

Abstract
Homogalacturonan (HG) is a pectic glycan in the plant cell wall that contributes to plant growth and development and cell wall structure and function, and interacts with other glycans and proteoglycans in the wall. HG is synthesized by the galacturonosyltransferase (GAUT) gene family. Two members of this family, GAUT1 and GAUT7, form a heteromeric enzyme complex in Arabidopsis thaliana. Here, we established a heterologous GAUT expression system in HEK293 cells and show that co-expression of recombinant GAUT1 with GAUT7 results in the production of a soluble GAUT1:GAUT7 complex that catalyzes elongation of HG products in vitro. The reaction rates, progress curves, and product distributions exhibited major differences dependent upon small changes in the degree of polymerization (DP) of the oligosaccharide acceptor. GAUT1:GAUT7 displayed >45-fold increased catalytic efficiency with DP11 acceptors relative to DP7 acceptors. Although GAUT1:GAUT7 synthesized high-molecular-weight polymeric HG (>100 kDa) in a substrate concentration–dependent manner typical of distributive (nonprocessive) glycosyltransferases with DP11 acceptors, reactions primed with short-chain acceptors resulted in a bimodal product distribution of glycan products that has previously been reported as evidence for a processive model of GT elongation. As an alternative to the processive glycosyltransfer model, a two-phase distributive elongation model is proposed in which a slow phase, which includes the de novo initiation of HG and elongation of short-chain acceptors, is distinguished from a phase of rapid elongation of intermediate- and long-chain acceptors. Upon reaching a critical chain length of DP11, GAUT1:GAUT7 elongates HG to high-molecular-weight products.
Keywords: plant cell wall, glycosyltransferase, protein complex, enzyme mechanism, processivity, galacturonosyltransferase, homogalacturonan, pectic glycan, pectin, polygalacturonide transferase
Introduction
Homogalacturonan (HG)3 is a plant cell wall polysaccharide and glycan component of more complex polysaccharides and proteoglycans that contributes to the structure and mechanical strength of the wall and has roles in plant growth, development, morphology, and response to biotic and abiotic stress (1–4). HG is a linear homopolymer of 1,4-linked α-d-galactopyranosyluronic acid (GalA) that may be partially methylesterified at O-6 and acetylated at O-2 and O-3. It is the simplest and most abundant glycan in the family of cell wall polysaccharides known as pectins, which include HG, rhamnogalacturonan I and rhamnogalacturonan II (1).
The pectic polysaccharides make contacts with each other (1), cellulose (5–7), hemicelluloses (1, 5), and proteoglycans (2) within the wall through covalent and noncovalent interactions. The presence of pectin provides a barrier to the deconstruction of cellulosic biomass and partially obstructs degradative enzymes from accessing cellulose and hemicelluloses during the processing and saccharification of plant biomass (1, 8–10). Reducing the content of pectin in woody and grass biofuel feedstocks has been shown to enhance biofuel production by increasing both biomass yield and the recovery of sugar (10–12). Our current understanding of pectin synthesis and structure, however, is insufficient to explain its diverse functions in the plant or how reductions to pectin synthesis correlate with phenotypes such as increased plant growth and reduced recalcitrance to deconstruction.
The first gene identified encoding an HG biosynthetic enzyme was galacturonosyltransferase 1 (GAUT1), a CAZy (13) family 8 glycosyltransferase (GT). The in vitro synthesis of HG or homogalacturonan:galacturonosyltransferase (HG:GalAT) activity was mapped to GAUT1 following MS sequencing of Arabidopsis thaliana solubilized membrane proteins (1, 14). Subsequent co-immunoprecipitation, MS sequencing, and bimolecular fluorescence complementation revealed that GAUT1 functions as a disulfide-linked heterocomplex with the homologous protein, GAUT7 (15). In vivo, GAUT1 is truncated at its N terminus by 167 residues and requires GAUT7 for localization in the Golgi (15). Based on these results, GAUT7 was proposed to be a noncatalytic membrane anchor for GAUT1 (14, 15). A more detailed study of the function of the GAUT1:GAUT7 complex and the mechanism of HG synthesis required the establishment of a recombinant protein expression system that could co-express GAUT1 and GAUT7 and properly post-translationally fold and process the active GT complex.
Progress in studying plant cell wall biosynthesis has been hampered by difficulties associated with expressing and biochemically characterizing the relevant glycosyltransferases (16, 17). Because plant cell wall biosynthetic GTs are typically large N-glycosylated proteins that contain hydrophobic transmembrane regions and may exist as multienzyme complexes, most successful purifications of such recombinant GTs have been achieved using eukaryotic expression systems, namely Nicotiana benthamiana (18–21), Pichia pastoris (16, 22, 23), and HEK293 cells (14, 24, 25), with the latter being a particularly successful host for recombinant expression and enzymatic characterization of soluble forms of eukaryotic GTs (26–32). Here, we show that the HEK293 cell system can be used to express the heteromeric, disulfide-linked GAUT1:GAUT7 complex from A. thaliana in sufficient quantities to enable a comprehensive characterization of its enzymatic properties.
Heteromeric and homomeric GT complexes have been identified in glycan biosynthetic pathways shared by model eukaryotic organisms, including Homo sapiens, Saccharomyces cerevisiae, and A. thaliana (33). GT complexes have diverse biological functions and appear in N- and O-linked glycan, proteoglycan, and glycolipid synthesis pathways (33). In addition to the GAUT1:GAUT7 complex, at least six examples of proven and putative GT complexes involved in the synthesis of plant cell wall glycans are known (34–36). Various hypotheses for the biological significance of GT complexes have been proposed, including enhancement of enzymatic activity, substrate channeling, and Golgi localization (33, 34).
Here we demonstrate that the GAUT1:GAUT7 complex can synthesize high-molecular-weight (MW) HG polysaccharides in vitro, and we expand upon the original model of the GAUT1:GAUT7 complex (15). We propose a two-phase model of polysaccharide elongation in which short-chain acceptors are elongated slowly, followed by a rapid elongation phase once the glycans reach an intermediate, critical degree of polymerization. This model reconciles data showing that HG extracted from plant cell walls consists of long polymers containing >100 GalA units (37) with reports that in vitro HG elongation occurs through a nonprocessive mechanism (37–40). The activity reported also provides a basis for the initiation of HG polysaccharides by the GAUT1:GAUT7 complex and suggests that GAUT7 has a previously unrecognized role in contributing to the synthesis of high-MW polysaccharides.
Results
GAUT1 and GAUT7 form a complex when co-expressed in HEK293F cells
The co-immunoprecipitation of GAUT1 with GAUT7 from A. thaliana solubilized membranes revealed that GAUT1 and GAUT7 function as a GAUT1:GAUT7 heterocomplex covalently linked by disulfide bonds (15). In an effort to obtain sufficient amounts of purified, soluble GAUT1:GAUT7 complex, recombinant GAUT1 and GAUT7 constructs were heterologously co-expressed in HEK293F suspension culture cells, and the GAUT1:GAUT7 complex was characterized following purification.
Individual GAUT1 and GAUT7 constructs lacking their N-terminal transmembrane domains (GAUT1Δ167 and GAUT7Δ43) were generated as secreted fusion proteins harboring chimeric N-terminal fusion tags. GAUT1 purified from A. thaliana membranes is N-terminally truncated by 167 residues in vivo (15). The fusion tags were composed of a signal sequence, His8 tag, AviTag, superfolder GFP, and tobacco etch virus (TEV) protease recognition site followed by the respective GAUT domains using a strategy previously employed for the expression of a large library of mammalian glycosylation enzyme expression constructs (32). The constructs were transfected into HEK293F cells, and the secreted fusion proteins were purified from the medium by Ni2+-Sepharose affinity chromatography.
Co-expression of GAUT1Δ167 and GAUT7Δ43 (hereafter referred to as GAUT1 and GAUT7) resulted in the production of secreted GAUT1:GAUT7 complex (Fig. 1A, lane 7), which required reducing conditions (+DTT) to separate GAUT1 and GAUT7 into monomers (Fig. 1A, lane 2). Treatment of the complex with peptide:N-glycosidase F (PNGase F) led to a reduction in the molecular weight of both GAUT1 and GAUT7, indicating that both proteins are N-glycosylated (Fig. 1A, lanes 3 and 5). From a 1-liter culture, a total of 47.5 mg of the enzyme complex was purified.
Figure 1.

Heterologous expression of the GAUT1:GAUT7 complex. A, Coomassie Blue–stained SDS-polyacrylamide gel of purified GAUT1Δ167 co-expressed with GAUT7Δ43 detected under reducing (+DTT, lanes 2–5) and nonreducing (−DTT, lanes 7–10) conditions. Incubation with TEV protease (+TEV, lanes 4 and 5 and lanes 9 and 10) removes N-terminal GFP/His8 tags, which electrophorese as ∼30 kDa bands post-cleavage. Incubation with PNGase F (+PNGase F, lanes 3, 5, 8, and 10) removes N-glycosylation. After 6 days of incubation, medium from transfected HEK293F cells was purified by nickel-affinity chromatography, protein concentration was quantified by UV-visible spectroscopy, and 4 μg of purified protein was separated by SDS-PAGE after overnight incubation with or without TEV and PNGase F. The locations of the GAUT1:GAUT7 complex after TEV and PNGase F treatment under nonreducing conditions and the GAUT1 and GAUT7 monomers under reducing conditions are designated on the right. B, Coomassie Blue–stained SDS-polyacrylamide gel of purified GAUT1 (lanes 3 and 5) compared with the GAUT1:GAUT7 complex (lanes 2 and 4). Proteins were detected under reducing (+DTT, lanes 2 and 3) and nonreducing (−DTT, lanes 4 and 5) conditions.
Expression of GAUT1 alone resulted in the secretion of a GAUT1 homocomplex that could be purified and identified by SDS-PAGE under nonreducing conditions (Fig. 1B, lane 5). However, heterologous expression of GAUT1 in the absence of GAUT7 also resulted in the formation of large amounts of truncation products and high-molecular weight aggregates (Fig. 1B, lanes 3 and 5). Expression of GAUT7 alone led to no detectable secretion of the GAUT7 fusion protein into the culture medium. These results suggest that co-expression of GAUT1 and GAUT7 and the formation of a disulfide-bonded heterocomplex is required for proper folding and secretion of the active GAUT1:GAUT7 complex.
Recombinant GAUT1:GAUT7 catalyzes the transfer of GalA residues from UDP-GalA onto homogalacturonan acceptors
The GAUT1:GAUT7 complex isolated from A. thaliana membranes has previously been shown to transfer GalA from UDP-GalA onto HG acceptors (HG:GalAT activity) (15). Here, we employed similar HG:GalAT acceptor–dependent activity assays to examine HG extension by the recombinant GAUT1:GAUT7 complex produced in HEK293F cells. The co-expressed complex, containing intact N-terminal tags and N-glycosylation, was used in all assays.
The activity of the purified enzyme was determined by radioactive incorporation of 14C-labeled GalA from UDP-[14C]GalA onto the nonreducing ends of an HG acceptor mix enriched for HG oligosaccharides with a degree of polymerization (DP) of 7–23 (15, 39, 41). To assess the stability of the enzyme, GAUT1:GAUT7 activity was assayed immediately after Ni2+-Sepharose affinity purification and also after 3 days of storage at −80 °C. GAUT1:GAUT7 retained 100% activity after storage at −80 °C (Fig. 2A). Single-thawed aliquots retained ∼40% activity after storage for 15 months. The enzyme complex has a pH optimum of 7.2 (Fig. 2B) and shows maximal activity in the presence of 0.1–1.0 mm MnCl2 (Fig. 2C). A lower level of activity was obtained when GAUT1:GAUT7 was assayed with CoCl2 (Fig. 2C). Manganese ion cofactors have been visualized in X-ray crystal structures of related GT8 enzymes, LgtC (42) and glycogenin (43), and make active-site contacts with UDP-sugar substrates. All of the other divalent and monovalent metal cations tested (NiSO4, FeCl2, CuSO4, CaCl2, ZnSO4, MgCl2, NaCl, and KCl) yielded less than 5% of the activity observed with MnCl2 (Fig. S1).
Figure 2.

