

Abstract
Atomically resolved crystal structures not only suffer from the inherent uncertainty in accurately locating H atoms but also are incapable of fully revealing the underlying forces enabling the formation of final structures. Therefore, the development and application of novel techniques to illuminate intermolecular forces in crystalline solids are highly relevant to understand the role of hydrogen atoms in structure adoption. Novel developments in 1H NMR MAS methodology can now achieve robust measurements of 1H chemical shift anisotropy (CSA) tensors which are highly sensitive to electrostatics. Herein, we use 1H CSA tensors, measured by MAS experiments and characterized using DFT calculations, to reveal the structure-driving factors between the two polymorphic forms of acetaminophen (aka Tylenol or paracetamol) including differences in hydrogen bonding and the role of aromatic interactions. We demonstrate how the 1H CSAs can provide additional insights into the static picture provided by diffraction to elucidate rigid molecules.
Graphical Abstract
Hydrogen atoms often mediate (de)stabilizing interactions, which drive structure, function, and dynamics in numerous chemical systems. While X-ray diffraction is an excellent source of information for systems of periodic structure, it fails to accurately locate hydrogen atoms, leaving important structural determinants, such as the hydrogen bond, poorly defined. Solid-state NMR spectroscopy is suitable for structural and dynamic insights in this regard; however, 1H NMR spectra in the solid state have historically exhibited insufficient resolution due to severe spectral broadening caused by strong homogeneous 1H–1H dipolar interactions. Recent developments in magic angle spinning (MAS) technology have dramatically improved the situation, achieving spinning frequencies (>100 kHz) that can provide atomic resolution even for the most rigid systems.1 Recent advances in methodology1 and applications2–7 have demonstrated the power of proton-detected techniques. Of notable interest for structural studies are techniques used to measure the 1H chemical shift anisotropy (CSA) tensor as a means to detect the magnetic tensorial contributions arising from the local electronic structure.8–11 These experiments quantify the CSA, ζ = δzz − δiso, and the asymmetry parameter η = (δxx‒δyy)/ζ; where δiso is the isotropic chemical shift and δii are the principal components of the CSA tensor ranked according to |δzz – δiso| ≥ |δxx – δiso| ≥ |δyy – δiso. Qualitatively, the CSA is then the magnetic tensorial value which deviates most from the isotropically averaged frequency (δiso), and η gives a sense for the shape of the full CSA tensor based on the other components. The CSA is of general interest for structural elucidation as it provides an orientation-dependent metric connecting 1H nuclei and their electronic landscape, which are typically detected independent of one another by neutron and X-ray diffraction. As such, the 1H CSA has the potential to resolve ambiguities from X-ray crystallography—an approach that was successfully demonstrated for the structural refinement of hydrogen bonded atomic positions in L-ascorbic acid.6 Due to the relative infancy of robust methods to detect the 1H CSA, limited experimental data has been measured to establish trends regarding different structural constraints. The influence of strong hydrogen bonding was explored in earlier work through computation and limited experimental results,12–17 which have been confirmed by recent measurements.18 Nevertheless, many open questions remain unaddressed regarding its sensitivity to weak hydrogen bonds, e.g., CH⋯O bonding, and other electrostatic forces such as π–π interactions, which also participate in structure formation. To explore such questions, determining the 1H CSAs for structurally distinct arrangements of the same molecule (i.e., polymorphic crystals) could establish trends without confounding influences from differences in covalent bonding. This approach is adopted herein by employing 2D CSA/CS correlation experiments under 90 kHz MAS to probe structural differences for two polymorphic forms of the drug acetaminophen (ACM) (Table 1).
Table 1.
