence suggests that the major pathway for nitrate reduction is through the oral microbiome, with lesser contribution from mammalian tissue xanthine oxidoreductase enzyme systems.
The physiological role for the nitrate–nitrite–NO pathway in the regulation of blood flow has been supported by the elegant studies by the Ahluwalia group (Kapil et al., 2010, 2013). As noted, antiseptic mouthwash use results in increased BP, accentuating the importance of our oral microbiome for the reduction of nitrate. The corollary is also true: supplementing with exogenous nitrate increases plasma nitrite levels, decreases BP and improves exercise performance (Larsen et al., 2006; Kapil et al., 2014; Mills et al., 2017). Thus, the chemical biology implicates a delicate interplay featuring many nitrogen oxidation states. This ‘nitrate–nitrite–NO pathway’ exists complementary to the traditional NOS‐derived NO‐signalling mechanism and postulates a large nitrate pool as a stable store of nitrite and nitrite, providing a stable vascular reserve of ephemeral NO.
Chemistry of NO
No discussion about nitrite or nitrate is possible without briefly remarking on the chemistry of NO, as it is the recognized bioactive downstream product of both salts. Critically, NO is a stable free radical in aqueous/physiological conditions and does not dimerize due to the delocalization of the unpaired electron over both the nitrogen and oxygen atoms. Thus, NO can only form a partial ON–NO bond (Fukuto et al., 2012). The exhibited stability of NO is paramount to the molecule's unique biology as interactions between diamagnetic (most biological substrates) and paramagnetic molecules (like NO) react very slowly. Simply put, NO diffuses freely until it encounters other paramagnetic molecules such as free radicals like superoxide (O2 −) and transition metals (Heinrich et al., 2013). The very high reaction rates and affinities for these molecules allow for specific signalling at very low NO concentrations. Finally, it is important to note that the reaction of aqueous NO with paramagnetic oxygen (i.e. autoxidation of NO) is a kinetically slow trimolecular reaction, which is second order in NO (Equation (1), 4k aq = ~8 × 106 M−2·s−1) (Ford et al., 1993). An interested reader in the relevant chemistry of NO should consult reviews by Fukuto et al. (2012; Heinrich et al., 2013) and Ford (2010) as well as other articles in this issue.
| (1) |
In vivo nitrate generation
The amounts of excreted nitrate greatly exceed that of what is typically ingested (Khatri et al., 2017), and levels found in the plasma are between 20 and 40 μM under fasting conditions (Khambata et al., 2015), implicating endogenous production. Nitrate is rapidly generated from the NO reaction with oxygenated‐haemoproteins in a process called NO‐dioxygenation (k 2 = 9 × 107 M−1·s−1, pH 7, 25°C) (Doyle and Hoekstra, 1981). Effectively, the oxy‐ferrous (Fe2+–O2) haem serves as an electron donor and catalyses the oxidation of NO (Equation (2)).
| (2) |
While protein environment affects the rate of NO‐dioxygenation, this reaction is nearly diffusion limited in most haemoproteins (Mishra and Meuwly, 2010). NO‐dioxygenation is viewed as a crucial detoxifying reaction to scrub cardiovascular tissue of excess toxic NO. The very short (>2 ms) intravascular half‐life of NO is due predominantly to these reactions, but the simultaneously generated nitrate functions as a stable nitrite source, which is then further harnessed for NO‐generation as discussed below. Another biological route to nitrate formation will also be considered later.
Nitrite bioactivation
Inorganic nitrite is the conjugate base of nitrous acid, which has a much higher pK a [3.11 at 37°C (da Silva et al., 2006)] than nitric acid (conjugate acid of nitrate, pK a = −1.3), consistent with nitrite protonation under certain physiological circumstances such as in the low pH of the stomach and, thus, increased lipid solubility. Nitrous acid generates NO via dehydration to N2O3, a strong nitrosating agent, followed by disproportionation (Equations (3) and (4)) (Fukuto et al., 2012). The standard reduction potential (E 0) at low pH is quite favourable and nitrite is a strong oxidant, where HNO2, H+/NO, E 0 = 0.98 V versus NHE (Bratsch, 1989), but this drops to 0.37 V at pH 7 (Ford, 2010). Thus, NO production from nitrite in this manner requires both relatively high concentrations of nitrite and acid. A few biological conditions support this possibility, including, critically, ischaemic events where reduced blood flow leads to tissue acidosis (Feelisch et al., 2008).
| (3) |
| (4) |
The reduction of nitrite to NO is catalysed at less acidic pH values by certain metalloproteins that provide an electron and facilitate proton donation as shown in Equation (5), where M is a redox‐active metal (e.g. iron) in the n oxidation state (Doyle et al., 1981b; Huang et al., 2005b; Li et al., 2008; Webb et al., 2008a).
| (5) |
The most prominent reductases contain porphyrin‐chelated iron‐cofactors known as haem. Haem readily cycles from the reduced ferrous (Fe2+) valance to the oxidized ferric (Fe3+) state and vice versa. Various haems are found in mammalian systems and are used in transporting and biosignalling processes, most notably as the oxygen‐binding component of haemoglobin (Hb) and myoglobin (Mb). As noted previously, at low partial pressures of oxygen (i.e. hypoxia), ferrous globins exhibit nitrite reductase (NiR) activity, binding and reducing nitrite while generating the met‐globin (ferric) and an equivalent of NO (with Hb, Equation (6)) (Lundberg et al., 2008).
| (6) |
An inner‐sphere electron transfer mechanism of nitrite reduction is supported by the work of Doyle and co‐workers (i.e. nitrite binding before electron transfer), where oxidation of ferrous deoxygenated‐Mb (deoxyMb) by alkyl nitrites, R–ONO, trigger facile homolytic cleavage of the RO–NO bond, resulting in the Mb–OR species and liberating NO (Doyle et al., 1981a, 1984). This chemical activation of nitrites by haemoproteins has been studied in great detail, and the binding mode appears to be relevant to the kinetics of NiR activity. Nitrite has relatively high nucleophilicity and can bind metal centres in different fashions, but only η 1‐coordination (i.e. one atom bound) is observed with haem (Ford, 2010). However, the manner of binding has been a subject of debate. Most ferric haem proteins and models favour N‐bound ‘nitro’, though the O‐bound ‘nitrito’ form is stabilized in native metmyoglobin and methaemoglobin, attributed to hydrogen‐bonding of the distal histidine, though the linkage isomers are close in energy (Yi et al., 2008). Mutation of the distal histidine H64 in metmyoglobin to a hydrophobic valine residue triggers linkage isomerization to the N‐nitro species, while also significantly slowing NiR activity by a factor of 16 (Yi et al., 2009). Nitrite reduction depends on protonation (Equation (5)), and the histidine delivers the requisite proton(s) via a hydrogen‐bonding network of water molecules, analogous to bacterial NiRs (Cutruzzolà et al., 2001; Perissinotti et al., 2008). Unlike bacterial reductases, mammalian globins have only one distal histidine and the ferrohaems are also thought to favour O‐nitrito coordination. O‐bound nitrite to ferrous globins allows for a single protonation step and facile loss of NO with concomitant formation of the ferric‐hydroxo complex (Figure 2), stabilized by the hydrogen‐bonding of the distal histidine to the metal‐bound oxygen of nitrite (Silaghi‐Dumitrescu, 2004). A crystallographic analysis claiming a solid state ferrous Mb further indicates an O‐nitrito configuration (Copeland et al., 2006), though disagreements still exist on the bonding mode as a computational study exploring NiR mechanisms favours N‐nitro coordination in ferrous Hb (deoxyHb) (Perissinotti et al., 2008). Regardless, while the ferrohaem may not follow the same binding scheme as the ferrihaem, in either case the distal histidine and its protic environment is paramount as its absence in H64V Mb appears to interrupt the stepwise proton transfer(s), resulting in decreased NiR activity (Yi et al., 2009). Finally, it is important to note that perturbation of the metal to other redox‐active 3d‐block metals cobalt or manganese (in Mb) greatly reduces the NiR activity. Iron is essential to nitrite reduction (Heinecke et al., 2012).
Figure 2.

NiR activity of haemoproteins in O‐nitrito nitrite coordination (i.e. Mb and Hb). Addition of a proton either directly from the histidine (shown) or water yields the nitrous acid bound species. After protonation and subsequent inner‐sphere electron transfer from the ferrohaem to the bound nitrous acid, NO dissociates leaving the hydroxo‐ferrihaem complex which can then be protonated to the aquomet form.
NiR activity of Hb
Physiological nitrite gradients from artery‐to‐vein (arterial, 176 ± 10 nM and venous, 143 ± 7 nM) (Dejam et al., 2005) are temporally associated with the formation of NO in the red cell (measured as an increase in venous iron‐nitrosyl‐Hb). Moreover, erythrocytic ferrous nitrosyl‐Hb is formed during intra‐arterial infusion of nitrite in the arm (Cosby et al., 2003). In this case, nitrite is reduced to NO by deoxyhaemoglobin, which also rapidly binds NO forming ferrous nitrosyl‐Hb [Equation (7), K NO T‐state = 9 × 109M−1, K NO R‐state = 1 × 1011 M−1 (Moore and Gibson, 1976); k NO T,R = 2.6 × 107 M−1·s−1 at 20°C, pH 7 (Cassoly and Gibson, 1975)]. These observations strongly support the model that Hb is the principal metalloprotein involved in reducing nitrite to NO in the vasculature.
| (7) |
Brooks initially characterized the reaction of deoxyhaemoglobin with nitrite under anaerobic conditions (Brooks, 1937). In the proposed mechanism (Equations (6) and (7)), equimolar amounts of methaemoglobin and ferrous nitrosyl‐Hb are produced and have been subsequently observed for Mb (Huang et al., 2005b). If the process is governed by a single reaction rate constant, this reaction should be a simple second‐order process, determined by the concentration of nitrite and deoxy‐haem. However, while the reaction of deoxyhaemoglobin with excess nitrite under stringently anaerobic conditions has confirmed Brooks' stoichiometry, the simultaneous formation kinetics of both nitrosylated and ferric Hb exhibit a sigmoidal shape rather than an exponential form, as observed with Mb (Figure 3), implying the reaction rate speeds up and slows down again (Huang et al., 2005b; Grubina et al., 2007). We discovered that this deviation from a simple second‐order reaction occurs because the reaction rates of nitrite with Hb are modulated by the allosteric conformation of the tetramer. The deoxygenated tense state or T‐state of Hb reacts much more slowly with nitrite than the relaxed or R‐state of Hb. Generally, the R‐state of Hb is stabilized when at least two of the four subunits of the Hb tetramer are either ferric or bound to a ligand such as O2, CO or NO due to changes in the proximal histidine. Thus, during anaerobic nitrite reduction (a case where there is no oxygen to determine R‐ and T‐state), the reaction rate increases as the R‐state is stabilized by the formation of nitrosyl‐Hb and ferric Hb, even though active ferrohaem centres are depleted by their generation. Given that each nitrite yields an oxidized haem and equivalent of NO, for every nitrite that is consumed, two Hb molecules can transition from the T‐ to the R‐state. This process is known as allosteric autocatalysis. The R‐state deoxyhaemoglobin that forms during the anaerobic reaction reduces nitrite faster as the T‐state is depleted (Huang et al., 2005a). The rate nonetheless slows again as more of the vacant R‐state haems are occupied by NO or converted to ferrihaem. The bimolecular rate constants of nitrite reduction for the T‐ and R‐states are 0.2 and 12 M−1·s−1 respectively (Huang et al., 2005b; Gladwin and Kim‐Shapiro, 2008). The observed rate of the reaction is given by Equation (8), where [R] and [T] represent the concentrations of the R‐ and T‐state available haems respectively.
| (8) |
Figure 3.
