

Abstract
During DNA replication or repair, the DNA polymerase cofactor, proliferating cell nuclear antigen (PCNA), homotrimerizes and encircles the replicating DNA, thereby acting as a DNA clamp that promotes DNA polymerase processivity. The formation of the PCNA trimer is also essential for targeting the replication-licensing protein, chromatin-licensing, and DNA replication factor 1 (CDT1), for ubiquitin-dependent proteolysis to prevent chromosomal DNA re-replication. CDT1 uses its PCNA-interacting peptide box (PIP box) to interact with PCNA, and the CRL4 E3 ubiquitin ligase subunit CDT2 is recruited through the formation of PCNA–CDT1 complexes. However, it remains unclear how CDT1 and many other PIP box–containing proteins are marked for degradation by the CRL4CDT2 ubiquitin ligase during DNA replication or damage. Here, using recombinant protein expression coupled with site-directed mutagenesis, we report that CDT2 and PCNA directly interact and this interaction depends on the presence of a highly conserved, C-terminal PIP box–like region in CDT2. Deletion or mutation of this region abolished the CDT2–PCNA interaction between CDT2 and PCNA both in vitro and in vivo. Moreover, PCNA-dependent CDT1 degradation in response to DNA damage and replication during the cell cycle requires an intact PIP box in CDT2. The requirement of the PIP boxes in both CDT2 and its substrate CDT1 suggests that the formation of the PCNA trimeric clamp around DNA during DNA replication and repair may bring together CDT1 and CRL4CDT2 ubiquitin E3 ligase to target CDT1 for proteolysis in a DNA synthesis–dependent manner.
Keywords: DNA damage response, DNA replication, DNA-protein interaction, ubiquitin ligase, cell cycle, CDT1, CRL4-CDT2, PCNA, PIP box, protein degradation
Introduction
Proliferating cell nuclear antigen (PCNA)2 is central to DNA replication and DNA repair (1, 2). It normally exists in a monomeric form in the absence of DNA replication or repair. During DNA replication or DNA repair synthesis, it is loaded onto the replicating DNA strands by the multisubunit replication factor C, the clamp loader, to form a homotrimeric DNA clamp that encircles the DNA. This trimeric form of PCNA acts as a processivity factor for DNA polymerases, such as DNA polymerase δ, during the leading and lagging strand DNA synthesis (1). The PCNA homotrimer also plays a key role in recruiting a large number of proteins onto the replicating DNA for replication-related processes or for replication-dependent proteolysis (2). The proteins that interact with PCNA contain a conserved PCNA-interacting peptide box (PIP) that mediates their interaction with PCNA. Recently, another PCNA interacting motif, the AlkB homologue 2 PCNA interacting motif (APIM), was found in more than 200 proteins that mediate their interaction with PCNA (1, 3). It was suggested that the PIP box–containing proteins are usually implicated in DNA replication, whereas the APIM-containing proteins are mainly important in the context of genotoxic stress (1, 3).
The PIP box in many proteins has been shown to be essential to promote their ubiquitin-dependent proteolysis in response to DNA replication or DNA repair synthesis (1, 2). The first example of these proteins is CDT1, a key replication licensing protein that is essential for licensing DNA replication origins for the initiation of DNA replication in S phase (2, 4, 5). Once DNA replication initiates, the CDT1 protein is rapidly degraded by the CRL4CDT2 ubiquitin E3 ligase complex (6–8). CDT1 is also triggered for rapid proteolysis by CRL4CDT2 if the cells are UV- or γ-irradiated (6, 7, 9). The degradation of CDT1 depends on the presence of a PIP box at its N terminus, which mediates the binding of CDT1 to PCNA (9–11). Interestingly, whereas CDT1 can bind to PCNA via its PIP box in vitro in the absence of DNA, CDT1 is only degraded by CRL4CDT2 in response to the onset of DNA replication or DNA damage (2, 6, 7, 9–11). The molecular mechanism by which CRL4CDT2 is recruited onto the PCNA–CDT1 complex during DNA replication or damage to target CDT1 for proteolysis remains unclear. In addition to CDT1, a large number of PIP box–containing proteins, such as CDK inhibitor p21 (Cip1/Waf1 or CDKN1A) and histone methyltransferase SET8 for the monomethylation of lysine 20 in histone H4 (H4K20), are degraded by the CRL4CDT2 ubiquitin E3 ligase complex in response to DNA replication or DNA damage via their PIP box–dependent interaction with PCNA (2, 12, 13). Loss or mutation of the PIP box in these proteins abolishes their degradation, even in the presence of ongoing DNA replication or DNA damage (10, 11).