Isotropic and Anisotropic Chemical Shift Parameters for Protons in the Monoclinic and Orthorhombic Polymorphs of Acetaminophen Measured from 90 kHz MAS NMR Using a 2D CSA/CS Pulse Sequence with and without (Denoted As *) 14N Decouplinga
| orthorhombic | OH | b | f,c | e | CH3 |
|---|---|---|---|---|---|
| δiso (ppm) | 9.4 | 7.1 | 4.7 | 6.3 | 1.8 |
| |ζ| (ppm) | 15.3 | 6.7 | 6.3 | 5.8 | 3.0 |
| η | 0.3 | 0.6 | 0.7 | 0.8 | 1.0 |
| |ζ| (ppm) w/o decoupling* | 16 | 7.2 | 6.7 | 7.1 | 3.7 |
| η w/o decoupling | 0.3 | 0.7 | 0.8 | 0.8 | 1.0 |
| monoclinic | OH | b | f,c | e | CH3 |
|---|---|---|---|---|---|
| δiso (ppm) | 9.4 | 8.0 | 6.8 | 5.8 | 1.1 |
| |ζ| (ppm) | 16.4 | 7.4 | 5.6 | 4.8 | 5.0 |
| η | 0.4 | 0.6 | 0.7 | 1.0 | 1.0 |
| |ζ| (ppm) w/o decoupling* | 16.0 | 7.2 | 5.6 | 4.8 | 5.0 |
| η w/o decoupling* | 0.5 | 0.6 | 0.7 | 1.0 | 1.0 |
The pulse sequence is given in the SI. CSA values obtained from DFT calculations are given in Tables S1 and S2.
The orthorhombic and monoclinic forms of acetaminophen are stable under ambient conditions. The structural differences (see Figure 1) between the two polymorphs make acetaminophen a good model to study polymorphism. In addition, acetaminophen contains chemically diverse functional groups, including a hydroxyl, amine, aromatic, and methyl group that exert varying intermolecular effects. Thermodynamically, the two polymorphs have an enantiotropic predicted transition point at −165 °C by DSC, above which the monoclinic form is favored due to a larger entropic contribution.19 While the thermodynamically stable monoclinic is easily isolated through evaporation of many solvents, pure crystallization of the metastable orthorhombic polymorph is more difficult. Many researchers have focused on finding ways to increase the scale of this polymorphic form through the use of different cooling strategies and by introducing functionalized surfaces or additives to the system.20–24 In the course of this study, we discovered a novel technique for consistently producing pure orthorhombic ACM from water at 0 °C, alluding to an Ostwald’s rule of stages scenario (see Supporting Information (SI) for details on crystal growth).
Figure 1.
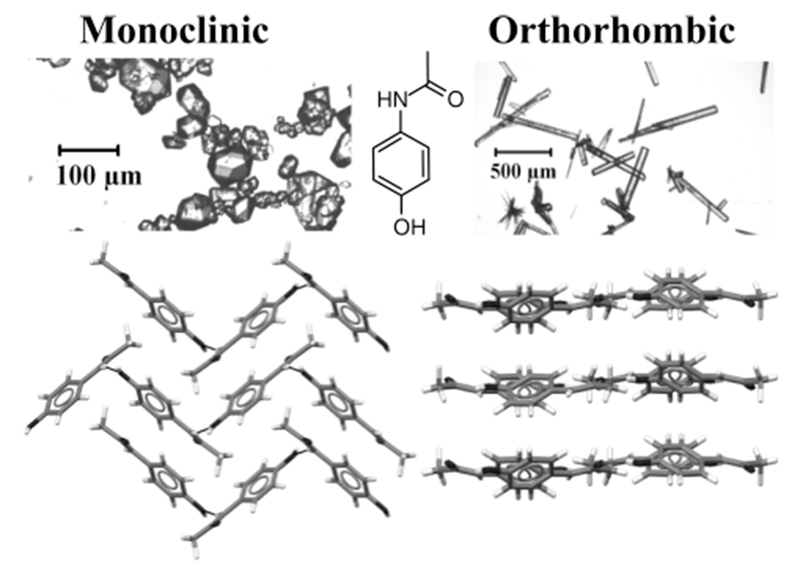
Crystal structures illustrating packing of monoclinic (left) and orthorhombic (right) polymorphs of acetaminophen. The lines display the intermolecular hydrogen bonding network, which is pleated with respect to the molecule for the monoclinic while flat for the orthorhombic in layered planes. FTIR and Raman spectra for both crystalline forms are given in SI Figures S1 and S2.
