

Abstract
The long noncoding RNA (lncRNA) NEAT1 (nuclear enriched abundant transcript 1) is the architectural component of nuclear paraspeckles, and it has recently gained considerable attention as it is abnormally expressed in pathological conditions such as cancer and neurodegenerative diseases. NEAT1 and paraspeckle formation are increased in cells upon exposure to a variety of environmental stressors and believed to play an important role in cell survival. The present study was undertaken to further investigate the role of NEAT1 in cellular stress response pathways. We show that NEAT1 is a novel target gene of heat shock transcription factor 1 (HSF1) and is up-regulated when the heat shock response pathway is activated by sulforaphane (SFN) or elevated temperature. HSF1 binds specifically to a newly identified conserved heat shock element in the NEAT1 promoter. In line with this, SFN induced the formation of NEAT1-containing paraspeckles via an HSF1-dependent mechanism. HSF1 plays a key role in the cellular response to proteotoxic stress by promoting the expression of a series of genes, including those encoding molecular chaperones. We have found that the expression of HSP70, HSP90, and HSP27 is amplified and sustained during heat shock in NEAT1-depleted cells compared with control cells, indicating that NEAT1 feeds back via an unknown mechanism to regulate HSF1 activity. This interrelationship is potentially significant in human diseases such as cancer and neurodegenerative disorders.
Keywords: long noncoding RNA (long ncRNA, lncRNA); heat shock factor protein 1 (HSF1); heat shock protein (HSP); gene expression; transcription regulation; heat shock response; NEAT1; paraspeckle; sulforaphane
Introduction
NEAT1 (nuclear enriched abundant transcript 1) is a highly abundant long noncoding RNA (lncRNA)2 that is essential for the formation of specific nuclear bodies called paraspeckles (1–3). There are two overlapping isoforms of NEAT1 transcribed from the same promoter: NEAT1_1 of 3.7 kb and NEAT1_2 of 22.3 kb (2–4). NEAT1_2 is indispensable for paraspeckle formation and is generated when the polyadenylation signal, and thus termination of the NEAT1_1 transcript, is suppressed by a heterogeneous nuclear ribonucleoprotein kinase-dependent mechanism (4). Unlike NEAT1_1, the 3′ end of NEAT1_2 is not polyadenylated but is processed by RNase P cleavage and subsequently stabilized through formation of a triple helical structure (3, 5, 6). Whereas NEAT1_1 is highly expressed in many tissues in mice, the expression patterns of NEAT1_2, and consequently the presence of paraspeckles, are more restricted (7). Recently, NEAT1 was found to be required for mammary gland development and lactation in mice (8). NEAT1 also has a critical role in corpus luteum formation (9). Even though the function of NEAT1 is still not fully understood, several reports have suggested that increased NEAT1 expression regulates the expression of certain genes by sequestering specific mRNAs and proteins into paraspeckles (10–12). NEAT1 expression is up-regulated in response to different cellular stresses, including viral infections, proteasome inhibition, oncogene-induced replication stress, and hypoxia (11–17). Emerging evidence suggests that NEAT1 plays a cytoprotective role, as cells deficient of NEAT1 display increased sensitivity toward stress-induced cell death (11, 15). In line with this, NEAT1 was found to be transcriptionally activated by HIF2α in response to hypoxia in cancer cells and, more recently, reported as a p53 target gene that prevents replication stress and DNA damage induced by mutagenic agents and oncogenes (13, 15, 18, 19). Interestingly, high levels of NEAT1 are associated with tumorigenic characteristics and poor clinical outcome in several human cancers (13, 15, 20).
Cells are constantly subjected to extrinsic and intrinsic stressors that might have detrimental effects unless neutralized by specific cytoprotective mechanisms. The heat shock response is a universal cellular defense mechanism toward agents causing proteotoxic stress (21, 22). Elevated temperatures, as well as wide range of oxidative and electrophilic agents, cause misfolding and damage of cellular proteins that will lead to cellular dysfunction or death unless repaired and/or removed. The heat shock transcription factor 1 (HSF1) plays a key role in this response mechanism (21–24). Under normal conditions, HSF1 is kept in an inactive form in the cytoplasm by a multichaperone complex consisting of Hsp90, Hsp70, Hsp40, and TriC (23, 25–29). Upon activation, HSF1 is released from the repressive complex, undergoes a series of post-translational modifications, and forms homotrimers that accumulate in the nucleus (21, 23, 30). Here, HSF1 stimulates the transcription of genes encoding proteins involved in repair and clearance of damaged proteins (21, 23, 31). HSF1 specifically binds to heat shock elements (HSE), inverted repeats of nGAAn, where “n” is any nucleotide, in the upstream regulatory regions of its target genes (32, 33). Among the best-studied target genes of HSF1 are those encoding protein chaperones, including Hsp70 and Hsp90, that restore proteostasis by regulating folding, activity, and degradation of proteins (34, 35). The heat shock response is attenuated when HSF1 is released from the promoters of its target genes and either degraded or re-engaged into the HSF1-repressive multichaperone complex by a negative feedback mechanism (21, 36).
Here, we report that the isothiocyanate compound sulforaphane (SFN) induces NEAT1 expression and paraspeckle formation in MCF7 cells. This is not dependent on the Keap1–NRF2 pathway but on binding and transcriptional activation of the NEAT1 promoter by HSF1. We have identified a HSE site in the NEAT1 promoter that is highly conserved among vertebrates. Moreover, we show that NEAT1 is up-regulated in response to heat shock demonstrating that up-regulation of NEAT1 is a general event in the heat shock response. Finally, we demonstrate that the expression of HSP70, HSP90, and HSP27 is enhanced and sustained in the heat shock response in NEAT1 knockdown cells, compared with control cells.
Results
SFN induces NEAT1 expression and paraspeckle formation
Several lines of evidence clearly point toward NEAT1 being a stress-induced lncRNA that is involved in cytoprotection (11, 13, 15). NEAT1 expression has recently been shown to be induced by hypoxia and confers protection to hypoxia-induced cell death in breast cancer cells (15). To further determine the role of NEAT1 in oxidative stress, MCF7 cells were treated with the isothiocyanate sulforaphane (SFN), which triggers an antioxidative response in cells by modifying thiol groups in several proteins, including Keap1 in the Keap1–NRF2 pathway (37, 38). NEAT1 expression was assessed by RT-qPCR using two different primer sets: one recognizing both isoforms, and one solely recognizing the long NEAT1_2 isoform (Fig. 1A). SFN potently and rapidly induced the expression of NEAT1 in MCF7 cells (Fig. 1A). Pretreatment of cells with N-acetylcysteine, a strong antioxidant and precursor of cellular GSH, counteracted the effect of SFN on NEAT1 expression (Fig. 1B).
Figure 1.

NEAT1 expression and paraspeckle formation are induced by SFN. A, MCF7 cells were treated with SFN (20 μm) for the indicated time points. RNA was isolated, and the expression of NEAT1 (both isoforms) and NEAT1_2 was determined by RT-qPCR. The mean value ± S.D. of three biological replicates in one experiment is presented as fold change relative to untreated cells. The results are representative of three independent experiments. B, MCF7 cells were pre-incubated with NAC (5 mm) and then treated with SFN for 6 h. NEAT1 expression was determined as described in A. C, MCF7 cells were left untreated or treated with SFN for 6 h, fixed, and subjected to RNA-FISH using probes recognizing the NEAT1_2 isoform. DAPI was used to visualize the nuclei. Bars, 10 μm. D, overall intensity of the dots in at least 250 cells was quantitated using the Volocity software. Mean values ± S.D. of three biological replicates are shown and presented as fold change relative to untreated cells. p values were calculated using ANOVA (A) or Student's t test (B and D) with p < 0.05 considered statistically significant. (***, p ≤ 0.001; **, p ≤ 0.01.)
