

Abstract
Doublecortin (DCX) is a protein needed for cortical development, and DCX mutations cause cortical malformations in humans. The microtubule-binding activity of DCX is well-described and is important for its function, such as supporting neuronal migration and dendrite growth during development. Previous work showed that microtubule binding is not sufficient for DCX-mediated promotion of dendrite growth and that domains in DCX's C terminus are also required. The more C-terminal regions of DCX bind several other proteins, including the adhesion receptor neurofascin and clathrin adaptors. We recently identified a role for DCX in endocytosis of neurofascin. The disease-associated DCX-G253D mutant protein is known to be deficient in binding neurofascin, and we now asked if disruption of neurofascin endocytosis underlies the DCX-G253D–associated pathology. We first demonstrated that DCX functions in endocytosis as a complex with both the clathrin adaptor AP-2 and neurofascin: disrupting either clathrin adaptor binding (DCX-ALPA) or neurofascin binding (DCX-G253D) decreased neurofascin endocytosis in primary neurons. We then investigated a known function for DCX, namely, increasing dendrite growth in cultured neurons. Surprisingly, we found that the DCX-ALPA and DCX-G253D mutants yield distinct dendrite phenotypes. Unlike DCX-ALPA, DCX-G253D caused a dominant-negative dendrite growth phenotype. The endocytosis defect of DCX-G253D thus was separable from its detrimental effects on dendrite growth. We recently identified Dcx-R59H as a dominant allele and can now classify Dcx-G253D as a second Dcx allele that acts dominantly to cause pathology, but does so via a different mechanism.
Keywords: microtubule, clathrin, endocytosis, dendrite, neurite outgrowth, axon initial segment, dominant allele of DCX, lissencephaly, multifunctional protein, neurofascin, clathrin adaptor, neuron
Introduction
The X-linked gene doublecortin (DCX) is a major genetic locus for type I lissencephaly, a neurodevelopmental defect causing mental retardation and untractable epilepsy (1–4). Lissencephaly brains show varying degrees of cortical malformations, which are thought to result from defects in neuronal migration (5). In addition, human and mouse brains show defects in major axon tracts (6–8). Cultured neurons derived from rodent models of Dcx loss also show dendrite growth defects (9, 10). Dendrite growth defects also occur in the hippocampus and cortex in vivo (11, 12). Consistent with a role in later stages of development, DCX localizes to axon and dendrite tips in post-migratory neurons (13–16).
On the molecular level, DCX binds microtubules (MTs)5 via two DC repeats (reviewed by Friocourt et al. (2)). Because most of the known human mutations in DCX are scattered across these repeats (17), the phenotypes associated with loss of DCX are attributed to impairment of MT-related functions, such as MT growth (18), MT bending (14), and plus-tip tracking (19). We showed recently that a Dcx allele with loss of MT binding in fact is incapable of promoting dendrite growth in culture (20). In addition, DCX also binds to and regulates MT motors (21). DCX thus plays important MT-based roles in developing neurons.
DCX also binds proteins with no direct links to MTs. The importance of these other binding interactions, such as with the cell adhesion molecule neurofascin (NF) and clathrin adaptors (22, 23), is currently not well-understood. Strikingly, the interaction with NF is disrupted in the patient allele Dcx-G253D (22). DCX-G253D has no striking defects in MT binding and might thus cause disease by disrupting non-MT interactions of DCX. We previously showed that localization of NF to the axon initial segment (AIS) is impaired when DCX is down-regulated or when DCX-G253D is expressed in cultured neurons (24). In addition, we demonstrated a novel function for DCX, namely promoting the endocytosis of NF (24). Based on these data, we proposed a model that DCX enhances AIS localization of NF by promoting its endocytosis from non-AIS regions, such as dendrites (24). We now show that a non-MT pool of DCX forms a complex with an endocytic cargo (NF) and the clathrin adaptor AP-2 and is thus able to function as a bona fide endocytic adaptor, linking endocytosing cargos to endocytic machinery. In fact, mutating either the AP-2–binding site (DCX-ALPA) or the NF-binding site (DCX-G253D) leads to decreased NF endocytosis in primary neurons and increased mislocalization of NF to dendrites. Surprisingly, DCX-ALPA does not impair dendrite growth at longer times of expression, whereas DCX-G253D impairs dendrite growth in a dominant manner. Therefore, the cellular defect in NF endocytosis does not underlie the dendrite growth defect of this allele. This raises the possibility that single Dcx mutant alleles have multiple cellular defects that differentially contribute to distinct pathological processes.
Results
A patient mutation in DCX, DCX-G253D, does not support NF endocytosis in neurons
The molecular and cellular defects caused by patient-associated mutations in DCX have been determined in a handful of cases (15, 17, 20, 21, 25) but are for the most part not known. The molecular defect of DCX-G253D is known: it does not bind the adhesion receptor NF (22) but still binds to microtubules (24). NF is highly enriched on the axon initial segment in vitro and in vivo with little detectable NF present on the dendrites. Down-regulation of DCX or expression of DCX-G253D (but not WT DCX) in primary neurons caused mislocalization of HA-tagged NF to dendrites (24). The localization of endogenous NF was also affected and was found at reduced levels at the AIS. This mislocalized dendritic pool of NF suggested to us that NF might normally be removed by endocytosis from dendrites by a mechanism that required binding of NF to DCX. To test whether DCX-G253D has a cellular defect in NF endocytosis, we turned to loss-of-function experiments using short hairpin plasmids targeting Dcx (shDcx). Endogenous levels of DCX were diminished by 75% on average after 3 days of expression of shDcx#2-GFP (see Fig. 1B′). shDcx#2-GFP expression significantly diminished signal intensity of endocytosed endogenous NF in endosomes in day in vitro 3 (DIV3) hippocampal neurons compared with untransfected neurons or neurons expressing shRandom-GFP control plasmid (Fig. 1, A, B, and E), similarly to our previous work (24). Line scans along a dendrite of untransfected cells (blue trace) compared with cells transfected with shRandom (Fig. 1A, green trace) or shDcx (Fig. 1B″, green trace) are shown for easier comparison of the intensity of NF-containing endosomes along dendrites. To test for specificity of the shDcx treatment, we created a shDcx#2-resistant version of WT Dcx (DCX-R2-FLAG) and tested how well it rescues the shDcx-mediated NF endocytosis defect. The NF endocytosis defect caused by shDcx could be fully rescued by co-expression of the resistant DCX-R2-FLAG protein (Fig. 1, C and E), demonstrating that the decreased NF endocytosis is a specific effect of down-regulating DCX. In contrast to WT DCX-R2-FLAG, the resistant DCX-R2-G253D-FLAG does not rescue the shDcx NF endocytosis defect (Fig. 1, D and E). DCX-R2-G253D-FLAG is expressed at similar levels to WT DCX-R2-FLAG in transfected neurons (Fig. 1D′) and by Western blots in HEK293 lysates (Fig. 2E, Lysate), so the failure of DCX-R2-G253D to rescue NF endocytosis after shDcx is not due to low expression of the mutant DCX. We also determined the level of overexpression of the WT and mutant rescue constructs compared with endogenous DCX in transfected neurons (see Fig. S1A). WT and mutant DCX are expressed on average ∼2.5-fold higher than endogenous DCX in untransfected cells. Levels of overexpressed WT and mutant DCX are comparable and not statistically significantly different from one another (Fig. S1A).
Figure 1.