The primary structural constraint in both forms is the hydrogen bond network composed of the hydroxyl group, which serves simultaneously as a donor and acceptor forming OH⋯O═C and NH⋯OH hydrogen bonds. For the orthorhombic form, this network lies in plane with the aromatic groups, forming stacked sheets, whereas in the monoclinic structure, it forms a two-dimensional ribbon causing buckled planes between the aromatic groups. Despite the different geometries, both IR and Raman spectroscopy, common techniques for bonding assessment, resulted in nearly identical spectra (see SI Figures S1 and S2). The 1H solid-state NMR spectra (Figures 2 and 3), on the other hand, are remarkably sensitive to the polymorphic changes. Given that the bond lengths, bond angles, and torsion angles are approximately the same for an individual molecule in each polymorph, we attribute the bulk of the chemical shift differences to intermolecular effects, the interpretation of which is discussed below.
Figure 2.
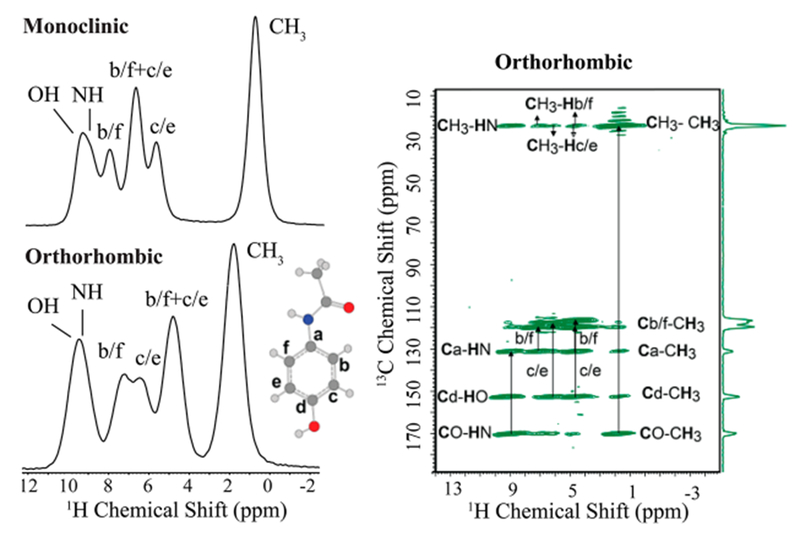
Left: 1H spin–echo NMR spectra collected under 90 kHz MAS of the two acetaminophen polymorphs with resonance assignments. Significant differences observed in the shape and frequency of spectral lines clearly demonstrate the sensitivity of 1H chemical shift to structural differences between the two crystalline forms. Right: two-dimensional 13C–1H HETCOR spectrum obtained under 70 kHz MAS confirming the resonance assignment of orthorhombic polymorph. A full discussion of the resonance assignment is also given in the SI.
Figure 3.
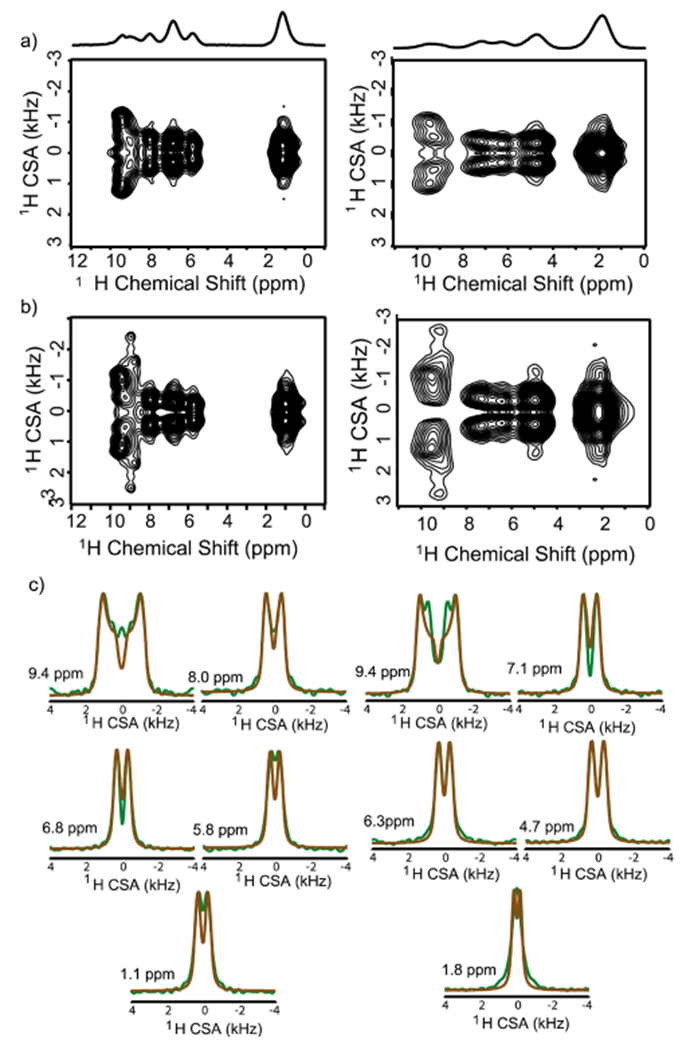
Two-dimensional 1H/1H CSA/CS correlation spectra obtained using the pulse sequence shown in SI Figure S3 under 90 kHz MAS with (a) and without (b) 14N decoupling for the orthorhombic (right) and monoclinic (left) polymorphs of acetaminophen. The influence of 14N–1H dipolar couplings are marked by the broadening and appearance of satellite peaks in panel b, with a greater magnitude for the orthorhombic. Slices from CSA (or the ω1) dimension and numerically simulated fits using SIMPSON25 are given in panel c.