Paraspeckles are dynamic ribonucleoprotein complexes that form around the NEAT1_2 isoform in the nucleus (4). To determine whether SFN-induced NEAT1 expression is associated with increased paraspeckle formation, we performed RNA-fluorescence in situ hybridization (RNA-FISH) on untreated and SFN-treated MCF7 cells using probes recognizing the long NEAT1_2 isoform. Whereas NEAT1_2-containing punctas appeared small and scarcely distributed in the nucleus of untreated MCF7 cells, SFN treatment potently increased the numbers and the overall signal intensity of the paraspeckles (Fig. 1, C and D).
SFN-induced NEAT1 expression is not dependent on NRF2
SFN stimulates several stress signaling pathways in cells, of which the Keap1–NRF2 pathway is the most prominent. To determine whether NRF2 is involved in SFN-induced NEAT1 expression, MCF7 cells were transfected with an siRNA toward NRF2 and stimulated with SFN for 6 h. The NRF2 protein accumulated after 6 h of SFN treatment, but its depletion did not interfere with the induction of NEAT1 (Fig. 2A). We also assessed the NEAT1 expression in control and NRF2-depleted cells after a prolonged treatment with SFN for 24 h. Elevated levels of NEAT1 were observed in both control and siNRF2-transfected cells (Fig. 2B). In contrast, SFN-mediated induction of NQO1 mRNA, a well-established target of NRF2, was severely reduced in NRF2-depleted cells (Fig. 2C). We conclude that SFN-induced NEAT1 expression is not dependent on the Keap1–NRF2 pathway.
Figure 2.

NEAT1 induction by SFN is not dependent on NRF2. A, MCF7 cells were transfected with an siRNA specifically targeting NRF2 (siNRF2) or control siRNA (siCtrl). Twenty four h post-transfection, cells were either left untreated or treated with SFN (20 μm) for 6 h. NEAT1 expression was determined by RT-qPCR as described in Fig. 1. Depletion of NRF2 expression in whole-cell extracts was verified by Western blotting analyses using an NRF2 antibody. The membrane was re-probed with an anti-actin antibody to ensure equal loading. B and C, MCF7 cells were transfected as described in A and subjected to a long-term treatment with SFN (10 μm) for 24 h. The expression of NEAT1 and NEAT1_2 (B) and NQO1 (C) was determined by RT-qPCR. Experiments were performed in triplicate, and the graph is representative of three independent experiments. (**, p ≤ 0.01; *, p, ≤ 0.05; ns, not significant.)
SFN-induced NEAT1 expression and paraspeckle formation are dependent on HSF1
SFNs, as well as other oxidants, have recently been shown to stimulate HSF1, the key transcription factor conferring cellular protection to agents causing protein misfolding (39, 40). We therefore sought to determine whether SFN-induced NEAT1 expression depends on a mechanism involving HSF1. SFN treatment indeed induced a mobility shift of HSF1, which is associated with its activation, and nuclear accumulation of the protein (Fig. 3, A and B). Consistent with the observed shift and nuclear translocation of HSF1, SFN potently induced the expression of the Hsp70 mRNA, a prominent target gene of HSF1 (Fig. 3C). We next transfected MCF7 cells with two different siRNAs specifically silencing HSF1 expression, and we determined the effect on SFN-induced NEAT1 expression. Both siRNAs significantly reduced the increase in NEAT1 levels observed after SFN treatment (Fig. 3D). The same was observed when HSF1 expression was silenced in SFN-treated HeLa cells (Fig. 3E). To determine whether SFN-induced paraspeckle formation depends on HSF1, we performed co-immuno-FISH analyses on control and HSF1-depleted cells using an HSF1 antibody and probes specifically binding to NEAT1_2. In line with the observations described above, SFN enhanced the nuclear staining of HSF1 (Fig. 4, A and B) and the formation of NEAT1_2 containing paraspeckles (Fig. 4, A and C). Importantly, SFN-induced paraspeckle formation was severely compromised in HSF1-depleted cells (Fig. 4, A and C). Taken together, our data clearly demonstrate that HSF1 is essential for increased NEAT1 expression and paraspeckle formation as a response to SFN-treatment in MCF7 cells.
Figure 3.

SFN-induced NEAT1 expression depends on HSF1. A and B, MCF7 cells were left untreated or treated with SFN (20 μm) for 6 h. HSF1 expression in WCE (A) and nuclear extracts (NE) (B) were determined by immunoblot analyses. Equal loading was verified by re-probing the membranes with actin (A) or lamin B (B) antibodies. C, cells were treated with SFN as described above, and HSP70 expression was determined by RT-qPCR. D, MCF7 cells were transfected with two different siRNAs targeting HSF1, siHSF1_#1, and siHSF1_#2, or a control siRNA. Forty eight hours post-transfection, cells were left untreated or treated with SFN for 6 h. NEAT1 expression was assessed by RT-qPCR. SiRNA-mediated HSF1 depletion was verified by immunoblot analyses. E, HeLa cells were transfected with siHSF1_#2 or control siRNA, and after 48 h SFN-induced NEAT1 expression was determined by RT-qPCR. HSF1 expression was determined by immunoblot analyses using actin as loading control. (*, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.)
Figure 4.

HSF1 depletion abrogates SFN-induced paraspeckle formation. A, MCF7 cells were transfected with an HSF1-specific siRNA or a control siRNA. After 48 h, cells were left untreated or treated with SFN for 6 h, fixed, and subjected to coimmuno-FISH analyses by confocal microscopy using an antibody recognizing HSF1 (red) and fluorescent probes binding to NEAT1_2 (green). Nuclei were visualized with DAPI (blue). All experiments were performed in triplicate. Bars, 10 μm. B and C, intensities of NEAT1_2 containing paraspeckles and nuclear HSF1 staining in at least 250 cells were quantitated using Volocity software. (***, p ≤ 0.001.)
NEAT1 is transcriptionally regulated by HSF1
Having established that SFN induces NEAT1 expression by an HSF1-dependent mechanism, we next asked whether SFN treatment leads to transcriptional activation of the NEAT1 gene. A luciferase reporter vector containing nucleotides −4040 to +144 of the NEAT1 upstream regulatory region was generated and transfected into MCF7 cells. Reporter gene assays were performed in extracts from untreated and SFN-treated cells. SFN significantly stimulated the NEAT1 promoter-driven luciferase activity (Fig. 5A). This stimulation was severely compromised upon co-transfection with an HSF1-directed siRNA, demonstrating that SFN-induced activation of the NEAT1 promoter depends on HSF1 (Fig. 5B). HSF1 binds to heat shock elements (HSE) within its target genes that are composed of alternating inverted repeats of 5 bp, nGAAn where “n” is any nucleotide (32, 33). We carefully inspected the NEAT1 promoter and identified three putative HSEs. One of these, located between nucleotides −445 and −431, specifically caught our attention as it is highly conserved between species (Fig. 5C). To determine whether this region is involved in SFN-activated NEAT1 transcription, a truncated construct of the NEAT1 promoter reporter vector was made containing nucleotides −470 to +144. We also made a mutated version where we introduced four point mutations in the predicted HSE core, and both constructs were transfected into MCF7 cells. SFN potently stimulated transcription from the truncated NEAT1 promoter (Fig. 5D). This stimulation was absolutely dependent on an intact HSE core, as point mutations in this region totally abolished the SFN-induced increase in NEAT1 promoter-driven luciferase activation. To analyze whether HSF1 can bind to the NEAT1 promoter in vivo, ChIP experiments were conducted on untreated and SFN-treated MCF7 cells using an antibody against HSF1 and RT-qPCR primers amplifying a 100-bp fragment of the NEAT1 promoter encompassing the HSE site. HSE-containing NEAT1 promoter fragments co-precipitated with the HSF1 antibody (Fig. 5E). Importantly, SFN robustly increased HSF1 binding to NEAT1 HSE fragments. Primers amplifying a GAPDH fragment and a region of the NEAT1 promoter upstream of the HSE site (“upstr”) were used as controls. Control ChIPs with IgG gave very high Ct values compared with that of the HSF1 antibody and, importantly, showed no differences upon SFN stimulation.