DCX-G253D fails to rescue NF endocytosis in primary neurons after DCX knockdown. A–D, endocytosis of endogenous NF (red) was determined in DIV3 hippocampal neurons expressing shRandom-GFP (A), shDcx#2-GFP (B), shDcx#2-GFP plus rescue plasmid WT DCX-R2-FLAG (blue) (C), and shDcx#2-GFP plus rescue plasmid DCX-R2-G253D-FLAG (blue) (D). Enlarged views of the red channel (endocytosed NF) are shown on the right in A–C. Green star, transfected cell; arrows in A–C, dendrites. An example of the brightness of NF-containing endosomes is shown as line scans for shRandom (A′) and shDcx (B″) for transfected (green line) and untransfected cells (blue line). Arrows, peaks corresponding to endosomes along the dendrite. B′, levels of endogenous DCX were plotted for untransfected cells (UT; black crosses) and cells expressing shDcx#2-GFP (red circles). For D, cell 1 (green arrows) corresponds to a transfected cell, and cell 2 (white arrows) corresponds to an untransfected cell. Corresponding green and red channels are shown on the right. Scale bars, 20 μm. D′, DCX-R2-FLAG (cross) and DCX-R2-G253D-FLAG (red square) are expressed at comparable levels in transfected neurons. n = 25 cells for each plasmid. Transfected cultures were also counterstained with antibody against DCX to evaluate the extent of overexpression compared with untransfected cells (Fig. S1). E, quantification of NF endocytosis levels for conditions shown in A–D. Quantification of one representative experiment is shown. Measurements are normalized to untransfected (UT) cells in the same field. n = 15–25 cells/condition/experiment. Error bars, S.E. **, p < 0.001 (ANOVA with post hoc test).
Figure 2.

DCX-ALPA is an AP-2–binding mutant. A, diagram of DCX domains indicating microtubule-binding (green boxes), phosphoneurofascin-binding (containing residue Gly-253), and AP-2–binding sites (YLPL at positions 350–353). 350YLPL353 was mutated to 350ALPA353 (ALPA). B–D, DCX-ALPA is deficient in binding AP-2 complex but still binds to HA-NF. HEK293 cells were transfected with the indicated plasmids, and immunoprecipitations were carried out as indicated. Immunoprecipitates were analyzed by Western blotting (WB) with the indicated antibodies. The lysate (5% of total) was probed by Western blotting as labeled. Arrows, the respective bands. All immunoprecipitations were carried out at least three times from independent lysates. E, DCX-G253D still binds to AP-2 complex. HEK293 cells were transfected with HA-μ2 together with WT DCX-FLAG (lane 1) or DCX-G253D-FLAG (lane 2). Immunoprecipitations (IP) were carried out with anti-HA antibody. Western blots were carried out with anti-FLAG antibody (top). Western blots of lysates are shown below. This immunoprecipitation was carried out at least two times from independent lysates. The blot in E represents additional lanes from the same experiment as C. The WT DCX lane is thus reused in E. Intervening irrelevant lanes were removed in B, C, and E. F, DCX-ALPA-GFP (green) remains stably associated with microtubules (blue, anti-tubulin) after detergent extraction in MT-stabilizing buffer BRB80. The panels on the right show each channel separately as well as a merged image. Scale bar, 20 μm. G, DCX-ALPA-GFP (green) is stably associated with microtubules in neurons after extraction in BRB80. MAP2 is counterstained in red.
Binding of DCX to AP-2 and creation of binding mutant
Because DCX reportedly binds the μ2 subunit of the endocytic clathrin adaptor AP-2 at its C-terminal YLPL motif (23) (residues 350–353; Fig. 2A), we set out to test the hypothesis that direct binding of DCX to AP-2 plays a functional role in NF endocytosis and AIS localization. We introduced two alanine substitutions at positions Tyr-350 and Leu-353 to create “DCX-ALPA” (Fig. 2A). DCX-ALPA (GFP- or FLAG-tagged) accumulates to similar levels as WT DCX-GFP/FLAG, indicating similar stability in the cell, when expressed in HEK293 cells (Fig. 2 (B–D), lysates). WT DCX-GFP consistently co-immunoprecipitates with the AP-2 subunit α-adaptin (Fig. 2B, lane 2), whereas DCX-ALPA-GFP does not (Fig. 2B, lane 3). In addition, the μ2 subunit of AP-2 (expressed with HA tag) co-immunoprecipitates with WT DCX, but not with DCX-ALPA (Fig. 2C).
We then tested whether DCX-ALPA is still capable of binding to its other binding partners, in particular NF (22) and MTs. HA-neurofascin (HA-NF) could be reproducibly recovered in a complex with DCX-GFP. DCX-ALPA-GFP also co-immunoprecipitates with HA-NF, albeit often at somewhat reduced levels (Fig. 2D). To test whether DCX-ALPA is still capable of binding to MTs, we performed in-cell MT-binding assays, as before (20, 24). COS cells (Fig. 2F) or cultured neurons (Fig. 2G) transfected with WT DCX-GFP or DCX-ALPA-GFP were detergent-extracted live in microtubule-stabilizing buffer (BRB80) to retain microtubules and the MT-associated proteins bound to them. DCX-ALPA-GFP is still found to decorate microtubules (Fig. 2, F and G), similar to WT DCX-GFP. DCX-ALPA can therefore be used to test the effects of losing clathrin adaptor interactions. Last, we also determined that DCX-G253D still binds HA-μ2 (Fig. 2E). We thus created two distinct DCX mutants that either lack NF binding or lack AP-2 binding.
A complex of NF, DCX, and AP-2 can be immunoprecipitated from transfected cells and brain
Binary complexes of DCX with AP-2 and of DCX with NF have been reported previously (22, 23) and were verified by us (see Fig. 2 (B–E), WT-DCX lanes), but it was not known whether DCX could bind both proteins simultaneously. We thus tested whether DCX, NF, and AP-2 exist in a immunoisolatable complex. We first tested for evidence of an endogenous complex of NF, DCX, and AP-2 in E18 rat brain. Immunoprecipitations were carried out with either anti-DCX (Fig. 3A (a)) or anti-NF antibodies (Fig. 3A (b)) from brain membrane fractions. Nonimmune IgG was used as a negative control. α-Adaptin could be co-immunoprecipitated when anti-DCX antibodies were used, verifying the endogenous complex of DCX and AP-2. α-Adaptin could also be co-immunoprecipitated when anti-NF antibodies were used (Fig. 3A (b)), but not when nonimmune IgG was used. The ability of anti-NF antibodies to co-immunoprecipitate AP-2 subunits is consistent with the existence of an endogenous complex of AP-2–DCX–NF in the brain.
Figure 3.

A complex of NF, DCX, and AP-2 can be immunoprecipitated from transfected cells and brain. A, α-adaptin is immunoprecipitated with anti-DCX antibodies from embryonic rat brain membrane fractions (a) as well as by anti-NF antibodies (b). Nonimmune IgG-coated beads were used as negative controls. Levels of endogenous α-adaptin are shown in the lysate blots (c). An intervening irrelevant lane was removed in a. B, α-adaptin is only co-immunoprecipitated with HA-NF if DCX-GFP is co-expressed. HEK293 cells were transfected with the indicated plasmids, and immunoprecipitations (IP) were carried out as labeled. Immunoprecipitates were analyzed by Western blotting (WB) with the indicated antibodies. The lysate (5% of total) was probed by Western blotting as labeled. C and D, freshly dissociated E18 rat cortex was homogenized and fractionated according to the diagram (shown in C) into a soluble (cytosolic and membrane proteins), MT-associated, and insoluble fraction. The insoluble (lane 1), soluble/membrane (lane 2), and microtubule-associated (lane 3) fractions were separated by SDS-PAGE and blotted against α-adaptin, DCX, and a membrane protein (NEEP21).
To test more directly whether DCX existed as a complex with NF and AP-2, we turned to HEK293 cells, which do not express DCX or NF endogenously. HEK293 cells were transfected with HA-NF and immunoprecipitations carried out with anti-HA antibody. Endogenous AP-2 is not co-immunoprecipitated (Fig. 3B, lane 3), demonstrating that HA-NF does not directly bind AP-2 adaptors. This is consistent with the absence of the adaptor-binding YRSLE motif in rodent NF (26). When HA-NF was co-transfected with Dcx-GFP, AP-2 could be recovered abundantly in the anti-HA immunoprecipitation (Fig. 3B, lane 2). AP-2 therefore is co-immunoprecipitated with HA-NF only when DCX-GFP is also expressed, consistent with the model that NF is endocytosed via an NF–DCX–AP-2 complex.