Biochemical characterization of HG:GalAT activity of the GAUT1:GAUT7 complex. A, reaction progress from 2 to 30 min. GAUT1:GAUT7 enzyme was assayed for HG:GalAT activity immediately after nickel-affinity purification (open circles) to identify starting activity, after storage at −80 °C for 3 days (boxes) to test the effects of freeze-thawing, and after storage at −80 °C for 15 months (triangles) to test the effects of long-term storage at −80 °C. Activity was measured in 30-μl reactions containing 100 nm GAUT1:GAUT7, 5 μm UDP-[14C]GalA, 1 mm total UDP-GalA, 10 μm HG acceptor mix, HEPES buffer, pH 7.2, 0.25 mm MnCl2, and 0.05% BSA. B, effect of pH on enzyme activity. C, effect of divalent cations on enzyme activity. D, Michaelis–Menten kinetics for the UDP-GalA donor. UDP-GalA concentration from 5 to 2000 μm was tested with an HG acceptor mix concentration of 100 μm. Kinetic constants were calculated by nonlinear regression using GraphPad Prism version 7. Dotted lines represent a 95% confidence interval. E, Michaelis–Menten kinetics for the HG acceptor mix (DP7–23). HG acceptor concentration from 0.01 to 50 μm was tested in reactions with 1 mm UDP-GalA. In B–E, all specific activity measurements were assayed in 5-min reactions. In B and C, error bars represent the S.D. from at least three independent experiments. In A, D, and E, data points represent results from individual experiments. Replicate data from kinetics experiments are displayed in Table S1.
Based on the assays outlined above, a standard reaction condition was defined: 5 min reactions containing 100 nm GAUT1:GAUT7, 1 mm UDP-GalA, 10 μm HG acceptor, and 0.25 mm MnCl2 in a pH 7.2 HEPES buffer containing 0.05% BSA. Using the standard conditions outlined, Michaelis–Menten kinetics were measured for the donor, UDP-GalA, and for the HG acceptor mixture with the nonvariable substrate held at saturating conditions. The GAUT1:GAUT7 complex shows a standard hyperbolic Michaelis-Menten curve for UDP-GalA with a Km of 151 μm (Fig. 2D). Substrate inhibition was observed at HG acceptor concentrations > 5 μm (Fig. 2E). Substrate inhibition at high concentrations of HG acceptor is expected if GAUT1:GAUT7 functions through an ordered kinetic mechanism in which the UDP-GalA donor binds to the active site prior to the HG acceptor (44, 45). This ordered substrate binding scheme has also been shown in crystal structures and inhibition assays for homologous GT8-family enzymes (42, 43, 46, 47). kcat values for the elongation of HG acceptors ranged from 0.92–1.99 s−1. Results from six independent assays are summarized in Table S1.
GAUT1:GAUT7 synthesizes high molecular weight polysaccharides in vitro by elongation of medium-chain HG acceptors (DP ≥ 11) using a distributive mechanism
The mechanism of HG backbone synthesis has not been clearly defined due to conflicting results from prior studies of this activity. HG:GalAT activity from detergent-solubilized microsomal membranes and from GAUT1:GAUT7 partially purified by immunoprecipitation has previously been shown to add between 1 to ∼30 GalA residues onto DP13–15 acceptors (14, 37–39), without detection of high-MW polymeric HG. These results suggested that GAUT1:GAUT7 uses a distributive mechanism, in which single GalA units are added during each catalytic event, followed by the release of the HG acceptor, and also that GAUT1:GAUT7 does not synthesize high-MW HG polymers in vitro. In contrast, intact Nicotiana tabacum microsomal membranes produce an HG-containing product of ∼105 kDa (39, 48). However, in those experiments, it was not shown whether the high-MW product was free HG polysaccharide or a more complex glycan or proteoglycan containing HG. The synthesis of high-MW HG polysaccharides has not yet been described from in vivo or in vitro experiments.
We used recombinant GAUT1:GAUT7 and HG acceptors enriched for homogeneous degrees of polymerization to measure the size of products synthesized by HG:GalATs. We reasoned that if the GAUT1:GAUT7 complex elongates HG acceptors by a distributive mechanism, the chain length of the products synthesized should be dependent on the ratio of donor to acceptor substrates (49, 50).
The degree of HG acceptor elongation by GAUT1:GAUT7 in vitro was determined by size-exclusion chromatography, high-percentage PAGE, and MALDI-TOF. Using 1 mm UDP-GalA and varying amounts of DP11 HG acceptor, reactions were performed under two conditions: 10 μm HG acceptor (100:1 molar excess of UDP-GalA donor) (Fig. 3, A and B) and 100 μm HG acceptor (10:1 molar excess of UDP-GalA donor) (Fig. 3, C and D). DP11 acceptors were rapidly elongated under both assay conditions. The products synthesized were larger at all time points when a 100:1 molar excess of UDP-GalA over HG acceptor was used (Fig. 3, A and B). The high-MW products are measured to be >100 kDa, but pectin MW may be overestimated relative to dextran standards due to the potential for aggregation or anomalous behavior in size-exclusion columns (51). As observed in high-percentage polyacrylamide gels in which individual bands corresponding to HG products up to DP30 could be distinguished, the DP11 acceptor was elongated to an estimated DP of 30–50 (<10 kDa) when incubated at a 10:1 donor/acceptor ratio (Fig. 3D). Comparison of these two reaction conditions showed that in vitro HG synthesis by GAUT1:GAUT7 matches the results expected for a distributive GT mechanism in which the narrow product distribution has a DP dependent on the available donor/acceptor ratio, as discussed for polysialyltransferases (49). Overnight incubation of GAUT1:GAUT7 with donor/acceptor ratios ranging from 10 to 10,000 further demonstrated that GAUT1:GAUT7 synthesizes high-MW products given available UDP-GalA (Fig. S2).
Figure 3.

Elongation of medium-chain-length HG acceptors (DP11–15) by GAUT1:GAUT7. A, size-exclusion chromatography of the products synthesized in a reaction containing 100 nm GAUT1:GAUT7, 1 mm UDP-GalA, and 10 μm DP11 acceptor. Reactions were incubated for the indicated times and injected into a Superose 12 column. Selected peaks are indicated. Arrows, peak retention volume of dextran standards (270-, 150-, 50-, and 12-kDa average MW). B, high-percentage polyacrylamide gel stained with a combination of alcian blue/silver. Reactions containing a 10 μm concentration of a DP11 acceptor were incubated, and aliquots representing 200 ng of acceptor oligosaccharide were separated on a 30% polyacrylamide gel. HG standards include the HG oligosaccharide acceptor mix (lane 1) and high-MW commercial polygalacturonic acid (PGA) (lane 2). Individual bands of discrete DP that can be counted in the HG standard are indicated to the left of the gel. C, size-exclusion chromatography of products synthesized in a reaction containing 100 μm DP11 acceptor. D, high-percentage PAGE of a reaction containing 100 μm DP11 acceptor. E, MALDI-TOF MS analysis of products produced by GAUT1:GAUT7 in reactions containing 100 μm DP15 HG acceptor and 1 mm UDP-GalA. Reaction products were reducing-end-labeled with 2-AB after the indicated reaction times. The series of ions (m/z) with a mass separation of 176 Da is consistent with the sequential addition of GalA to the DP15 acceptor.
Labeling of the reducing ends of HG acceptors with the fluorescent tag 2-aminobenzamide (2-AB) has been used to detect HG elongation using high-performance anion-exchange chromatography (40). Fluorescent labeling of oligosaccharides has also been shown to enhance detection of oligosaccharides by MALDI-TOF (24). We therefore used MALDI-TOF to test the pattern of elongation of a DP15 fluorescently labeled HG acceptor in reactions incubated from 5 min up to 4 h. Over time, products up to DP30 were detected (Fig. 3E). The apparent Poisson distribution of HG products formed over time is also typical of a distributive mechanism of synthesis (24). However, it should be noted that, as observed in polyacrylamide gels (Fig. 3D), products larger than DP30 were also synthesized under these reaction conditions, indicating that the apparent Poisson distribution observed by MALDI-TOF was partially due to the loss of higher-MW signals. Longer-chain oligosaccharides may be poorly ionized or may have precipitated when exposed to acidic conditions during the labeling procedure.
Elongation of short-chain HG acceptors (DP ≤ 7) occurs with low efficiency relative to longer-chain acceptors (DP ≥ 11)
Prior assays of HG:GalAT activity from N. tabacum and Petunia axillaris solubilized membranes demonstrated a preference of the enzyme for exogenous HG acceptors of DP ≥ 10–12, but also indicated that acceptors as short as DP5 can be elongated (38, 39). To more clearly define the acceptor specificity of GAUT1:GAUT7, we compared the rates of elongation of homogenous HG acceptors enriched for DPs of 3, 7, 11, and 15.
GAUT1:GAUT7 elongates HG acceptors at least as small as DP3 (Fig. 4A). GalA transfer is most rapid using longer-chain acceptors (DP ≥ 11). The rate of transfer to DP11 and DP15 acceptors was approximately equal from 5 to 60 min. Activity was detected using shorter acceptors (DP7 and DP3), but the initial rates of synthesis were low relative to the longer-DP acceptors. The highest rates of synthesis for longer-chain acceptors were detected in the initial linear phase of the reaction, as expected for a standard steady-state reaction progress curve. A lag phase was observed during the elongation of DP7 and DP3 acceptors, and longer incubation periods were required to detect above-background levels of activity.
Figure 4.
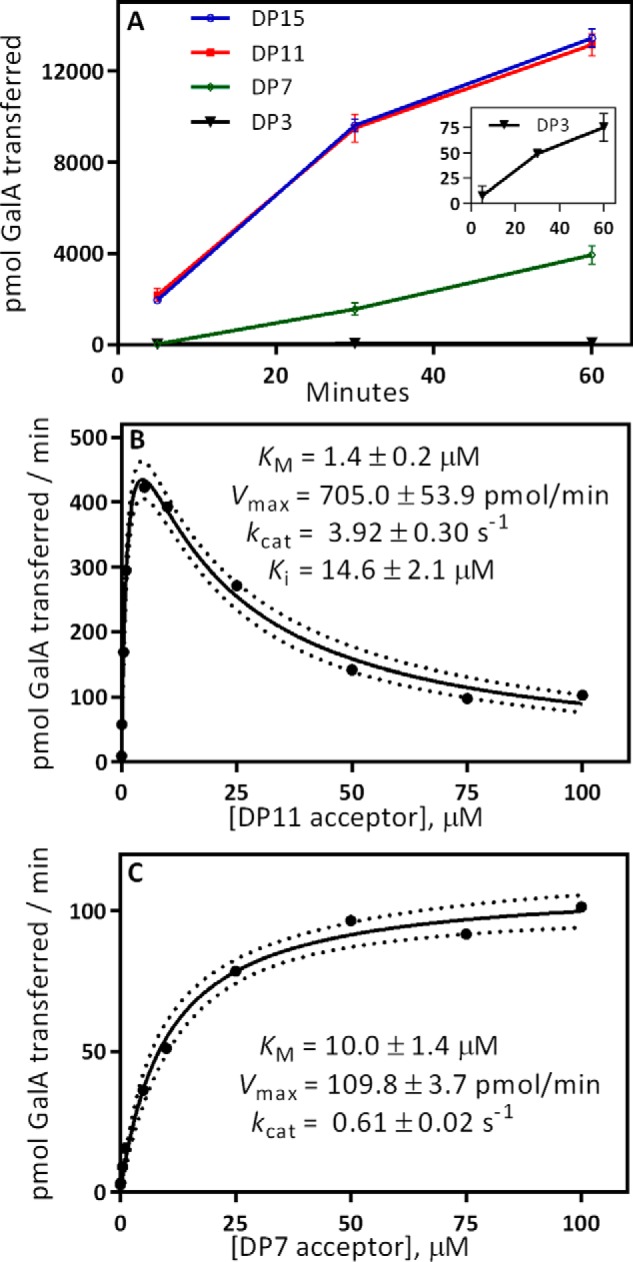
Comparison of acceptor elongation activity using HG acceptors of different degrees of polymerization. A, GAUT1:GAUT7 elongation activity from 5 to 60 min. Activity was measured in 30-μl reactions containing 100 nm GAUT1:GAUT7, 5 μm UDP-[14C]GalA, 1 mm total UDP-GalA, and 10 μm purified, homogeneous DP acceptor, as indicated. Error bars, S.D. from three independent experiments. Inset, activity using a DP3 acceptor. B, Michaelis–Menten kinetics for the DP11 acceptor. HG acceptor concentration from 0.01 to 100 μm was tested in reactions with 1 mm UDP-GalA. Kinetic constants were calculated by nonlinear regression with substrate inhibition using GraphPad Prism version 7. Dotted lines represent a 95% confidence interval. C, Michaelis–Menten kinetics for the DP7 acceptor. HG acceptor concentration from 0.01 to 100 μm was tested in reactions with 1 mm UDP-GalA. Kinetic constants were calculated by nonlinear regression using the standard Michaelis–Menten equation. In B and C, data points represent results from individual experiments. Results from replicate kinetics experiments are displayed in Table S1.