Figure 5.

HSF1 binds to and transcriptionally activates the NEAT1 promoter. A, MCF7 cells were transfected with a luciferase reporter vector containing 4040 bp of the NEAT1 upstream region (pNEAT1-luc) or empty control vector. After 24 h, cells were left untreated or treated with SFN (20 μm) for 8 h, and luciferase assays were performed. The experiments were performed in triplicate, and mean values ± S.D. are shown. The result is representative of three independent experiments. B, MCF7 cells were co-transfected with pNEAT1-luc and siHSF1_#2 as described under “Experimental procedures.” Cells were left untreated or treated with SFN for 8 h, and luciferase assays were performed. C, sequence conservation within NEAT1 upstream regions is illustrated by PhyloP Basewise Conservation scores from 100 vertebrates (USCS Genome Browser). An alignment of conserved HSE core sequences from human, rhesus, mouse, dog, and elephant is shown. D, truncated mutant of the NEAT1 promoter luciferase reporter construct encompassing the putative HSE site was generated and transfected into MCF7 cells along with a version harboring four point mutations within the HSE consensus sequence. SFN-induced luciferase activity was measured 24 h post-transfection. E, MCF7 cells were left untreated or treated with SFN (20 μm) for 6 h, and ChIP assays were performed using an anti-HSF1 antibody. qPCR was performed with primers flanking the HSE site. Primers flanking a region further upstream in the NEAT1 promoter (“upstr”), as well as primers amplifying a region of the GAPDH promoter, were used as negative controls. The relative amount of immunoprecipitated DNA compared with input DNA for each primer set is shown for the HSF1 ChIP. The values obtained by the IgG ChIP was less than 0.003% for the HSF1 and control primers. The result is representative of three independent ChIP experiments, where qPCR reactions were done as triplicates. (***, p ≤ 0.001; **, p ≤ 0.01; ns, not significant.)
NEAT1 is induced by heat shock
Having established that NEAT1 levels are enhanced by an HSF1-dependent mechanism upon SFN treatment, we next sought to determine whether NEAT1 is induced as response to heat shock (HS). MCF7 cells were incubated at 43 °C for 30 min and either harvested directly or after recovery at 37 °C for the indicated periods. HSF1 was rapidly activated during HS as assessed by a mobility shift in Western blotting (Fig. 6A). This was accompanied by increased expression of the Hsp70 mRNA (Fig. 6B). Importantly, HS rapidly and transiently stimulated the expression of NEAT1 (Fig. 6C). This indicates that elevated NEAT1 expression is a general mechanism in the heat shock response pathway.
Figure 6.

NEAT1 is induced by heat shock. A and B, MCF7 cells were subjected to heat shock by incubation at 43 °C for 30 min and then returned to 37 °C to recover for the indicated time periods. Activation of HSF1 was verified by shifted migration in Western blotting analyses (A) and by induction of HSP70 mRNA expression (B). C, cells were treated as above, and expression of NEAT1 and NEAT1_2 was assessed by RT-qPCR. (***, p ≤ 0.001; **, p ≤ 0.01; *, p ≤ 0.05.)
Proliferation is compromised and expression of HSF1 target genes is amplified in NEAT1-depleted cells
Elevated NEAT1 levels and paraspeckle formation in response to cellular stress are widely observed and believed to play a prosurvival role by regulating the expression of specific genes. To start unraveling the function of NEAT1 in the heat shock response, we measured the sensitivity of control and NEAT1-depleted cells to heat shock by cell confluence proliferation assays. MCF7 cells were transfected with NEAT1-specific GapmeR antisense oligonucleotides (ASOs), which generally reduced the NEAT1 expression by 70–80% for up to 120 h or a control GapmeR. Cell confluence was then monitored for 96 h using the IncuCyte® live cell analysis system. After the first 48 h, half of the cells were subjected to heat shock for 30 min and then returned to IncuCyte system for another 48 h. Strikingly, NEAT1 depletion severely decreased the confluency of MCF7 cells, indicating that NEAT1 is necessary for their proliferation or survival (Fig. 7A). The proliferation rate was not further decreased after heat shock compared with cells kept at 37 °C over the whole-monitoring period (Fig. 7, A and B). Taken together, this suggests that NEAT1 is generally required for the proliferation or survival of MCF7 cells and that an additional stress, such as heat shock, does not further affect the already growth-inhibited cells. Control-transfected cells generally recovered well after heat shock with only a slight reduction in confluency (Fig. 7, A and B).
Figure 7.
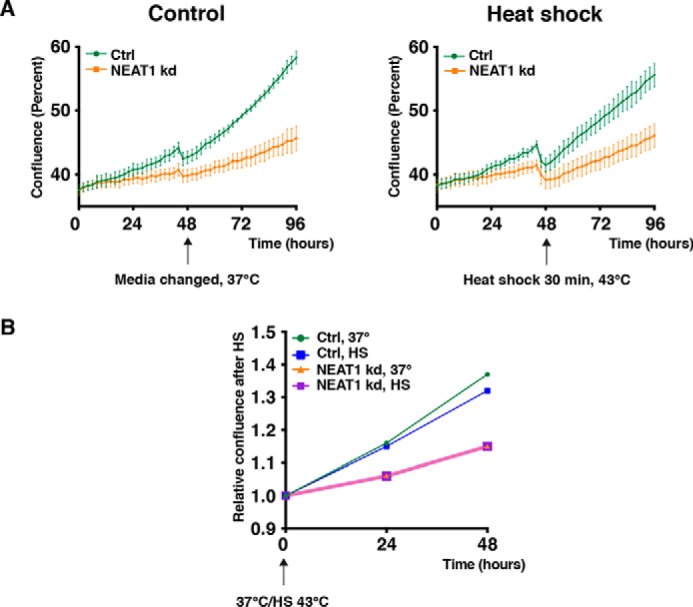
Proliferation is compromised in NEAT1-depleted cells. A, MCF7 cells were transfected with two LNA-GapmeR antisense oligos targeting NEAT1 or a negative control oligo and immediately placed in a IncuCyte® live cell analysis system for cell confluence monitoring. After 48 h, cells were removed from the incubator, and for half of the cells the medium was changed at 37 °C, whereas the other half was subjected to heat shock at 43 °C for 30 min. All the cells were then returned to the IncuCyte® live cell analysis system and monitored for another 48 h. Confluency (%) was calculated using the IncuCyte® S3 software. Mean values ± S.D. of 15 images (three images from each well of five wells in total) are shown. The results are representative for three independent experiments. B, relative confluency of cells over the last 48 h of the experiment described in A is shown.