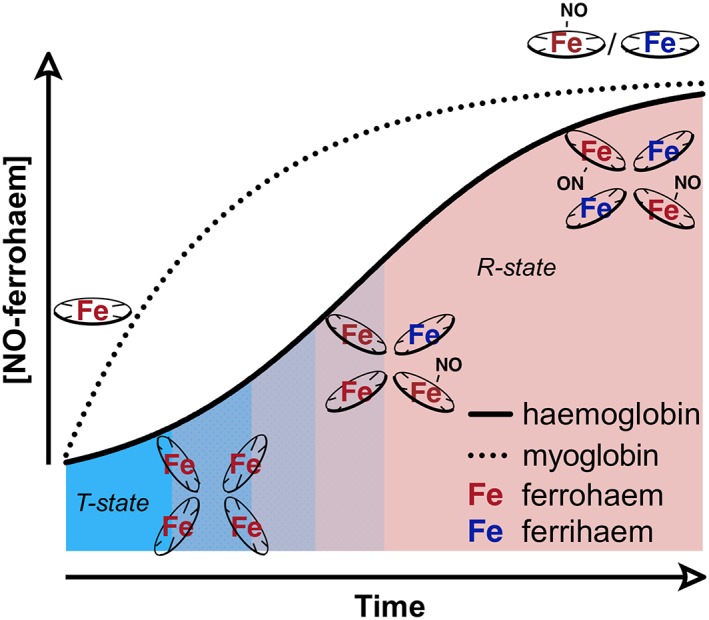
Simulated data representing the NiR activity of deoxymyoglobin and deoxyhaemoglobin under anoxic conditions, specifically tracing the formation of nitrosylated ferrohaem (Equation (6)) over time. Mb, which has a constant k NiR, behaves in an exponential manner. Hb exhibits autocatalysis and a sigmoidal shape: tetrameric T‐state (blue zone) deoxyHbII reduces nitrite, generating a methaem (3+) and an equivalent of NO. The NO binds a vacant ferrohaem on the same or different tetramer to form a nitrosylated ferrohaem species. Each of these new species stabilizes the R‐state (red zone), which has a higher bimolecular rate constant than the T‐state, resulting in an increase in apparent rate and propagating nitrite reduction. As the vacant reactive ferrohaem sites are filled, the rate of nitrosylated‐ferrohaem formation drops off.
While the autocatalytic reaction is observed in vitro and caused by NO binding to Hb, the regulation of R‐ and T‐states in vivo are determined by oxygen concentrations and Hb oxygen affinity. Therefore, the fastest rate of nitrite reduction to NO paradoxically occurs when the most deoxyhaems are available on the R‐state tetramer: the observed k NiR' is highest in arterial blood, but vacant deoxyhaem sites necessary for NiR activity are maximized in venous blood. This optimized nitrite reduction scenario arises when oxygen is unloaded from R‐state Hb in the circulation at about 40–60% oxygen saturation (around the P 50) and naturally occurs at the point of arterial‐to‐capillary transit (Gladwin et al., 2009). Further, physiological studies show the onset of hypoxic vasodilation to occur at these oxygen tensions (Ross et al., 1962). Thus, the Hb maximal reductase activity occurring at the natural P 50 is also consistent with a role in hypoxic vasodilation. At lower oxygen tensions, oxygen‐dependent NOS enzymes function less effectively, consistent with the hypothesis of nitrite serving as a complementary NO‐generating pathway. It should be noted that this chemistry is somewhat complicated by nitrite interaction with oxyhaemoglobin, discussed in detail below.
The disparity of bimolecular rate constants between the T‐ and R‐states has been attributed to a difference in redox potential of the haem between the states (Huang et al., 2005b). R‐state Hb has a lower redox potential than T‐state Hb (E 1/2 vs. NHE: HbAR = 42 mV, HbAT = 154 mV), meaning the R‐state is more readily oxidizable (Bonaventura et al., 2013). Canonically, as an exogenous ligand (e.g. oxygen) binds T‐state ferrohaem, the pentacoordinate high spin iron (II) (which is displaced from the porphyrin ring), is rendered low spin, decreasing the iron radius, pulling it flush with the porphyrin ring and shifting the proximal histidine and protein backbone. This ‘proximal pull’ is the basis of the T‐to‐R‐state transition and is responsible for the decrease in redox potential (Bonaventura et al., 2013). Thus, the lower redox potential R‐state increases the probability of nitrite reduction. Furthermore, this R‐state potential is analogous to that of Mb (Taboy et al., 2002). Sickle cell Hb (HbS) has a higher NiR activity than healthy adult Hb (HbA), correlating to its more favourable potential than HbA (Grubina et al., 2008). It is important to note that redox potential is not solely responsible for the effect. A study comparing deoxyhaemoglobin and other modified haemoglobins indicated faster nitrite reduction even as the redox potential increased (Bonaventura et al., 2008). Bonaventura et al. (2013) ascribe the shift in the kinetics of the reaction to ligand affinity changes driven by the steric and electronic factors of the haem active site, as redox potential is responsible solely for the thermodynamic driving potential. But, as mentioned above, the T‐to‐R state transition triggers physical rearrangement as well as a concomitant decrease in redox potential in HbA. These changes together can be responsible for the increased k NiR, as the more open R‐state pocket is a better NiR catalyst (i.e. lowers the activation energy of the reaction) and binds nitrite more readily.
The importance of a favourable proton‐donating environment and a consequence of the tetrameric nature of Hb is illustrated by the proton dependence of nitrite reduction from pH 6.0 to 8.0 (Doyle et al., 1981b). As expected, nitrous acid formation in the distal pocket is implicated as the catalysed species leading to NO formation. However, the authors observed rate order [H+] dependence of 0.88 instead of an ideal first‐order dependence, deviating from Equation (6). They noted this may result from conformation changes in Hb, and the unexpected sub‐unity proton dependence has indeed since been ascribed to the redox Bohr effect (Huang et al., 2005b). Allosteric proton binding results in a more stabilized T‐state and consequently a diminished haem redox potential, decreasing NiR activity. As a nitrite protonation step is essential for reduction, the smaller contribution from the Bohr effect works in the opposite direction, resulting in a less than first‐order proton dependence on the rate, whereas a close agreement to a slope of 1.00 has been observed for all other haemoproteins apart from Hb (Gladwin et al., 2009).
The ultimate practicality of NiR activity of Hb in RBCs depends on two essential matters. First, the basal levels of nitrite must be sufficient to be physiologically relevant to hypoxic vasodilation. Nitrite is found in plasma (120 nM) but is pooled in even greater amounts in the erythrocytes themselves (300 nM) (Dejam et al., 2005). As such, Hb is proposed as a dominant NiRs in blood, responsible for hypoxic vasodilation (Cosby et al., 2003; Huang et al., 2005b; Crawford et al., 2006), though it does not act alone. Several metalloenzymes in the vasculature have been evaluated for nitrite reduction, notably eNOS and XO (Webb et al., 2008a, vide infra). However, these do not appear to generate NO at physiological pH and oxygen tensions. In mice, Mb contributes to nitrite reduction at low oxygen, with the Mb‐knockout mice showing reduced nitrite‐mediated hypoxic vasodilation and cardioprotection (Hendgen‐Cotta et al., 2008).
The second matter is the question of how NO escapes from the RBC. NO generated in a partially oxygenated environment (where NiR activity is maximized) would be expected to be consumed by the rapid dioxygenation activity of oxyhaemoglobin (Equation (2)) or captured by the ferrous deoxyhaemoglobin (Equation (7)). And yet, even slightly higher than physiological amounts of exogenously supplemented nitrite (2.5 μM) stimulate vasorelaxation, implying that the NO must be escaping from the RBC and stimulating sGC in the smooth muscle (Cosby et al., 2003). Moreover, NO generation has been confirmed directly by experiments with partially deoxygenated RBCs and nitrite (Huang et al., 2005b; Crawford et al., 2006; Shiva et al., 2011; Liu et al., 2015), as well as with aortic ring dilation experiments (Cosby et al., 2003; Isbell et al., 2007). In addition, experiments involving inhibition of platelet activation [a known effect of NO (Radomski et al., 1987; Loscalzo, 2001)] strongly support the notion that nitrite can export NO activity. Physiologically, relevant concentrations of nitrite inhibit platelet activation in the presence – but not absence – of RBCs, and this activity is inhibited by NO scavengers (Srihirun et al., 2012; Wajih et al., 2016, 2017). Numerous mechanisms have been proposed: nitrite reduction being limited to RBC membrane compartments, production of another species such as a nitrosothiol (RSNO) or N2O3 formation (Fernandez and Ford, 2003; Basu et al., 2007; Wajih et al., 2016).
Ferrihaem‐nitrite disproportionation
Erythrocytic Hb consumes NO via Equations (2) and (7), yet NO is exported from RBCs, suggesting a relatively stable intermediate NOx species is formed. One proposed species is N2O3, generated by rapid reaction of radical NO2 • with NO. N2O3 possesses NO+NO2 − character and thus in the presence of a nucleophile (e.g. a thiolate, Equation (9)) results in nitrosation (Heinrich et al., 2013) and reforms nitrite. Under aqueous conditions, N2O3 hydrolyzes to two equivalents of nitrous acid (Grätzel et al., 1970) but may persist in hydrophobic pockets (e.g. the RBC membrane); N2O3 is small and uncharged, facilitating diffusion, and exhibits homolytic scission (regenerating NO). Thus, N2O3 formation is an attractive supposition for NO escaping from the RBC.
| (9) |
The manner in which N2O3 is generated is disputed. It can result from autoxidation of NO, transiently generating NO2 • (Equation (10), back reaction of Equation (4)). Though as discussed previously, the second‐order dependence on NO (Equation (1)) implies N2O3 formation from NO autoxidation is too slow to be significant physiologically, although it has been postulated to occur in hydrophobic environments where NO and oxygen levels will be significantly higher (Liu et al., 1998; Vrancken et al., 2016) and conditions of nitrosative stress (Thomas et al., 2008).
| (10) |
The oxidation of nitrite to NO2 • is known to occur via metal catalysis (Thomas et al., 2008). Specifically regarding RBCs, an EPR‐silent NO‐modified Hb intermediate was initially hypothesized (Nagababu et al., 2003) but was later determined to be nitrite‐bound methaemoglobin (Basu et al., 2007). Consistent with nitrite binding to methaems, the O‐nitrito‐bound nitrite (vide supra) favours electron delocalization imparting more iron (II) character (Basu et al., 2007). The newly formed Fe2+‐bound NO2 • has significant enough radical character to react with even low amounts of NO, yielding N2O3 (Figure 4, path a). Even though nitrite affinity for ferrihaem is 1–2 orders of magnitude lower than NO, experiments on the complex in a glass matrix indicate that NO entering the distal pocket does not displace the nitrite. In fact, a ferrohaem product is formed, consistent with the formation of N2O3 (Navati and Friedman, 2009). Pertinent to hypoxic NO generation from nitrite, this N2O3 liberation by Hb represents ‘ferrihaem‐nitrite disproportionation’ or a ‘nitrite anhydrase’ mechanism.