Reaction kinetics were measured using increasing amounts of DP11 (Fig. 4B) and DP7 (Fig. 4C) HG acceptors. The reaction kinetics using the DP11 HG acceptor appear similar to the results observed using the HG acceptor mix (Fig. 2E), with inhibition observed at concentrations above 5 μm. Because activity with the DP7 acceptor was near background level when measured at 5 min, a 30-min incubation time was used. Standard hyperbolic Michaelis–Menten kinetics were observed under these conditions. The catalytic efficiency, defined as kcat/Km, of a DP11 acceptor was 45-fold higher than a DP7 acceptor, representing a strong preference for longer-chain acceptors. This measurement may underestimate the difference in catalytic efficiency between DP7 and DP11 acceptors because the low initial rates and the lag phase observed only with short-chain acceptors makes it difficult to calculate true initial rates of synthesis.
Elongation of short-chain HG acceptors and de novo synthesis in the absence of exogenous oligosaccharide acceptors leads to a bimodal product distribution with minimal observable intermediates
As shown in Fig. 4, initial rates of synthesis using short-chain HG acceptors (DP ≤ 7) are low relative to longer-chain acceptors. To test whether the DP7 acceptor is elongated by a different reaction mechanism distinct from longer-chain acceptors, the degree of DP7 acceptor elongation was measured by size-exclusion chromatography, high-percentage PAGE, and MALDI-TOF. Reactions were carried out using 100 μm acceptor (10:1 molar excess of UDP-GalA donor) (Fig. 5, A and B).
Figure 5.

Elongation of short-chain HG acceptors and de novo initiation of high-molecular-weight HG polysaccharides. A, size-exclusion chromatography of the products synthesized in a reaction containing 100 nm GAUT1:GAUT7, 1 mm UDP-GalA, and 100 μm DP7 HG acceptor. Reactions were incubated for the indicated times and injected into a Superose 12 column. Selected peaks are indicated. Arrows, peak retention times of dextran standards. B, high-percentage polyacrylamide gel stained with a combination of alcian blue/silver. Reactions containing 100 μm DP7 acceptor were incubated for the indicated times, and an aliquot representing 200 ng of acceptor oligosaccharide was removed and separated on a 30% polyacrylamide gel. HG oligosaccharide standards (lane 1) and PGA (lane 2) are described in the legend to Fig. 3B. C, detection of HG polysaccharides synthesized de novo by ELISA. Reactions (30-μl total volume) containing 100 nm GAUT1:GAUT7 and 1 mm UDP-GalA were incubated for 5 min or 12 h. At the indicated time points, aliquots containing a 1- or 0.1-μl reaction volume ( or of the total reaction volume) were boiled and spotted onto a 96-well plate. Reaction products were detected by anti-HG antibodies CCRC-M38, CCRC-M131, and JIM5 but showed no reactivity toward anti-xylan antibody CCRC-M149. D, anti-HG antibody ELISA controls. In C and D, error bars represent the S.D. from a total of four replicate measurements from two independent experiments. E, high-percentage polyacrylamide gel of products synthesized de novo by GAUT1:GAUT7 or in the presence of DP3 or DP7 acceptors, stained with a combination of alcian blue/silver. Reactions (30-μl total volume) containing 100 μm DP7, 100 μm DP3, or no acceptor (de novo synthesis) were incubated for 24 h, and then 5 μl was removed for separation on a 30% polyacrylamide gel.
Unlike the results observed using a DP11 HG acceptor (Fig. 3), a bimodal distribution of high-MW and short-chain products was synthesized during the elongation of a DP7 acceptor, even under conditions of a low molar ratio of UDP-GalA to acceptor. Polysaccharides of intermediate molecular weights were not observed. When detecting low-MW products, the addition of only 1–2 GalA residues was observed at all time points (Fig. 5B and Fig. S3B). Unlike DP11 elongation, a Poisson distribution of HG oligomers was not observed. Instead, the intensity of high MW products increased with reaction time from 1 to 12 h. A similar bimodal product distribution was observed with donor/acceptor ratios ranging from 10 to 200 (Fig. S4).
Having identified that GAUT1:GAUT7 can elongate short-chain acceptors as small as DP3, the complex was tested for the ability to initiate HG synthesis de novo in the absence of exogenously added acceptors. Prior attempts to identify de novo initiation of HG using detergent-solubilized membrane fractions yielded no measurable activity, leading to the conclusion that the enzyme only functions in the elongation step of HG biosynthesis (1).
Three monoclonal antibodies previously shown to react with HG (CCRC-M38, CCRC-M131, and JIM5) (52) were used in ELISAs to determine whether GAUT1:GAUT7 could synthesize HG de novo. Following incubation with 1 mm UDP-GalA, all three anti-HG antibodies reacted with GAUT1:GAUT7-synthesized product in a time- and concentration-dependent manner (Fig. 5C). Control ELISAs confirmed that the anti-HG antibodies detected HG acceptor controls but had no reactivity toward GAUT1:GAUT7 itself or toward UDP-GalA (Fig. 5D). To the best of our knowledge, this is the first evidence that GAUT1:GAUT7 can synthesize HG de novo.
The de novo synthesis product synthesized in overnight reactions was of high MW, similar to the products of overnight reactions with DP7 and DP3 acceptors (Fig. 5E). The de novo synthesis product is of a more uniform MW than the relatively polydisperse product formed following elongation of DP7 acceptors. The polysaccharides synthesized de novo were sensitive to digestion by the HG-specific enzyme, endopolygalacturonase (Fig. S5). These results demonstrate that in addition to elongation of acceptors of all degrees of polymerization, GAUT1:GAUT7 can initiate the synthesis of high-MW HG de novo.
A two-phase model of distributive elongation is favored over a processive model for HG synthesis
During in vitro HG polymerization by GAUT1:GAUT7, small differences in the chain length of HG acceptors appeared to affect the mechanism of elongation. We investigated whether short- and long-chain acceptors are elongated using distinct mechanisms by further defining the effect of acceptor DP on product size distribution. GAUT1:GAUT7 was incubated for 12 h in the presence of a low donor/acceptor ratio (10:1) of intermediately sized HG acceptors ranging from DP7 to DP11 (Fig. 6A). As previously observed, elongation of the DP11 acceptor resulted in products ranging in size from DP11 to a DP of ∼30–50. The majority of the starting acceptor was incorporated into larger-sized HG products. In contrast, all acceptors of DP ≤ 10 were elongated relatively poorly, as indicated by the appreciable amount of acceptors remaining unelongated even following a 12-h incubation. Quantitation using fluorescently tagged acceptors indicated that <3% of the original DP7 acceptor was elongated to high-MW products in overnight reactions (Fig. S7).
Figure 6.

Nonprocessive elongation of HG acceptors. A, high-percentage polyacrylamide gel separation of products synthesized by GAUT1:GAUT7 stained with a combination of alcian blue/silver. Reactions containing 100 nm GAUT1:GAUT7, 1 mm UDP-GalA, and 100 μm acceptor of DP7–11 were incubated for 12 h. An aliquot representing 200 ng of acceptor oligosaccharide was removed and separated on a 30% polyacrylamide gel. T0 reactions containing preboiled enzyme are displayed in Fig. S6. HG standards are described in the legend to Fig. 3B. Lane 3, a mixture of HG DP7–11 standards, 50 ng each. B, competition assay to test for a processive elongation mechanism. UDP-GalA (1 mm) and either no acceptor or HG DP7 acceptor (100 μm) were added during a preincubation phase (0–15 min) to allow formation of enzyme:acceptor processive elongation complexes. Additional acceptors were added during the reaction phase (15–20 min), as indicated. Samples were boiled after 20 min. In the T0 sample (lane 2), the reaction was boiled immediately upon the addition of DP7 acceptor. In the DP7 control reaction (lane 3), no additional acceptor was added at 15 min. For competition assay samples (lanes 4 and 5), reactions containing 100 nm GAUT1:GAUT7 were preincubated (0 min) with 1 mm UDP-GalA and 100 μm DP7 acceptor. After 15 min, DP11 acceptor was added (100 μm = 10:1 donor/acceptor ratio; 10 μm = 100:1 donor/acceptor ratio). For DP11 standard reaction controls (lanes 6 and 7), no acceptor was added during the preincubation phase. DP11 acceptor was added after 15 min. For simultaneous incubation reaction controls (lanes 8 and 9), no acceptor was added during the preincubation phase. A mix of 100 μm DP7 and the indicated DP11 acceptor was added after 15 min. DP11 standard (lane 10) shows the DP11 acceptor and the presence of background bands due to minor impurities.
Product size was inversely related to the size of the HG acceptor, with high-MW polymeric HG observed following incubation with smaller acceptors. Contrary to the DP10 and DP11 acceptors, elongation of DP7 and DP8 acceptors resulted in a bimodal product distribution, with an intermediate distribution for DP9 (Fig. 6A).
Three observations appeared to support the hypothesis that GAUT1:GAUT7 uses distinct processive and distributive mechanisms, depending on the chain length of the acceptor, and that short-chain acceptors were elongated by a processive mechanism. First, a bimodal product distribution formed over time (Fig. 5, B and E). The archetypal processive model for GT activity proposes that a single acceptor molecule remains tightly bound without dissociating from the enzyme until after many rounds of elongation, leading to the formation of high-MW products with minimal observable intermediates (49, 50, 53). Second, a lag phase was observed during early points in the progress curve (Fig. 4A). The lag phase has been proposed to be a feature common to processive enzymes due to the low affinity of short-chain acceptors that are not capable of filling a requisite number of “acceptor subsites” within an extended active-site domain (50, 54). Third, the amount of acceptors within the starting pool used during the reaction was low, measured at <3% (Fig. S7). In traditional measures of processivity, if <10% of the total starting acceptor pool is elongated, then it has been assumed that each acceptor has only associated with the enzyme in a single priming event (53, 55). All three of these observations were suggestive of the processive model but did not directly demonstrate the existence of tight enzyme–acceptor binding complexes demanded by that model. Because these results were inconsistent with the rapid elongation of DP ≥ 11 acceptors in a nonprocessive manner, we considered that the processive model may not accurately describe the elongation of HG by GAUT1:GAUT7.
To directly test for evidence of a processive elongation mechanism in the presence of low-DP acceptors, a competition assay was designed. GAUT1:GAUT7 was preincubated with UDP-GalA and a DP7 acceptor for 15 min at a 1000-fold excess of acceptor over enzyme. We reasoned that 15 min was a sufficient preincubation time to allow all enzyme molecules to bind to the acceptor and to begin processive synthesis. Following preincubation, a DP11 acceptor was added, and incubation was continued for another 5 min (Fig. 6B, lanes 4 and 5). If the DP7 HG served as an acceptor for processive catalysis, the DP11 acceptor would not have been able to compete for binding to GAUT1:GAUT7 or to serve as an acceptor. The results show, however, that the DP11 HG acceptor was able to compete for enzyme binding and was elongated during the 5-min reaction period.
The DP11 acceptor was elongated to the same chain lengths as two control reactions: a standard 5-min reaction with no competing DP7 acceptor added during the preincubation period (Fig. 6B, lanes 6 and 7) and a 5-min reaction in which DP7 and DP11 acceptors were added at the same time (lanes 8 and 9). These results argue against the hypothesis that short-chain acceptors are synthesized by a processive mechanism, in which the growing acceptor should have formed tightly binding complexes with the enzyme, making it unavailable for binding to DP11 acceptors. Furthermore, there was no effect on the size distribution of the products synthesized by elongation of DP11 acceptors in the presence of DP7. This result is consistent with the enzyme having a strong preference for binding to longer-chain acceptors and releasing the acceptor following each round of GalA transfer.