AP-2 is in a complex with DCX that is not bound to microtubules
DCX is best known as a MT-binding protein, but there are also populations of DCX not bound to MTs (13, 20, 27). In previous work, we found that DCX-R89G, a patient allele that lacks MT binding, is still able to support NF endocytosis in PC12 cells (24). These observations suggested that DCX might bind AP-2 while not bound to MTs. Consistent with this idea, AP-2 subunits were identified in an MS experiment of proteins co-immunoprecipitating with membrane-associated DCX.6 To test more directly the notion that it is the non-MT pool of DCX that primarily associates with AP-2 in cells, we used detergent extraction in MT-stabilizing buffer to fractionate freshly dissociated neurons into solubilized (lane 2), MT-bound (lane 3), and insoluble fractions (lane 1) (27). Briefly, the solubilized pool includes cytosolic and membrane proteins. The pellet was subsequently treated with calcium to depolymerize MTs, thus releasing MT-bound proteins into the supernatant. The final pellet contains insoluble proteins (Fig. 3C). A neuronal transmembrane protein (NEEP21 (28)) was used to verify that membrane-bound proteins were completely solubilized in the first extraction step (lane 2) and were not carried over into the calcium-released MT fraction. Western blotting revealed that DCX is localized primarily in the solubilized (lane 2; ∼30%) and microtubule-bound (lane 3; ∼70%) fractions (Fig. 3D). AP-2 fractionated almost entirely in the solubilized fraction (lane 2), and virtually none (<2%) was detected with the microtubule-bound DCX fraction (lane 3; Fig. 3D), indicating that microtubule-bound DCX does not co-fractionate with AP-2.
DCX-ALPA does not rescue the NF endocytosis defect caused by down-regulation of DCX
To test directly whether NF endocytosis requires interaction of DCX with AP clathrin adaptors, we again turned to loss-of-function experiments using shDcx. If DCX promotes the endocytosis of NF via direct interaction between DCX and AP-2, DCX-ALPA should not be able to rescue the shDcx#2 NF endocytosis defect. When DCX-ALPA resistant to shDcx#2 down-regulation (“DCX-R2-ALPA-FLAG”) was co-expressed with shDcx#2-GFP, NF endocytosis was not rescued (Fig. 4, A and C). The lack of rescue was not due to lower expression levels for DCX-R2-ALPA-FLAG (Fig. 4B and Fig. S1A). DCX-mediated endocytosis of NF in neurons thus depends on an intact YLPL clathrin adaptor binding motif. We note that shDcx or overexpression of WT or mutant DCX does not change the staining pattern for AP-2 in neurons (data not shown). In addition, we previously showed that overexpression of DCX does not change the endocytosis of other cargos (such as transferrin or L1) in PC12 cells (24).
Figure 4.
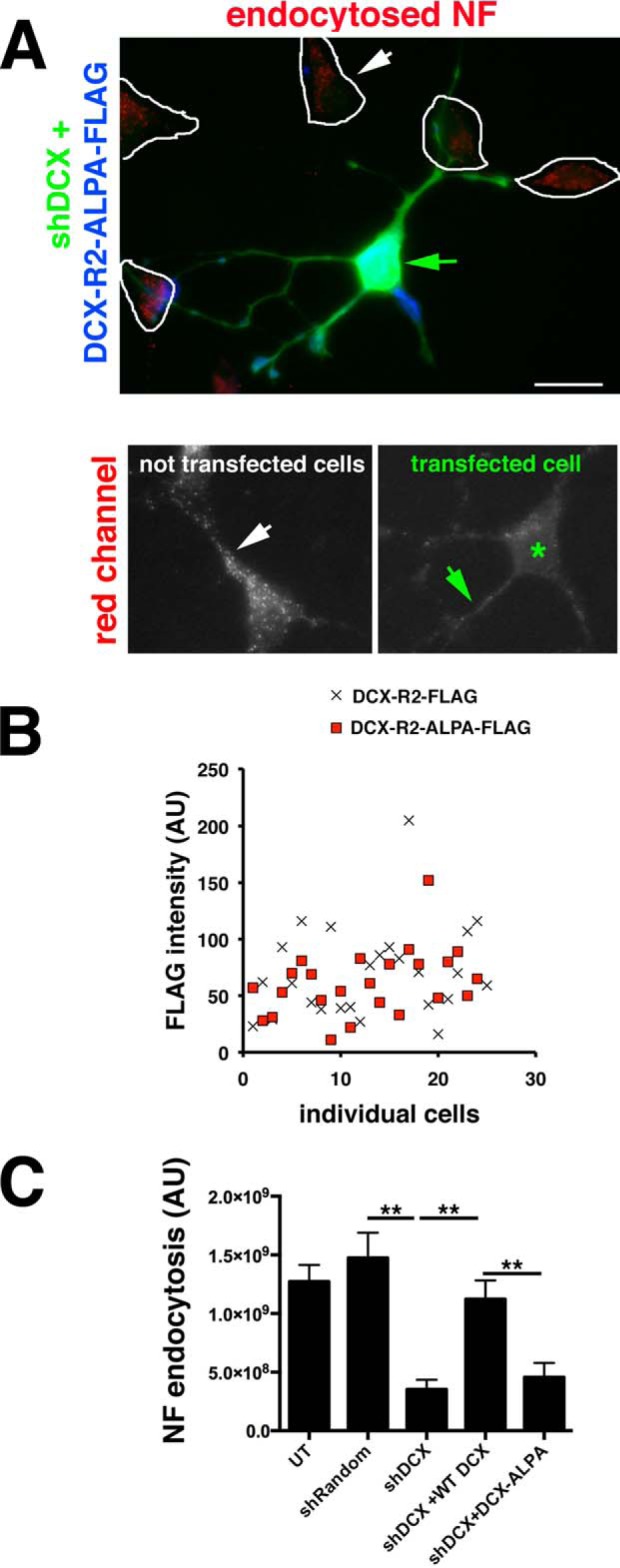
DCX-ALPA does not rescue the NF endocytosis defect caused by down-regulation of DCX. A, endocytosis of endogenous NF (red) was determined in DIV3 hippocampal neurons expressing shDcx#2-GFP plus rescue plasmid Dcx-R2-ALPA-FLAG (blue). Enlarged views of the red channel are shown below. Green star, transfected cell; arrows, dendrites. Controls are the same as in Fig. 1 and are not shown again. Scale bar, 20 μm. B, DCX-R2-FLAG (cross) and DCX-R2-ALPA-FLAG (red square) are expressed at comparable levels in transfected neurons. n = 25 cells for each plasmid. Transfected cultures were also counterstained with antibody against DCX to compare the extent of overexpression compared with untransfected cell (Fig. S1). C, quantification of NF endocytosis levels. n = 25 cells/condition/experiment. One representative experiment is shown. Error bars, S.E. **, p < 0.001 (ANOVA with post hoc test).
Co-expression of DCX-ALPA interferes with HA-NF accumulation on the AIS
We previously found that DCX-G253D impairs AIS localization of HA-NF (24). If a complex of DCX–NF–AP-2 is responsible for NF endocytosis and thus promotes NF localization to the AIS, DCX-ALPA and DCX-G253D should phenocopy each other. We thus tested whether expressing DCX-ALPA impairs HA-NF AIS localization similarly to what we found for DCX-G253D (24). Acutely expressed HA-NF accumulated 5–6-fold higher on the AIS than on dendrites (AIS polarity index (PI) = 5–6 (Fig. 5, A and D)). Co-expression of HA-NF and WT DCX-GFP for 20 h did not significantly alter AIS PI of HA-NF (Fig. 5, B and D). Co-expression of HA-NF with DCX-ALPA-GFP, on the other hand, significantly reduced AIS PI of HA-NF (Fig. 5, C and D). This decrease in AIS PI is due to increased levels of HA-NF on the somatodendritic surface, similarly to what was observed for DCX-G253D. DCX-ALPA and DCX-G253D therefore have the same cellular phenotypes with respect to NF endocytosis and NF surface distribution, consistent with a simultaneous requirement for binding of DCX to NF and AP-2 in a complex. Levels of overexpression of WT DCX, DCX-G253D, and DCX-ALPA were comparable in this experimental assay (Fig. S1B), and thus differences in AIS PI are not due to different levels of accumulation of the different constructs.
Figure 5.