A critical feature of the 2D radiofrequency pulse sequence employed is that it simultaneously recouples the CSA and the through-space heteronuclear dipolar couplings, encoding in the indirect dimension a line shape that contains both contributions. To achieve CSA-only line shapes, heteronuclear decoupling must be applied to all other abundant nuclei in the sample, which is required for 14N nuclei in ACM. Due to the very large magnitude of the 14N quadrupolar interaction, 14N decoupling can be difficult to accomplish experimentally. However, it was recently shown that, under conditions of fast spinning and moderately high RF field strengths, the 14N decoupling becomes quite efficient.8 A comparison of the 2D spectra acquired with (Figure 3a) and without (Figure 3b) 14N decoupling, however, can be utilized for spectral assignment and qualitative structural assessment. For the second-most downfield peak in both forms, the indirect dimension clearly marks the NH resonance with the appearance of satellite peaks in the undecoupled spectrum. In addition, the CSA values for all other protons in the orthorhombic form are perturbed under heteronuclear dipolar evolution, while the values in the monoclinic, except the NH and OH peaks, are virtually unaffected. The apparent stronger 14N–1H dipolar couplings in the orthorhombic form, particularly in the aromatic region, are direct evidence of its denser and/or more rigid structure. Indeed, simulations including the heteronuclear 14N–1H dipolar couplings based on static distances in the crystal structures for the nearest 14N neighbor of each proton demonstrate that the line-shapes should be similarly influenced for both polymorphs in the aromatic region (see SI Figures S6 and S7). This implies that the monoclinic must undergo increased molecular/librational motion on the NMR time scale to reduce the 14N–1H dipolar couplings. A measure of the CSA with and without 14N decoupling provides a qualitative indication of the dynamic features (or structural disorder) between the two forms, showing that the monoclinic undergoes increased dynamics. While molecular motion also averages the CSA, thereby narrowing the line shape in the observed CSA spectra, without precise, a priori knowledge of the geometry of the electrostatic field causing the CSA, comparing the extent of motional averaging between the two polymorphs is difficult from the CSA alone.