The competition assay argues against the hypothetical processive elongation model because the results suggest that GAUT1:GAUT7 and HG acceptors do not form tight enzyme–acceptor binding complexes. An alternative, two-phase model proposes that acceptors of all sizes are elongated by a distributive mechanism and that the bimodal product distribution results from large differences in catalytic efficiency between shorter- and longer-chain acceptors. Small acceptors are inefficiently elongated by the enzyme, as evidenced by the >45-fold difference in catalytic efficiency between DP7 and DP11 acceptors. Acceptors are only rapidly elongated after reaching a critical DP, estimated to be DP11 (Fig. 6A). The relative inefficiency of short-chain acceptors results in a slow rate of synthesis during the early phase of chain elongation. In reactions containing only DP7 acceptors, high-MW products are observed because there is an effective increase in the donor/acceptor ratio for the small number of acceptors that reach the critical DP, causing them to become rapidly elongated.
Synthesis of high-MW HG depends upon GAUT1:GAUT7 complex formation and electrostatic interactions
HG is a negatively charged polymer. It has been shown for other charged polymers, including polysialic acid (49) and DNA (56), that electrostatic substrate–enzyme interactions affect product size distribution and the mechanism by which enzymes maintain contact with their charged substrates. We hypothesized that electrostatic HG–enzyme interactions may be involved in acceptor binding and elongation by GAUT1:GAUT7. Prior evaluation of a homology model of the GAUT1 GT8 domain using LgtC from N. meningitidis as a template identified a patch of positively charged residues near the active site of GAUT1 that could create an extended acceptor binding groove (57). We predicted that incubation with NaCl would disrupt these interactions and limit the ability of GAUT1:GAUT7 to maintain the contacts with the HG acceptor needed for efficient transfer. We tested whether the addition of NaCl affected product formation by GAUT1:GAUT7.
The presence of 100 mm NaCl in reactions containing a 1000:1 ratio of UDP-GalA to DP11 HG acceptor inhibited the synthesis of high-MW HG products, even under long reaction times (Fig. 7A). In reactions containing a 10:1 donor/acceptor ratio and 0, 50, and 100 mm NaCl, elongation of a DP7 acceptor was likewise inhibited with increasing salt concentration (Fig. 7B). Individual bands of intermediate-sized products (DP > 20) were detectable following reactions containing NaCl, leading to the loss of the bimodal product distribution. This result also argues against DP7 being elongated processively. Short-chain elongation products were visible in both reactions, but the addition of NaCl to the reaction appears to prevent high-MW product formation.
Figure 7.
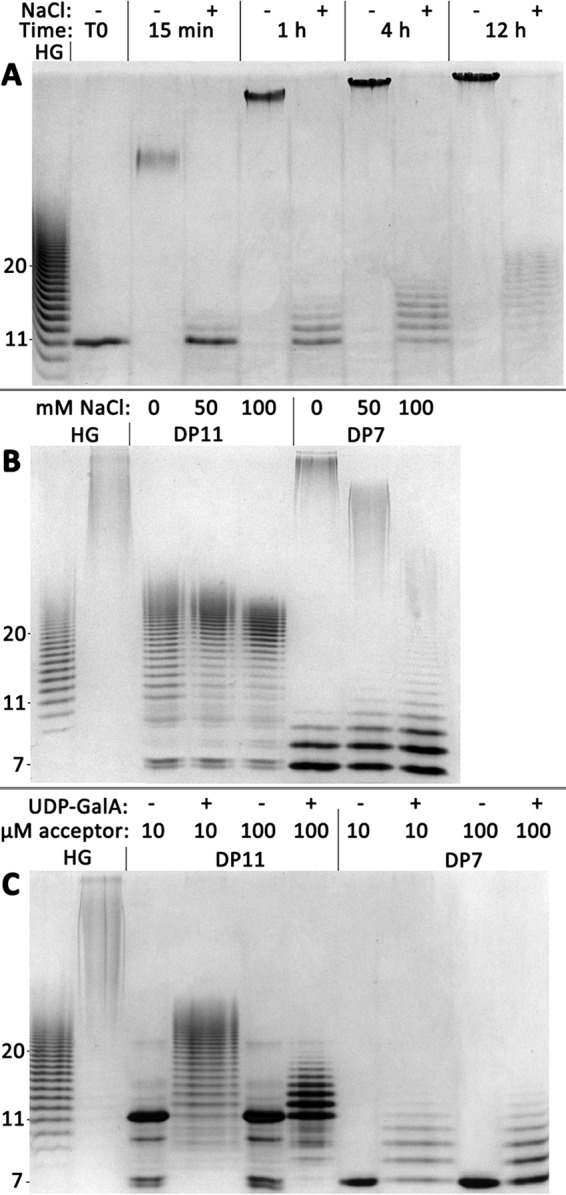
The addition of NaCl or incubation of GAUT1 alone prevents high-MW HG synthesis. A, reactions containing 100 nm GAUT1:GAUT7, 1 mm UDP-GalA, and 1 μm DP11 acceptor (1000:1 donor/acceptor ratio) were incubated for the indicated times with or without the addition of 100 mm NaCl. B, reactions containing 100 nm GAUT1:GAUT7, 1 mm UDP-GalA, and 100 μm DP11 or DP7 acceptor (10:1 donor/acceptor ratio) were incubated with 0, 50, or 100 mm NaCl for 12 h. C, reactions containing 100 nm GAUT1, 1 mm UDP-GalA, and 10 or 100 μm DP11 or DP7 acceptor (100:1 or 10:1 donor/acceptor ratio, respectively) were incubated for 12 h.
Synthesis of intermediate-sized HG upon elongation of a 10:1 donor/acceptor ratio of DP11 acceptor was only weakly affected by the presence of NaCl (Fig. 7B). For both DP7 and DP11 acceptors, product size was limited to approximately DP30–50 when GAUT1/GAUT7 was assayed with 100 mm NaCl. We propose that the electrostatic interactions guide the rapid elongation phase by orienting longer-chain acceptors within the active site for GalA transfer to the nonreducing end. The NaCl concentrations used here are within the physiological range and may not otherwise be expected to have such a strong inhibitory effect, as many glycosyltransferases are regularly purified, stored, and assayed under similar NaCl concentrations (32, 58).
The possibility that previously unidentified structural domains are required for the synthesis of high-MW HG opens the possibility that GAUT7 also contributes to the function of the complex. The activity of GAUT1 expressed and purified in the absence of GAUT7 was tested. Upon incubation of GAUT1 with DP11 and DP7 acceptors (Fig. 7C), high-MW product was not observed. The size of the products synthesized by GAUT1 was limited to DP of ∼30–50, even in reactions containing a large excess of donor (10 μm acceptor, 100:1 donor/acceptor ratio). The consistency of this result with the reduced product sizes observed following incubation of GAUT1:GAUT7 with NaCl suggests that GAUT7 also contributes to an acceptor-binding domain/pocket required for high-MW polymerization. Due to poor expression of GAUT1 and GAUT7 as individual enzymes, the nature of the contribution of GAUT7 to HG synthesis remains to be more thoroughly investigated.
Discussion
Soluble expression of plant cell wall glycosyltransferases using the HEK293F cell system
Plant cell wall matrix polysaccharides are synthesized in the secretory pathway by the coordinated efforts of an estimated 200 glycosyltransferases that generate the backbone and side-chain linkages of pectins and hemicelluloses, including xylans and xyloglucan. A study published in 2009 (16) identified a total of nine plant cell wall GTs that had in vitro activities verified by expression in a heterologous system. In the years since that publication, numerous additional plant cell wall GT in vitro activities have been demonstrated and mapped to individual genes or multigene families (18–24, 58–65). Similar to activities assayed from native plant membranes, the use of recombinant enzymes produced in microsomal membranes from sources such as P. pastoris or N. benthamiana may suffer from difficulties including low protein yields and unknown concentrations of the protein of interest (61). Here, the HEK293F cell expression system was used for robust co-expression of a disulfide-linked enzymatically active GT complex in a soluble secreted form. In addition to the expression of the GAUT1:GAUT7 heterocomplex, the HEK293F cell system has been used to express the plant cell wall GTs xylan synthase-1 (Xys1) and fucosyltransferase 1 (Fut1) (24, 25).
A model for HG synthesis by GAUT1:GAUT7
The results described here expand upon previous reports of HG synthesis by demonstrating that the GAUT1:GAUT7 complex can synthesize high-MW polysaccharides in vitro using a distributive mechanism. Previously, intact and partially solubilized N. tabacum membranes were shown to incorporate [14C]GalA into high-MW polysaccharides with an apparent mass > 100 kDa (39, 48). It was uncertain whether the large size of the polysaccharide products detected in cellular membrane preparations was due to the initiation of polymeric HG or due to incorporation of [14C]GalA into large endogenous acceptors of an unknown size. We demonstrate here that as long as a sufficient concentration of UDP-GalA is available, recombinant GAUT1:GAUT7 is capable of producing high-MW polysaccharides in vitro.
A hypothetical model of distributive HG elongation is depicted in Fig. 8 as a series of five steps. Binding of UDP-GalA (step 1) is followed by HG acceptor binding (step 2). The binding of longer-chain acceptors is enhanced by structural features of the GAUT1:GAUT7 complex, including charged interactions within an extended acceptor-binding groove that also appears to require the presence of GAUT7. GalA is transferred to the nonreducing end of the HG acceptor (step 3). The HG acceptor, elongated by a single GalA residue, departs from the active site (step 4), followed by departure of UDP (step 5). The conformational state of the enzyme is then reset for the next round of glycosyl transfer. Numbered GalA units in the acceptor molecule (center images) represent subsites within the proposed extended acceptor binding domain.
Figure 8.

A proposed model for HG synthesis by GAUT1:GAUT7. Distributive elongation of a DP11 acceptor by the GAUT1:GAUT7 complex is shown as a series of five steps, with GAUT1 in red and GAUT7 in green. A patch of positively charged residues proposed to be necessary for the binding and orientation of growing acceptor chains is indicated in blue. In the center images, individual GalA units are numbered 1–11 to visualize binding of the acceptor along an extended acceptor-binding groove. Numbered steps (circled) are outlined under “Discussion.” A, slow phase of HG synthesis, representing the initiation of new polysaccharide chains and the elongation of short-chain acceptors. Short-chain acceptors are inefficiently elongated and cannot fill subsites along the extended acceptor-binding groove. B, fast phase of HG synthesis, representing the elongation of HG acceptors with chain lengths longer than the critical DP, estimated to be DP11.
The two-phase model is more complete than traditional descriptions of distributive glycosyltransfer mechanisms because it accommodates the observation of distinct, acceptor size–dependent slow and rapid elongation phases. This phenomenon may be common to GTs but would not be observed unless the reaction progress is independently assayed with a wide range of oligosaccharide acceptors. Elongation of short-chain acceptors exhibits several characteristics that have previously been proposed to be common to processive GTs, including a bimodal product distribution, the lack of intermediate-sized products, a lag phase during the early time points of the reaction progress, and large proportions of the starting acceptors remaining unelongated (49, 50, 53, 55, 66). However, here we show that HG acceptors of DP ≥ 11 are elongated in vitro by a distributive mechanism with greater catalytic efficiency than smaller HG acceptors.
For several polymerases that favor longer acceptors, a model has been proposed in which oligosaccharides must be elongated to a certain minimum length before the transferase exhibits maximum activity. Longer acceptors can fill acceptor-binding subsites within the active site groove (50, 54, 67). This “acceptor subsites” model is compatible with the slower rates of synthesis that have been observed with DP ≤ 7 HG acceptors. Only acceptors longer than the critical DP can efficiently bind to the active site and be rapidly elongated. The nearly identical rates of transfer to DP11 or DP15 HG acceptors support an HG acceptor size of DP11 as being sufficient for maximum activity. The model that we present expands upon the hypothesis presented during the analysis of the processive bacterial galactan polymerase GlfT2 (50) because we argue that acceptor subsites and kinetic lag phases are features that are not necessarily limited to processive polymerases.