The CRL4 ubiquitin E3 ligase complex belongs to the cullin-RING (CRLs) ubiquitin E3 ligase family (14). The CRL4 core complex, consisting of CUL4A or CUL4B, DDB1, and RBX1 (ROC1), utilizes a specific set of WDR proteins including the DCAFs (the DDB1–CUL4-associated factors or CDWs), as substrate-specific subunits to interact with specific substrates (7–9, 15, 16). We have previously identified CDT2 (also called L2DTL, DTL, or DCAF2) as the specific subunit of the CRL4CDT2 ubiquitin E3 ligase that targets CDT1 for proteolysis in response to DNA damage or replication (4, 6, 7, 9). In this process, the PIP box of CDT1 interacts with PCNA, which recruits CDT1 to the sites of ongoing DNA replication or repair DNA synthesis, to target the CDT1 protein for ubiquitin-dependent degradation via the CRL4CDT2 ubiquitin E3 ligase complex (2, 9, 10). It has been shown that CDT2 binds to the PCNA–CDT1 binary complex and specific mutations in PCNA such as aspartic acid residue 122 (Asp-122) affects CDT2 recruitment to the PCNA–CDT1 binary complex during DNA replication or repair (12, 17). However, because ubiquitin E3 ligase usually directly interacts with its protein substrate (14), it remains unclear whether a specific region of CDT2 is required for its interaction with the CDT1–PCNA protein complex during DNA replication or DNA damage to target CDT1 and other PIP box proteins, such as p21 or SET8, for degradation. Furthermore, it remains unclear why CDT1 is only degraded when PCNA is loaded onto the replicating DNA during S phase or in cells that are exposed to DNA damage, although the PIP box of CDT1 interacts with PCNA even in the absence of DNA (2, 7, 10, 18). Here, we report that CDT2 directly interacts with PCNA through a unique sequence in CDT2 to target CDT1 for degradation during DNA synthesis in S phase or in response to DNA damage.
Results
CDT2 and PCNA form protein complexes in vivo
Because PCNA interacts with proteins for DNA replication and repair, we tried to isolate PCNA-interacting proteins from human 293 cells by expressing a 3×FLAG–3×HA–PCNA protein in which the triple FLAG and triple HA tags were fused in-frame to the N terminus of PCNA in the retroviral vector, pMSCV-puro. The cells stably expressed the 3×FLAG–3×HA–PCNA protein was selected by puromycin resistance. The 3×FLAG–3×HA–PCNA protein complex was subsequently purified by sequential anti-FLAG antibody and then anti-HA antibody affinity chromatography. The proteins associated with the purified PCNA complexes were resolved in protein gel. Protein bands were excised, digested with trypsin, and derivative peptides were identified by an Orbitrap XL MS system (7, 19). We found that the ectopically expressed 3×FLAG–3×HA–PCNA protein complex contained a significant amount of endogenous PCNA (Fig. 1A), suggesting that 3×FLAG–3×HA–PC likely formed oligomers with the endogenous PCNA protein. We also identified several peptides corresponding to DNMT1 and UHRF1 (Fig. 1A and Table 1), which are known to form a UHRF1–DNMT1–PCNA complex for methylating DNA during DNA replication (1, 20, 21). In addition, our analysis also revealed the presence of a few peptides from CDT2, a component of the CRL4CDT2 ubiquitin E3 ligase complex that targets many PCNA-interacting proteins for ubiquitin-dependent proteolysis (Fig. 1A, Table 1) (2, 6–9).
Figure 1.