The 1H CSA is known to be quite sensitive to hydrogen bonding as the nuclear and electronic coordinates become distorted in hydrogen bond formation,26 resulting in a less shielded nucleus, larger isotropic CS values, and an increase in the magnitude of the CSA.12–15 Typically the CSA in strong hydrogen bonds is negative by the convention reported here.27–29 Unfortunately, due to the γ-encoded symmetry-based recoupling scheme used, only the magnitude, not the sign, of the CSA could be measured. As such, we performed DFT calculations to support the experimental results (see computational details and Figure S4 in the SI). Between the two forms, the hydrogen bond donor/acceptor distances in the primary hydrogen bond network are only slightly shorter for the monoclinic. For NH⋯Ohydroxyl, the heavy atom distances are 2.97 and 2.93 Å for the orthorhombic and monoclinic, respectively, while the OH⋯O donor/acceptor distances are 2.73 Å (ortho) and 2.66 Å (mono). The hydroxyl protons have very similar isotropic CS values, which is consistent with the IR data, despite the 0.13 Å closer donor-acceptor distance. The CSA, however, is 0.9 ppm larger for the monoclinic OH, suggesting a slightly stronger bond strength. The DFT calculations confirm that the CSA for the two positions is due to hydrogen bonding as they are both negative (see Tables S1 and S2 in SI). The result demonstrates that the 1H CSA is capable of measuring minute hydrogen bonding differences. More substantial differences in hydrogen bonding are found for the aromatic H atoms. The aromatic protons are well-resolved between the two forms with the largest CSA and most deshielded isotropic chemical shift corresponding to position b in both polymorphs. This is closest to the intramolecular carbonyl (Caro⋯OC═O distances of 2.28 Å in ortho, and 2.31 Å for mono) and within the limits for CH⋯O hydrogen bonding.30 Monoclinic ACM has an additional intermolecular contact (2.89 Å) to a carbonyl oxygen which could explain its 0.8 ppm downfield CS and 0.7 ppm larger CSA. In addition, the sign of the computed CSA is negative for the monoclinic and positive for the orthorhombic, supporting a stronger hydrogen bond argument in the monoclinic (see Table S1 in the SI). A similar effect is found for position e where both forms have Caro⋯OC═O distances at 2.85 Å (ortho) and 2.92 Å (mono), but in the orthorhombic a nitrogen is also nearby at 2.86 Å, likely contributing to its downfield shift (both resonances have negative computed CSA values). For both the b and e aromatic positions, the CS and CSA indicate that a proton in one form but not the other is involved in a ternary bond complex, which causes the differences in the NMR parameters.
The aromatic protons are also influenced by ring current effects, which exert a strong influence on nuclear shielding. The interactions of neighboring ring currents are the source for the substantial CS differences at the overlapping f and c positions between the two polymorphs. The influence of aromatic rings on the CSA tensor depends on the orientation of the ring with respect to the proton: the shielding field is perpendicular, while the deshielding is parallel to the plane of the ring. The atypical upfield 4.7 ppm isotropic value of the f and c protons in the orthorhombic form suggests that the protons point toward the face of other rings in the shielded orientation. This is exactly what is found in the orthorhombic, with f and c positioned at distances of 2.70 and 2.81 Å to the centroid of neighboring rings. This effect leads to the largest CS difference in the system where the same positions in the monoclinic are 2 ppm downfield and have no close contacts, but also have a 0.7 ppm smaller CSA. A similar effect is found for the methyl group where, despite CH3 rotations, finite CSA contributions can be measured and the isotropic values between the two forms also differ by 0.7 ppm. For the monoclinic, the CH3 group faces a neighboring aromatic group with the methyl carbon being 3.8 Å from the aromatic centroid. In the orthorhombic, the methyl group is relatively isolated from any neighboring groups.