To further analyze the role of NEAT1 in the heat shock response, we assayed the expression of the HSF1 target genes encoding Hsp70, Hsp90, and Hsp27 in control and NEAT1-depleted cells. MCF7 cells were transfected with two different GapmeR ASOs, which either targeted both isoforms of NEAT1 or solely the long NEAT1_2 isoform. Transfected cells were exposed to heat shock, and HSP70, HSP90, and HSP27 expression was assessed by RT-qPCR. Interestingly, the expression of all target genes was repeatedly amplified and sustained in cells where NEAT1 was silenced, compared with cells transfected with a control GapmeR (Fig. 8). Moreover, the background expression in unstressed cells was slightly enhanced. Of note, a stronger effect on the HSF1 target genes was observed for cells transfected with the GapmeR targeting both isoforms of NEAT1, compared with those transfected with the GapmeR only silencing the NEAT1_2 isoform. Taken together, our data suggest that NEAT1 depletion, by some mechanism, potentiates the HSF1 activity by either creating additional proteotoxic stress in the cells or by regulating the turnover or the activity of the HSF1 protein.
Figure 8.

NEAT1 knockdown amplifies the expression of HSF1 target genes upon heat shock. MCF7 cells were transfected with two different LNA-GapmeR NEAT1 antisense oligos either targeting both isoforms of NEAT1 or solely the long NEAT1_2 isoform and a negative control oligo. After 48 h, cells were subjected to heat shock and recovered for the indicated time periods. The expression of Hsp70, Hsp90, and Hsp27 was determined by RT-qPCR. Knockdown of NEAT1 and NEAT1_2 was verified by RT-qPCR. (***, p ≤ 0.001; **, p ≤ 0.01; *, p ≤ 0.05.)
Discussion
High-throughput RNA-Seq has demonstrated that most cells express a plethora of long noncoding transcripts (41, 42). During the last few years, huge efforts have been made to reveal their biological function, and many of them now appear as important contributors to gene regulation at different levels. NEAT1 is the architectural component of nuclear ribonucleoprotein complexes called paraspeckles, and it has recently gained considerable attention as several reports have shown that the transcript is abnormally expressed in human diseases, including cancer (13, 15, 20). The function of NEAT1 remains elusive, but emerging evidence suggests that NEAT1 and paraspeckles have a role in cytoprotection. Here, we show that NEAT1 is induced at the transcriptional level by the isothiocyanate compound SFN. This is accompanied by increased paraspeckle formation. SFN mimics oxidative stress in cells by modifying thiol groups in cellular proteins and induces antioxidative response pathways of which Keap1–NRF2 is the most prominent (37, 38). We demonstrate that SFN-induced NEAT1 expression is not dependent on NRF2. In contrast, depletion of HSF1 severely abrogates SFN-induced NEAT1 expression and paraspeckle formation. Several reports have shown that SFN and other sulfhydryl-reactive compounds can stimulate the heat shock response pathway in cells by activating HSF1 (39, 40, 43, 44). The mechanism for how SFN activates HSF1 is somewhat obscure, but previous studies have shown that oxidative compounds might promote the DNA-binding activity of HSF1 by modifying cysteine residues in the DNA-binding domain (45, 46). SFN has also been shown to modify Hsp90 and thereby disrupt complex formation between Hsp90 and its protein partners (47, 48). Recently, Naidu et al. (44) reported that phenethyl isothiocyanate indeed modified cysteine residues within Hsp90 leading to dissociation and activation of HSF1.
Our results show that HSF1 accumulates in the nucleus upon SFN treatment and binds to the NEAT1 promoter in vivo. We have identified a conserved HSE in the NEAT1 promoter that is critical for SFN-induced transcriptional activation of the NEAT1 gene. Intriguingly, this site overlaps with a recently reported NF-κB–binding site, which is necessary for lipopolysaccharide-induced NEAT1 expression in lung cancer cells (49). An overlapping NF-κB- and HSF1-binding site has been identified previously in the promotor of the gene encoding MHC class I chain-related protein A (MICA) (50). Here, HSF1 and NF-κB bind mutually exclusive to the site, and overexpression of a truncated version of HSF1 containing only the DNA-binding domain outcompetes NF-κB binding and abolishes TNFα-induced MICA expression. The question as to whether the overlapping HSF1/NF-κB site in the NEAT1 promoter represents a regulatory hub, coordinating outputs from different signaling pathways, remains to be resolved.
In this study, we show that NEAT1, as well as being induced by SFN, is also induced upon heat shock. This clearly suggests that NEAT1 up-regulation is a general phenomenon in the heat shock response. This is supported by a study by Hirose et al. (11), demonstrating that NEAT1 expression and paraspeckle formation are induced by inhibition of the 26S proteasome by MG132 or bortezomib. Proteasome inhibition causes a proteotoxic stress in the cells as proteins that are destined for degradation form aggregates in both the cytoplasm and the nucleus (11, 51). Activation of HSF1 to induce expression of molecular chaperones is a general cellular response mechanism to proteasome inhibition (52–54). Thus, we envision that NEAT1 induction upon proteasome inhibition might be mediated by HSF1-mediated transcriptional activation of the NEAT1 promoter.
Several reports have shown that NEAT1 depletion sensitizes cells to a variety of stressors. Thus, we hypothesized that knockdown of NEAT1 expression would make cells more susceptible to heat shock. However, we repeatedly observed that transient transfection with NEAT1 antisense oligos by itself dramatically reduced the proliferation of MCF7 cells and that this tendency was not reinforced by heat shock. This shows that MCF7 cells cultivated in vitro are highly dependent on NEAT1. To further dissect the function of NEAT1 in the heat shock response, we knocked down NEAT1 expression by antisense oligos and assessed the effect on the expression of three HSF1 target genes encoding Hsp70, Hsp90, and Hsp27. Interestingly, knockdown of NEAT1 amplified and prolonged the expression of these target genes. The mechanism for this is still obscure. NEAT1 depletion abrogates the formation of paraspeckles (4). This might lead to mislocalization of paraspeckle-associated proteins that disturbs proteostasis in the cells and thereby contribute to the activation of HSF1. Alternatively, NEAT1 might regulate the turnover of the HSF1 protein or activity by a negative feedback mechanism. Interestingly, the effect of NEAT1 depletion on HSF1 target genes was significantly stronger when cells were transfected with a GapmeR targeting both isoforms compared with one only reducing NEAT1_2 expression. This indicates that the short NEAT1_1 isoform has an important function in the regulation of the heat shock response.
HSF1 plays a critical role in the cellular defense to proteotoxic stress. Many neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), Huntington's disease, and Alzheimer's disease are associated with the formation of protein aggregates (31, 55). Loss of HSF1 expression or activity is frequently observed in these diseases (55–58). Our results demonstrate that HSF1 activates the expression of NEAT1 during the heat shock response. Interestingly, several reports have shown that NEAT1 is abnormally expressed in ALS and Huntington's disease (59–61). Moreover, mislocalization of two paraspeckle proteins, FUS (Fused in sarcoma) and TDP-43 (TAR DNA-binding protein-43) is well-known to be associated with ALS (62). It has been speculated that NEAT1 expression and paraspeckle formation might have a protective role in neuronal cells in early stages of ALS and Huntington's disease (60, 61, 63). In line with this, Hirose et al. (11) showed that mouse embryonic fibroblasts from NEAT1 knockout cells displayed an increased sensitivity to proteasome inhibitors causing formation of protein aggregates, compared with WT cells. The cross-talk between NEAT1, paraspeckle formation, subcellular localization of FUS and TDP-43, and HSF1 in these devastating diseases should be a focus of future research.
Constitutive activation of HSF1 and abnormal expression of NEAT1 are both frequently observed in human cancers (13, 15, 20, 64–66). There is clear evidence that both HSF1 and NEAT1 have cytoprotective roles in tumors and are associated with poor prognosis. In this study, we demonstrate that NEAT1 is a novel target gene of HSF1. It remains to be determined whether there is any correlation between HSF1 activation and NEAT1 expression in cancer.