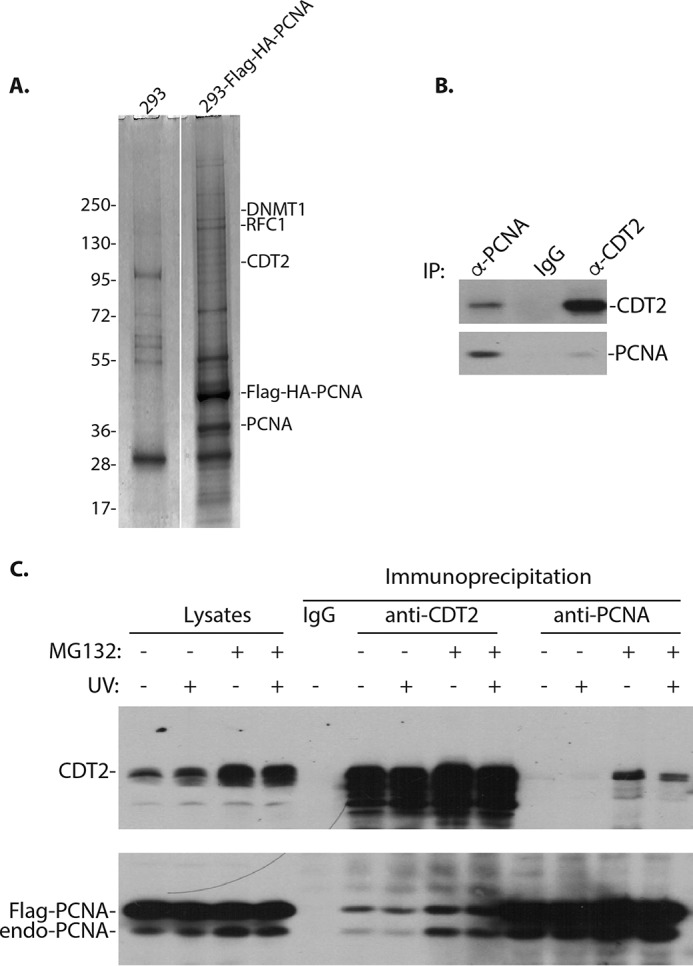
The interaction between CDT2 and PCNA is stabilized by MG132. A, isolated PCNA protein complexes contain CDT2, DNMT1, and RFC1 (see Table 1 for additional information). The 3×FLAG–3×HA–PCNA protein complexes were isolated from 293 cells stably express the tagged PCNA by sequential anti-FLAG M2 and anti-HA antibody affinity chromatography. 293 cells with an empty vector were used as a control. The proteins associated with the PCNA complexes were resolved in protein gel, excised, trypsinized, and derivative peptides were fractionated by nanoliter LC and identified by MS. The positions of some proteins associated with the PCNA complexes are shown. B, endogenous PCNA and CDT2 proteins interact. 293 cell lysates were immunoprecipitated (IP) with anti-CDT2 or anti-PCNA antibodies, followed by Western blot analysis. Nonspecific IgG was used as a control. Experiments were repeated three independent times with the same conclusion and one example is shown. C, 293 cells were transfected with expression constructs of CDT2 and FLAG-tagged PCNA for 48 h. The cells were treated with MG132 (10 μg/ml) for 4 h in the presence or absence of UV irradiation as indicated. The CDT2 and PCNA proteins complexes were immunoprecipitated by anti-CDT2 and PCNA antibodies and Western blotted as indicated.
Table 1.
Partial list of peptides identified by mass spectrometry
The 3×FLAG–3×HA-tagged PCNA protein complex was purified by anti-FLAG antibody affinity chromatography from 293 cells by anti-FLAG affinity chromatography, eluted with 3×FLAG peptide, re-purified with anti-HA affinity chromatography. The proteins in the 3×FLAG–3×HA-tagged PCNA protein complexes were fractionated in protein SDS gel. Protein bands were excised, trypsinized, and fractionated by nanoliter liquid chromatography. The peptides were identified by mass spectrometry analyses in an Orbitrap XL mass spectrometry system with the Protein Discovery software. Three independent mass spectrometry analyses were performed for the identity of a specific peptide/protein in repeated purification samples. Only one preparation sample is shown.