Figure 4.
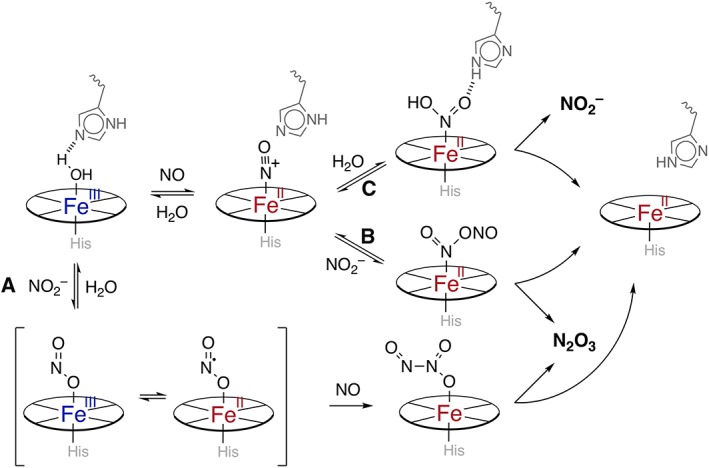
Various mechanisms of non‐enzymically induced redox cycling ferrihaem (blue‐centres) to ferrohaem (red‐centres). The top path represents reductive nitrosylation, where NO displaces water from the aquomet‐globin generating a nitrosyl‐ferrihaem/nitrosonium‐ferrohaem (shown). A nucleophile such as hydroxide/water (path c) generates nitrite, whereas when nitrite is the nucleophile (path b), N2O3 is generated. Importantly, other nucleophiles such as thiolates can participate in this chemistry. Similarly, the nitrite anhydrase mechanism (path a) represents a situation where nitrite binds the methaem first. The subsequent nitrito‐ferrihaem complex (here depicted as O‐nitrito, but the N‐nitro is possible) exhibits some radical NO2 •‐ferrohaem character, which can readily react with NO to generate diffusible N2O3.
An alternate pathway advanced by Fernandez and Ford involves nitrite acting as a general base (Fernandez and Ford, 2003). The inner‐sphere mechanism suggests nitrite directly attacks the nitrosylated‐ferrihaem species yielding the Fe2+–N2O3 as seen in Figure 4, path b (Fernandez et al., 2004; Ford, 2010). This mechanism was favoured in part by the authors due to the relatively high nucleophilicity of nitrite. They could not preclude the possibility of an outer‐sphere mechanism involving nitrite oxidation by nitrosylated‐ferrihaem and subsequent reaction of the generated NO2 • radical with the ferrous‐nitrosyl species. Unlike ‘ferrihaem‐nitrite disproportionation’, this mechanism is effectively reductive nitrosylation (Figure 4, path c, vide infra) where the nucleophilic hydroxide/water is replaced by nitrite. Both mechanisms have been shown to be energetically feasible computationally, and both lead to the same products while redox cycling the globin (Hopmann et al., 2011). Either mechanism would be possible under the appropriate conditions. Thus, Hb nitrite anhydrase activity to yield N2O3 feasibly explains erythrocytic NO export as well as formation of S‐nitrosothiols (Equation (9)), important signalling agents themselves (Nagababu et al., 2003; Basu et al., 2007; Thomas et al., 2008).
The nitrite oxyhaemoglobin reaction
Nitrite also reacts with oxyhaemoglobin generating nitrate and methaemoglobin. The kinetics follow a complex reaction profile. In excess nitrite, a slower lag or initiation phase is observed that then accelerates to a rapid autocatalytic propagation phase (Kim‐Shapiro et al., 2005). Though there are various proposed mechanisms, experiential evidence suggest the rate‐limiting initiation reaction generates hydrogen peroxide, methaem and nitrate with an estimated bimolecular rate constant between 0.2 and 0.4 M−1·s−1 (Equation (11)) (Keszler et al., 2008).
| (11) |
The methaemoglobin and peroxide then form a ferryl (FeIV=O)–Hb radical, propagating NO2 formation from the reaction with additional nitrite (Equation (12)). During this autocatalytic propagative phase, NO2 • reacts with additional oxyhaem to regenerate ferrylhaem‐radical and more nitrate. The proposed elementary steps are shown in their entirety in Keszler et al. (2008).
| (12) |
Grubina et al. (2007) also observed at varying oxygen tensions both the deoxy‐nitrite reduction and this oxy‐reaction occurring simultaneously, but the former (Equation (6)) quenched the oxy‐reaction intermediates. One possibility is the interaction of NO with NO2 • (the back reaction of Equation (4)) generating N2O3. Another possibility provides a supporting mechanism of NO escape from RBCs. In this scenario, the stable ferrous nitrosyl‐Hb is oxidized under the propagation conditions by NO2 •, generating a far more labile ferric nitrosyl‐Hb. This process is called ‘oxidative denitrosylation’, simultaneously liberating NO and limiting the oxy‐pathway by consuming NO2 • intermediates, promoting the reductive nitrite pathway to NO.
Physiological nitrite generation
Nitrite is clearly an important NO source under hypoxic conditions, but where does it originate? As our dietary intake of nitrite is limited, most of our nitrite is generated from our microbiome as discussed in the context of the nitrate–nitrite–NO pathway above and survives passage through the stomach and GI tract into circulation. However, other processes exist which yield biological nitrite and maintain nitrite homeostasis. In the absence of ferrous haemoproteins, nitrite is the chief oxidation product of NO in aqueous solutions with oxygen (Ignarro et al., 1993), but this is kinetically limited. Most ferric haemoproteins can undergo ‘reductive nitrosylation’ or ‘autoreduction’ when exposed to excess NO (Figure 4, path c), generating nitrite. Reductive nitrosylation first involves production of a nitrosyl‐ferrihaem. The NO‐moiety is electrophilic as the complex has nitrosonium‐ferrohaem character and is subject to nucleophilic attack by water/hydroxide, determined after studies on rate dependence on pH and NO concentration (Hoshino et al., 1996). Reductive nitrosylation depends on the concentration of OH− and is considered the opposite of NiR activity where the dependence of the redox reaction is on the concentration of the proton. Importantly, unlike Mb, Hb undergoes reductive nitrosylation at low pH values (~6), vital for broader physiological relevance. Additionally, other nucleophiles can react with ferric nitrosylglobins to generate nitrosated species [e.g. S‐nitrosoglutathione (Reichenbach et al., 2001)].
Physiological nitrite is also generated by oxidation of NO by ceruloplasmin, a protein that is present in micromolar concentrations in human plasma (Shiva et al., 2006). This hexacopper oxidase catalyses the oxidation of NO to nitrite via reduction from Cu2+ to Cu1+ and is also implicated in RSNO formation as the resulting nitrosonium cation is incredibly electrophilic and reactive. The principal nucleophile in plasma is water yielding nitrite, competing with NO‐dioxygenation reaction of globins. After myocardial infarction, ceruloplasmin induction has been observed (Singh, 1992), and mice where the protein is knocked out sustain greater I/R injury than controls (Shiva et al., 2006). Such observations are consistent with the paradigm that nitrite improves outcomes of hypoxic insults, and thus, it has been posited that ceruloplasmin modulates response to ischaemia by generating nitrite. Vrancken et al. evaluated species differences in ceruloplasmin levels and activity and found correlations between ceruloplasmin levels and plasma RSNO during NO donor exposure (Vrancken et al., 2013). While they did not see differences in nitrite levels, the oxidation of NO to NO+ would be expected to form both RSNO and nitrite, so the results of this study remain equivocal.
Other haem‐based mammalian NiRs
There are numerous other mammalian enzymes that reduce nitrite under appropriate conditions. Other haem enzymes produce NO from nitrite including the canonical NO‐generating eNOS, but under anoxic conditions (Vanin et al., 2007). eNOS may play a role in the regulation of NiR activity in RBCs during ischaemic events as the enzyme is localized within the RBC membrane, thus NO generation is more likely to diffuse away from the cell instead of undergoing autocapture (Webb et al., 2008a). Recently discovered globins such as neuroglobin (Ngb) (Petersen et al., 2008; Tiso et al., 2011) and cytoglobin (Cygb) (Li et al., 2012; Corti et al., 2016) exhibit NiR activity under physiologically relevant conditions, though their respective primary functions are unknown. As with the oxygen‐carrying globins, these haem proteins follow Equation (5) for nitrite reduction, where the reductant is the ferrous protein. However, significant differences exist.
Unlike Hb and Mb, the distal histidine in Ngb and Cygb is bound to the iron, which means these proteins do not have an immediately available coordination site for nitrite to bind. However, unlike hexacoordinate cytochromes, the distal histidine is labile and thus interconverts from hexacoordinate to pentacoordinate rapidly. The interconversion is rate limiting: the NiR activity of Ngb increases significantly upon mutation of the distal histidine to a non‐hydrogen bonding, non‐coordinating amino acid as reflected by a ~2200‐fold increase in rate constant (k NiR, Table 1) from the wild‐type to H64L (Tiso et al., 2011). The difference stands in stark contrast to that of Mb, the rate constant of which decreases upon mutation to H64V due to the loss of hydrogen‐bonding environment (vide supra). Mb already contains an available coordination site, whereas in Ngb, removal of the coordinated distal histidine provides a binding site for nitrite, resulting in a large increase in NiR activity upon mutation. Further, the rate‐limiting dissociation of the distal histidine confirms an inner‐sphere nitrite reduction. Although these studies suggest that Ngb has redox‐regulated NiR activity, also recently demonstrated for Cygb, more work is needed to evaluate the physiological roles the six‐coordinate globins may play, as functional NiRs and NO signalling molecules.
Table 1.
Kinetic parameters of NO production from nitrite reduction by representative haemoproteins (top) and molybdenum–molybdopterin containing proteins (bottom).
| k NiR (M−1·s−1) | T (°C) | Reference | |
|---|---|---|---|
| HbT | 0.12 | 25 | Huang et al. (2005b) |
| 0.2 | 37 | Gladwin and Kim‐Shapiro (2008) | |
| HbR | 6 | 25 | Huang et al. (2005b) |
| 12 | 37 | Gladwin and Kim‐Shapiro (2008) | |
| Mb | 5.5 | 25 | Yi et al. (2009) |
| 12 | 37 | Shiva et al. (2007) | |
| Mb (H64V) | 0.35 | 25 | Yi et al. (2009) |
| NgbS–S | 0.12 | 25 | Tiso et al. (2011) |
| 0.26 | 37 | Tiso et al. (2011) | |
| NgbS–H | 0.062 | 25 | Tiso et al. (2011) |
| Ngb (H64L) | 259 | 25 | Tiso et al. (2011) |
| CygbS–S (monomer) | 32.3 | 25 | Reeder and Ukeri (2018) |
| CygbS–H | 0.63 | 25 | Reeder and Ukeri (2018) |
| CygbS–S (dimer) | 0.26 | 25 | Reeder and Ukeri (2018) |
| CBS | 0.66 | 37 | Carballal et al. (2016) |
| Cytochrome c (horse) | 0.07 | 25 | Li et al. (2012) |
| Kinetic parameters | ||||||
|---|---|---|---|---|---|---|
| Type, substrate | k cat (s−1) | K m (nitrite, mM) | Rate of NO formation: V max (nmol·s−1·mg−1) | T (°C) | Reference | |
| XO | Bovine, aldehyde | 0.693 | 0.585 | – | 25 | Maia and Moura (2011) |
| Bovine, NADHa | – | 22.9 | 62 | 37 | Millar et al. (1998) | |
| Bovine, NADH | 0.28 | 2.25 | 0.92 | 37 | Li et al. (2001) | |
| AO | Rat, aldehyde | 1.89 | 9.7 | – | 25 | Maia et al. (2015) |
| Rat NADH | ≥0.331 | ≥3.99 | – | 25 | Maia et al. (2015) | |
| Rat, NADH | – | 2.7 | 8.5a | 37 | Li et al. (2009) | |
| SO | Human, sulfite | 0.002 | 1.6 | 0.0361 | 37 | Wang et al. (2015) |
| Human, phenosafranine | 1.9 | 80 | 34 | 37 | Wang et al. (2015) | |
| mARC–1 | human, b 5 r/b 5 | 0.1 | 9.5 | 3.6 | 37 | Sparacino‐Watkins et al. (2014) |
All bimolecular rate constants are reported for human haemoproteins at pH 7.4. Kinetic parameters of Mo‐proteins are determined at pH 7.4 unless noted. Regarding the kinetic parameters of the Mo‐containing enzymes, the actual nitrite reduction rate will depend on abundance of the protein. See text and appropriate references for details.
pH 7.2.