Experimental procedures
Cell culture and treatments
MCF7 (ATCC® HTB-22TM) and HeLa (ATCC® CCL-2TM) cells were purchased from American Type Culture Collection and maintained in minimal essential medium (Sigma) supplemented with 10% fetal bovine serum (Biochrom) and 1% penicillin/streptomycin (Sigma). MCF7 cells were cultured in the presence of 0.01 mg/ml insulin (Sigma). All cells were grown at 37 °C in humidified condition containing 5% CO2. SFN (catalog no. S4441) and N-acetylcysteine (NAC, catalog no. A9165) were purchased from Sigma. SFN was added to the cells at a final concentration of 20 μm for short-term treatments up to 8 h and at a final concentration of 10 μm for long-term treatment (24 h). When included, NAC (5 mm) was added to the media 1 h before SFN treatment. To induce a cellular heat shock response, cells were incubated at 43 °C for 30 min and then either harvested directly or returned to 37 °C for recovery.
Plasmid constructions
The human NEAT1 promoter (−4040/+144) was cloned from genomic DNA by performing two PCR amplification reactions using PrimeSTAR®GXL DNA polymerase (Takara Bio Inc, R050Q) generating fragments of 1756 bp (primers NP1.1F/NP2.1R) and 2414 bp (nested PCR, outer primer set NP2.1F/NP3.1R; inner primer set NP2.2F/NP3.2R). The 1756-bp fragment was digested with NheI (provided in primer) and HindIII (internal) and cloned into corresponding sites in pGL3-Basic (Promega). This was followed by insertion of the 2414-bp fragment into the HindIII site using internal HindIII sites. The resulting pNEAT1(−4040/+144)-luc plasmid was verified by sequencing. pNEAT1(−470/+144)-luc was generated from a promoter construct containing the 2414-bp PCR product (pNEAT1(−2384 bp/+144)-luc) by cutting with KpnI and PstI followed by religation. pNEAT1(−470/+144)-HSEmut-luc was made by site-directed mutagenesis according to the QuikChange II site-directed mutagenesis kit protocol (Agilent Technologies). All primer sequences are provided in Table 1.
Table 1.
Primer and siRNA/ASO sequences
F is forward, R is reverse.
RNAi
siNRF2 (siGENOME SMART pool human NFE2L2, DM-003-755-02) was purchased from Dharmacon, and siHSF1_#1 (Silencer® Select, s6950), siHSF1_#2 (Silencer® Select, s6952), and Silencer® Select Negative Control No. 2 were obtained from ThermoFisher Scientific. Locked nucleic acid-GapmeR NEAT1 antisense oligos and control GapmeRs were purchased from Exiqon. All sequences are provided in Table 1. Cells were transfected using Lipofectamine 2000 according to the reverse transfection protocol provided by the manufacturer (ThermoFisher Scientific). Successful knockdown was verified by RT-qPCR or Western blotting analyses.
Reverse transcription and quantitative PCR
Cells were lysed in TRI Reagent®, and total RNA was isolated with Direct-zol RNA MiniPrep (Zymo Research) according to the manufacturer. RNA concentration was measured by NanoDrop 2000 (ThermoFisher Scientific), and cDNA synthesis of total RNA was performed with SuperScriptTM IV reverse transcriptase (ThermoFisher Scientific). 2.5 μm of random hexamer primer (ThermoFisher Scientific) and ∼250 ng of template were used for the reaction. Total RNA was denatured at 65 °C for 5 min, and cDNA was synthesized at 50 °C for 10 min. Quantitative PCR was run on a LightCycler 96 (Roche Applied Science) with the SYBR Green Reaction Mix FastStart Essential DNA Green Master (Roche Applied Science) and 0.25 μm forward and reverse primer. (Thermal cycle conditions used are as follows: 95 °C for 10 min and 40 cycles of 95 °C for 10 s, 60 °C 10 s, and 72 °C for 10 s.) All primers sequences are provided in Table 1. Experiments were done in triplicates, and the ΔΔCq method was used for fold change calculations. GAPDH was used as reference gene.
Immunoblotting
Whole-cell extracts (WCE) were made by lysing cells directly in 2× NuPAGE LDS Sample Buffer (ThermoFisher Scientific). Nuclear extracts (NE) were isolated using the NE-PERTM nuclear and cytoplasmic extraction kit (ThermoFisher Scientific) according to the manufacturer's instruction. In brief, cells were resuspended in Cytoplasmic Extraction Reagent I and II, and nuclei were pelleted by centrifugation at 16,000 × g. The pellet was resuspended in ice-cold Nuclear Extraction Reagent, vortexed for 1 min, and incubated on ice for 10 min. This step was repeated three more times before centrifugation at 16,000 × g for 10 min. Proteins were resolved on SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Equal loading of proteins was verified by probing the membranes with an antibody recognizing actin (WCE) or lamin B (NE). The following primary antibodies were used, all at 1:000 dilution: rabbit anti-NRF2 (Abcam, catalog no. ab62359); rabbit anti-HSF1 (Cell Signaling, catalog no. 4356); rabbit anti-lamin B (Proteintech, catalog no. 12987-1-AP); and mouse anti-actin (Millipore, MAB1501). The blots were detected with IRDye®-conjugated secondary antibodies (LI-COR Biosciences) at a 1:10,000 dilution (800CW goat anti-rabbit, catalog no. 926-32211; 680LT goat anti-mouse, catalog no. 926-68020), and the Odyssey® CLx IR Imaging System.
RNA-fluorescence in situ hybridization and immunofluorescence staining
Stellaris® NEAT1 RNA FISH probes recognizing the NEAT1_2 isoform (VSMF-2251-5, Quasar® 670-conjugated) were purchased from LGC Biosearch Technologies. Preparation of cells, hybridization, and mounting were performed according to the Stellaris® RNA-FISH Probes manual. In brief, cells were seeded onto circular coverslips in 12-well dishes and allowed to attach for 2–3 days. They were fixed with 4% freshly made formaldehyde at room temperature, and permeabilized with 70% ethanol. Hybridization was done at 37 °C in a humidifying chamber overnight. For co-immuno-FISH experiments, the hybridization was performed as described above, and cells were subsequently incubated in 1% RNase-free BSA for 30 min and then stained with anti-HSF1 antibody for 1 h (1:50, Cell Signaling, catalog no. 4356A). Cells were incubated with goat anti-rabbit Alexa 488-conjugated secondary antibody (1:500, ThermoFisher Scientific, catalog no. A11070) and mounted using Vectashield® antifade mounting medium containing DAPI (Vector Laboratories, H-1200). Images were generated using a Zeiss LSM780 confocal microscope (Carl Zeiss Microscopy GmbH, Jena, Germany). In all samples, Z-stack (five slices, 2.5 μm total height) images were taken at ×40 magnification. For all images, the middle Z-slice was positioned at DAPI's best focus. The same treatment and setting were applied to all replicates, and for each slide at least 10 pictures were taken for Volocity analysis. The Volocity software (PerkinElmer Life Sciences, version 6.3) was used to measure signal intensities for both NEAT1_2 and HSF1 signals. At least 250 cells in each group of treatments were analyzed by Volocity software. The mean intensity of NEAT1_2 or nuclear HSF1 signals in the SFN-treated group was normalized against control.