| Protein identification | Number of peptides | NCBI accession number |
|---|---|---|
| PCNA | 42 | NM_002592.2 |
| UHRF1 | 7 | NM_001290052.1 |
| DNA polymerase α | 5 | NM_001330360.1 |
| RFC1 | 4 | NM_002913.4 |
| DNMT1 | 4 | NM_001130823.1 |
| CDT2 (DTL) | 3 | NM_016448.3 |
| MCM5 | 3 | NM_006739.3 |
The interaction between CDT2 and PCNA is destabilized by the 26S proteosome
Our independent immunoprecipitation and Western blotting confirmed that the endogenous CDT2 protein indeed interacts with PCNA in vivo (Fig. 1B). This is consistent with our previous observation that CDT2 interacts with PCNA, and both CDT2 and PCNA are required for CDT1 degradation in response to DNA damage (9). To test how the interaction between CDT2 and PCNA is regulated, we transiently co-transfected 293 cells with expression constructs of human CDT2 and the FLAG-tagged PCNA. Immunoprecipitation of CDT2 revealed that CDT2 interacted with both exogenous and endogenous PCNA (Fig. 1C). PCNA also reciprocally associated with CDT2. Our studies further revealed that treatment of cells with MG132, an inhibitor of the 26S proteosome (22), significantly enhanced the interaction between CDT2 and PCNA (Fig. 1C). This enhanced interaction was in part due to the increased levels of CDT2 and PCNA proteins after MG132 treatment (Fig. 1C), suggesting that the stability of both CDT2 and PCNA proteins are regulated by ubiquitin-dependent proteolysis. These experiments raised the possibility that the interaction between CDT2 and PCNA is destabilized by the 26S proteosome.
Purified recombinant CDT2 directly interacts with PCNA
To determine how CDT2 interacts with PCNA, we used the purified recombinant GST–CDT2 protein isolated from bacteria and tried to determine whether it can directly interact with the recombinant PCNA protein in vitro (Fig. 2). Because both CDK inhibitors p21 and CDT1 contain a PIP box that is known to interact with PCNA directly (Fig. 2A) (2), we also used the GST–p21 and GST–CDT1 as a positive control (Fig. 2B). Incubation of these GST–proteins isolated from bacteria with the recombinant human PCNA in a bacterial lysate revealed that PCNA was associated with GST–CDT2, GST–p21, and GST–CDT1, but not with the GST protein control. To further confirm these observations, we also used the GST–CDT2 protein purified from baculoviral expressed SF9 cells for the binding of PCNA, using baculoviral expressed GST–p21 as a positive control. We also used GST–p27 (Kip1, CDKN1B), a CDK inhibitor that does not contain a PIP box, and GST–CUL4A, a component of the CRL4 core complex (6, 7), as negative controls. Our studies confirmed that both GST–CDT2 and GST–p21 proteins bind to PCNA, whereas GST–p27 and GST–CUL4A were negative for the interaction with PCNA (Fig. 2C). Our studies thus indicate that CDT2 can directly and independently bind to PCNA in the absence of CDT1 or other components of the CRL4 complex.
Figure 2.

Recombinant CDT2 independently interacts with PCNA in vitro. A, CDK inhibitor p21 and CDT1 contain the PIP box that interacts with PCNA. The consensus PIP box: QXXψXXθθ (ψ: hydrophobic amino acid residues, Leu, Val, Ile, Met; θ: aromatic amino acid residues, Phe and Tyr; X: any amino acid residues). B, GST–p21, GST–CDT2, GST–CDT1, and control GST proteins were purified from bacteria. The GST proteins were incubated with a bacterial lysate containing recombinant human PCNA protein for 4 h at 4 °C. The protein complexes were isolated by GSH-Sepharose and Western blotted with anti-PCNA and GST antibodies. C, GST–p21, GST–p27, GST–CUL4A, and GST–CDT2 were isolated from Sf9 cells infected with baculoviral constructs of GST proteins. The GST proteins (1 μg each) were incubated with bacterial lysates containing expressed recombinant human PCNA protein as in A and the proteins pulled down by the GSH-Sepharose were analyzed by anti-PCNA and anti-GST antibodies.