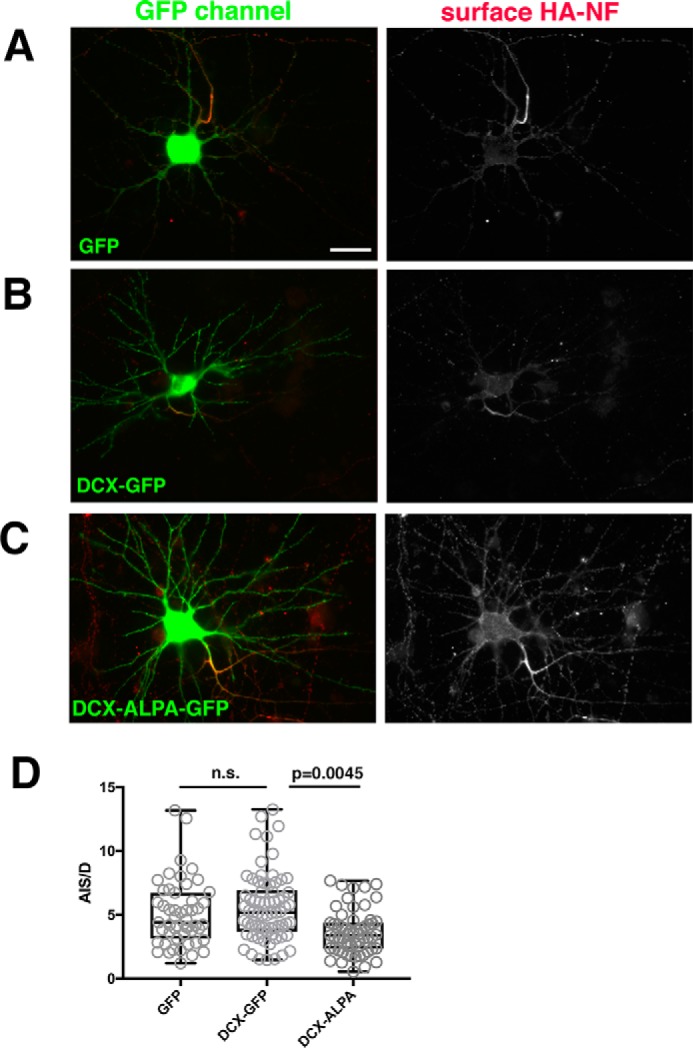
Co-expression of DCX-ALPA interferes with HA-NF accumulation on the AIS. A–C, HA-NF (red) accumulates predominantly at the surface of the AIS in cells expressing GFP as a control (A) or DCX-GFP (B), but HA-NF mislocalizes to the somatic and dendritic surface when DCX-ALPA-GFP is expressed (C). The right panels show single-channel images of the HA-NF surface distributions (DIV9 neurons 20 h post-transfection). Scale bar, 20 μm. Transfected cultures were also counterstained with antibody against DCX to compare the extent of overexpression compared with control cells expressing GFP (Fig. S1). D, surface distribution of HA-NF was quantified by determining the surface intensity of HA-NF on the AIS versus dendrites (AIS/D PI) for neurons expressing GFP, DCX-GFP, or DCX-ALPA-GFP. ***, p < 0.0001; n.s., not significant. One representative experiment is shown (of a total of three independent experiments). n = 51 cells for GFP, 70 cells for DCX-GFP, and 59 cells for DCX-ALPA. Whisker plots show the median as a line, 25–75% as a box, and the data range as whiskers (Kruskal–Wallis test; p = 0.0045 for DCX-GFP versus DCX-ALPA; p = 0.0014 for GFP versus DCX-ALPA).
AP-2 binding of DCX is required for supporting DCX-dependent dendrite growth in culture at DIV3 but not DIV5
Previous work showed that loss of DCX impairs dendrite development in cultured neurons (9, 10). Because reduction of DCX levels also impairs endocytosis (24), we investigated whether DCX-dependent dendrite growth depends on the binding of DCX to AP-2 (i.e. on DCX-mediated endocytosis). Because knockdown of DCX gave variable effects on dendrite growth in our hands (24), we made use of an overexpression “gain-of-function” assay developed by the Reiner laboratory (10) and previously used by us to characterize several DCX patient alleles (20): overexpression of WT DCX increases dendrite number. This assay allowed us to test whether the DCX mutants still possess WT DCX activity. Overexpression of WT DCX increased the number of dendrites in young cultured cortical neurons compared with GFP controls (Fig. 6, A–D). DCX-ALPA-GFP failed to increase dendrite number (Fig. 6A) at DIV3 compared with WT-DCX controls, suggesting that the clathrin adaptor–binding activity of DCX contributes to the DCX-dependent augmentation of dendrite growth. When dendrite growth was assessed after 5 days of DCX-ALPA expression (DIV5), growth of dendrites was comparable with WT DCX and no longer showed a defect (Fig. 6, B and G). Overexpression levels were quantified and found to be similar for WT DCX and DCX-ALPA (Fig. S1, C and D). AP-2 binding capacity of DCX is thus only transiently required for DCX-mediated dendrite growth promotion and does not cause a lasting decrease in dendrite growth.
Figure 6.

Divergent phenotypes of DCX-ALPA and DCX-G253D on dendrite growth. A–D, number of dendrites at DIV3 and DIV5 of at least 15-μm length from center of soma was determined for neurons expressing GFP, WT DCX-GFP, or DCX-ALPA-GFP (A and B) or DCX-G253D-GFP (C and D) for DIV3 (A and C) and DIV5 (B and D). One representative experiment is shown (of a total of three independent experiments). n = 50–75 cells/condition in each experiment. The axon was excluded from counts. Transfected cultures were also counterstained with antibody against DCX to compare the extent of overexpression compared with control cells expressing GFP (Fig. S1). For parametric data, we used one-way ANOVA followed by Fisher's least significant difference test. For nonparametric data sets, we used Kruskal–Wallis followed by uncorrected Dunn's multiple-comparison test. Whisker plots show the median as a line, 25–75% as a box, and the data range as whiskers. *, p < 0.05; **, p < 0.001; ****, p < 0.00001; n.s., not significant. Measurements from individual cells are also shown. E–H, representative examples of DIV5 neurons transfected with GFP (E), WT DCX (F), DCX-ALPA (G), or DCX-G253D (H). Dendrites were traced in the right-hand panels in black. The axon is traced in red.
The patient allele DCX-G253D does not increase dendrite growth in culture at DIV3 and becomes dominant-negative at DIV5
Because DCX-ALPA and DCX-G253D showed identical loss-of-function cellular phenotypes with respect to NF endocytosis and NF surface distribution (Figs. 1 and 4), we predicted that DCX-G253D would phenocopy the transient dendrite growth phenotype of DCX-ALPA. Indeed, DCX-G253D phenocopied the DCX-ALPA with respect to dendrite growth at DIV3 (Fig. 6C), consistent with the model that DCX-mediated endocytosis (as probed with DCX-ALPA) plays a role in augmentation of dendrite growth at DIV3 (Fig. 6A). Surprisingly however, at DIV5, expression of DCX-G253D became detrimental to dendrite growth, and neurons expressing DCX-G253D had fewer dendrites than GFP-expressing control neurons (Fig. 6, D and H). Overall, neurons expressing DCX-G253D for 5 days had thinner and shorter dendrites and looked generally to be less robust. Overexpression levels were quantified and found to be similar for WT DCX and DCX-G253D (Fig. S1, E and F). DCX-G253D is thus a dominant missense allele for dendrite growth, a phenotype not dependent on clathrin adaptor binding and promoting endocytosis. We previously identified another dominant missense patient allele, DCX-R59H, whose toxicity we attributed to the formation of large aggregates that triggered cellular stress responses and autophagy (20). Unlike DCX-R59H, DCX-G253D does not form aggregates (Fig. 6F), so its mechanism of toxicity is distinct from that of DCX-R59H.