Reporter gene assays
Sub-confluent MCF7 cells in 12-well plates were transfected with 150 ng of luciferase reporter plasmids using Lipofectamine®2000 reagent (ThermoFisher Scientific) according to the manual provided by the manufacturer. After 24 h, cells were either left untreated or treated with SFN (20 μm) for 8 h. Cells were harvested, and luciferase assays were performed using the Dual-Light® luciferase and β-galactosidase reporter gene assay system (ThermoFisher Scientific). Of note, cells were initially co-transfected with luciferase reporter plasmids and an expression vector for β-gal, but as SFN repeatedly interfered with the β-gal activity in the cells, the expression vector was omitted from the transfections, and only the luciferase activity was included in the analyses. Co-transfections with siRNA and plasmid DNA were performed in two steps using Lipofectamine®2000. First, siRNAs were introduced into the cells by reverse transfection. After 48 h, plated cells were re-transfected with plasmid DNA and left for another 24 h.
Chromatin immunoprecipitation (ChIP) assays
MCF7 cells were seeded at a density of 6 million cells per 10-cm dish the day before use. The cells were left untreated or treated with SFN (20 μm) for 6 h before harvesting. Two 10-cm dishes were used per condition. The “iDeal ChIP-seq kit for Transcription Factors” (Diagenode, C01010055) was used for harvest and ChIP according to the manufacturer's instruction. The two dishes for each treatment were combined, and the approximate cell number was estimated to be 15 million cells. Volumes of buffers used in the kit were adjusted to this. Cells were fixed for 15 min. Sonication was performed in ice-cold water on a Bioruptor UCD-200 (Diagenode) for 3 × 10 minutes using 30-s on/off pulses. Samples run on an agarose gel showed a majority of DNA with size from 100 to 400 bp after shearing. For immunoprecipitation, 10 μl of anti-HSF1 antibody (Cell Signaling, 4356) or 1 μl of IgG (provided with the kit) was used with 200 μl of sheared chromatin. Two μl (1%) of input chromatin was set aside. The eluate had a volume of 25 μl, which was diluted 1:10 before 5 μl was used in a qPCR. qPCR was performed in triplicate on a LightCycler 96 (Roche Applied Science). The relative amount of immunoprecipitated (IP) DNA compared with input DNA was calculated using the “percent input method” as follows: Because the input chromatin was 1%, a dilution factor of 100 (6644 cycles, log2 of 100) was subtracted to adjust input Ct value to 100%. To calculate the percentage of specific chromatin co-immunoprecipitated with the HSF1 antibody or the IgG control, the triplicate average Ct values, Ct(IP), for the specific qPCR primers (HSE, “upstream,” and GAPDH) were used in the equation: 100·2∧(adjusted input − Ct(IP)). Primer sequences are given in Table 1.
Cell confluence proliferation assay
MCF7 cells were transfected in solution with indicated LNA-GapmeR antisense oligos and seeded in 96-well plates at an initial confluency of ∼30% (20,000 cells per well) and immediately placed in an IncuCyte® S3 live-cell analysis system, which is a fully equipped automated microscope for cell confluence monitoring. Three phase-contrast images were acquired from each well at 120-min intervals over a period of 96 h, using a ×20 objective. For each condition, five wells were monitored. Data were analyzed using the IncuCyte® S3 software.
Statistics
GraphPad software (Prism version 7, Mac OS X) was used to analyze quantitative data. Statistical significance was evaluated with unpaired Student's t test or one-way ANOVA followed by the Dunnett's multiple comparison test. The data were considered statistically significant when p ≤ 0.05. For all experiments, significance is expressed as ***, p ≤ 0.001, **, p ≤ 0.01, and *, p ≤ 0.05. The error bars indicate ± S.D. in all figures. All the experiments were performed at least three times.
Author contributions
S. L., I. A. R., A. H., I. M., E. K., and M. P. formal analysis; S. L., I. A. R., A. H., L. T. K., I. M., E. K., and M. P. investigation; S. L., I. A. R., A. H., L. T. K., I. M., and M. P. methodology; S. L. and M. P. writing-original draft; E. K. and M. P. conceptualization; M. P. supervision; M. P. project administration.
Acknowledgments
We thank Kenneth Bowitz Larsen for help with the image analyses and Hanne Britt Brenne for technical advice.
This work was supported by Helse Nord RHF (Northern Norway Regional Health Authority). The authors declare that they have no conflicts of interest with the contents of this article.
- lncRNA
- long noncoding RNA
- WCE
- whole-cell extract
- qPCR
- quantitative PCR
- SFN
- sulforaphane
- HSE
- heat shock element
- ANOVA
- analysis of variance
- IP
- immunoprecipitated
- HS
- heat shock
- DAPI
- 4′,6-diamidino-2-phenylindole
- oligo
- oligonucleotide
- ALS
- amyotrophic lateral sclerosis
- NAC
- N-acetylcysteine
- RNA-FISH
- RNA-fluorescence in situ hybridization
- NE
- nuclear extract
- ASO
- antisense oligonucleotide
- MICA
- MHC class I chain-related protein A
- FUS
- Fused in sarcoma
- LNA
- locked nucleic acid.
References
- 1. Clemson C. M., Hutchinson J. N., Sara S. A., Ensminger A. W., Fox A. H., Chess A., and Lawrence J. B. (2009) An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 33, 717–726 10.1016/j.molcel.2009.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hutchinson J. N., Ensminger A. W., Clemson C. M., Lynch C. R., Lawrence J. B., and Chess A. (2007) A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8, 39 10.1186/1471-2164-8-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sunwoo H., Dinger M. E., Wilusz J. E., Amaral P. P., Mattick J. S., and Spector D. L. (2009) MEN ϵ/β nuclear-retained noncoding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 19, 347–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Naganuma T., Nakagawa S., Tanigawa A., Sasaki Y. F., Goshima N., and Hirose T. (2012) Alternative 3′-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. EMBO J. 31, 4020–4034 10.1038/emboj.2012.251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown J. A., Valenstein M. L., Yario T. A., Tycowski K. T., and Steitz J. A. (2012) Formation of triple-helical structures by the 3′-end sequences of MALAT1 and MENβ noncoding RNAs. Proc. Natl. Acad. Sci. U.S.A. 109, 19202–19207 10.1073/pnas.1217338109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wilusz J. E., Freier S. M., and Spector D. L. (2008) 3′ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 135, 919–932 10.1016/j.cell.2008.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakagawa S., Naganuma T., Shioi G., and Hirose T. (2011) Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J. Cell Biol. 193, 31–39 10.1083/jcb.201011110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Standaert L., Adriaens C., Radaelli E., Van Keymeulen A., Blanpain C., Hirose T., Nakagawa S., and Marine J. C. (2014) The long noncoding RNA Neat1 is required for mammary gland development and lactation. RNA 20, 1844–1849 10.1261/rna.047332.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakagawa S., Shimada M., Yanaka K., Mito M., Arai T., Takahashi E., Fujita Y., Fujimori T., Standaert L., Marine J. C., and Hirose T. (2014) The lncRNA Neat1 is required for corpus luteum formation and the establishment of pregnancy in a subpopulation of mice. Development 141, 4618–4627 10.1242/dev.110544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen L. L., and Carmichael G. G. (2009) Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Mol. Cell 35, 467–478 10.1016/j.molcel.2009.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hirose T., Virnicchi G., Tanigawa A., Naganuma T., Li R., Kimura H., Yokoi T., Nakagawa S., Bénard M., Fox A. H., and Pierron G. (2014) NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Mol. Biol. Cell 25, 169–183 10.1091/mbc.e13-09-0558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Imamura K., Imamachi N., Akizuki G., Kumakura M., Kawaguchi A., Nagata K., Kato A., Kawaguchi Y., Sato H., Yoneda M., Kai C., Yada T., Suzuki Y., Yamada T., Ozawa T., et al. (2014) Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Mol. Cell 53, 393–406 10.1016/j.molcel.2014.01.009 [DOI] [PubMed] [Google Scholar]