The NMR results show that the 1H CSA is quite sensitive to differences in the electrostatic fields imposed by different polymorphic arrangements, which help to illuminate the static picture provided by X-ray crystallography. The CSA response to hydrogen bonding, both weak and strong, differentiates the two polymorphs; IR spectroscopy was much less sensitive in this regard. In addition, the rather strong influence of ring current effects on the CSA provides an excellent metric for π–π interactions. In full consideration of the data, a justification for the previously reported free energy differences can be proposed. First, the NH⋯OH⋯O hydrogen bonding network is similar in strength, slightly favoring the monoclinic as evidenced by the CSA. In addition, CHaro⋯O/N bonding was found in both forms, with varying strengths, the strongest of which, based on deshielding and CSA magnitude, involved an intra/intermolecular ternary complex for the CHb resonance in monoclinic ACM. Despite the comparatively weak nature of CH⋯O hydrogen bonds, it has been shown that they are of structural significance in many crystal structures31 and are stronger when the acceptor oxygen is also involved in a stronger hydrogen bond,32,33 as is the case here. The CSA parameters then suggest that the overall hydrogen bond formation in the monoclinic is potentially a bit stronger than orthorhombic ACM. Additional contributions from the aromatic groups were also identified. In the orthorhombic form, the rings within a layer are sandwiched by two other rings on opposite ends, which is reflected in the substantial difference between the CSA parameters for CHf and CHc positions. This could account for the stronger contribution of the 14N–1H dipolar couplings in the undecoupled spectra, caused by a more rigid structure. The lack of such a constraint in the monoclinic ACM provides it with more degrees of freedom, consistent with previous findings.19
In this contribution, we have demonstrated that polymorphic differences are captured by the 1H CSA, including changes in hydrogen bond structures, aromatic π electrons constraints, and methyl group interactions. Interpretations for differences imposed by the polymorphic changes on the CSA values were provided based on the reported crystal structures. The results demonstrate the remarkable potential of 1H ultrafast MAS NMR to significantly augment crystal structures obtained by diffraction techniques through the direct measures of hydrogen atoms across a range of chemically distinct groups. In addition, this study has enabled a rationale for the stabilization of the monoclinic structure and the basis for the formation of the metastable orthorhombic forms of ACM.
EXPERIMENTAL SECTION
All ACM 2D CSA/CS data were acquired on a JNM-ECZ600R spectrometer (JEOL RESONANCE Inc., Japan) at 600 MHz 1H Larmor frequency using a JEOL RESONANCE Inc. 0.75 mm double-resonance MAS probe under 90 kHz ± 20 Hz MAS at room temperature (25 °C). Six scans were used with a recycle delay of 120 s and 32 t1 points in all 2D experiments. Resonance assignments, supporting NMR experiments, description of DFT calculations and NMR simulations can be found in the SI along with details on sample preparation.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of Health (RO1 GM106180 A.J.M. and GM084018 to A.R.) and funds to purchase the 600 MHz and to upgrade the 400 MHz solid-state NMR spectrometers to A.R. This material is based upon the work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE #1256260 (to J.T.D.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.7b01650.