Discussion
DCX plays important roles via microtubule binding (18, 17, 29), but it also binds a number of other proteins (2). The roles played by these other binding interactions of DCX are still not well-established, but it stands to reason that they also contribute to the overall function of DCX. We previously showed that DCX regulates correct NF surface distribution and promotes NF endocytosis (24). NF surface distribution thus depends on DCX-dependent endocytosis of NF from the somatodendritic surface. We additionally showed that the patient mutation DCX-G253D, which is deficient in NF binding, does not support NF endocytosis in PC12 cells. These observations suggested the hypothesis that the inability of DCX-G253D to endocytose NF forms the molecular basis for DCX-G253D pathology in human patients. In a separate paper (20), we showed that several DCX patient mutations are deficient in promoting dendrite outgrowth in culture, but whether DCX-dependent endocytosis is involved in regulating dendrite growth is not known. To answer these questions, we turned to cultured hippocampal neurons, an experimental system where cell biological assays can be carried out at a mechanistic level. As we argued before (20), the mild phenotypes of Dcx KO mice and the difficulty of taking molecular analysis into an in vivo context make experiments in cultured primary neurons a good first step to gain initial mechanistic insights. These insights can then be followed up with future in vivo experiments.
In this work, we generated a new DCX mutant (DCX-ALPA), which is incapable of binding the endocytic clathrin adaptor AP-2. Analysis of the two mutants DCX-G253D and DCX-ALPA led to the conclusion that a membrane-bound pool of DCX binds simultaneously to cargo (neurofascin/NF) and ubiquitous endocytic machinery (the clathrin adaptor AP-2) to mediate NF endocytosis. We then compared the ability of WT DCX and the two mutants (DCX-G253D and DCX-ALPA) to support AIS localization of NF and dendrite growth. We found that the two mutants phenocopy each other with respect to reduced NF endocytosis, resulting in altered NF surface distribution. In contrast, they do not phenocopy each other with respect to DCX-mediated dendrite growth in cultured neurons; DCX-ALPA causes a transient delay in dendrite outgrowth, whereas DCX-G253D becomes dominant-negative with longer times of expression, resulting in stunted dendrites. Therefore, the dominant-negative dendrite phenotype of DCX-G253D is not due to the NF endocytosis defect observed at the cellular level.
Membrane-bound DCX plays a role as a cargo-specific endocytic adaptor
A previous paper identified AP-2 as an interactor of DCX (23). The functional significance of this reported interaction remained unclear for over 10 years because no cargo was known that depended on DCX for endocytosis. Because we identified such a DCX-dependent cargo (namely NF (24)), we set out to test the hypothesis that DCX mediates the endocytosis of NF via AP-2 clathrin adaptors. Our results show that an AP-2 binding–deficient DCX protein (DCX-ALPA) is unable to rescue shDcx-mediated NF endocytosis defects, and a complex between NF, DCX, and AP-2 can be immune-isolated from heterologous cells and from brain, supporting our hypothesis.
AP-2 is a ubiquitous clathrin adaptor that mediates receptor-mediated endocytosis (30). Many AP-2–dependent endocytic cargos directly bind to the μ2 subunit of AP-2 (31). In contrast, other cargos do not directly bind to AP-2 (31–33) but bind specific monomeric nonclassic adaptors that bridge the cargo and AP-2 (32). In many cases, the interaction of the cargo with the intermediary adaptors are subject to regulation such that cargo internalization is not constitutive but subject to signaling (32, 34) that allows an additional layer of regulation. Our data suggest that DCX allows regulated endocytosis of subsets of cargos and is thus a new member of this class of nonclassic endocytic adaptors: DCX binds both cargo (i.e. NF) and classic clathrin adaptors, such as AP-2 (i.e. intermediary adaptor); DCX does not mediate endocytosis of transferrin or L1 CAM (23) (i.e. is cargo-specific); and only NF phosphorylated on the FIGQY motif downstream of NGF signaling binds to DCX (22) (i.e. regulated). DCX thus joins the ranks of NUMB and DAB2 as genes with neurodevelopmental importance that mediate regulated endocytosis of specific cargos by linking as an intermediary with AP-2 (reviewed by Yap and Winckler (33)).
Functional roles for DCX as an endocytic adaptor?
Our experiments provide several pieces of evidence at the molecular and cellular level that DCX is capable of mediating NF endocytosis, but the functional significance of this activity is not understood. NF is a cell adhesion molecule involved in both axon outgrowth in development (35) and directing inhibitory synapse formation to the AIS (36, 37). Later in development, it plays critical roles in the formation and maintenance of nodes of Ranvier (38, 39). The early roles in neurite outgrowth in particular are not well-studied, but might be the roles most likely modulated by DCX binding. Interestingly, levels of NF are changed in callosal axons in Dcx KO mice (6), supporting a possible functional connection between NF sorting and DCX.
We observed only a small and transient defect in dendrite growth in culture when the AP-2–binding site of DCX was mutated. It is possible that the culture growth conditions (in terms of substrate and intercellular interactions) are not conducive to uncovering a larger role for DCX-mediated endocytosis or for DCX-regulated NF trafficking. An additional explanation for the mild dendrite phenotype comes to mind: DCX has been implicated in multiple neurodevelopmental processes, including neuronal migration, axon guidance in response to netrin toward the midline, and dendrite growth (1, 6). It is thus possible that different binding modalities of DCX underlie its roles in these distinct pathways. Interestingly, earlier work showed that the Dcx-303X truncated allele (which lacks the AP-2–binding site) is completely incapable of supporting the migration of cerebellar granule neurons from a cell aggregate in culture (40). This is in contrast to our own findings that DCX-303X shows only a small defect in dendrite growth (20). DCX-303X thus behaves as a complete loss-of-function allele with respect to the migration assay but as a mild hypomorph with respect to dendrite growth. Future work in vivo will be needed to shed light on the question of whether the clathrin-binding site of DCX might be differentially important in migration, axon guidance, and dendrite growth during brain development.
The DCX patient allele, DCX-G253D, is a dominant missense allele for dendrite growth
The molecular binding defects of many of the identified DCX patient alleles have been characterized for some time. Several of them appear largely normal for microtubule binding (17). It is currently mostly unknown what defects these microtubule binding–competent DCX patient alleles might have that cause disease. Recently, investigators have made great strides in the task of identifying cellular pathways (6, 9, 13, 14) and cellular defects for several alleles (21, 27, 40). In the current work, we identify a cellular defect for DCX-G253D: the inability of DCX-G253D to bind to neurofascin (22) leads to endocytosis defects of neurofascin in primary neurons and alterations of the surface distribution of NF. Both of these phenotypes of DCX-G253D are loss-of-function defects. With respect to dendrite growth, DCX-G253D shows an unexpected dominant phenotype, with neurons having worse dendrite growth than GFP-expressing controls. Because DCX is an X-linked gene, it is not known which alleles are dominant in patients, but we have now identified two patient alleles with dominant-negative phenotypes, Dcx-G253D (in this work) and Dcx-R59H (20). These observations raise the possibility that other alleles might also be dominant alleles. Loss-of-function alleles have also been identified (20, 27), so some patients carry loss-of-function alleles, whereas others carry dominant alleles. Furthermore, because DCX has multiple distinct functions that may be differentially required at different stages for executing different growth and neuronal maturation steps, it makes some sense that the same allele could be loss-of-function for one process but dominant for a different process. This is nevertheless a surprising finding and one that we are not aware of having many precedents in the literature. In light of our findings, it is important to keep in mind that the use of the Dcx knockout mouse might not be the appropriate model for understanding all aspects of the human disease.
Lack of NF endocytosis is not the cause of DCX-G253D dendrite pathology
The identification of a complex of NF–DCX–AP-2 allowed us to ask about the functional roles of NF endocytosis mediated by DCX because we could disrupt either the NF–DCX interaction (DCX-G253D) or the DCX–AP-2 interaction (DCX-ALPA) separately. If the NF–DCX–AP-2 complex is important, the two DCX mutants are predicted to phenocopy each other. As expected, neither mutant supports NF endocytosis in primary neurons. Contrary to our expectation, however, the two mutants have distinct dendrite growth defects, thus uncovering that the cellular NF endocytosis defect of DCX-G253D is not the cause of the dominant dendrite phenotype at DIV5. It is currently unknown how DCX-G253D causes a dominant defect in dendrite growth/maintenance and how this relates to its dysfunction in human brain development. We do not think that loss of NF binding underlies the dendrite growth defect in cultured neurons because down-regulation of NF itself does not result in dendrite growth phenotypes in culture.7 What other binding interactions might be aberrant in this allele are not known, and more work is needed to identify additional binding partners. It is also unclear whether NF binding is important for DCX function in other pathways (such as axon growth or neuronal migration). Previous work showed changes in NF levels in corpus callosum axons from Dcx KO mouse brain by MS (6), so it remains to be tested whether axon growth toward or across the midline requires NF–DCX interactions.