- 13. Adriaens C., Standaert L., Barra J., Latil M., Verfaillie A., Kalev P., Boeckx B., Wijnhoven P. W., Radaelli E., Vermi W., Leucci E., Lapouge G., Beck B., van den Oord J., Nakagawa S., et al. (2016) p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat. Med. 22, 861–868 10.1038/nm.4135 [DOI] [PubMed] [Google Scholar]
- 14. Beeharry Y., Goodrum G., Imperiale C. J., and Pelchat M. (2018) The Hepatitis Delta Virus accumulation requires paraspeckle components and affects NEAT1 level and PSP1 localization. Sci. Rep. 8, 6031 10.1038/s41598-018-24500-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choudhry H., Albukhari A., Morotti M., Haider S., Moralli D., Smythies J., Schödel J., Green C. M., Camps C., Buffa F., Ratcliffe P., Ragoussis J., Harris A. L., and Mole D. R. (2015) Tumor hypoxia induces nuclear paraspeckle formation through HIF-2α dependent transcriptional activation of NEAT1 leading to cancer cell survival. Oncogene 34, 4482–4490 10.1038/onc.2014.378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ma H., Han P., Ye W., Chen H., Zheng X., Cheng L., Zhang L., Yu L., Wu X., Xu Z., Lei Y., and Zhang F. (2017) The long noncoding RNA NEAT1 exerts antihantaviral effects by acting as positive feedback for RIG-I signaling. J. Virol. 91, e02250–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Q., Chen C. Y., Yedavalli V. S., and Jeang K. T. (2013) NEAT1 long noncoding RNA and paraspeckle bodies modulate HIV-1 posttranscriptional expression. MBio 4, e00596–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Idogawa M., Ohashi T., Sasaki Y., Nakase H., and Tokino T. (2017) Long non-coding RNA NEAT1 is a transcriptional target of p53 and modulates p53-induced transactivation and tumor-suppressor function. Int. J. Cancer 140, 2785–2791 10.1002/ijc.30689 [DOI] [PubMed] [Google Scholar]
- 19. Mello S. S., Sinow C., Raj N., Mazur P. K., Bieging-Rolett K., Broz D. K., Imam J. F. C., Vogel H., Wood L. D., Sage J., Hirose T., Nakagawa S., Rinn J., and Attardi L. D. (2017) Neat1 is a p53-inducible lincRNA essential for transformation suppression. Genes Dev. 31, 1095–1108 10.1101/gad.284661.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chakravarty D., Sboner A., Nair S. S., Giannopoulou E., Li R., Hennig S., Mosquera J. M., Pauwels J., Park K., Kossai M., MacDonald T. Y., Fontugne J., Erho N., Vergara I. A., Ghadessi M., et al. (2014) The oestrogen receptor α-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat. Commun. 5, 5383 10.1038/ncomms6383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gomez-Pastor R., Burchfiel E. T., and Thiele D. J. (2018) Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 19, 4–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li J., Labbadia J., and Morimoto R. I. (2017) Rethinking HSF1 in stress, development, and organismal health. Trends Cell Biol. 27, 895–905 10.1016/j.tcb.2017.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anckar J., and Sistonen L. (2011) Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu. Rev. Biochem. 80, 1089–1115 10.1146/annurev-biochem-060809-095203 [DOI] [PubMed] [Google Scholar]
- 24. Dayalan Naidu S., and Dinkova-Kostova A. T. (2017) Regulation of the mammalian heat shock factor 1. FEBS J. 284, 1606–1627 10.1111/febs.13999 [DOI] [PubMed] [Google Scholar]
- 25. Abravaya K., Myers M. P., Murphy S. P., and Morimoto R. I. (1992) The human heat shock protein hsp70 interacts with HSF, the transcription factor that regulates heat shock gene expression. Genes Dev. 6, 1153–1164 10.1101/gad.6.7.1153 [DOI] [PubMed] [Google Scholar]
- 26. Ali A., Bharadwaj S., O'Carroll R., and Ovsenek N. (1998) HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol. Cell. Biol. 18, 4949–4960 10.1128/MCB.18.9.4949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neef D. W., Jaeger A. M., Gomez-Pastor R., Willmund F., Frydman J., and Thiele D. J. (2014) A direct regulatory interaction between chaperonin TRiC and stress-responsive transcription factor HSF1. Cell Rep. 9, 955–966 10.1016/j.celrep.2014.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shi Y., Mosser D. D., and Morimoto R. I. (1998) Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 12, 654–666 10.1101/gad.12.5.654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zou J., Guo Y., Guettouche T., Smith D. F., and Voellmy R. (1998) Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94, 471–480 10.1016/S0092-8674(00)81588-3 [DOI] [PubMed] [Google Scholar]
- 30. Vujanac M., Fenaroli A., and Zimarino V. (2005) Constitutive nuclear import and stress-regulated nucleocytoplasmic shuttling of mammalian heat-shock factor 1. Traffic 6, 214–229 10.1111/j.1600-0854.2005.00266.x [DOI] [PubMed] [Google Scholar]
- 31. Labbadia J., and Morimoto R. I. (2015) The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435–464 10.1146/annurev-biochem-060614-033955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kroeger P. E., and Morimoto R. I. (1994) Selection of new HSF1 and HSF2 DNA-binding sites reveals difference in trimer cooperativity. Mol. Cell. Biol. 14, 7592–7603 10.1128/MCB.14.11.7592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trinklein N. D., Chen W. C., Kingston R. E., and Myers R. M. (2004) Transcriptional regulation and binding of heat shock factor 1 and heat shock factor 2 to 32 human heat shock genes during thermal stress and differentiation. Cell Stress Chaperones 9, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Radons J. (2016) The human HSP70 family of chaperones: where do we stand? Cell Stress Chaperones 21, 379–404 10.1007/s12192-016-0676-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schopf F. H., Biebl M. M., and Buchner J. (2017) The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 18, 345–360 10.1038/nrm.2017.20 [DOI] [PubMed] [Google Scholar]
- 36. Huang C., Wu J., Xu L., Wang J., Chen Z., and Yang R. (2018) Regulation of HSF1 protein stabilization: an updated review. Eur. J. Pharmacol. 822, 69–77 10.1016/j.ejphar.2018.01.005 [DOI] [PubMed] [Google Scholar]