Crystallization conditions for the polymorphs of acetaminophen, IR and Raman spectroscopic characterization of the two forms, details of solid-state NMR experiments, tables of data for solid-state NMR experiments, and details of computational work (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Nishiyama Y Fast Magic-Angle Sample Spinning Solid-State NMR at 60–100 kHz for Natural Abundance Samples. Solid State Nucl. Magn. Reson 2016, 78, 24–36. [DOI] [PubMed] [Google Scholar]
- (2).Fricke P; Chevelkov V; Zinke M; Giller K; Becker S; Lange A Backbone Assignment of Perdeuterated Proteins by Solid-State NMR Using Proton Detection and Ultrafast Magic-Angle Spinning. Nat. Protoc 2017, 12, 764–782. [DOI] [PubMed] [Google Scholar]
- (3).Stanek J; Andreas LB; Jaudzems K; Cala D; Lalli D; Bertarello A; Schubeis T; Akopjana I; Kotelovica S; Tars K; et al. NMR Spectroscopic Assignment of Backbone and Side-Chain Protons in Fully Protonated Proteins: Microcrystals, Sedimented Assemblies, and Amyloid Fibrils. Angew. Chem., Int. Ed 2016, 55, 15504–15509. [DOI] [PubMed] [Google Scholar]
- (4).Asami S; Reif B Proton-Detected Solid-State NMR Spectroscopy at Aliphatic Sites: Application to Crystalline Systems. Acc. Chem. Res 2013, 46, 2089–2097. [DOI] [PubMed] [Google Scholar]
- (5).Agarwal V; Penzel S; Szekely K; Cadalbert R; Testori E; Oss A; Past J; Samoson A; Ernst M; Bockmann A; et al. De Novo 3D Structure Determination from Sub-Milligram Protein Samples by Solid-State 100 kHz MAS NMR Spectroscopy. Angew. Chem., Int. Ed 2014, 53, 12253–12256. [DOI] [PubMed] [Google Scholar]
- (6).Miah HK; Cresswell R; Iuga D; Titman JJ 1H CSA Parameters by Ultrafast MAS NMR: Measurement and Applications to Structure Refinement. Solid State Nucl. Magn. Reson 2017, DOI: 10.1016/j.ssnmr.2017.02.002. [DOI] [PubMed] [Google Scholar]
- (7).Jain MG; Lalli D; Stanek J; Gowda C; Prakash S; Schwarzer TS; Schubeis T; Castiglione K; Andreas LB; Madhu PK; et al. Selective 1H- 1H Distance Restraints in Fully Protonated Proteins by Very Fast Magic-Angle Spinning Solid-State NMR. J. Phys. Chem. Lett 2017, 8, 2399–2405. [DOI] [PubMed] [Google Scholar]
- (8).Pandey MK; Nishiyama Y Determination of NH Proton Chemical Shift Anisotropy with 14N-1H Heteronuclear Decoupling Using Ultrafast Magic Angle Spinning Solid-State NMR. J. Magn. Reson 2015, 261, 133–140. [DOI] [PubMed] [Google Scholar]
- (9).Zhang R; Mroue KH; Ramamoorthy A Proton Chemical Shift Tensors Determined by 3D Ultrafast MAS Double-Quantum NMR Spectroscopy. J. Chem. Phys 2015, 143, 144201–144201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hou G; Gupta R; Polenova T; Vega AJ A Magic-Angle-Spinning NMR Spectroscopy Method for the Site-Specific Measurement of Proton Chemical-Shift Anisotropy in Biological and Organic Solids. Isr. J. Chem 2014, 54, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wu CH; Ramamoorthy A; Gierasch LM; Opella SJ Simultaneous Characterization of the Amide 1H Chemical Shift, 1H-15N Dipolar, and 15N Chemical Shift Interaction Tensors in a Peptide Bond by Three-Dimensional Solid-State NMR Spectroscopy. J. Am. Chem. Soc 1995, 117, 6148–6149. [Google Scholar]
- (12).Parker LL; Houk AR; Jensen JH Cooperative Hydrogen Bonding Effects Are Key Determinants of Backbone Amide Proton Chemical Shifts in Proteins. J. Am. Chem. Soc 2006, 128, 9863–9872. [DOI] [PubMed] [Google Scholar]
- (13).Sharma Y; Kwon OY; Brooks B; Tjandra N An Ab Initio Study of Amide Proton Shift Tensor Dependence on Local Protein Structure. J. Am. Chem. Soc 2002, 124, 327–335. [DOI] [PubMed] [Google Scholar]
- (14).Rohlfing CM; Allen LC; Ditchfield R Proton Chemical Shift Tensors in Hydrogen-Bonded Dimers of RCOOH and ROH. J. Chem. Phys 1983, 79, 4958–4966. [Google Scholar]
- (15).Suzuki Y; Takahashi R; Shimizu T; Tansho M; Yamauchi K; Williamson MP; Asakura T Intra- and Intermolecular Effects on 1H Chemical Shifts in a Silk Model Peptide Determined by High-Field Solid State 1H NMR and Empirical Calculations. J. Phys. Chem. B 2009, 113, 9756–9761. [DOI] [PubMed] [Google Scholar]