One intriguing caveat suggesting NiR activity in the native mammalian six‐coordinate globins is the presence of two well‐conserved cysteines which form intramolecular disulfide bonds (Cys46 and 55 in Ngb, Cys38 and 83 in Cygb), resulting in either reduced (S–H) or oxidized (S–S) forms. In humans, these disulfides affect the position of the E‐helix where the distal histidine is located and modulate the haem ligand binding equilibrium: K His (k on /k off) shifts from ~3000 in NgbS–H to 280 in NgbS–S (Hamdane et al., 2004) and ~2000 to 0.48 in the respective Cygbs (Beckerson et al., 2015). Access to the distal binding site is necessary for nitrite reduction, reflected by a twofold increase in the bimolecular rate constant k NiR in Ngb (Tiso et al., 2011) and a 50‐fold increase in Cygb (Reeder and Ukeri, 2018) (Table 1) in proteins with the intramolecular disulfide. This bond may play a pivotal role in the otherwise unknown activity of these two globins, rendering them redox‐sensitive to their local environments, as well as creating efficient NiRs under reducing (hypoxic) cellular conditions, controlling NO signalling. It is worth noting that Reeder and Ukeri (2018) point out that the intermolecular disulfide bond of Cygb (dimer) has a k NiR about 140‐fold less than that of the intramolecular disulfide monomer.
Cystathionine β‐synthase (CBS), a hydrogen sulfide generating enzyme and vital enzyme in sulfur amino acid metabolism, also exhibits NiR activity and has been proposed as a potential NO source in vivo (Carballal et al., 2016). Mammalian CBS contains a haem ancillary to the active pocket, but its purpose is relatively unknown; it is suspected to allosterically regulate the active site as ferrous binding of exogenous ligands, such as NO, results in abolition of CBS activity (Carballal et al., 2016). Further, the haem has an unusual electronic structure with a low reduction potential (−350 mV vs. NHE). Consistent with this low potential, Carballal and co‐workers observed that dissociation of the iron‐bound cysteine had little effect on the rate of nitrite reduction, implying that this haem participated in rapid outer‐sphere electron transfer to nitrite. Coupled with the observation that NO binding at this haem is weaker than that of Hb or Mb [K NO is 3–5 orders of magnitude lower in CBS (Vicente et al., 2014)], a significant rate of nitrite reduction (Table 1), and an inability to form an oxygen‐bound ferrous species (thus excluding NO‐dioxygenase activity), CBS may function as a NiR in vivo.
Nitrite reduction by molybdenum–molybdopterin containing proteins
Other mammalian metal enzymes exhibit NiR activity, specifically enzymes containing molybdenum (Mo)‐bound molybdopterin cofactors. Examples include XO, aldehyde oxidase (AO), sulfite oxidase (SO) and mitochondrial amidoxime‐reducing component–1 and 2 (mARC‐1 and mARC‐2). These typically catalyse oxygen atom transfer reactions, where the molybdenum core cycles between Mo6+ and Mo4+ (Maia and Moura, 2015). However, each of these Mo‐enzymes exhibit one‐electron oxidations (Mo4+ to Mo5+) with nitrite, generating an equivalent of NO (Millar et al., 1998; Li et al., 2009; Maia and Moura, 2011; Sparacino‐Watkins et al., 2014; Wang et al., 2015; Maia et al., 2015). These enzymes are broadly distributed throughout human tissues and have been implicated as a major source of nitrite‐induced vasorelaxation especially as XO is present in the endothelium, as well as in the membrane of RBCs (Webb et al., 2008a). Additionally, XO does not bind NO. However, inhibition of XO does not inhibit nitrite‐dependent vasodilation in humans, implying that it is not necessarily the primary NiR in humans (Dejam et al., 2007). Mo‐enzyme NiR activity is inhibited by oxygen and is only pertinent at hypoxic or anoxic conditions (Maia and Moura, 2015). The Michalis–Menten kinetic parameters have been assessed by several laboratories under various conditions, and a sampling is included in Table 1, bottom. Though the relatively high K m values are 1–3 orders of magnitude higher than the concentration of nitrite in tissue [1–20 μM (Shiva, 2013)], only nanomolar amounts of NO are needed to carry out vascular functions, meaning Mo‐enzymes can act as NiRs at low oxygen tensions in addition to their primary functions.
Mechanistically speaking, the Mo5+ species has been observed via EPR, indicating that the enzymes are reducing nitrite and not simply catalysing a disproportionation of nitrous acid (Equation (13)) (Yang et al., 2015). Metal involvement of NiR activity has been confirmed as tungsten‐substituted mARC‐1 abolishes all nitrite reduction (Sparacino‐Watkins et al., 2014), and oxipurinol (a specific Mo‐binding XO inhibitor) similarly prevented NO generation (Okamoto et al., 2008).
| (13) |
Computational studies by Yang et al. (2015) on the postulated mARC active site favour a hydroxyl radical transfer mechanism, in which protonation of the metal‐bound oxygen of nitrite is required for facile NO release and is consistent with the pH dependence of Mo‐enzyme‐catalysed NO formation.
Nitrite and nitrate therapeutics
The discussion above highlights the diverse chemistry underlying how nitrite may regulate NO signalling, especially in low pH and oxygen environments. I/R injury is characterized by the latter and, in many cases, by low NO bioavailability. Thus, nitrite therapy may improve NO signalling in a targeted manner and avoid unwanted, off‐target, systemic effects associated with other NO‐releasing drugs. Indeed, to date, several preclinical animal model studies have demonstrated that nitrite administered during the ischaemia phase or very soon thereafter affords protection (see more detail in Lundberg et al., 2009, 2015; Calvert and Lefer, 2010; Vitturi and Patel, 2011). Importantly, protection has been observed in all major organ systems including the brain, heart, kidney, liver and lungs with a range of mechanisms from antioxidative, anti‐inflammatory, improvement of blood flow and angiogenesis to preventing cell death. While data derived from animal models are largely supportive of nitrite‐therapeutics, early phase clinical studies are less clear. Most available human data to date have evaluated effects of nitrite on acute myocardial infarction. Some researchers did not observe any protective effects of nitrite (Siddiqi et al., 2014). However, Jones et al. (2015) did observe a reduction in infarct size, a reduction in major adverse cardiac events and a reduction in inflammatory endpoints (Jones et al., 2017) in nitrite treated patients, especially the subgroup with very low coronary perfusion at the time of catheterization. Differences between nitrite dosing may underlie the outcome variability in these studies. Moreover, a similar effect of nitrite‐based protection against I/R injury has been proposed in liver transplant patients receiving inhaled NO versus placebo. In this case, inhaled NO gas‐derived circulating nitrite was proposed to limit I/R injury and improve the rate of allograft function recovery, especially in patients more prone to I/R injury (Lang et al., 2014). Thus, the therapeutic efficacy of nitrite may be most pronounced and observed in patients with the greatest degree of I/R injury.
In addition to acute disease states, nitrite‐therapy is being tested in chronic diseases associated with NO insufficiency, particularly vascular disease such as atherosclerosis, diabetes and systemic and pulmonary hypertension. A number of initial studies suggest positive outcomes for selected endpoints (e.g. BP, improved vascular remodelling and reductions in neutrophil numbers) (Greenway et al., 2012; Bir et al., 2014; Jones et al., 2017). A recently completed study of oral nitrite showed reductions in BP and inhibition of platelet activation with oral doses of 20 mg in normal volunteers (Hughan et al., 2017). Targeted delivery of nitrite via inhalation is being evaluated in clinical trials for treating pulmonary hypertension (Sparacino‐Watkins et al., 2012; Rix et al., 2015; Simon et al., 2016). Inhaled and oral nitrite are being studied in patients with heart failure with preserved ejection fraction (HFpEF) and have been shown in open label studies to reduce pulmonary and left heart filling pressures and to improve exercise cardiac output (Borlaug et al., 2015, 2016; Simon et al., 2016). However, a recently completed placebo‐controlled trial of inhaled nitrite for HFpEF patients was presented at the American Heart Association meetings and reported no efficacy of inhaled nitrite on exercise capacity (Reddy et al., 2017). Our research group is currently performing a placebo‐controlled trial of oral nitrite for patients with pulmonary hypertension and HFpEF that is currently enrolling (ClinicalTrials.gov, NCT03015402). At this time, additional randomized placebo‐controlled studies are clearly required to determine therapeutic efficacy for cardiovascular diseases.
Additionally, nitrate from the diet (vide supra) as an NO‐repleting therapeutic agent has been evaluated. Numerous, small, blinded, placebo‐controlled studies with healthy volunteers or hypertensive patients demonstrate that dietary nitrate can lower BP and platelet reactivity and improve exercise performance and cellular energetics; all endpoints associated with increased NO bioavailability (Lundberg et al., 2009, 2018; Kapil et al., 2014; Gee and Ahluwalia, 2016; Carlström et al., 2018). Animal studies have shown that dietary nitrate prevents I/R injury, improves ischaemic angiogenic signalling with revascularization and prevents metabolic syndrome, all expectedly modulated via the formation of nitrite and subsequent stimulation of NO‐dependent signalling (Carlström et al., 2010; Hendgen‐Cotta et al., 2012). Thus, nitrate clinical trials have been proposed for improving vascular‐related endpoints in active coronary artery disease (Rathod et al., 2016; Schwarz et al., 2016). A number of placebo‐controlled randomized trials have evaluated beetroot juice that has high nitrate concentrations compared with placebo nitrate‐depleted juice in patients with diabetes and HFpEF. In patients with Type 2 diabetes, there were no improvements in exercise capacity, endothelial function, glucose homeostasis or other measured endpoints (Gilchrist et al., 2013; Shepherd et al., 2015, 2016). Similar studies in HFpEF patients showed mixed results with one trial reporting a significant improvement in BP and exercise endurance after a week of beetroot treatment (Eggebeen et al., 2016), while a second trial found that there was no added benefit observed for any outcomes when comparing beetroot juice to placebo in either hypertensive or HFpEF patients undergoing exercise training (P ≥ 0.14) (Shaltout et al., 2017).