Processive polymerization mechanisms may require enzymes that physically constrain and enclose the growing acceptor chain, such as cellulose synthase (68), DNA-binding enzymes (56, 69), or, putatively, multi-transmembrane-channel-forming GTs, such as the xyloglucan backbone synthase CSLC4 (70, 71). Many type II GTs have a single GT domain and have no obvious structural basis for confining the growing glycan chain within a processive tunnel. Rather than inferring a processive model by induction from end-point product distribution data, the competition assay that we present (Fig. 6) is part of a recent push to find direct assays to test for the formation of enzyme:acceptor complexes that would serve as positive evidence for the archetypal processive model (50). The competition assay showed that longer, more efficient acceptors can compete for binding to the enzyme following a preincubation period sufficient for formation of processive enzyme:acceptor complexes. The results of this assay show that GAUT1:GAUT7, at least in vitro, does not remain tightly bound to the HG polymer during elongation, a key feature that distinguishes distributive from processive polymerases.
A two-phase model with distinct differences in elongation rates between short-chain and longer-chain acceptors has been described previously for the nonprocessive in vitro activity of K92 polysialyltransferase from Escherichia coli, which has a similar critical acceptor chain length, approximately DP10–12 (72). Heparosan synthase from Pasteurella multocida provides a similar example of a nonprocessive transferase in which a slow initiation phase is observed (73). Two-phase elongation provides an alternative hypothesis for the interpretation of bimodal or polydisperse product distributions that may be observed in studies of GT activity. As an example, levansucrase from Bacillus subtilis was determined to have a processive activity on the basis of a bimodal product distribution, which was lost upon the addition of DP16 acceptors (66). If levansucrase functions similarly to GAUT1:GAUT7, then longer-chain acceptors are more efficiently elongated. The bimodal product distribution would result from large differences in catalytic efficiency and acceptor preference based on chain length. The buildup of inefficient short-chain acceptors and rapid elongation as soon as the acceptor reaches a critical DP can explain non-Poissonian product distributions for GTs that have not been directly shown to be processive.
In vitro polymer initiation and extension in the absence of a glycan primer has been demonstrated for at least four bacterial capsular polysaccharides (74–77), including two negatively charged polysaccharides: Neisseria meningitidis serogroup X and serogroup A. The de novo synthesis products are both synthesized as high-MW polysaccharides with relatively homogeneous product dispersity compared with acceptor-primed reactions (75, 76). For the de novo initiation of HG polysaccharides, a proposed candidate for the primer is a molecule of UDP-GalA, similar to elongation of β-1,4-linked GlcNAc oligomers synthesized by hyaluronan synthase that retain UDP at the reducing end (78, 79). Alternatively, the primer could be monomeric GalA formed following hydrolysis of UDP-GalA. The large size of the products synthesized de novo, containing several hundred GalA units per reducing end, have prevented the primer for de novo synthesis from being identified in the current study.
Anionic polymers, such as HG, may have electrostatic enzyme–acceptor interactions that aid polymerization by promoting the proper binding and orientation of growing acceptor chains. The proposed positively charged acceptor-binding groove was identified using a homology model of the GT8 domain of GAUT1 (57). Future structural and acceptor binding studies will be necessary to confirm the existence of this extended binding groove. Basic residues located in the acceptor-binding pocket influence processivity and product dispersity in a polysialyltransferase from N. meningitidis (49). Incubation with NaCl disrupts polymer sliding in enzymes that bind to DNA, such as the endonuclease BamHI (56). In the case of the nonprocessive activity of GAUT1:GAUT7, strong inhibition of activity by NaCl may be due to disruption of electrostatic interactions that contribute to the ability of acceptors to bind to the charged acceptor-binding groove.
The model described in this report provides a basis for the synthesis of high MW HG polysaccharides. GAUT1:GAUT7 uses a distributive mechanism in which rapid elongation requires that the acceptor reach an intermediate chain length, approximately DP11. The significance of this acceptor size, including comparisons with other GTs that function using multiphase elongation mechanisms, will require further investigation. The relative inefficiency of elongation of short-chain acceptors may provide a mechanism by which HG polymer size and chain initiation are regulated in vivo. This model is consistent with previous reports that HG:GalAT activity is nonprocessive (37–40). Whereas the precise role of GAUT7 remains uncertain, GAUT1 appears to be unable to synthesize high-MW products in the absence of GAUT7. Proper folding and secretion of the complex requires co-expression of GAUT1 with GAUT7, but GAUT7 also appears to have a functional role in contributing to HG polymer synthesis. The results reported here provide a comprehensive characterization of the in vitro distributive activity of a heterologously expressed plant cell wall polysaccharide biosynthetic GT complex, with implications for investigations of kinetic mechanisms and processivity of GT heterocomplexes.
Experimental procedures
Cloning, expression, and purification of GAUT1 and GAUT7 in HEK293F cells
Gateway expression vectors for transient expression in HEK293F cells, and cloning and expression methods, were adapted from other publications (32). Constructs to generate truncated forms of A. thaliana GAUT1 (UniProt Q9LE59, residues 168–673) and GAUT7 (UniProt Q9ZVI7, residues 44–619) were amplified from previously cloned vector templates containing A. thaliana GAUT1 and GAUT7 sequences (14) using Phusion High Fidelity Polymerase (Thermo Scientific). Two rounds of PCR, using gene-specific and Gateway attB site–containing universal primers, were performed. PCR products were introduced into Gateway pDONR221 entry vectors using BP Clonase (Invitrogen). Coding sequences were recombined into a Gateway-adapted version of the mammalian destination vector pGEn2 using LR Clonase (Invitrogen) to generate final chimeric fusion protein sequences containing an N-terminal signal sequence, His8 tag, AviTag, “superfolder” GFP, and a TEV protease recognition site, followed by the truncated GAUT coding regions. Primers used for entry vector cloning are listed below, with universal attB-site sequences in lowercase and gene-specific sequences in capital letters: GAUT1Δ167F (5′-aacttgtactttcaaggcCGGGCAAATGAGTTAGTTCAGC-3′), GAUT1R (5′-acaagaaagctgggtcctaTTCATGAAGGTTGCAACGACG-3′), GAUT7Δ43F (5′-aacttgtactttcaaggcCACAATGGCTTTCACTCTCCTG-3′), GAUT7R (5′-acaagaaagctgggtcctaAGGATTCACGTTACAGTCAC-3′), Universal PrimerF (5′-ggggacaagtttgtacaaaaaagcaggctctgaaacttgtactttcaaggc-3′), and Universal PrimerR (5′-ggggaccactttgtacaagaaaagctgggtc-3′).
Sequences were verified following cloning into entry and expression vectors. Expression plasmids were purified using Purelink HiPure Plasmid Gigaprep kits (Invitrogen). HEK293F suspension culture cells grown to a cell density of 2.5 × 106 in Freestyle 293 expression medium (Thermo Fisher Scientific) were used for transfection. Transfection of DNA, either GAUT1Δ167 or GAUT1Δ167 co-transfected with GAUT7Δ43, was done at a total concentration of 3 μg/ml total culture volume with polyethyleneimine (9 μg/ml). Cells were incubated in a humidified shaking 37 °C CO2 incubator at 150 rpm for 24 h before 1:1 dilution in Freestyle 293 medium supplemented to a final concentration of 2.2 mm valproic acid (Sigma). Medium containing secreted protein was collected after a 6-day total incubation time.
Secreted proteins were purified from suspension culture medium by nickel-affinity purification using HisTrap HP (GE Healthcare) columns connected to an ÄKTA FPLC system (GE Healthcare). Vacuum-filtered culture medium was injected into HisTrap HP columns at 1 ml/min, washed with column buffer, and eluted using a gradient from 20–300 mm imidazole (20 column volumes). Fractions containing protein were exchanged into a storage buffer containing 50 mm HEPES, pH 7.2, 0.25 mm MnCl2, and 20% glycerol using a PD-10 desalting column (GE Healthcare) and concentrated using a 30-kDa MW cutoff Amicon Ultra centrifugal filter unit (Millipore).
The concentration of nickel-affinity purified proteins was determined by UV-visible spectroscopy (Nanodrop) using a 10-cm path length cuvette, and purification was confirmed by SDS-PAGE on a 4–15% gradient gel (Bio-Rad).
Metal-depleted GAUT1:GAUT7 was generated by overnight dialysis against HEPES, pH 7.2, buffer containing Chelex-100. Activity was measured under standard conditions with the addition of a 0.25 mm concentration of the following metals: MnCl2, CoCl2, NiSO4, FeCl2, CuSO4, CaCl2, ZnSO4, MgCl2, NaCl, KCl, or no metal.
Synthesis of UDP-[14C]GalA and HG oligosaccharide acceptors
UDP-d-[14C]galactopyranosyluronic acid was synthesized enzymatically from UDP-d-[14C]glucopyranosyluronic acid (PerkinElmer Life Sciences) as described (15, 80). The batch-specific activity value of 249 mCi/mmol was used to convert cpm readings from scintillation counting to reported pmol values in activity assay figures. Nonradiolabeled UDP-d-galactopyranosyluronic acid was purchased from CarboSource Services.
The HG acceptor mix, enriched for HG oligosaccharides of DP7–23, was generated by partial digestion of polygalacturonic acid with endopolygalacturonase, and the purity was confirmed by HPAEC-PAD as described (39). HG acceptors enriched for homogeneous degrees of polymerization of 7–15 were purified by HPAEC-PAD as described (39). Trigalacturonic acid (DP3) was purchased from Sigma.
Deglycosylation, fusion tag removal, and protein gel electrophoresis
Recombinant TEV protease and PNGase F were expressed as N-terminal His/GFP fusion proteins in E. coli and purified by nickel-affinity chromatography as described (26). Recombinant proteins were incubated at a 1:10 ratio of TEV and PNGase F relative to the GAUT1:GAUT7 complex overnight at room temperature to cleave the fusion tags and N-glycan structures.
Protein samples (4 μg) were mixed with Laemmli sample buffer (81) containing 25 mm DTT to reduce the samples. DTT was omitted in nonreduced samples. Samples were boiled for 10 min and resolved by 4–15% gradient Tris-glycine SDS-PAGE gel. Electrophoresis was performed in a running buffer containing 25 mm Tris, 192 mm glycine, and 0.1% (w/v) SDS using 150-mV constant voltage.
HG:GalAT activity radiolabeled filter assays
Unless otherwise noted, HG:GalAT activity was measured under standard conditions in 30-μl reactions containing 100 nm GAUT1:GAUT7, 5 μm UDP-[14C]GalA, 1 mm total UDP-GalA, 10 μm HG acceptor, HEPES buffer, pH 7.2, 0.25 mm MnCl2, and 0.05% BSA. Reactions were incubated at 30 °C, and 5 min was used as a standard time for linear range specific activity comparisons. As indicated, reactions were modified from the standard conditions to include acceptors enriched for a homogeneous degree of polymerization or higher concentrations of acceptors to modify the donor/acceptor ratio or were conducted in the absence of exogenous acceptors for samples labeled “de novo” synthesis.
HG:GalAT activity was measured using a filter assay as described (82) with modifications. Reactions were terminated by the addition of 400 mm NaOH (5 μl). Reactions were spotted onto 2 × 2-cm squares of Whatman 3MM chromatography paper coated with cetylpyridinium chloride. Filters were air-dried for 5 min prior to three 15-min rounds of washing in a 4-liter bath containing 150 mm NaCl, for a total washing period of 45 min. Filters were air-dried for at least 2 h prior to scintillation counting. Scintillation counting was performed using a PerkinElmer Life Sciences Tri-Carb 2910 TR liquid scintillation counter, 14C program, 1-min count time/sample. Background cpm was measured in each assay using T0 samples, in which NaOH was added to the reaction mixture prior to the addition of enzyme. Background cpm was subtracted from net cpm readings for all reaction samples, and cpm was converted to total pmol transferred.
Alcian blue–stained PAGE
Reactions were incubated under standard conditions described above, except UDP-[14C]GalA was omitted from the reaction buffer. Reactions were terminated by boiling. Aliquots containing an estimated 200–500 ng of total polysaccharide were analyzed.