- (16).Tjandra N; Bax A Solution NMR Measurement of Amide Proton Chemical Shift Anisotropy in 15N-Enriched Proteins. Correlation with Hydrogen Bond Length. J. Am. Chem. Soc 1997, 119, 8076–8082. [Google Scholar]
- (17).Yao L; Grishaev A; Cornilescu G; Bax A The Impact of Hydrogen Bonding on Amide 1H Chemical Shift Anisotropy Studied by Cross-Correlated Relaxation and Liquid Crystal NMR Spectroscopy. J. Am. Chem. Soc 2010, 132, 10866–10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Miah HK; Bennett DA; Iuga D; Titman JJ Measuring Proton Shift Tensors with Ultrafast MAS NMR. J. Magn. Reson 2013, 235, 1–5. [DOI] [PubMed] [Google Scholar]
- (19).Sacchetti M Thermodynamic Analysis of DSC Data for Acetaminophen Polymorphs. J. Therm. Anal. Calorim 2001, 63, 345–350. [Google Scholar]
- (20).Agnew LR; McGlone T; Wheatcroft HP; Robertson A; Parsons AR; Wilson CC Continuous Crystallization of Paracetamol (Acetaminophen) Form II: Selective Access to a Metastable Solid Form. Cryst. Growth Des 2017, 17, 2418–2427. [Google Scholar]
- (21).Kaur Bhangu S; Ashokkumar M; Lee J Ultrasound Assisted Crystallization of Paracetamol: Crystal Size Distribution and Polymorph Control. Cryst. Growth Des 2016, 16, 1934–1941. [Google Scholar]
- (22).Mikhailenko MA Growth of Large Single Crystals of the Orthorhombic Paracetamol. J. Cryst. Growth 2004, 265, 616–618. [Google Scholar]
- (23).Al-Zoubi N; Nikolakakis I; Malamataris S Crystallization Conditions and Formation of Orthorhombic Paracetamol from Ethanolic Solution. J. Pharm. Pharmacol 2002, 54, 325–333. [DOI] [PubMed] [Google Scholar]
- (24).Delmas T; Shah UV; Roberts MM; Williams DR; Heng JYY Crystallisation of the Orthorhombic Form of Acetaminophen: Combined Effect of Surface Topography and Chemistry. Powder Technol 2013, 236, 24–29. [Google Scholar]
- (25).Bak M; Rasmussen JT; Nielsen NC SIMPSON: A General Simulation Program for Solid-State NMR Spectroscopy. J. Magn. Reson 2000, 147, 296–330. [DOI] [PubMed] [Google Scholar]
- (26).Steiner T The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed 2002, 41, 48–76. [DOI] [PubMed] [Google Scholar]
- (27).Van Hecke P; Weaver JC; Neff BL; Waugh JS Determination of the Proton Chemical Shielding Tensor in Anhydrous A-oxalic Acid by Multiple Pulse NMR. J. Chem. Phys 1974, 60, 1668–1670. [Google Scholar]
- (28).Wu G; Freure CJ; Verdurand E Proton Chemical Shift Tensors and Hydrogen Bond Geometry: A 1H-2H Dipolar NMR Study of the Water Molecule in Crystalline Hydrates. J. Am. Chem. Soc 1998, 120, 13187–13193. [Google Scholar]
- (29).Kibalchenko M; Lee D; Shao L; Payne MC; Titman JJ; Yates J R Distinguishing Hydrogen Bonding Networks in α-D-Galactose Using NMR Experiments and First Principles Calculations. Chem. Phys. Lett 2010, 498, 270–276. [Google Scholar]
- (30).Veljkovic DZ; Janjic GV; Zaric SD Are C-H/O Interactions Linear? The Case of Aromatic CH Donors. CrystEngComm 2011, 13, 5005. [Google Scholar]
- (31).Taylor R It Isn’t, It Is: The C-H⋯X (X = O, N, F, Cl) Interaction Really Is Significant in Crystal Packing. Cryst. Growth Des 2016, 16, 4165–4168. [Google Scholar]
- (32).Dragelj JL; Stanković IM; Božinovski DM; Meyer T; Veljković DZ; Medaković VB; Knapp E-W; Zarić SD C-H/O Interactions of Aromatic CH Donors within Proteins: A Crystallographic Study. Cryst. Growth Des 2016, 16, 1948–1957. [Google Scholar]
- (33).Dragelj JL; Janjić GV; Veljković DŽ; Zarić SD Crystallographic and Ab Initio Study of Pyridine CH-O Interactions: Linearity of the Interactions and Influence of Pyridine Classical Hydrogen Bonds. CrystEngComm 2013, 15, 10481. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.