While these ongoing studies explore the potential for nitrate‐therapy in diseased populations, it is important to highlight that some studies have reported no effect of dietary nitrate leading to the concept of ‘nitrate‐responders’ versus ‘non‐responders’. The existence of these two populations suggest that oral microbiome diversity, abundance of nitrate‐reducing bacteria and presence of metabolites that inhibit saliva nitrate transport or reduction are potential variables that underlie discrepant responses (Burleigh et al., 2018). Frequency of mouthwash use and type are obvious factors, but more recent work identifies other lifestyle variables such as smoking: smoking‐derived thiocyanate competes with and inhibits nitrate transport into the salivary glands (Bailey et al., 2016), and smokers have oral dysbiosis characterized by lower nitrate‐reductase activity (Ahmed et al., 2017). Moreover, as discussed previously, stomach pH is suggested to be an important variable in connecting nitrite from our oral microbiome to the activation of NO signalling systemically. Such factors must be allowed for in the design of any clinical trial.
Concluding remarks
Nitrate and nitrite represent a relatively new paradigm in intravascular NO signalling, where nitrite is a stable intermediate reduced when needed under physiological and pathophysiological hypoxia and nitrate can resupply the nitrite pool. Consistent with this paradigm, nitrate is protective against I/R injuries and several other cardiovascular disorders in animal models (Lundberg et al., 2011; Kapil et al., 2014; Hezel et al., 2016; Mills et al., 2017), and changes in plasma nitrite are protective down to 200 nM (Duranski et al., 2005). Moreover, nitrate supplementation in human studies show pronounced beneficial effects on BP and overall vascular health as thoroughly reviewed by Kapil et al. (2014). These observations are consistent with the nitrate–nitrite–NO postulation and nitrite chemistries described throughout this text and largely summarized in Figure 5, where nitrate is ingested or produced endogenously and converted to nitrite, the chief vascular reserve for NO and other NOx species. Nitrite is rapidly converted to NO by multiple enzymes; in erythrocytes, this occurs during deoxygenation of Hb from artery‐to‐vein transit, where the NO in the RBC can form nitrate, generate N2O3 or bind ferrohaem and subsequently undergo oxidative denitrosylation regenerating NO. The chemical biology described in this review expounds the physiological and pharmacological observations around this pathway, illuminating an essential endogenous source of NO delivery, especially when the conventional NOS route is compromised under hypoxic conditions.
Figure 5.
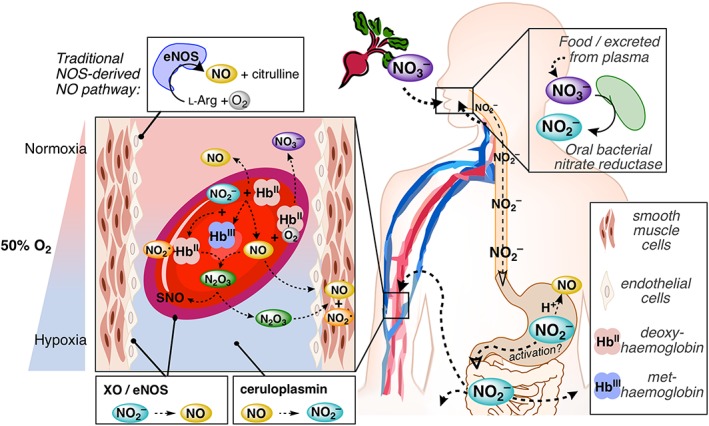
The nitrate–nitrite–NO pathway and simplified nitrite chemical biology in the vasculature. The nitrate–nitrite–NO path is a complementary route for NO generation in the vasculature that occurs under hypoxic conditions, when canonical NO generation at normoxia from eNOS (upper left) is less effective. Nitrate is ingested (upper right) from various foods including beetroot and leafy greens. Oral bacteria in the salivary glands reduce nitrate to nitrite, which is subsequently swallowed, traversing the oesophagus and into the acidic gastric fluids and into GI tract (hollow arrowheads). Some of the nitrite (as nitrous acid) may dehydrate and disproportionate (Equations (3) and (4) in text) to generate NO in the stomach. Orally swallowed nitrite has been suggested to be bioactivated in the stomach under non‐enzymic acidic conditions. Nitrite may also escape the GI tract and enter the circulation (bottom right). Numerous reactions occur in RBCs (centre left), especially at the point of artery‐to‐vein transport (Hb is ~50% O2‐saturated or P 50) where the nitrite reduction rate by deoxyHb is maximized. NiR activity yields metHb and an equivalent of NO (Equation (6)). metHb reacts with nitrite to form a radical NO2 •‐bound ferrohaem, which reacts rapidly with NO to generate N2O3 (Figure 4, path a), responsible for RSNO formation (Equation (9)). N2O3 is one mechanism by which NO is proposed to escape from the RBC and generate NO (Equation (4)) in the smooth muscle. NO may also be autocaptured by deoxyHb generating ferrous nitrosyl‐Hb (Equation (7), not shown) and subsequently released via oxidative nitrosylation (Equations (11) and (12), not shown), or NO may react with oxyHb to liberate nitrate (Equation (2)). The nitrate is secreted from the plasma into the salivary glands (upper right), starting the process anew. XO and eNOS found in both endothelial cells and the surface of RBCs have been suspected to generate NO from nitrite, but only under hypoxic and more acidic conditions (bottom left). Finally, nitrite is generated by the copper plasma protein ceruloplasmin from NO, preventing dioxygenation to nitrate or reductive nitrosylation by ferrihaems (not shown).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018) and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Author contributions
A.W.D. prepared the initial and final drafts and all figures. R.P.P. critically read the manuscript and contributed the section on nitrite therapeutics. D.B.K.‐S. and M.T.G. contributed to the preparation and editing of the full manuscript.
Conflict of interest
M.T.G. is a co‐inventor of pending patent applications and planned patents directed to the use of recombinant neuroglobin and haem‐based molecules as antidotes for CO poisoning, which have recently been licensed by Globin Solutions, Inc. M.T.G. is a shareholder, advisor and director in Globin Solutions, Inc. Additionally, and unrelated to CO poisoning, Dr. Gladwin is a co‐inventor on patents directed to the use of nitrite salts in cardiovascular diseases, which have been licensed by United Therapeutics and Hope Pharmaceuticals, and is a co‐investigator in a research collaboration with Bayer Pharmaceuticals to evaluate riociguate as a treatment for patients with SCD.
Acknowledgements
We thank Dr. Jesús Tejero (University of Pittsburgh) for his careful reading of this manuscript and suggestions. Studies in the Gladwin laboratory are supported by National Institutes of Health grants 2R01HL098032, 1R01HL125886, 5P01HL103455 and T32HL110849; the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania.
DeMartino, A. W. , Kim‐Shapiro, D. B. , Patel, R. P. , and Gladwin, M. T. (2019) Nitrite and nitrate chemical biology and signalling. British Journal of Pharmacology, 176: 228–245. 10.1111/bph.14484.
References
- Ahluwalia A, Gladwin M, Coleman GD, Hord N, Howard G, Kim‐Shapiro DB et al (2016). Dietary nitrate and the epidemiology of cardiovascular disease: report from a National Heart, Lung, and Blood Institute Workshop. J Am Heart Assoc 5: e003402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed KA, Nichols AL, Honavar J, Dransfield MT, Matalon S, Patel RP (2017). Measuring nitrate reductase activity from human and rodent tongues. Nitric Oxide Biol Chem 66: 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017). The concise guide to pharmacology 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DM, Rasmussen P, Overgaard M, Evans KA, Bohm AM, Seifert T et al (2017). Nitrite and S‐nitrosohemoglobin exchange across the human cerebral and femoral circulation clinical perspective: relationship to basal and exercise blood flow responses to hypoxia. Circulation 135: 166–176. [DOI] [PubMed] [Google Scholar]
- Bailey SJ, Blackwell JR, Wylie LJ, Holland T, Winyard PG, Jones AM (2016). Improvement in blood pressure after short‐term inorganic nitrate supplementation is attenuated in cigarette smokers compared to non‐smoking controls. Nitric Oxide Biol Chem 61: 29–37. [DOI] [PubMed] [Google Scholar]
- Basu S, Grubina R, Huang J, Conradie J, Huang Z, Jeffers A et al (2007). Catalytic generation of N2O3 by the concerted nitrite reductase and anhydrase activity of hemoglobin. Nat Chem Biol 3: 785–794. [DOI] [PubMed] [Google Scholar]
- Beckerson P, Reeder BJ, Wilson MT (2015). Coupling of disulfide bond and distal histidine dissociation in human ferrous cytoglobin regulates ligand binding. FEBS Lett 589: 507–512. [DOI] [PubMed] [Google Scholar]
- Bir SC, Pattillo CB, Pardue S, Kolluru GK, Shen X, Giordano T et al (2014). Nitrite anion therapy protects against chronic ischemic tissue injury in db/db diabetic mice in a NO/VEGF‐dependent manner. Diabetes 63: 270–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaventura C, Henkens R, Alayash AI, Banerjee S, Crumbliss AL (2013). Molecular controls of the oxygenation and redox reactions of hemoglobin. Antioxid Redox Signal 18: 2298–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaventura C, Henkens R, Alayash AI, Crumbliss AL (2008). Allosteric effects on oxidative and nitrosative reactions of cell‐free hemoglobins. IUBMB Life 59: 498–505. [DOI] [PubMed] [Google Scholar]
- Borlaug BA, Koepp KE, Melenovsky V (2015). Sodium nitrite improves exercise hemodynamics and ventricular performance in heart failure with preserved ejection fraction. J Am Coll Cardiol 66: 1672–1682. [DOI] [PubMed] [Google Scholar]
- Borlaug BA, Melenovsky V, Koepp KE (2016). Inhaled sodium nitrite improves rest and exercise hemodynamics in heart failure with preserved ejection fraction. Circ Res 119: 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratsch SG (1989). Standard electrode potentials and temperature coefficients in water at 298.15 K. J Phys Chem Ref Data Monogr 18: 1–21. [Google Scholar]
- Brooks J (1937). The action of nitrite on haemoglobin in the absence of oxygen. Proc R Soc Lond B 123: 368–382. [Google Scholar]
- Burleigh MC, Liddle L, Monaghan C, Muggeridge DJ, Sculthorpe N, Butcher JP et al (2018). Salivary nitrite production is elevated in individuals with a higher abundance of oral nitrate‐reducing bacteria. Free Radic Biol Med 120: 80–88. [DOI] [PubMed] [Google Scholar]
- Calvert JW, Lefer DJ (2010). Clinical translation of nitrite therapy for cardiovascular diseases. Nitric Oxide 22: 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballal S, Cuevasanta E, Yadav PK, Gherasim C, Ballou DP, Alvarez B et al (2016). Kinetics of nitrite reduction and peroxynitrite formation by ferrous heme in human cystathionine β‐synthase. J Biol Chem 291: 8004–8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlström M, Larsen FJ, Nyström T, Hezel M, Borniquel S, Weitzberg E et al (2010). Dietary inorganic nitrate reverses features of metabolic syndrome in endothelial nitric oxide synthase‐deficient mice. Proc Natl Acad Sci 107: 17716–17720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlström M, Lundberg JO, Weitzberg E (2018). Mechanisms underlying blood pressure reduction by dietary inorganic nitrate. Acta Physiol Oxf Engl 224: e13080. [DOI] [PubMed] [Google Scholar]