- 37. Dinkova-Kostova A. T., Holtzclaw W. D., Cole R. N., Itoh K., Wakabayashi N., Katoh Y., Yamamoto M., and Talalay P. (2002) Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. U.S.A. 99, 11908–11913 10.1073/pnas.172398899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kensler T. W., Egner P. A., Agyeman A. S., Visvanathan K., Groopman J. D., Chen J. G., Chen T. Y., Fahey J. W., and Talalay P. (2013) Keap1-nrf2 signaling: a target for cancer prevention by sulforaphane. Top. Curr. Chem. 329, 163–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gan N., Wu Y. C., Brunet M., Garrido C., Chung F. L., Dai C., and Mi L. (2010) Sulforaphane activates heat shock response and enhances proteasome activity through up-regulation of Hsp27. J. Biol. Chem. 285, 35528–35536 10.1074/jbc.M110.152686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Y., Ahn Y. H., Benjamin I. J., Honda T., Hicks R. J., Calabrese V., Cole P. A., and Dinkova-Kostova A. T. (2011) HSF1-dependent upregulation of Hsp70 by sulfhydryl-reactive inducers of the KEAP1/NRF2/ARE pathway. Chem. Biol. 18, 1355–1361 10.1016/j.chembiol.2011.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rinn J. L., and Chang H. Y. (2012) Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–166 10.1146/annurev-biochem-051410-092902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schonrock N., Jonkhout N., and Mattick J. S. (2016) Seq and you will find. Curr. Gene Ther. 16, 220–229 10.2174/1566523216666160524144159 [DOI] [PubMed] [Google Scholar]
- 43. Dayalan Naidu S., Kostov R. V., and Dinkova-Kostova A. T. (2015) Transcription factors Hsf1 and Nrf2 engage in crosstalk for cytoprotection. Trends Pharmacol. Sci. 36, 6–14 10.1016/j.tips.2014.10.011 [DOI] [PubMed] [Google Scholar]
- 44. Naidu S. D., Suzuki T., Yamamoto M., Fahey J. W., and Dinkova-Kostova A. T. (2018) Phenethyl isothiocyanate, a dual activator of transcription factors NRF2 and HSF1. Mol. Nutr. Food Res. 62, e1700908 10.1002/mnfr.201700908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ahn S. G., and Thiele D. J. (2003) Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 17, 516–528 10.1101/gad.1044503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu M., Kim H. E., Li C. R., Kim S., Kwak I. J., Lee Y. J., Kim S. S., Moon J. Y., Kim C. H., Kim D. K., Kang H. S., and Park J. S. (2008) Two distinct disulfide bonds formed in human heat shock transcription factor 1 act in opposition to regulate its DNA binding activity. Biochemistry 47, 6007–6015 10.1021/bi702185u [DOI] [PubMed] [Google Scholar]
- 47. Gibbs A., Schwartzman J., Deng V., and Alumkal J. (2009) Sulforaphane destabilizes the androgen receptor in prostate cancer cells by inactivating histone deacetylase 6. Proc. Natl. Acad. Sci. U.S.A. 106, 16663–16668 10.1073/pnas.0908908106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Y., Karagöz G. E., Seo Y. H., Zhang T., Jiang Y., Yu Y., Duarte A. M., Schwartz S. J., Boelens R., Carroll K., Rüdiger S. G., and Sun D. (2012) Sulforaphane inhibits pancreatic cancer through disrupting Hsp90-p50(Cdc37) complex and direct interactions with amino acids residues of Hsp90. J. Nutr. Biochem. 23, 1617–1626 10.1016/j.jnutbio.2011.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou W., Chen X., Hu Q., Chen X., Chen Y., and Huang L. (2018) Galectin-3 activates TLR4/NF-κB signaling to promote lung adenocarcinoma cell proliferation through activating lncRNA-NEAT1 expression. BMC Cancer 18, 580 10.1186/s12885-018-4461-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lin D., Lavender H., Soilleux E. J., and O'Callaghan C. A. (2012) NF-κB regulates MICA gene transcription in endothelial cell through a genetically inhibitable control site. J. Biol. Chem. 287, 4299–4310 10.1074/jbc.M111.282152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goldberg A. L. (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899 10.1038/nature02263 [DOI] [PubMed] [Google Scholar]
- 52. Holmberg C. I., Illman S. A., Kallio M., Mikhailov A., and Sistonen L. (2000) Formation of nuclear HSF1 granules varies depending on stress stimuli. Cell Stress Chaperones 5, 219–228 10.1379/1466-1268(2000)005%3C0219:FONHGV%3E2.0.CO%3B2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim D., Kim S. H., and Li G. C. (1999) Proteasome inhibitors MG132 and lactacystin hyperphosphorylate HSF1 and induce hsp70 and hsp27 expression. Biochem. Biophys. Res. Commun. 254, 264–268 10.1006/bbrc.1998.9840 [DOI] [PubMed] [Google Scholar]
- 54. Zhou M., Wu X., and Ginsberg H. N. (1996) Evidence that a rapidly turning over protein, normally degraded by proteasomes, regulates hsp72 gene transcription in HepG2 cells. J. Biol. Chem. 271, 24769–24775 10.1074/jbc.271.40.24769 [DOI] [PubMed] [Google Scholar]
- 55. Neef D. W., Jaeger A. M., and Thiele D. J. (2011) Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat. Rev. Drug Discov. 10, 930–944 10.1038/nrd3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen H. J., Mitchell J. C., Novoselov S., Miller J., Nishimura A. L., Scotter E. L., Vance C. A., Cheetham M. E., and Shaw C. E. (2016) The heat shock response plays an important role in TDP-43 clearance: evidence for dysfunction in amyotrophic lateral sclerosis. Brain 139, 1417–1432 10.1093/brain/aww028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gomez-Pastor R., Burchfiel E. T., Neef D. W., Jaeger A. M., Cabiscol E., McKinstry S. U., Doss A., Aballay A., Lo D. C., Akimov S. S., Ross C. A., Eroglu C., and Thiele D. J. (2017) Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington's disease. Nat. Commun. 8, 14405 10.1038/ncomms14405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang P., Wander C. M., Yuan C. X., Bereman M. S., and Cohen T. J. (2017) Acetylation-induced TDP-43 pathology is suppressed by an HSF1-dependent chaperone program. Nat. Commun. 8, 82 10.1038/s41467-017-00088-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nishimoto Y., Nakagawa S., Hirose T., Okano H. J., Takao M., Shibata S., Suyama S., Kuwako K., Imai T., Murayama S., Suzuki N., and Okano H. (2013) The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol. Brain 6, 31 10.1186/1756-6606-6-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shelkovnikova T. A., Robinson H. K., Troakes C., Ninkina N., and Buchman V. L. (2014) Compromised paraspeckle formation as a pathogenic factor in FUSopathies. Hum. Mol. Genet. 23, 2298–2312 10.1093/hmg/ddt622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sunwoo J. S., Lee S. T., Im W., Lee M., Byun J. I., Jung K. H., Park K. I., Jung K. Y., Lee S. K., Chu K., and Kim M. (2017) Altered expression of the long noncoding RNA NEAT1 in Huntington's disease. Mol. Neurobiol. 54, 1577–1586 10.1007/s12035-016-9928-9 [DOI] [PubMed] [Google Scholar]
- 62. Gao F. B., Almeida S., and Lopez-Gonzalez R. (2017) Dysregulated molecular pathways in amyotrophic lateral sclerosis-frontotemporal dementia spectrum disorder. EMBO J. 36, 2931–2950 10.15252/embj.201797568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shelkovnikova T. A., Kukharsky M. S., An H., Dimasi P., Alexeeva S., Shabir O., Heath P. R., and Buchman V. L. (2018) Protective paraspeckle hyper-assembly downstream of TDP-43 loss of function in amyotrophic lateral sclerosis. Mol. Neurodegener. 13, 30 10.1186/s13024-018-0263-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dai C., and Sampson S. B. (2016) HSF1: guardian of proteostasis in cancer. Trends Cell Biol. 26, 17–28 10.1016/j.tcb.2015.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mendillo M. L., Santagata S., Koeva M., Bell G. W., Hu R., Tamimi R. M., Fraenkel E., Ince T. A., Whitesell L., and Lindquist S. (2012) HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150, 549–562 10.1016/j.cell.2012.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Santagata S., Hu R., Lin N. U., Mendillo M. L., Collins L. C., Hankinson S. E., Schnitt S. J., Whitesell L., Tamimi R. M., Lindquist S., and Ince T. A. (2011) High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 108, 18378–18383 10.1073/pnas.1115031108 [DOI] [PMC free article] [PubMed] [Google Scholar]