In summary, we identify here a dominant missense patient allele of DCX, namely DCX-G253D. This DCX allele has separable phenotypes for NF endocytosis and dendrite growth, which raises the possibility that single DCX mutant alleles could have multiple cellular defects that differentially contribute to distinct pathological processes. We previously identified a different patient allele, Dcx-R59H, as a dominant allele, but the pathways impaired by the two dominant alleles are distinct (20). DCX-R59H causes profound aggregation of multiple cellular proteins, including the actin-binding protein spinophilin, which is also a DCX binding partner, and disruption of actin and microtubule elements (20). DCX-G253D acts dominantly without aggregate formation (this work) and thus has a distinct mode of action. More work is needed to identify what cellular pathways are impaired by DCX-G253D to cause this dominant effect.
Experimental Procedures
Reagents
Antibodies
Anti-DCX (rabbit polyclonal against N terminus) (catalogue no. 4604, lot 3, Cell Signaling) recognizes a single band at 45 kDa from fetal rat brain. Rabbit polyclonal antibody, against the C terminus (catalogue no. 18724, lot GR55971-1, Abcam), recognizes a single band at 45 kDa from mouse brain lysate. Rabbit polyclonal antibody (anti-FLAG F7425, lot 064M4757V) and mouse polyclonal antibody (catalogue no. F1804, lot SLBJ4607V) were from Sigma. Anti-GFP, mouse monoclonal antibody (catalogue no. A11120, lot 877587), was from Molecular Probes; rabbit monoclonal antibody (2956S, lot 4, from Cell Signaling), recognizes a single band at 27 kDa from HCC827 cell lysate. GFP booster was from Chromotek. Anti-tubulin mouse DM1a was from Sigma, and rat monoclonal antibody (catalogue no. yol1/34, lot G0714) was from Santa Cruz Biotechnology. Anti-Map2 chicken antibody (RRID AB_2138173) was from EnCor Biotechnology. This antibody shows a single band on Western blots from rat and mouse brain lysates (EnCor Biotechnology).
Anti-neurofascin was from the UC Davis/National Institutes of Health NeuroMab Facility (clone A12/18). Anti-AP-2 α-adaptin (mouse) (clone 8, catalogue no. 610501) was from BD Biosciences. Anti-HA was MMS-101P (Covance Inc.) (mouse); clone Y-11, sc-805, was from Santa Cruz Biotechnology (rabbit; discontinued). Secondary antibodies were Alexa dye–coupled antibodies (Molecular Probes) and horseradish peroxidase (HRP)-coupled secondary antibodies (Amersham Biosciences/GE Healthcare).
Plasmids
DCX plasmid was obtained from mouse WT Dcx-GFP and FLAG-Dcx (24, 41). HA-NF was from Kizhatil et al. (22). HA-μ2-AP-2 plasmid was from Kim and Ryan (42). shRNA targeting the rat Dcx sequences, shDcx-GFP#2, was obtained from Super Array Biosciences (24).
The shDcx-associated phenotypes were specific to depletion of DCX and not due to off-target effects because several independent shDcx plasmids gave identical phenotypes with respect to NF endocytosis (24), and the phenotypes can be rescued by co-expression of a resistant WT Dcx plasmid (this work).
Generation of DCX mutants
Point mutations
Point mutations were introduced using the site-directed QuikChange mutagenesis kit from Stratagene. All mutations were confirmed by sequencing. The Dcx-ALPA mutant corresponds to Dcx-Y350A/L353A (6). Multiple versions of WT and mutant Dcx constructs were created, encoding either GFP or FLAG tags. No differences in behaviors were apparent, and the most convenient tag was used in different experiments.
Dcx-R2-FLAG
Full-length mouse cDNA of Dcx resistant to shDcx#2 was from Yap et al. (24). The same silent point mutations were introduced into Dcx-ALPA and Dcx-G253D to generate Dcx-R2-ALPA-FLAG and FLAG-Dcx-R2-G253D-FLAG.
Neuronal cultures
Neuronal cultures were prepared from E18 rat hippocampi, as approved in animal care and use committee protocol 3422 of the University of Virginia. Hippocampi from all pups in one litter were combined and thus contained male and female animals. Cells were plated on poly-l-lysine coverslips and incubated with Dulbecco's modified Eagle's medium with 10% horse serum. After 4 h, the cells were transferred into serum-free medium supplemented with B27 (Gibco BRL) and cultured for 9–11 DIV. Transfections were carried out with Lipofectamine 2000 at DIV7/8. Alternatively, neurons were electroporated after dissociation using Nucleofection (Lonza) and then cultured for 3 or 5 days.
COS and HEK293 cells
COS and HEK293 cells were maintained in Dulbecco's modified Eagle's medium + 10% fetal bovine serum, and all transfections were conducted using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
Immunocytochemistry
Cells were fixed in 2% paraformaldehyde, 3% sucrose, PBS in 50% conditioned medium at room temperature for 30 min, quenched in 10 mm glycine, PBS for 10 min. The fixation conditions used do not introduce holes into the overwhelming majority of cells (43). Coverslips were then blocked in 5% horse serum, 1% BSA, PBS with or without 0.05% Triton X-100 for 30 min. Antibodies were diluted in 1% BSA, PBS and incubated for 1–2 h. Coverslips were mounted in Vectashield (Vector Laboratories) and viewed on a Zeiss Axiophot microscope with a 40× objective. Images were captured with the Orca-ER cooled CCD camera (Hamamatsu) using Openlab software (ImproVision) and processed identically in Adobe Photoshop.
Endocytosis assay
Neurons expressing short hairpin plasmids for 3 days were incubated with mouse anti-neurofascin (anti-NF) antibodies for 30 min at 37 °C and washed several times, and all antibody remaining on the surface was stripped by treatment with pH 2.0 minimum essential medium for 2 min before fixation in 2% paraformaldehyde, 3% sucrose, PBS, pH 7.4. Internalized anti-NF antibody was detected by applying Alexa 568 goat anti-mouse antibody after permeabilization. Permeabilization was achieved with 0.2% Triton X-100 for 10 min at room temperature as described previously (24).
Immunoprecipitation
From HEK293 cells
For immunoprecipitations, transfected HEK293 cells were lysed for 45 min on ice in buffer containing 20 mm Tris-HCl (pH 7.4), 2 mm EDTA, 2 mm EGTA, 0.1 mm DTT, 100 mm KCl, 10 mm NaF, 1 mm phenylmethylsulfonyl fluoride, 1% Triton X-100, and Complete Protease Inhibitor mixture. Lysed cells were centrifuged at 12,000 rpm for 15 min, and the supernatants were then precleared with protein G–Sepharose (GE Healthcare) at 4 °C for 3 h. Precleared lysates were immunoprecipitated with mouse anti-HA or mouse anti-GFP for 1 h, followed by overnight incubation at 4 °C with protein G–Sepharose. Immunoprecipitates were washed five times with lysis buffer containing 0.1% Triton X-100 and eluted by 150 mm Tris-HCl (pH 6.8), 2% SDS, 5% β-mercaptoethanol, and 8% glycerol at 95 °C. Samples were subjected to SDS-PAGE and Western blot analysis.
From brain membrane fraction
E18 rat brains were homogenized in 3 volumes of buffer containing 20 mm Hepes (pH 7.4), 2 mm EDTA, 2 mm EGTA, 0.1 mm DTT, 100 mm KCl, 10 mm NaF, 1 mm phenylmethylsulfonyl fluoride, 0.32 m sucrose, and Halt protease and phosphatase inhibitors on ice and centrifuged at 1000 rpm for 10 min to remove nuclei. The postnuclear supernatant was further centrifuged at 14,000 rpm for 50 min, and the pellet obtained was lysed in homogenization buffer containing 1% Triton X-100 for 45 min on ice. This was followed by centrifugation at 12,000 rpm for 15 min, and the supernatant was used for immunoprecipitations as described above.