- Cassoly R, Gibson QH (1975). Conformation, co‐operativity and ligand binding in human hemoglobin. J Mol Biol 91: 301–313. [DOI] [PubMed] [Google Scholar]
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S et al (2013). Cardioprotection by S‐nitrosation of a cysteine switch on mitochondrial complex I. Nat Med 19: 753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland DM, Soares AS, West AH, Richter‐Addo GB (2006). Crystal structures of the nitrite and nitric oxide complexes of horse heart myoglobin. J Inorg Biochem 100: 1413–1425. [DOI] [PubMed] [Google Scholar]
- Corti P, Xue J, Tejero J, Wajih N, Sun M, Stolz DB et al (2016). Globin X is a six‐coordinate globin that reduces nitrite to nitric oxide in fish red blood cells. Proc Natl Acad Sci 113: 8538–8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S et al (2003). Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med 9: 1498–1505. [DOI] [PubMed] [Google Scholar]
- Crawford JH, Isbell TS, Huang Z, Shiva S, Chacko BK, Schechter AN et al (2006). Hypoxia, red blood cells, and nitrite regulate NO‐dependent hypoxic vasodilation. Blood 107: 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutruzzolà F, Brown K, Wilson EK, Bellelli A, Arese M, Tegoni M et al (2001). The nitrite reductase from Pseudomonas aeruginosa: essential role of two active‐site histidines in the catalytic and structural properties. Proc Natl Acad Sci 98: 2232–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalsgaard T, Simonsen U, Fago A (2007). Nitrite‐dependent vasodilation is facilitated by hypoxia and is independent of known NO‐generating nitrite reductase activities. Am J Physiol‐Heart Circ Physiol 292: H3072–H3078. [DOI] [PubMed] [Google Scholar]
- Dejam A, Hunter CJ, Pelletier MM, Hsu LL, Machado RF, Shiva S et al (2005). Erythrocytes are the major intravascular storage sites of nitrite in human blood. Blood 106: 734–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejam A, Hunter CJ, Tremonti C, Pluta RM, Hon YY, Grimes G et al (2007). Nitrite infusion in humans and nonhuman primates: endocrine effects, pharmacokinetics, and tolerance formation. Circulation 116: 1821–1831. [DOI] [PubMed] [Google Scholar]
- Doyle MP, Hoekstra JW (1981). Oxidation of nitrogen oxides by bound dioxygen in hemoproteins. J Inorg Biochem 14: 351–358. [DOI] [PubMed] [Google Scholar]
- Doyle MP, LePoire DM, Pickering RA (1981a). Oxidation of hemoglobin and myoglobin by alkyl nitrites inhibition by oxygen. J Biol Chem 256: 12399–12404. [PubMed] [Google Scholar]
- Doyle MP, Pickering RA, da Conceição J (1984). Structural effects in alkyl nitrite oxidation of human hemoglobin. J Biol Chem 259: 80–87. [PubMed] [Google Scholar]
- Doyle MP, Pickering RA, DeWeert TM, Hoekstra JW, Pater D (1981b). Kinetics and mechanism of the oxidation of human deoxyhemoglobin by nitrites. J Biol Chem 256: 12393–12398. [PubMed] [Google Scholar]
- Duranski MR, Greer JJM, Dejam A, Jaganmohan S, Hogg N, Langston W et al (2005). Cytoprotective effects of nitrite during in vivo ischemia‐reperfusion of the heart and liver. J Clin Invest 115: 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggebeen J, Kim‐Shapiro DB, Haykowsky M, Morgan TM, Basu S, Brubaker P et al (2016). One week of daily dosing with beetroot juice improves submaximal endurance and blood pressure in older patients with heart failure and preserved ejection fraction. JACC Heart Fail 4: 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feelisch M, Fernandez BO, Bryan NS, Garcia‐Saura MF, Bauer S, Whitlock DR et al (2008). Tissue processing of nitrite in hypoxia: an intricate interplay of nitric oxide generating and scavenging systems. J Biol Chem 283: 33927–33934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez BO, Ford PC (2003). Nitrite catalyzes ferriheme protein reductive nitrosylation. J Am Chem Soc 125: 10510–10511. [DOI] [PubMed] [Google Scholar]
- Fernandez BO, Lorkovic IM, Ford PC (2004). Mechanisms of ferriheme reduction by nitric oxide: nitrite and general base catalysis 1. Inorg Chem 43: 5393–5402. [DOI] [PubMed] [Google Scholar]
- Ford PC (2010). Reactions of NO and nitrite with heme models and proteins. Inorg Chem 49: 6226–6239. [DOI] [PubMed] [Google Scholar]
- Ford PC, Wink DA, Stanbury DM (1993). Autoxidation kinetics of aqueous nitric oxide. FEBS Lett 326: 1–3. [DOI] [PubMed] [Google Scholar]
- Fukuto JM, Carrington SJ, Tantillo DJ, Harrison JG, Ignarro LJ, Freeman BA et al (2012). Small molecule signaling agents: the integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species. Chem Res Toxicol 25: 769–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchgott RF, Bhadrakom S (1953). Reactions of strips of rabbit aorta to epinephrine, isopropylarterenol, sodium nitrite and other drugs. J Pharmacol Exp Ther 108: 129–143. [PubMed] [Google Scholar]
- Gee LC, Ahluwalia A (2016). Dietary nitrate lowers blood pressure: epidemiological, pre‐clinical experimental and clinical trial evidence. Curr Hypertens Rep 18: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist M, Winyard PG, Aizawa K, Anning C, Shore A, Benjamin N (2013). Effect of dietary nitrate on blood pressure, endothelial function, and insulin sensitivity in type 2 diabetes. Free Radic Biol Med 60: 89–97. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Grubina R, Doyle MP (2009). The new chemical biology of nitrite reactions with hemoglobin: R‐state catalysis, oxidative denitrosylation, and nitrite reductase/anhydrase. Acc Chem Res 42: 157–167. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Kim‐Shapiro DB (2008). The functional nitrite reductase activity of the heme‐globins. Blood 112: 2636–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grätzel M, Taniguchi S, Henglein A (1970). Pulsradiolytische Untersuchung der NO‐Oxydation und des Gleichgewichts N2O3 ⇄ NO + NO2 in wäßriger Lösung. Berichte Bunsenges. Für Phys Chem 74: 488–492. [Google Scholar]
- Greenway FL, Predmore BL, Flanagan DR, Giordano T, Qiu Y, Brandon A et al (2012). Single‐dose pharmacokinetics of different oral sodium nitrite formulations in diabetes patients. Diabetes Technol Ther 14: 552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubina R, Basu S, Tiso M, Kim‐Shapiro DB, Gladwin MT (2008). Nitrite reductase activity of hemoglobin S (Sickle) provides insight into contributions of heme redox potential versus ligand affinity. J Biol Chem 283: 3628–3638. [DOI] [PubMed] [Google Scholar]
- Grubina R, Huang Z, Shiva S, Joshi MS, Azarov I, Basu S et al (2007). Concerted nitric oxide formation and release from the simultaneous reactions of nitrite with deoxy‐ and oxyhemoglobin. J Biol Chem 282: 12916–12927. [DOI] [PubMed] [Google Scholar]
- Haldane J, Makgill RH, Mavrogordato AE (1897). The action as poisons of nitrites and other physiologically related substances. J Physiol 21: 160–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdane D, Kiger L, Dewilde S, Green BN, Pesce A, Uzan J et al (2004). Coupling of the heme and an internal disulfide bond in human neuroglobin. Micron 35: 59–62. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinecke JL, Yi J, Pereira JCM, Richter‐Addo GB, Ford PC (2012). Nitrite reduction by CoII and MnII substituted myoglobins: towards understanding necessary components of Mb nitrite reductase activity. J Inorg Biochem 107: 47–53. [DOI] [PubMed] [Google Scholar]
- Heinrich TA, da Silva RS, Miranda KM, Switzer CH, Wink DA, Fukuto JM (2013). Biological nitric oxide signalling: chemistry and terminology. Br J Pharmacol 169: 1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendgen‐Cotta UB, Luedike P, Totzeck M, Kropp M, Schicho A, Stock P et al (2012). Dietary nitrate supplementation improves revascularization in chronic ischemia. Circulation 126: 1983–1992. [DOI] [PubMed] [Google Scholar]
- Hendgen‐Cotta UB, Merx MW, Shiva S, Schmitz J, Becher S, Klare JP et al (2008). Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia‐reperfusion injury. Proc Natl Acad Sci 105: 10256–10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hezel M, Peleli M, Liu M, Zollbrecht C, Jensen BL, Checa A et al (2016). Dietary nitrate improves age‐related hypertension and metabolic abnormalities in rats via modulation of angiotensin II receptor signaling and inhibition of superoxide generation. Free Radic Biol Med 99: 87–98. [DOI] [PubMed] [Google Scholar]
- Hopmann KH, Cardey B, Gladwin MT, Kim‐Shapiro DB, Ghosh A (2011). Hemoglobin as a nitrite anhydrase: modeling methemoglobin‐mediated N2O3 formation. Chem A Eur J 17: 6348–6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino M, Maeda M, Konishi R, Seki H, Ford PC (1996). Studies on the reaction mechanism for reductive nitrosylation of ferrihemoproteins in buffer solutions. J Am Chem Soc 118: 5702–5707. [Google Scholar]
- Huang KT, Keszler A, Patel N, Patel RP, Gladwin MT, Kim‐Shapiro DB et al (2005a). The reaction between nitrite and deoxyhemoglobin: reassessment of reaction kinetics and stoichiometry. J Biol Chem 280: 31126–31131. [DOI] [PubMed] [Google Scholar]
- Huang Z, Shiva S, Kim‐Shapiro DB, Patel RP, Ringwood LA, Irby CE et al (2005b). Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest 115: 2099–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughan KS, Wendell SG, Delmastro‐Greenwood M, Helbling N, Corey C, Bellavia L et al (2017). Conjugated linoleic acid modulates clinical responses to oral nitrite and nitrate novelty and significance. Hypertension 70: 634–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter CJ, Dejam A, Blood AB, Shields H, Kim‐Shapiro DB, Machado RF et al (2004). Inhaled nebulized nitrite is a hypoxia‐sensitive NO‐dependent selective pulmonary vasodilator. Nat Med 10: 1122–1127. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Fukuto JM, Griscavage JM, Rogers NE, Byrns RE (1993). Oxidation of nitric oxide in aqueous solution to nitrite but not nitrate: comparison with enzymatically formed nitric oxide from L‐arginine. Proc Natl Acad Sci U S A 90: 8103–8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro LJ, Gruetter CS (1980). Requirement of thiols for activation of coronary arterial guanylate cyclase by glyceryl trinitrate and sodium nitrite possible involvement of S‐nitrosothiols. Biochim Biophys Acta BBA ‐ Gen Subj 631: 221–231. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Lippton H, Edwards JC, Baricos WH, Hyman AL, Kadowitz PJ et al (1981). Mechanism of vascular smooth muscle relaxation by organic nitrates, nitrites, nitroprusside and nitric oxide: evidence for the involvement of S‐nitrosothiols as active intermediates. J Pharmacol Exp Ther 218: 739–749. [PubMed] [Google Scholar]