PAGE and visualization by a combination of alcian blue and silver nitrate staining was performed as described with modifications (14). Samples were mixed with a loading buffer (final concentration 0.1 m Tris, pH 6.8, 0.01% phenol red, and 10% glycerol), loaded onto a stacking gel (5% acrylamide (Bio-Rad), 0.64 m Tris, pH 6.8), and separated over a 30% acrylamide resolving gel (0.38 m Tris, pH 8.8, 30% acrylamide) at 17.5 mA for 60 min. The gel was stained for 20 min with 0.1% alcian blue (Sigma) in 40% ethanol and washed with at least three changes of water until background staining was eliminated. Silver staining and developer was performed using a silver staining kit (Bio-Rad). Staining was terminated by the addition of 5% acetic acid.
Size-exclusion chromatography of HG:GalAT products
Scaled up reactions with a total volume of 400 μl were incubated, using concentrations of reagents consistent with small-scale, nonradioactive, standard condition HG:GalAT assays. At each indicated time point, a 50-μl aliquot was removed, and the reaction was stopped by boiling. Denatured enzyme was removed from the sample by centrifugation at 12,000 rpm for 5 min, and the polysaccharide sample was frozen at −20 °C until analysis. For T0 samples, an equivalent aliquot was removed prior to the addition of UDP-GalA to the reaction.
Size-exclusion chromatography (SEC) was performed using a Superose 12 10/300GL column connected to a Dionex system at a flow rate of 0.5 ml/min in 50 mm ammonium formate buffer. The refractive index of polysaccharide products was measured. Dextran standards (270, 150, 50, and 12 kDa) (Sigma) were used as molecular mass standards. The peak retention volume of each dextran standard is indicated by an arrow at the top of each SEC figure. The molecular weight estimations of pectins may be overestimations due to anomalous behavior of pectins compared with dextrans during SEC (51).
For 2-AB–labeled polysaccharides measured by fluorescence detection, reactions were incubated for 12 h, polysaccharide reducing ends were chemically labeled with 2-AB (described below), and reaction products were injected onto the SEC column under the same conditions as above. An RF 2000 fluorescence detector, under high sensitivity (x16) settings, was used for detection. Quantitation of high-MW polysaccharide was performed by fluorescence signal integration of product and acceptor peaks in Chromeleon version 6.80 software (Dionex).
MALDI of 2-AB–labeled HG oligosaccharide products
Following incubation under the indicated reaction conditions and times, HG products were incubated with 0.2 m 2-AB and 1 m sodium cyanoborohydride in 10% acetic acid to chemically label the reducing ends of HG oligosaccharides, as described (24, 40). Samples were dialyzed four times against water in a 3500 MW cutoff tubing (VWR Scientific) and recovered by lyophilization. Retention of HG oligosaccharides during dialysis has been described (51).
Nafion 117 solution (Sigma) was applied to a Bruker MSP 96 ground steel target and air-dried, as described (27, 83). Following 2-AB labeling, reaction samples were mixed 1:1 with a 20 mg/ml 2,5-dihydroxybenzoic acid matrix solution in 50% methanol. Positive- or negative-ion MALDI-TOF MS spectra were acquired using an LT Bruker LT Microflex spectrometer.
ELISA of HG:GalAT activity using anti-HG monoclonal antibodies
Monoclonal antibodies directed against plant cell wall polysaccharides were characterized previously (52). Antibodies directed against HG (CCRC-M38, CCRC-M131, and JIM5) and xylan (CCRC-M149) were used in this study. Antibody epitope, immunoreactivity, and supplier information is available at WallMabDB (http://www.wallmabdb.net).4 Following incubation under standard reaction conditions unless otherwise noted, aliquots containing either 1 μl ( of the reaction) or 0.1 μl ( of the reaction) were diluted to a final volume of 50 μl, and reactions were terminated by boiling. ELISAs were performed as described previously (84). The 50-μl diluted reaction sample was incubated in a 96-well plate (Costar 3598) and evaporated to dryness overnight. Nonspecific spots on the plate were blocked by incubation for 1 h in 0.1 m Tris-buffered saline (TBS) containing 1% nonfat dry milk (Publix) (200 μl). Primary antibodies (50 μl), diluted 10-fold from the hybridoma supernatant, were dispensed into each well and incubated for 1 h. All wells were washed with 300 μl of wash buffer (0.1 m TBS containing 0.1% nonfat dry milk) for a total of three washes. Secondary antibodies were diluted to 1:5000 in wash buffer. For CCRC series antibodies, anti-mouse (Sigma, A4416), and for JIM series antibodies, anti-rat (Sigma, A9037) secondary antibodies conjugated with horseradish peroxidase (50 μl) were incubated for 1 h. Plates were washed with wash buffer for a total of five washes. 3,3′,5,5′-Tetramethylbenzidine peroxidase substrate (Vector Laboratories) was incubated in each well (50 μl) for 20 min, and the reaction was stopped by the addition of 0.25 m sulfuric acid. OD values were measured using a plate reader at 450 nm with background readings at 655 nm subtracted. Boiled enzyme reaction buffer controls contain reaction mixtures incubated under the same conditions, except GAUT1:GAUT7 enzyme was inactivated by boiling prior to the addition to the reaction mixture.
Endopolygalacturonase digestion
Following incubation of the HG:GalAT reaction, the mixture was adjusted to pH 4.2 by the addition of 1 m sodium acetate buffer, pH 4.2 (3 μl) and 2 m acetic acid (15 μl). The mixture was incubated overnight at 30 °C with 20 milliunits of endopolygalacturonase-I (Aspergillus niger, EC 3.2.1.15) (85); 1 unit = 1 μmol of reducing sugar produced per min, as determined by a p-hydroxybenzoic acid hydrazide reducing sugars assay (Sigma). The endopolygalacturonase (EPG) reaction was terminated by the addition of 1 m NaOH (30 μl). The final reaction mixture was assayed using the filter assay, as described above. Control samples labeled Boiled EPG (Fig. S5) were incubated with EPG that was deactivated by boiling for 1 h prior to use.
Modeling of Michaelis–Menten kinetics
Standard Michaelis–Menten kinetics and substrate inhibition kinetics were modeled using formulas for nonlinear regression analysis using GraphPad Prism version 7 for Windows (GraphPad Software, La Jolla, CA). Kinetic parameters (Vmax, Km, and Ki) were calculated for each independent experiment, measured using 8–12 different concentrations of the variable substrate spanning a range of 0.1–10 times Km. Replicate experiments were calculated as separate data sets, using the standard Michaelis–Menten equation (Equation 1) and the substrate inhibition equation (Equation 2).
| (Eq. 1) |
| (Eq. 2) |
Author contributions
R. A. A., K. W. M., and D. M. conceptualization; R. A. A. data curation; R. A. A. and D. M. investigation; R. A. A., S. P., J.-Y. Y., M. A. A., B. R. U., K. W. M., and D. M. methodology; R. A. A. and D. M. writing-original draft; S. P., M. A. A., B. R. U., and D. M. supervision; S. P., J.-Y. Y., M. A. A., B. R. U., and K. W. M. writing-review and editing; J.-Y. Y., M. A. A., and K. W. M. resources; D. M. funding acquisition; D. M. project administration.
Supplementary Material
Acknowledgments
We thank Z. A. Wood for technical support and guidance with enzyme kinetics experiments, M. G. Hahn for providing monoclonal antibodies, M. A. O'Neill for technical support, and the members of the Mohnen laboratory, including L. Tan, A. K Biswal, and K. A. Engle, for training and helpful research discussions. The BioEnergy Science Center and the Center for Bioenergy Innovation are United States Department of Energy Bioenergy Research Centers supported by the Office of Biological and Environmental Research in the Department of Energy's Office of Science. We also acknowledge the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the United States Department of Energy through Grant DE-FG02–12ER16324 for funding structural studies of pectin. Generation of the CCRC series of monoclonal antibodies used in this work was supported by National Science Foundation Plant Genome Program Grant DBI-0421683. Distribution of JIM antibodies used in this work was supported in part by National Science Foundation Grants DBI-0421683 and RCN 009281.
This work was supported primarily by BioEnergy Science Center Grant DE-PS02-06ER64304 and partially by the Center for Bioenergy Innovation. The research was also partially funded by the Department of Energy–funded Center for Plant and Microbial Complex Carbohydrates Grant DE-SC0015662 (DE-FG02-93ER20097), by United States Department of Agriculture AFRI Grant 2010-65115-20396, and National Institutes of Health Grants P41GM103390 and P01GM107012. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1 and Figs. S1–S7.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- HG
- homogalacturonan
- HPAEC-PAD
- high-performance anion-exchange chromatography coupled with pulsed electrochemical detection
- GalA
- α-d-galactopyranosyluronic acid
- GalAT
- galacturonosyltransferase
- PNGase F
- peptide:N-glycosidase F
- DP
- degree of polymerization
- 2-AB
- 2-aminobenzamide
- TEV
- tobacco etch virus
- SEC
- size-exclusion chromatography
- MW
- molecular weight
- EPG
- endopolygalacturonase.