Polarity index
Distal axon, AIS (ankG-positive), and dendrite fluorescence intensities were measured along a 1-pixel-wide line in ImageJ after background subtraction and normalized to length (as described (2, 24)). The AIS polarity of HA-NF was determined by dividing average AIS intensity of HA-NF surface staining by the average intensity of surface HA-NF on dendrites. GFP, DCX-GFP, DCX-G253D, and DCX-ALPA were transfected in parallel in three independent repeats together with HA-NF, and AIS/D PI was quantified for each set. The data for DCX-G253D were reported in our previous work (24). The data for DCX-ALPA are reported in this work.
Detergent extractions
Detergent extractions of live cells were carried out in BRB80 buffer (80 mm Pipes, 1 mm MgCl2, 1 mm EGTA, pH 6.8) or PBS++ (BioWhittaker) with 0.15% Triton X-100 at 37 °C for 3 min (24, 25, 17). Cells were washed once in BRB80 or PBS++ buffer and fixed.
Microtubule fractionation
Freshly dissociated cortical neurons from E18 rat brain cortex were washed with BRB80 solution containing 15% glycerol and then permeabilized with the same solution added with 0.5% Nonidet P-40 at 37 ºC for 15 min. Cells were harvested and centrifuged at 700 × g for 5 min at room temperature. Supernatants were collected as free tubulin-soluble/membrane fractions. The remaining pellets were resuspended in permeabilization solution containing 50 mm CaCl2 at 37 ºC for 15 min. After centrifugation at 700 × g for 5 min at room temperature, supernatants were collected as microtubule fractions.
Dendrite quantification
Dendrites were quantified on images of transfected cells fixed on DIV3 and DIV5 and counterstained with DCX antibody. Because dendrites were short at this time point, the number of dendrites that measured at least 15 μm in length from the center of the soma was counted, as described previously (20).
Quantification of extent of overexpression
Experiments using GFP- or FLAG-tagged DCX (WT or mutants) were counterstained with antibody against DCX to evaluate the extent of overexpression, compared with control cells expressing GFP or untransfected cells. ImageJ was used to quantify somatic DCX fluorescence in 25–40 cells/condition. Three different expression modes were separately quantified: at DIV3 or DIV5 after electroporation and after Lipofectamine transfection at DIV8, to parallel the experimental paradigms used in Figs. 1, 4, 5, and 6. These results are shown in Fig. S1.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism software. The statistical test used is indicated in the figure legends. All experiments were repeated in at least three independent cultures, and quantification of at least 50 cells per experiment and condition was carried out for 2–3 experiments.
Author contributions
C. C. Y. and B. W. conceptualization; C. C. Y., X.-q. F., and J. S. L. resources; C. C. Y., L. D., K. K., and M. R. data curation; C. C. Y., L. D., K. K., M. R., and B. W. formal analysis; C. C. Y. and B. W. supervision; C. C. Y. and B. W. funding acquisition; C. C. Y., L. D., and B. W. validation; C. C. Y., L. D., K. K., M. R., X.-q. F., J. S. L., and B. W. investigation; C. C. Y. and L. D., visualization; C. C. Y., L. D., K. K., M. R., X.-q. F., J. S. L., and B. W. methodology; C. C. Y. and B. W. writing-original draft; C. C. Y. and B. W. project administration.
Supplementary Material
Acknowledgments
We thank Max Vakulenko for initiating the endocytosis studies on DCX-ALPA. We thank all members of the Winckler laboratory for constructive engagement throughout the duration of this work and critical reading of the manuscript.
This work was supported by National Institutes of Health Grant R01NS081674 (to B. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
C. C. Yap, unpublished results.
C. C. Yap and L. Digilio, unpublished results.
- MT
- microtubule
- NF
- neurofascin
- AIS
- axon initial segment
- sh
- short hairpin
- DIV
- day(s) in vitro
- E18
- embryonic day 18
- PI
- polarity index
- ANOVA
- analysis of variance.
References
- 1. Liu J. S. (2011) Molecular genetics of neuronal migration disorders. Curr. Neurol. Neurosci. Rep. 11, 171–178 10.1007/s11910-010-0176-5 [DOI] [PubMed] [Google Scholar]
- 2. Friocourt G., Marcorelles P., Saugier-Veber P., Quille M.-L., Marret S., and Laquerrière A. (2011) Role of cytoskeletal abnormalities in the neuropathology and pathophysiology of type I lissencephaly. Acta Neuropathol. 121, 149–170 10.1007/s00401-010-0768-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dixon-Salazar T. J., and Gleeson J. G. (2010) Genetic regulation of human brain development: lessons from Mendelian diseases. Ann. N.Y. Acad. Sci. 1214, 156–167 10.1111/j.1749-6632.2010.05819.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. LoTurco J. J., and Bai J. (2006) The multipolar stage and disruptions in neuronal migration. Trends Neurosci. 29, 407–413 10.1016/j.tins.2006.05.006 [DOI] [PubMed] [Google Scholar]
- 5. Bai J., Ramos R. L., Ackman J. B., Thomas A. M., Lee R. V., LoTurco J. J. (2003) RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat. Neurosci. 6, 1277–1283 10.1038/nn1153 [DOI] [PubMed] [Google Scholar]
- 6. Fu X., Brown K. J., Yap C. C., Winckler B., Jaiswal J. K., Liu J. S. (2013) Doublecortin (dcx) family proteins regulate filamentous actin structure in developing neurons. J. Neurosci. 33, 709–721 10.1523/JNEUROSCI.4603-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kappeler C., Dhenain M., Phan Dinh Tuy F., Saillour Y., Marty S., Fallet-Bianco C., Souville I., Souil E., Pinard J. M., Meyer G., Encha-Razavi F., Volk A., Beldjord C., Chelly J., and Francis F. (2007) Magnetic resonance imaging and histological studies of corpus callosal and hippocampal abnormalities linked to doublecortin deficiency. J. Comp. Neurol. 500, 239–254 10.1002/cne.21170 [DOI] [PubMed] [Google Scholar]
- 8. Koizumi H., Tanaka T., and Gleeson J. G. (2006) Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron 49, 55–66 10.1016/j.neuron.2005.10.040 [DOI] [PubMed] [Google Scholar]
- 9. Deuel T. A. S., Liu J. S., Corbo J. C., Yoo S. Y., Rorke-Adams L. B., and Walsh C. A. (2006) Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron 49, 41–53 10.1016/j.neuron.2005.10.038 [DOI] [PubMed] [Google Scholar]
- 10. Cohen D., Segal M., and Reiner O. (2008) Doublecortin supports the development of dendritic arbors in primary hippocampal neurons. Dev. Neurosci. 30, 187–199 10.1159/000109862 [DOI] [PubMed] [Google Scholar]
- 11. Bazelot M., Simonnet J., Dinocourt C., Bruel-Jungerman E., Miles R., Fricker D., and Francis F. (2012) Cellular anatomy, physiology and epileptiform activity in the CA3 region of Dcx knockout mice: a neuronal lamination defect and its consequences. Eur. J. Neurosci. 35, 244–256 10.1111/j.1460-9568.2011.07962.x [DOI] [PubMed] [Google Scholar]
- 12. Martineau F. S., Sahu S., Plantier V., Buhler E., Schaller F., Fournier L., Chazal G., Kawasaki H., Represa A., Watrin F., and Manent J. B. (2018) Correct laminar positioning in the neocortex influences proper dendritic and synaptic development. Cereb. Cortex 28, 2976–2990 10.1093/cercor/bhy113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tint I., Jean D., Baas P. W., and Black M. M. (2009) Doublecortin associates with microtubules preferentially in regions of the axon displaying actin-rich protrusive structures. J. Neurosci. 29, 10995–11010 10.1523/JNEUROSCI.3399-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jean D. C., Baas P. W., and Black M. M. (2012) A novel role for doublecortin and doublecortin-like kinase in regulating growth cone microtubules. Hum. Mol. Genet. 21, 5511–5527 10.1093/hmg/dds395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schaar B. T., Kinoshita K., and McConnell S. K. (2004) Doublecortin microtubule affinity is regulated by a balance of kinase and phosphatase activity at the leading edge of migrating neurons. Neuron 41, 203–213 10.1016/S0896-6273(03)00843-2 [DOI] [PubMed] [Google Scholar]