- Ingram TE, Pinder AG, Bailey DM, Fraser AG, James PE (2009). Low‐dose sodium nitrite vasodilates hypoxic human pulmonary vasculature by a means that is not dependent on a simultaneous elevation in plasma nitrite. Am J Physiol‐Heart Circ Physiol 298: H331–H339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isbell TS, Gladwin MT, Patel RP (2007). Hemoglobin oxygen fractional saturation regulates nitrite‐dependent vasodilation of aortic ring bioassays. Am. J. Physiol.‐Heart Circ. Physiol. 293: H2565–H2572. [DOI] [PubMed] [Google Scholar]
- Jansson EÅ, Huang L, Malkey R, Govoni M, Nihlén C, Olsson A et al (2008). A mammalian functional nitrate reductase that regulates nitrite and nitric oxide homeostasis. Nat Chem Biol 4: 411–417. [DOI] [PubMed] [Google Scholar]
- Jeffers A, Xu X, Huang KT, Cho M, Hogg N, Patel RP et al (2005). Hemoglobin mediated nitrite activation of soluble guanylyl cyclase. Comp Biochem Physiol A Mol Integr Physiol 142: 130–135. [DOI] [PubMed] [Google Scholar]
- Jones DA, Khambata RS, Andiapen M, Rathod KS, Mathur A, Ahluwalia A (2017). Intracoronary nitrite suppresses the inflammatory response following primary percutaneous coronary intervention. Heart 103: 508–516. [DOI] [PubMed] [Google Scholar]
- Jones DA, Pellaton C, Velmurugan S, Rathod KS, Andiapen M, Antoniou S et al (2015). Randomized phase 2 trial of intracoronary nitrite during acute myocardial infarction novelty and significance. Circ Res 116: 437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapil V, Haydar SMA, Pearl V, Lundberg JO, Weitzberg E, Ahluwalia A (2013). Physiological role for nitrate‐reducing oral bacteria in blood pressure control. Free Radic Biol Med 55: 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapil V, Milsom AB, Okorie M, Maleki‐Toyserkani S, Akram F, Rehman F et al (2010). Inorganic nitrate supplementation lowers blood pressure in humans: role for nitrite‐derived NO. Hypertension 56: 274–281. [DOI] [PubMed] [Google Scholar]
- Kapil V, Weitzberg E, Lundberg JO, Ahluwalia A (2014). Clinical evidence demonstrating the utility of inorganic nitrate in cardiovascular health. Nitric Oxide 38: 45–57. [DOI] [PubMed] [Google Scholar]
- Keszler A, Piknova B, Schechter AN, Hogg N (2008). The reaction between nitrite and oxyhemoglobin, a mechanistic study. J Biol Chem 283: 9615–9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khambata RS, Ghosh SM, Ahluwalia A (2015). “Repurposing” of xanthine oxidoreductase as a nitrite reductase: a new paradigm for therapeutic targeting in hypertension. Antioxid Redox Signal 23: 340–353. [DOI] [PubMed] [Google Scholar]
- Khatri J, Mills CE, Maskell P, Odongerel C, Webb AJ (2017). It is rocket science – why dietary nitrate is hard to ‘beet’! Part I: twists and turns in the realization of the nitrate–nitrite–NO pathway. Br J Clin Pharmacol 83: 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim‐Shapiro DB, Gladwin MT, Patel RP, Hogg N (2005). The reaction between nitrite and hemoglobin: the role of nitrite in hemoglobin‐mediated hypoxic vasodilation. J Inorg Biochem 99: 237–246. [DOI] [PubMed] [Google Scholar]
- Koch CD, Gladwin MT, Freeman BA, Lundberg JO, Weitzberg E, Morris A (2017). Enterosalivary nitrate metabolism and the microbiome: intersection of microbial metabolism, nitric oxide and diet in cardiac and pulmonary vascular health. Free Radic Biol Med 105: 48–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov AV, Costantino G, Sobhian B, Szalay L, Umar F, Nohl H et al (2005). Mechanisms of vasodilatation induced by nitrite instillation in intestinal lumen: possible role of hemoglobin. Antioxid Redox Signal 7: 515–521. [DOI] [PubMed] [Google Scholar]
- Lang JD, Smith AB, Brandon A, Bradley KM, Liu Y, Li W et al (2014). A randomized clinical trial testing the anti‐inflammatory effects of preemptive inhaled nitric oxide in human liver transplantation. PloS One 9: e86053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen FJ, Ekblom B, Sahlin K, Lundberg JO, Weitzberg E (2006). Effects of dietary nitrate on blood pressure in healthy volunteers. N Engl J Med 355: 2792–2793. [DOI] [PubMed] [Google Scholar]
- Lauer T, Preik M, Rassaf T, Strauer BE, Deussen A, Feelisch M et al (2001). Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc Natl Acad Sci 98: 12814–12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Cui H, Kundu TK, Alzawahra W, Zweier JL (2008). Nitric oxide production from nitrite occurs primarily in tissues not in the blood: critical role of xanthine oxidase and aldehyde oxidase. J Biol Chem 283: 17855–17863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Cui H, Liu X, Zweier JL (2005). Xanthine oxidase catalyzes anaerobic transformation of organic nitrates to nitric oxide and nitrosothiols characterization of this mechanism and the link between organic nitrate and guanylyl cyclase activation. J Biol Chem 280: 16594–16600. [DOI] [PubMed] [Google Scholar]
- Li H, Hemann C, Abdelghany TM, El‐Mahdy MA, Zweier JL (2012). Characterization of the mechanism and magnitude of cytoglobin‐mediated nitrite reduction and nitric oxide generation under anaerobic conditions. J Biol Chem 287: 36623–36633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Kundu TK, Zweier JL (2009). Characterization of the magnitude and mechanism of aldehyde oxidase‐mediated nitric oxide production from nitrite. J Biol Chem 284: 33850–33858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Samouilov A, Liu X, Zweier JL (2001). Characterization of the magnitude and kinetics of xanthine oxidase‐catalyzed nitrate reduction ‐ evaluation of its role in nitric oxide generation in anoxic tissues. J Biol Chem 276: 24482–24489. [DOI] [PubMed] [Google Scholar]
- Li H, Samouilov A, Liu X, Zweier JL (2003). Characterization of the magnitude and kinetics of xanthine oxidase‐catalyzed nitrate reduction: evaluation of its role in nitrite and nitric oxide generation in anoxic tissues. Biochemistry (Mosc) 42: 1150–1159. [DOI] [PubMed] [Google Scholar]
- Liu C, Wajih N, Liu X, Basu S, Janes J, Marvel M et al (2015). Mechanisms of human erythrocytic bioactivation of nitrite. J Biol Chem 290: 1281–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Miller MJS, Joshi MS, Thomas DD, Lancaster JR (1998). Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc Natl Acad Sci 95: 2175–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscalzo J (2001). Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ Res 88: 756–762. [DOI] [PubMed] [Google Scholar]
- Lundberg JO, Carlström M, Larsen FJ, Weitzberg E (2011). Roles of dietary inorganic nitrate in cardiovascular health and disease. Cardiovasc Res 89: 525–532. [DOI] [PubMed] [Google Scholar]
- Lundberg JO, Carlström M, Weitzberg E (2018). Metabolic effects of dietary nitrate in health and disease. Cell Metab 28: 9–22. [DOI] [PubMed] [Google Scholar]
- Lundberg JO, Gladwin MT, Ahluwalia A, Benjamin N, Bryan NS, Butler A et al (2009). Nitrate and nitrite in biology, nutrition and therapeutics. Nat Chem Biol 5: 865–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg JO, Gladwin MT, Weitzberg E (2015). Strategies to increase nitric oxide signalling in cardiovascular disease. Nat Rev Drug Discov 14: 623–641. [DOI] [PubMed] [Google Scholar]
- Lundberg JO, Weitzberg E, Gladwin MT (2008). The nitrate–nitrite–nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov 7: 156–167. [DOI] [PubMed] [Google Scholar]
- Maher AR, Milsom AB, Gunaruwan P, Abozguia K, Ahmed I, Weaver RA et al (2008). Hypoxic modulation of exogenous nitrite‐induced vasodilation in humans. Circulation 117: 670–677. [DOI] [PubMed] [Google Scholar]
- Maia L, Moura J (2011). Nitrite reduction by xanthine oxidase family enzymes: a new class of nitrite reductases. J Biol Inorg Chem 16: 443–460. [DOI] [PubMed] [Google Scholar]
- Maia L, Moura J (2015). Nitrite reduction by molybdoenzymes: a new class of nitric oxide‐forming nitrite reductases. J Biol Inorg Chem 20: 403–433. [DOI] [PubMed] [Google Scholar]
- Maia LB, Pereira V, Mira L, Moura JJG (2015). Nitrite reductase activity of rat and human xanthine oxidase, xanthine dehydrogenase, and aldehyde oxidase: evaluation of their contribution to no formation in vivo. Biochemistry (Mosc) 54: 685–710. [DOI] [PubMed] [Google Scholar]
- Millar TM, Stevens CR, Benjamin N, Eisenthal R, Harrison R, Blake DR (1998). Xanthine oxidoreductase catalyses the reduction of nitrates and nitrite to nitric oxide under hypoxic conditions. FEBS Lett 427: 225–228. [DOI] [PubMed] [Google Scholar]
- Mills CE, Khatri J, Maskell P, Odongerel C, Webb AJ (2017). It is rocket science – why dietary nitrate is hard to ‘beet’! Part II: further mechanisms and therapeutic potential of the nitrate–nitrite–NO pathway. Br J Clin Pharmacol 83: 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Meuwly M (2010). Atomistic simulation of NO dioxygenation in group I truncated hemoglobin. J Am Chem Soc 132: 2968–2982. [DOI] [PubMed] [Google Scholar]
- Mittall CK, Arnold WP, Murad F (1978). Characterization of protein inhibitors of guanylate cyclase activation from rat heart and bovine lung. J Biol Chem 253: 1266–1271. [PubMed] [Google Scholar]
- Modin A, Björne H, Herulf M, Alving K, Weitzberg E, Lundberg JON (2001). Nitrite‐derived nitric oxide: a possible mediator of ‘acidic–metabolic’ vasodilation. Acta Physiol Scand 171: 9–16. [DOI] [PubMed] [Google Scholar]
- Montenegro MF, Sundqvist ML, Larsen FJ, Zhuge Z, Carlström M, Weitzberg E et al (2017). Blood pressure–lowering effect of orally ingested nitrite is abolished by a proton pump inhibitor novelty and significance. Hypertension 69: 23–31. [DOI] [PubMed] [Google Scholar]
- Moore EG, Gibson QH (1976). Cooperativity in the dissociation of nitric oxide from hemoglobin. J Biol Chem 251: 2788–2794. [PubMed] [Google Scholar]
- Nagababu E, Ramasamy S, Abernethy DR, Rifkind JM (2003). Active nitric oxide produced in the red cell under hypoxic conditions by deoxyhemoglobin‐mediated nitrite reduction. J Biol Chem 278: 46349–46356. [DOI] [PubMed] [Google Scholar]
- Navati MS, Friedman JM (2009). Reactivity of glass‐embedded met hemoglobin derivatives toward external NO: implications for nitrite‐mediated production of bioactive NO. J Am Chem Soc 131: 12273–12279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Eger BT, Nishino T, Pai EF, Nishino T (2008). Mechanism of inhibition of xanthine oxidoreductase by allopurinol: crystal structure of reduced bovine milk xanthine oxidoreductase bound with oxipurinol. Nucleosides Nucleotides Nucleic Acids 27: 888–893. [DOI] [PubMed] [Google Scholar]
- Patel RP, Hogg N, Kim‐Shapiro DB (2011). The potential role of the red blood cell in nitrite‐dependent regulation of blood flow. Cardiovasc Res 89: 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perissinotti LL, Marti MA, Doctorovich F, Luque FJ, Estrin DA (2008). A microscopic study of the deoxyhemoglobin‐catalyzed generation of nitric oxide from nitrite anion. Biochemistry (Mosc) 47: 9793–9802. [DOI] [PubMed] [Google Scholar]