References
- 1. Atmodjo M. A., Hao Z., and Mohnen D. (2013) Evolving views of pectin biosynthesis. Annu. Rev. Plant Biol. 64, 747–779 10.1146/annurev-arplant-042811-105534 [DOI] [PubMed] [Google Scholar]
- 2. Tan L., Eberhard S., Pattathil S., Warder C., Glushka J., Yuan C., Hao Z., Zhu X., Avci U., Miller J. S., Baldwin D., Pham C., Orlando R., Darvill A., Hahn M. G., Kieliszewski M. J., and Mohnen D. (2013) An Arabidopsis cell wall proteoglycan consists of pectin and arabinoxylan covalently linked to an arabinogalactan protein. Plant Cell 25, 270–287 10.1105/tpc.112.107334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cosgrove D. J. (2016) Catalysts of plant cell wall loosening. F1000Res 5, F1000 10.12688/f1000research.7180.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Voxeur A., and Höfte H. (2016) Cell wall integrity signaling in plants: “To grow or not to grow that's the question”. Glycobiology 26, 950–960 10.1093/glycob/cww029 [DOI] [PubMed] [Google Scholar]
- 5. Dick-Pérez M., Zhang Y., Hayes J., Salazar A., Zabotina O. A., and Hong M. (2011) Structure and interactions of plant cell-wall polysaccharides by two- and three-dimensional magic-angle-spinning solid-state NMR. Biochemistry 50, 989–1000 10.1021/bi101795q [DOI] [PubMed] [Google Scholar]
- 6. Wang T., Zabotina O., and Hong M. (2012) Pectin-cellulose interactions in the Arabidopsis primary cell wall from two-dimensional magic-angle-spinning solid-state nuclear magnetic resonance. Biochemistry 51, 9846–9856 10.1021/bi3015532 [DOI] [PubMed] [Google Scholar]
- 7. Wang T., and Hong M. (2016) Solid-state NMR investigations of cellulose structure and interactions with matrix polysaccharides in plant primary cell walls. J. Exp. Bot. 67, 503–514 10.1093/jxb/erv416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lionetti V., Francocci F., Ferrari S., Volpi C., Bellincampi D., Galletti R., D'Ovidio R., De Lorenzo G., and Cervone F. (2010) Engineering the cell wall by reducing de-methyl-esterified homogalacturonan improves saccharification of plant tissues for bioconversion. Proc. Natl. Acad. Sci. U.S.A. 107, 616–621 10.1073/pnas.0907549107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chung D., Pattathil S., Biswal A. K., Hahn M. G., Mohnen D., and Westpheling J. (2014) Deletion of a gene cluster encoding pectin degrading enzymes in Caldicellulosiruptor bescii reveals an important role for pectin in plant biomass recalcitrance. Biotechnol. Biofuels 7, 147 10.1186/s13068-014-0147-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Biswal A. K., Atmodjo M. A., Pattathil S., Amos R. A., Yang X., Winkeler K., Collins C., Mohanty S. S., Ryno D., Tan L., Gelineo-Albersheim I., Hunt K., Sykes R. W., Turner G. B., Ziebell A., et al. (2018) Working towards recalcitrance mechanisms: increased xylan and homogalacturonan production by overexpression of GAlactUronosylTransferase12 (GAUT12) causes increased recalcitrance and decreased growth in Populus. Biotechnol. Biofuels 11, 9 10.1186/s13068-017-1002-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Biswal A. K., Hao Z., Pattathil S., Yang X., Winkeler K., Collins C., Mohanty S. S., Richardson E. A., Gelineo-Albersheim I., Hunt K., Ryno D., Sykes R. W., Turner G. B., Ziebell A., Gjersing E., et al. (2015) Downregulation of GAUT12 in Populus deltoides by RNA silencing results in reduced recalcitrance, increased growth and reduced xylan and pectin in a woody biofuel feedstock. Biotechnol. Biofuels 8, 41 10.1186/s13068-015-0218-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Biswal A. K., Atmodjo M. A., Li M., Baxter H. L., Yoo C. G., Pu Y., Lee Y. C., Mazarei M., Black I. M., Zhang J. Y., Ramanna H., Bray A. L., King Z. R., LaFayette P. R., Pattathil S., et al. (2018) Sugar release and growth of biofuel crops are improved by downregulation of pectin biosynthesis. Nat. Biotechnol. 36, 249–257 10.1038/nbt.4067 [DOI] [PubMed] [Google Scholar]
- 13. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., and Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sterling J. D., Atmodjo M. A., Inwood S. E., Kumar Kolli V. S., Quigley H. F., Hahn M. G., and Mohnen D. (2006) Functional identification of an Arabidopsis pectin biosynthetic homogalacturonan galacturonosyltransferase. Proc. Natl. Acad. Sci. U.S.A. 103, 5236–5241 10.1073/pnas.0600120103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Atmodjo M. A., Sakuragi Y., Zhu X., Burrell A. J., Mohanty S. S., Atwood J. A. 3rd, Orlando R., Scheller H. V., and Mohnen D. (2011) Galacturonosyltransferase (GAUT)1 and GAUT7 are the core of a plant cell wall pectin biosynthetic homogalacturonan:galacturonosyltransferase complex. Proc. Natl. Acad. Sci. U.S.A. 108, 20225–20230 10.1073/pnas.1112816108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petersen B. L., Egelund J., Damager I., Faber K., Jensen J. K., Yang Z., Bennett E. P., Scheller H. V., and Ulvskov P. (2009) Assay and heterologous expression in Pichia pastoris of plant cell wall type-II membrane anchored glycosyltransferases. Glycoconj. J. 26, 1235–1246 10.1007/s10719-009-9242-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Culbertson A. T., and Zabotina O. A. (2015) The glycosyltransferases involved in synthesis of plant cell wall polysaccharides: present and future. JSM Enzymol. Protein Sci. 1, 1004 [Google Scholar]
- 18. Wu Y., Williams M., Bernard S., Driouich A., Showalter A. M., and Faik A. (2010) Functional identification of two nonredundant Arabidopsis α(1,2)fucosyltransferases specific to arabinogalactan proteins. J. Biol. Chem. 285, 13638–13645 10.1074/jbc.M110.102715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Geshi N., Johansen J. N., Dilokpimol A., Rolland A., Belcram K., Verger S., Kotake T., Tsumuraya Y., Kaneko S., Tryfona T., Dupree P., Scheller H. V., Höfte H., and Mouille G. (2013) A galactosyltransferase acting on arabinogalactan protein glycans is essential for embryo development in Arabidopsis. Plant J. 76, 128–137 [DOI] [PubMed] [Google Scholar]
- 20. Liwanag A. J., Ebert B., Verhertbruggen Y., Rennie E. A., Rautengarten C., Oikawa A., Andersen M. C., Clausen M. H., and Scheller H. V. (2012) Pectin biosynthesis: GALS1 in Arabidopsis thaliana is a β-1,4-galactan β-1,4-galactosyltransferase. Plant Cell 24, 5024–5036 10.1105/tpc.112.106625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chiniquy D., Sharma V., Schultink A., Baidoo E. E., Rautengarten C., Cheng K., Carroll A., Ulvskov P., Harholt J., Keasling J. D., Pauly M., Scheller H. V., and Ronald P. C. (2012) XAX1 from glycosyltransferase family 61 mediates xylosyltransfer to rice xylan. Proc. Natl. Acad. Sci. U.S.A. 109, 17117–17122 10.1073/pnas.1202079109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Knoch E., Dilokpimol A., Tryfona T., Poulsen C. P., Xiong G., Harholt J., Petersen B. L., Ulvskov P., Hadi M. Z., Kotake T., Tsumuraya Y., Pauly M., Dupree P., and Geshi N. (2013) A β-glucuronosyltransferase from Arabidopsis thaliana involved in biosynthesis of type II arabinogalactan has a role in cell elongation during seedling growth. Plant J. 76, 1016–1029 10.1111/tpj.12353 [DOI] [PubMed] [Google Scholar]
- 23. Basu D., Liang Y., Liu X., Himmeldirk K., Faik A., Kieliszewski M., Held M., and Showalter A. M. (2013) Functional identification of a hydroxyproline-o-galactosyltransferase specific for arabinogalactan protein biosynthesis in Arabidopsis. J. Biol. Chem. 288, 10132–10143 10.1074/jbc.M112.432609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Urbanowicz B. R., Peña M. J., Moniz H. A., Moremen K. W., and York W. S. (2014) Two Arabidopsis proteins synthesize acetylated xylan in vitro. Plant J. 80, 197–206 10.1111/tpj.12643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Urbanowicz B. R., Bharadwaj V. S., Alahuhta M., Peña M. J., Lunin V. V., Bomble Y. J., Wang S., Yang J. Y., Tuomivaara S. T., Himmel M. E., Moremen K. W., York W. S., and Crowley M. F. (2017) Structural, mutagenic and in silico studies of xyloglucan fucosylation in Arabidopsis thaliana suggest a water-mediated mechanism. Plant J. 91, 931–949 10.1111/tpj.13628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meng L., Forouhar F., Thieker D., Gao Z., Ramiah A., Moniz H., Xiang Y., Seetharaman J., Milaninia S., Su M., Bridger R., Veillon L., Azadi P., Kornhaber G., Wells L., Montelione G. T., Woods R. J., Tong L., and Moremen K. W. (2013) Enzymatic basis for N-glycan sialylation: structure of rat α2,6-sialyltransferase (ST6GAL1) reveals conserved and unique features for glycan sialylation. J. Biol. Chem. 288, 34680–34698 10.1074/jbc.M113.519041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Praissman J. L., Live D. H., Wang S., Ramiah A., Chinoy Z. S., Boons G. J., Moremen K. W., and Wells L. (2014) B4GAT1 is the priming enzyme for the LARGE-dependent functional glycosylation of α-dystroglycan. eLife 3, 10.7554/eLife.03943 10.7554/eLife.03943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Praissman J. L., Willer T., Sheikh M. O., Toi A., Chitayat D., Lin Y. Y., Lee H., Stalnaker S. H., Wang S., Prabhakar P. K., Nelson S. F., Stemple D. L., Moore S. A., Moremen K. W., Campbell K. P., and Wells L. (2016) The functional O-mannose glycan on α-dystroglycan contains a phospho-ribitol primed for matriglycan addition. eLife 5, e14473 10.7554/eLife.14473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Subedi G. P., Johnson R. W., Moniz H. A., Moremen K. W., and Barb A. (2015) High yield expression of recombinant human proteins with the transient transfection of HEK293 cells in suspension. J. Vis. Exp. e53568 10.3791/53568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Halmo S. M., Singh D., Patel S., Wang S., Edlin M., Boons G. J., Moremen K. W., Live D., and Wells L. (2017) Protein O-linked mannose β-1,4-N-acetylglucosaminyl-transferase 2 (POMGNT2) is a gatekeeper enzyme for functional glycosylation of α-dystroglycan. J. Biol. Chem. 292, 2101–2109 10.1074/jbc.M116.764712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sheikh M. O., Halmo S. M., Patel S., Middleton D., Takeuchi H., Schafer C. M., West C. M., Haltiwanger R. S., Avci F. Y., Moremen K. W., and Wells L. (2017) Rapid screening of sugar-nucleotide donor specificities of putative glycosyltransferases. Glycobiology 27, 206–212 10.1093/glycob/cww114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moremen K. W., Ramiah A., Stuart M., Steel J., Meng L., Forouhar F., Moniz H. A., Gahlay G., Gao Z., Chapla D., Wang S., Yang J. Y., Prabhakar P. K., Johnson R., Rosa M. D., Geisler C., Nairn A. V., Seetharaman J., Wu S. C., Tong L., Gilbert H. J., LaBaer J., and Jarvis D. L. (2018) Expression system for structural and functional studies of human glycosylation enzymes. Nat. Chem. Biol. 14, 156–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kellokumpu S., Hassinen A., and Glumoff T. (2016) Glycosyltransferase complexes in eukaryotes: long-known, prevalent but still unrecognized. Cell Mol. Life Sci. 73, 305–325 10.1007/s00018-015-2066-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oikawa A., Lund C. H., Sakuragi Y., and Scheller H. V. (2013) Golgi-localized enzyme complexes for plant cell wall biosynthesis. Trends Plant Sci. 18, 49–58 10.1016/j.tplants.2012.07.002 [DOI] [PubMed] [Google Scholar]
- 35. Zeng W., Lampugnani E. R., Picard K. L., Song L., Wu A. M., Farion I. M., Zhao J., Ford K., Doblin M. S., and Bacic A. (2016) Asparagus IRX9, IRX10, and IRX14A are components of an active xylan backbone synthase complex that forms in the Golgi apparatus. Plant Physiol. 171, 93–109 10.1104/pp.15.01919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jiang N., Wiemels R. E., Soya A., Whitley R., Held M., and Faik A. (2016) Composition, assembly, and trafficking of a wheat xylan synthase complex. Plant Physiol. 170, 1999–2023 10.1104/pp.15.01777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yasui K. J., J; Ohashi T., and Ishimizu T. (2009) In vitro synthesis of polygalacturonic acid. in Pectin and Pectinases (Schols H. A., Visser R. G. F, Voragen A. G. J., eds) pp. 167–175, Wagenigen Academic Publishers, Wagenigen, The Netherlands [Google Scholar]
- 38. Akita K., Ishimizu T., Tsukamoto T., Ando T., and Hase S. (2002) Successive glycosyltransfer activity and enzymatic characterization of pectic polygalacturonate 4-α-galacturonosyltransferase solubilized from pollen tubes of Petunia axillaris using pyridylaminated oligogalacturonates as substrates. Plant Physiol. 130, 374–379 10.1104/pp.005587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Doong R. L., and Mohnen D. (1998) Solubilization and characterization of a galacturonosyltransferase that synthesizes the pectic polysaccharide homogalacturonan. Plant J. 13, 363–374 10.1046/j.1365-313X.1998.00042.x [DOI] [Google Scholar]