- 16. Friocourt G., Koulakoff A., Chafey P., Boucher D., Fauchereau F., Chelly J., and Francis F. (2003) Doublecortin functions at the extremities of growing neuronal processes. Cereb. Cortex 13, 620–626 10.1093/cercor/13.6.620 [DOI] [PubMed] [Google Scholar]
- 17. Sapir T., Horesh D., Caspi M., Atlas R., Burgess H. A., Wolf S. G., Francis F., Chelly J., Elbaum M., Pietrokovski S., and Reiner O. (2000) Doublecortin mutations cluster in evolutionarily conserved functional domains. Hum. Mol. Genet. 9, 703–712 10.1093/hmg/9.5.703 [DOI] [PubMed] [Google Scholar]
- 18. Moores C. A., Perderiset M., Francis F., Chelly J., Houdusse A., and Milligan R. A. (2004) Mechanism of microtubule stabilization by doublecortin. Mol. Cell 14, 833–839 10.1016/j.molcel.2004.06.009 [DOI] [PubMed] [Google Scholar]
- 19. Bechstedt S., and Brouhard G. J. (2012) Doublecortin recognizes the 13-protofilament microtubule cooperatively and tracks microtubule ends. Dev. Cell 23, 181–192 10.1016/j.devcel.2012.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yap C. C., Digilio L., McMahon L., Roszkowska M., Bott C. J., Kruczek K., and Winckler B. (2016) Different doublecortin (DCX) patient alleles show distinct phenotypes in cultured neurons. J. Biol Chem. 291, 26613–26626 10.1074/jbc.M116.760777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu J. S., Schubert C. R., Fu X., Fourniol F. J., Jaiswal J. K., Houdusse A., Stultz C. M., Moores C. A., and Walsh C. A. (2012) Molecular basis for specific regulation of neuronal Kinesin-3 motors by doublecortin family proteins. Mol. Cell 47, 707–721 10.1016/j.molcel.2012.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kizhatil K., Wu Y.-X., Sen A., and Bennett V. (2002) A new activity of doublecortin in recognition of the phospho-FIGQY tyrosine in the cytoplasmic domain of neurofascin. J. Neurosci. 22, 7948–7958 10.1523/JNEUROSCI.22-18-07948.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Friocourt G., Chafey P., Billuart P., Koulakoff A., Vinet M. C., Schaar B. T., McConnell S. K., Francis F., and Chelly J. (2001) Doublecortin interacts with μ subunits of clathrin adaptor complexes in the developing nervous system. Mol. Cell. Neurosci. 18, 307–319 10.1006/mcne.2001.1022 [DOI] [PubMed] [Google Scholar]
- 24. Yap C. C., Vakulenko M., Kruczek K., Motamedi B., Digilio L., Liu J. S., Winckler B. (2012) Doublecortin (DCX) mediates endocytosis of neurofascin independently of microtubule binding. J. Neurosci. 32, 7439–7453 10.1523/JNEUROSCI.5318-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim M. H., Cierpicki T., Derewenda U., Krowarsch D., Feng Y., Devedjiev Y., Dauter Z., Walsh C. A., Otlewski J., Bushweller J. H., and Derewenda Z. S. (2003) The DCX-domain tandems of doublecortin and doublecortin-like kinase. Nat. Struct. Biol. 10, 324–333 10.1038/nsb918 [DOI] [PubMed] [Google Scholar]
- 26. Kamiguchi H., Long K. E., Pendergast M., Schaefer A. W., Rapoport I., Kirchhausen T., and Lemmon V. (1998) The neural cell adhesion molecule L1 interacts with the AP-2 adaptor and is endocytosed via the clathrin-mediated pathway. J. Neurosci. 18, 5311–5321 10.1523/JNEUROSCI.18-14-05311.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Toriyama M., Mizuno N., Fukami T., Iguchi T., Toriyama M., Tago K., Itoh H. (2012) Phosphorylation of doublecortin by protein kinase A orchestrates microtubule and actin dynamics to promote neuronal progenitor cell migration. J. Biol. Chem. 287, 12691–12702 10.1074/jbc.M111.316307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yap C. C., Digilio L., McMahon L., and Winckler B. (2017) The endosomal neuronal proteins Nsg1/NEEP21 and Nsg2/P19 are itinerant, not resident proteins of dendritic endosomes. Sci. Rep. 7, 10481 10.1038/s41598-017-07667-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taylor K. R., Holzer A. K., Bazan J. F., Walsh C. A., and Gleeson J. G. (2000) Patient mutations in doublecortin define a repeated tubulin-binding domain. J. Biol. Chem. 275, 34442–34450 10.1074/jbc.M007078200 [DOI] [PubMed] [Google Scholar]
- 30. Boehm M., and Bonifacino J. S. (2001) Adaptins: the final recount. Mol. Biol. Cell 12, 2907–2920 10.1091/mbc.12.10.2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Traub L. M., and Bonifacino J. S. (2013) Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb. Perspect. Biol. 5, a016790 10.1101/cshperspect.a016790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reider A., and Wendland B. (2011) Endocytic adaptors: social networking at the plasma membrane. J. Cell Sci. 124, 1613–1622 10.1242/jcs.073395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yap C. C., and Winckler B. (2015) Adapting for endocytosis: roles for endocytic sorting adaptors in directing neural development. Front. Cell Neurosci. 9, 119 10.3389/fncel.2015.00119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gurevich V. V., and Gurevich E. V. (2014) Extensive shape shifting underlies functional versatility of arrestins. Curr. Opin. Cell Biol. 27, 1–9 10.1016/j.ceb.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Volkmer H., Leuschner R., Zacharias U., and Rathjen F. G. (1996) Neurofascin induces neurites by heterophilic interactions with axonal NrCAM while NrCAM requires F11 on the axonal surface to extend neurites. J. Cell Biol. 135, 1059–1069 10.1083/jcb.135.4.1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kriebel M., Metzger J., Trinks S., Chugh D., Harvey R. J., Harvey K., and Volkmer H. (2011) The cell adhesion molecule neurofascin stabilizes axo-axonic GABAergic terminals at the axon initial segment. J. Biol. Chem. 286, 24385–24393 10.1074/jbc.M110.212191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ebel J., Beuter S., Wuchter J., Kriebel M., and Volkmer H. (2014) Organisation and control of neuronal connectivity and myelination by cell adhesion molecule neurofascin. Adv. Neurobiol. 8, 231–247 10.1007/978-1-4614-8090-7_10 [DOI] [PubMed] [Google Scholar]
- 38. Zonta B., Desmazieres A., Rinaldi A., Tait S., Sherman D. L., Nolan M. F., and Brophy P. J. (2011) A critical role for neurofascin in regulating action potential initiation through maintenance of the axon initial segment. Neuron 69, 945–956 10.1016/j.neuron.2011.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rasband M. N. (2010) The axon initial segment and the maintenance of neuronal polarity. Nat. Rev. Neurosci. 11, 552–562 10.1038/nrn2852 [DOI] [PubMed] [Google Scholar]
- 40. Tanaka T., Serneo F. F., Higgins C., Gambello M. J., Wynshaw-Boris A., and Gleeson J. G. (2004) Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J. Cell Biol. 165, 709–721 10.1083/jcb.200309025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tsukada M., Prokscha A., Oldekamp J., and Eichele G. (2003) Identification of neurabin II as a novel doublecortin interacting protein. Mech. Dev. 120, 1033–1043 10.1016/S0925-4773(03)00177-1 [DOI] [PubMed] [Google Scholar]
- 42. Kim S. H., and Ryan T. A. (2009) Synaptic vesicle recycling at CNS snapses without AP-2. J. Neurosci. 29, 3865–3874 10.1523/JNEUROSCI.5639-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wisco D., Anderson E. D., Chang M. C., Norden C., Boiko T., Fölsch H., and Winckler B. (2003) Uncovering multiple axonal targeting pathways in hippocampal neurons. J. Cell Biol. 162, 1317–1328 10.1083/jcb.200307069 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.