- Petersen MG, Dewilde S, Fago A (2008). Reactions of ferrous neuroglobin and cytoglobin with nitrite under anaerobic conditions. J Inorg Biochem 102: 1777–1782. [DOI] [PubMed] [Google Scholar]
- Piknova B, Park JW, Kwan (Jeff) Lam K, Schechter AN (2016). Nitrate as a source of nitrite and nitric oxide during exercise hyperemia in rat skeletal muscle. Nitric Oxide 55–56: 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piknova B, Park JW, Swanson KM, Dey S, Noguchi CT, Schechter AN (2015). Skeletal muscle as an endogenous nitrate reservoir. Nitric Oxide 47: 10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinder A, Pittaway E, Morris K, James P (2009). Nitrite directly vasodilates hypoxic vasculature via nitric oxide‐dependent and ‐independent pathways. Br J Pharmacol 157: 1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluta RM, Oldfield EH, Bakhtian KD, Fathi AR, Smith RK, Devroom HL et al (2011). Safety and feasibility of long‐term intravenous sodium nitrite infusion in healthy volunteers. PloS One 6: e14504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomski MW, Palmer RMJ, Moncada S (1987). Comparative pharmacology of endothelium‐derived relaxing factor, nitric oxide and prostacyclin in platelets. Br J Pharmacol 92: 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathod KS, Jones DA, Van‐Eijl TJA, Tsang H, Warren H, Hamshere SM et al (2016). Randomised, double‐blind, placebo‐controlled study investigating the effects of inorganic nitrate on vascular function, platelet reactivity and restenosis in stable angina: protocol of the NITRATE‐OCT study. BMJ Open 6: e012728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy YNV, Lewis GD, Shah SJ, LeWinter M, Semigran M, Davila‐Roman VG et al (2017). INDIE‐HFpEF (inorganic nitrite delivery to improve exercise capacity in heart failure with preserved ejection fraction): rationale and design. Circ Heart Fail 10: e003862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeder BJ, Ukeri J (2018). Strong modulation of nitrite reductase activity of cytoglobin by disulfide bond oxidation: implications for nitric oxide homeostasis. Nitric Oxide 72: 16–23. [DOI] [PubMed] [Google Scholar]
- Reichenbach G, Sabatini S, Palombari R, Palmerini CA (2001). Reaction mechanism between nitric oxide and glutathione mediated by Fe (III) myoglobin. Nitric Oxide 5: 395–401. [DOI] [PubMed] [Google Scholar]
- Rix PJ, Vick A, Attkins NJ, Barker GE, Bott AW, Alcorn H et al (2015). Pharmacokinetics, pharmacodynamics, safety, and tolerability of nebulized sodium nitrite (AIR001) following repeat‐dose inhalation in healthy subjects. Clin Pharmacokinet 54: 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JM, Fairchild HM, Weldy J, Guyton AC (1962). Autoregulation of blood flow by oxygen lack. Am J Physiol‐Leg Content 202: 21–24. [DOI] [PubMed] [Google Scholar]
- Schwarz K, Singh S, Parasuraman SK, Bruce M, Shepstone L, Feelisch M et al (2016). A randomized double‐blind placebo‐controlled crossover trial of sodium nitrate in patients with stable angina INAS. Future Cardiol 12: 617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaltout HA, Eggebeen J, Marsh AP, Brubaker PH, Laurienti PJ, Burdette JH et al (2017). Effects of supervised exercise and dietary nitrate in older adults with controlled hypertension and/or heart failure with preserved ejection fraction. Nitric Oxide Biol Chem 69: 78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd AI, Gilchrist M, Winyard PG, Jones AM, Hallmann E, Kazimierczak R et al (2015). Effects of dietary nitrate supplementation on the oxygen cost of exercise and walking performance in individuals with type 2 diabetes: a randomized, double‐blind, placebo‐controlled crossover trial. Free Radic Biol Med 86: 200–208. [DOI] [PubMed] [Google Scholar]
- Shepherd AI, Wilkerson DP, Fulford J, Winyard PG, Benjamin N, Shore AC et al (2016). Effect of nitrate supplementation on hepatic blood flow and glucose homeostasis: a double‐blind, placebo‐controlled, randomized control trial. Am J Physiol Gastrointest Liver Physiol 311: G356–G364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiva S (2013). Nitrite: a physiological store of nitric oxide and modulator of mitochondrial function. Redox Biol 1: 40–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiva S, Rassaf T, Patel RP, Gladwin MT (2011). The detection of the nitrite reductase and NO‐generating properties of haemoglobin by mitochondrial inhibition. Cardiovasc Res 89: 566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L et al (2007). Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med 204: 2089–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiva S, Wang X, Ringwood LA, Xu X, Yuditskaya S, Annavajjhala V et al (2006). Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat Chem Biol 2: 486–493. [DOI] [PubMed] [Google Scholar]
- Siddiqi N, Neil C, Bruce M, MacLennan G, Cotton S, Papadopoulou S et al (2014). Intravenous sodium nitrite in acute ST‐elevation myocardial infarction: a randomized controlled trial (NIAMI). Eur Heart J 35: 1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silaghi‐Dumitrescu R (2004). Linkage isomerism in nitrite reduction by cytochrome cd1 nitrite reductase. Inorg Chem 43: 3715–3718. [DOI] [PubMed] [Google Scholar]
- da Silva G, Kennedy EM, Dlugogorski BZ (2006). Ab initio procedure for aqueous‐phase pK a calculation: the acidity of nitrous acid. J Phys Chem A 110: 11371–11376. [DOI] [PubMed] [Google Scholar]
- Simon MA, Vanderpool RR, Nouraie M, Bachman TN, White PM, Sugahara M et al (2016). Acute hemodynamic effects of inhaled sodium nitrite in pulmonary hypertension associated with heart failure with preserved ejection fraction. JCI Insight 1: e89620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh TK (1992). Serum ceruloplasmin in acute myocardial infarction. Acta Cardiol 47: 321–329. [PubMed] [Google Scholar]
- Sparacino‐Watkins CE, Lai Y‐C, Gladwin MT (2012). Nitrate–nitrite–nitric oxide pathway in pulmonary arterial hypertension therapeutics. Circulation 125: 2824–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparacino‐Watkins CE, Tejero J, Sun B, Gauthier MC, Thomas J, Ragireddy V et al (2014). Nitrite reductase and nitric‐oxide synthase activity of the mitochondrial molybdopterin enzymes mARC1 and mARC2. J Biol Chem 289: 10345–10358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srihirun S, Sriwantana T, Unchern S, Kittikool D, Noulsri E, Pattanapanyasat K et al (2012). Platelet inhibition by nitrite is dependent on erythrocytes and deoxygenation. PLoS One 7: e30380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taboy CH, Bonaventura C, Crumbliss AL (2002). [18] ‐ Anaerobic oxidations of myoglobin and hemoglobin by spectroelectrochemistry In: Sen CK, Packer L. (eds). Methods in Enzymology. Academic Press: New York, pp. 187–209. [DOI] [PubMed] [Google Scholar]
- Thomas DD, Ridnour LA, Isenberg JS, Flores‐Santana W, Switzer CH, Donzelli S et al (2008). The chemical biology of nitric oxide: Implications in cellular signaling. Free Radic Biol Med 45: 18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiso M, Tejero J, Basu S, Azarov I, Wang X, Simplaceanu V et al (2011). Human neuroglobin functions as a redox‐regulated nitrite reductase. J Biol Chem 286: 18277–18289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanin AF, Bevers LM, Slama‐Schwok A, van Faassen EE (2007). Nitric oxide synthase reduces nitrite to NO under anoxia. Cell Mol Life Sci 64: 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente JB, Colaço HG, Mendes MIS, Sarti P, Leandro P, Giuffrè A (2014). NO• binds human cystathionine β‐synthase quickly and tightly. J Biol Chem 289: 8579–8587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villacorta L, Gao Z, Schopfer FJ, Freeman BA, Chen YE (2016). Nitro‐fatty acids in cardiovascular regulation and diseases: characteristics and molecular mechanisms. Front Biosci Landmark Ed 21: 873–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitturi DA, Patel RP (2011). Current perspectives and challenges in understanding the role of nitrite as an integral player in nitric oxide biology and therapy. Free Radic Biol Med 51: 805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrancken K, Schroeder HJ, Longo LD, Power GG, Blood AB (2013). Role of ceruloplasmin in nitric oxide metabolism in plasma of humans and sheep: a comparison of adults and fetuses. Am J Physiol ‐ Regul Integr Comp Physiol 305: R1401–R1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrancken K, Schroeder HJ, Longo LD, Power GG, Blood AB (2016). Postprandial lipids accelerate and redirect nitric oxide consumption in plasma. Nitric Oxide 55–56: 70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajih N, Basu S, Jailwala A, Kim HW, Ostrowski D, Perlegas A et al (2017). Potential therapeutic action of nitrite in sickle cell disease. Redox Biol 12: 1026–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajih N, Liu X, Shetty P, Basu S, Wu H, Hogg N et al (2016). The role of red blood cell S‐nitrosation in nitrite bioactivation and its modulation by leucine and glucose. Redox Biol 8: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Krizowski S, Fischer‐Schrader K, Niks D, Tejero J, Sparacino‐Watkins C et al (2015). Sulfite oxidase catalyzes single‐electron transfer at molybdenum domain to reduce nitrite to nitric oxide. Antioxid Redox Signal 23: 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb A, Bond R, McLean P, Uppal R, Benjamin N, Ahluwalia A (2004). Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia–reperfusion damage. Proc Natl Acad Sci U S A 101: 13683–13688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AJ, Milsom AB, Rathod KS, Chu WL, Qureshi S, Lovell MJ et al (2008a). Mechanisms underlying erythrocyte and endothelial nitrite reduction to nitric oxide in hypoxia: role for xanthine oxidoreductase and endothelial nitric oxide synthase. Circ Res 103: 957–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AJ, Patel N, Loukogeorgakis S, Okorie M, Aboud Z, Misra S et al (2008b). Acute blood pressure lowering, vasoprotective, and antiplatelet properties of dietary nitrate via bioconversion to nitrite. Hypertension 51: 784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Giles LJ, Ruppelt C, Mendel RR, Bittner F, Kirk ML (2015). Oxyl and hydroxyl radical transfer in mitochondrial amidoxime reducing component‐catalyzed nitrite reduction. J Am Chem Soc 137: 5276–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Heinecke J, Tan H, Ford PC, Richter‐Addo GB (2009). The distal pocket histidine residue in horse heart myoglobin directs the O‐binding mode of nitrite to the heme iron. J Am Chem Soc 131: 18119–18128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Safo MK, Richter‐Addo GB (2008). The nitrite anion binds to human hemoglobin via the uncommon O‐nitrito mode. Biochemistry (Mosc) 47: 8247–8249. [DOI] [PubMed] [Google Scholar]