- 40. Ishii T. (2002) A sensitive and rapid bioassay of homogalacturonan synthase using 2-aminobenzamide-labeled oligogalacturonides. Plant Cell Physiol. 43, 1386–1389 10.1093/pcp/pcf150 [DOI] [PubMed] [Google Scholar]
- 41. Scheller H. V., Doong R. L., Ridley B. L., and Mohnen D. (1999) Pectin biosynthesis: a solubilized α1,4-galacturonosyltransferase from tobacco catalyzes the transfer of galacturonic acid from UDP-galacturonic acid onto the non-reducing end of homogalacturonan. Planta 207, 512–517 10.1007/s004250050511 [DOI] [Google Scholar]
- 42. Persson K., Ly H. D., Dieckelmann M., Wakarchuk W. W., Withers S. G., and Strynadka N. C. (2001) Crystal structure of the retaining galactosyltransferase LgtC from Neisseria meningitidis in complex with donor and acceptor sugar analogs. Nat. Struct. Biol. 8, 166–175 10.1038/84168 [DOI] [PubMed] [Google Scholar]
- 43. Gibbons B. J., Roach P. J., and Hurley T. D. (2002) Crystal structure of the autocatalytic initiator of glycogen biosynthesis, glycogenin. J. Mol. Biol. 319, 463–477 10.1016/S0022-2836(02)00305-4 [DOI] [PubMed] [Google Scholar]
- 44. Cornish-Bowden A. (2012) Fundamentals of Enzyme Kinetics, pp. 210–214, Wiley-VCH, Weinheim, Germany [Google Scholar]
- 45. Piszkiewicz D. (1977) Kinetics of Chemical and Enzyme-catalyzed Reactions, pp. 97–99 and 140–144, Oxford University Press, New York [Google Scholar]
- 46. Chaikuad A., Froese D. S., Berridge G., von Delft F., Oppermann U., and Yue W. W. (2011) Conformational plasticity of glycogenin and its maltosaccharide substrate during glycogen biogenesis. Proc. Natl. Acad. Sci. U.S.A. 108, 21028–21033 10.1073/pnas.1113921108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ly H. D., Lougheed B., Wakarchuk W. W., and Withers S. G. (2002) Mechanistic studies of a retaining α-galactosyltransferase from Neisseria meningitidis. Biochemistry 41, 5075–5085 10.1021/bi012031s [DOI] [PubMed] [Google Scholar]
- 48. Doong R. L., Liljebjelke K., Fralish G., Kumar A., and Mohnen D. (1995) Cell-free synthesis of pectin (identification and partial characterization of polygalacturonate 4-α-galacturonosyltransferase and its products from membrane preparations of tobacco cell-suspension cultures). Plant Physiol. 109, 141–152 10.1104/pp.109.1.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Keys T. G., Fuchs H. L., Ehrit J., Alves J., Freiberger F., and Gerardy-Schahn R. (2014) Engineering the product profile of a polysialyltransferase. Nat. Chem. Biol. 10, 437–442 10.1038/nchembio.1501 [DOI] [PubMed] [Google Scholar]
- 50. Levengood M. R., Splain R. A., and Kiessling L. L. (2011) Monitoring processivity and length control of a carbohydrate polymerase. J Am. Chem. Soc. 133, 12758–12766 10.1021/ja204448t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mort A. J. M., Moerschbacher B. M., Pierce M. L., and Maness N. O. (1991) Problems encountered during the extraction, purification, and chromatography of pectic fragments, and some solutions to them. Carbohydr. Res. 215, 219–227 10.1016/0008-6215(91)84022-7 [DOI] [Google Scholar]
- 52. Pattathil S., Avci U., Baldwin D., Swennes A. G., McGill J. A., Popper Z., Bootten T., Albert A., Davis R. H., Chennareddy C., Dong R., O'Shea B., Rossi R., Leoff C., Freshour G., et al. (2010) A comprehensive toolkit of plant cell wall glycan-directed monoclonal antibodies. Plant Physiol. 153, 514–525 10.1104/pp.109.151985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bloom L. B., and Goodman M. F. (2001) Polymerase processivity: measurement and mechanisms. in eLS, pp. 1–6, Wiley, Chichester, UK [Google Scholar]
- 54. Fiebig T., Litschko C., Freiberger F., Bethe A., Berger M., and Gerardy-Schahn R. (2018) Efficient solid-phase synthesis of meningococcal capsular oligosaccharides enables simple and fast chemoenzymatic vaccine production. J. Biol. Chem. 293, 953–962 10.1074/jbc.RA117.000488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bambara R. A., Fay P. J., and Mallaber L. M. (1995) Methods of analyzing processivity. Methods Enzymol. 262, 270–280 10.1016/0076-6879(95)62023-0 [DOI] [PubMed] [Google Scholar]
- 56. Nardone G., George J., and Chirikjian J. G. (1986) Differences in the kinetic properties of BamHI endonuclease and methylase with linear DNA substrates. J. Biol. Chem. 261, 12128–12133 [PubMed] [Google Scholar]
- 57. Yin Y., Chen H., Hahn M. G., Mohnen D., and Xu Y. (2010) Evolution and function of the plant cell wall synthesis-related glycosyltransferase family 8. Plant Physiol. 153, 1729–1746 10.1104/pp.110.154229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Culbertson A. T., Chou Y. H., Smith A. L., Young Z. T., Tietze A. A., Cottaz S., Fauré R., and Zabotina O. A. (2016) Enzymatic activity of xyloglucan xylosyltransferase 5. Plant Physiol. 171, 1893–1904 10.1104/pp.16.00361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dilokpimol A., Poulsen C. P., Vereb G., Kaneko S., Schulz A., and Geshi N. (2014) Galactosyltransferases from Arabidopsis thaliana in the biosynthesis of type II arabinogalactan: molecular interaction enhances enzyme activity. BMC Plant Biol. 14, 90 10.1186/1471-2229-14-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Basu D., Tian L., Wang W., Bobbs S., Herock H., Travers A., and Showalter A. M. (2015) A small multigene hydroxyproline-O-galactosyltransferase family functions in arabinogalactan-protein glycosylation, growth and development in Arabidopsis. BMC Plant Biol. 15, 295 10.1186/s12870-015-0670-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rennie E. A., Hansen S. F., Baidoo E. E., Hadi M. Z., Keasling J. D., and Scheller H. V. (2012) Three members of the Arabidopsis glycosyltransferase family 8 are xylan glucuronosyltransferases. Plant Physiol. 159, 1408–1417 10.1104/pp.112.200964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lee C., Teng Q., Zhong R., and Ye Z. H. (2012) Arabidopsis GUX proteins are glucuronyltransferases responsible for the addition of glucuronic acid side chains onto xylan. Plant Cell Physiol. 53, 1204–1216 10.1093/pcp/pcs064 [DOI] [PubMed] [Google Scholar]
- 63. Vuttipongchaikij S., Brocklehurst D., Steele-King C., Ashford D. A., Gomez L. D., and McQueen-Mason S. J. (2012) Arabidopsis GT34 family contains five xyloglucan α-1,6-xylosyltransferases. New Phytol. 195, 585–595 10.1111/j.1469-8137.2012.04196.x [DOI] [PubMed] [Google Scholar]
- 64. Ogawa-Ohnishi M., and Matsubayashi Y. (2015) Identification of three potent hydroxyproline O-galactosyltransferases in Arabidopsis. Plant J. 81, 736–746 10.1111/tpj.12764 [DOI] [PubMed] [Google Scholar]
- 65. Laursen T., Stonebloom S. H., Pidatala V. R., Birdseye D. S., Clausen M. H., Mortimer J. C., and Scheller H. V. (2018) Bifunctional glycosyltransferases catalyze both extension and termination of pectic galactan oligosaccharides. Plant J. 94, 340–351 10.1111/tpj.13860 [DOI] [PubMed] [Google Scholar]
- 66. Raga-Carbajal E., Carrillo-Nava E., Costas M., Porras-Dominguez J., López-Munguia A., and Olvera C. (2016) Size product modulation by enzyme concentration reveals two distinct levan elongation mechanisms in Bacillus subtilis levansucrase. Glycobiology 26, 377–385 10.1093/glycob/cwv112 [DOI] [PubMed] [Google Scholar]
- 67. Forsee W. T., Cartee R. T., and Yother J. (2006) Role of the carbohydrate binding site of the Streptococcus pneumoniae capsular polysaccharide type 3 synthase in the transition from oligosaccharide to polysaccharide synthesis. J. Biol. Chem. 281, 6283–6289 10.1074/jbc.M511124200 [DOI] [PubMed] [Google Scholar]
- 68. Morgan J. L., Strumillo J., and Zimmer J. (2013) Crystallographic snapshot of cellulose synthesis and membrane translocation. Nature 493, 181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Breyer W. A., and Matthews B. W. (2001) A structural basis for processivity. Protein Sci. 10, 1699–1711 10.1110/ps.10301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Davis J., Brandizzi F., Liepman A. H., and Keegstra K. (2010) Arabidopsis mannan synthase CSLA9 and glucan synthase CSLC4 have opposite orientations in the Golgi membrane. Plant J 64, 1028–1037 10.1111/j.1365-313X.2010.04392.x [DOI] [PubMed] [Google Scholar]
- 71. Pauly M., and Keegstra K. (2016) Biosynthesis of the plant cell wall matrix polysaccharide xyloglucan. Annu. Rev. Plant Biol. 67, 235–259 10.1146/annurev-arplant-043015-112222 [DOI] [PubMed] [Google Scholar]
- 72. Vionnet J., and Vann W. F. (2007) Successive glycosyltransfer of sialic acid by Escherichia coli K92 polysialyltransferase in elongation of oligosialic acceptors. Glycobiology 17, 735–743 10.1093/glycob/cwm032 [DOI] [PubMed] [Google Scholar]
- 73. Chavaroche A. A., Springer J., Kooy F., Boeriu C., and Eggink G. (2010) In vitro synthesis of heparosan using recombinant Pasteurella multocida heparosan synthase PmHS2. Appl. Microbiol. Biotechnol. 85, 1881–1891 10.1007/s00253-009-2214-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. DeAngelis P. L., and White C. L. (2002) Identification and molecular cloning of a heparosan synthase from Pasteurella multocida type D. J. Biol. Chem. 277, 7209–7213 10.1074/jbc.M112130200 [DOI] [PubMed] [Google Scholar]
- 75. Fiebig T., Berti F., Freiberger F., Pinto V., Claus H., Romano M. R., Proietti D., Brogioni B., Stummeyer K., Berger M., Vogel U., Costantino P., and Gerardy-Schahn R. (2014) Functional expression of the capsule polymerase of Neisseria meningitidis serogroup X: a new perspective for vaccine development. Glycobiology 24, 150–158 10.1093/glycob/cwt102 [DOI] [PubMed] [Google Scholar]
- 76. Fiebig T., Freiberger F., Pinto V., Romano M. R., Black A., Litschko C., Bethe A., Yashunsky D., Adamo R., Nikolaev A., Berti F., and Gerardy-Schahn R. (2014) Molecular cloning and functional characterization of components of the capsule biosynthesis complex of Neisseria meningitidis serogroup A: toward in vitro vaccine production. J. Biol. Chem. 289, 19395–19407 10.1074/jbc.M114.575142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Litschko C., Romano M. R., Pinto V., Claus H., Vogel U., Berti F., Gerardy-Schahn R., and Fiebig T. (2015) The capsule polymerase CslB of Neisseria meningitidis serogroup L catalyzes the synthesis of a complex trimeric repeating unit comprising glycosidic and phosphodiester linkages. J. Biol. Chem. 290, 24355–24366 10.1074/jbc.M115.678094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Weigel P. H., West C. M., Zhao P., Wells L., Baggenstoss B. A., and Washburn J. L. (2015) Hyaluronan synthase assembles chitin oligomers with -GlcNAc(α1→)UDP at the reducing end. Glycobiology 25, 632–643 10.1093/glycob/cwv006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Weigel P. H., Baggenstoss B. A., and Washburn J. L. (2017) Hyaluronan synthase assembles hyaluronan on a [GlcNAc(β1,4)]n-GlcNAc(α1→)UDP primer and hyaluronan retains this residual chitin oligomer as a cap at the nonreducing end. Glycobiology 27, 536–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liljebjelke K., Adolphson R., Baker K., Doong R. L., and Mohnen D. (1995) Enzymatic synthesis and purification of uridine diphosphate [14C]galacturonic acid: a substrate for pectin biosynthesis. Anal. Biochem. 225, 296–304 10.1006/abio.1995.1158 [DOI] [PubMed] [Google Scholar]
- 81. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 82. Sterling J. D., Lemons J. A., Forkner I. F., and Mohnen D. (2005) Development of a filter assay for measuring homogalacturonan: α-(1,4)-galacturonosyltransferase activity. Anal. Biochem. 343, 231–236 10.1016/j.ab.2005.05.037 [DOI] [PubMed] [Google Scholar]
- 83. Jacobs A., and Dahlman O. (2001) Enhancement of the quality of MALDI mass spectra of highly acidic oligosaccharides by using a nafion-coated probe. Anal. Chem. 73, 405–410 10.1021/ac001222i [DOI] [PubMed] [Google Scholar]
- 84. Pattathil S., Avci U., Miller J. S., and Hahn M. G. (2012) Immunological approaches to plant cell wall and biomass characterization: glycome profiling. Methods Mol. Biol. 908, 61–72 [DOI] [PubMed] [Google Scholar]
- 85. Benen J. A., Kester H. C., and Visser J. (1999) Kinetic characterization of Aspergillus niger N400 endopolygalacturonases I, II and C. Eur. J. Biochem. 259, 577–585 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.