CDT2 contains a conserved C-terminal motif that interacts with PCNA
To locate the region in CDT2 that mediates its interaction with PCNA, we cloned various regions of CDT2 into the GST fusion constructs and monitored the interaction between GST–CDT2 mutants and PCNA. CDT2 contains an N-terminal WD40 repeat domain (50–400 amino acid residues, full-length of CDT2: 730 amino acid residues) that is required for the CUL4/DDB1 binding and a carboxyl-half with no clearly defined function. We found that the deletion in the N-terminal WD40 repeat region of CDT2 did not affect the interaction between CDT2 and PCNA (Fig. 3, A and B). However, deletion of the C-terminal region of CDT2 abolished its interaction with PCNA (Fig. 3). Further mapping of the C-terminal half of CDT2 revealed that the extreme C-terminal region is required for its interaction with PCNA (Fig. 3, A and C).
Figure 3.

Localization of the region in CDT2 for PCNA interaction. A, schematic illustration of the deletion mutants of the CDT2 protein and their ability to interact with PCNA. B and C, the C-terminal region (600–730) of CDT2 interacts with PCNA. GST-CDT2 and various N-terminal or C-terminal deletion constructs were expressed in bacteria and purified. They were incubated with bacterial lysates recombinant PCNA. The interactions were analyzed in GSH-Sepharose pulldown and blotting with anti-PCNA and GST antibodies.
Examination of the C terminus of CDT2 revealed that it contains a highly conserved motif, MRKICTYF (amino acid residues 706–713), which resembles that of the canonical PIP box in p21 and CDT1, QXXψXXθθ (ψ: Leu, Val, Ile, Met; θ: Phe and Tyr; X: any amino acid residues; Figs. 2A and 4A) (2). Although the methionine residue (Met-706) in the CDT2 motif is different from the starting glutamine (Gln) in the PIP box of p21 and CDT1, we have noticed that methionine is also present in the PIP box of human DNA polymerase η that mediates its interaction with PCNA (Fig. 4A) (2). We therefore tested the possibility that this PIP-like motif in CDT2 may mediate the interaction between CDT2 and PCNA. We found that deletion of this C-terminal motif (ΔPIP) in CDT2 is sufficient to abolish the interaction between various fragments of CDT2 and PCNA (Fig. 4B). We also tested whether tyrosine 712 (Tyr-712) and phenylalanine 713 (Phe-713) in the PIP-like motif are critical for PCNA binding (10), as it is known that similar aromatic amino acid residues in the PIP box of p21 and CDT1 are essential for the binding of p21 and CDT1 to PCNA (2). We found that conversion of Tyr-712 and Phe-713 to alanine (YF/AA) in CDT2 again completely abolished the interaction between CDT2 and PCNA (Fig. 4, B and C). We conclude that CDT2 contains a PIP-like motif at its C terminus that mediates its direct interaction with PCNA in vitro.
Figure 4.

CDT2 contains a PIP box–like motif at its C terminus for PCNA interaction. A, the C-terminal region (amino acid residues 705–713) of CDT2 contains a conserved MXXIXXYF motif that resembles the PIP box motif in p21, CDT1, and DNA polymerase η. The conversion of Tyr-712 and Phe-713 to alanine (Ala) is shown. B, deletion or mutation of the C-terminal PIP box–like motif abolishes the interaction between CDT2 and PCNA. Deletion of the PIP (ΔPIP) or conversion of Tyr-712 and Phe-713 to alanine (YF/AA) in various N-terminal deletion mutants of CDT2 was expressed in bacteria, and their binding to PCNA was monitored. C, full-length GST–CDT2, CDT2 YF/AA mutant, and CUL4A were expressed in baculovirus-infected Sf9 cells and their interaction with PCNA was analyzed by GSH-Sepharose pulldown and blotting with anti-PCNA and GST antibodies.
The PIP box–like motif in CDT2 abolishes the interaction with PCNA and stabilizes CDT2 protein in vivo
To examine the effects of the PIP mutants of CDT2 on the interaction with PCNA in vivo, the FLAG-CDT2 and YF/AA mutant were transfected into 293 cells and their binding to PCNA was analyzed in the presence or absence of MG132 (Fig. 5A). As we observed in Fig. 1C, the protein level of FLAG–CDT2 (WT, WT) was relatively low, which was stabilized by MG132 (Fig. 5A). The binding between CDT2 and PCNA is also relatively weak in the absence of MG132 but the interaction was enhanced by MG132. However, mutation of the PIP box–like motif in CDT2 was sufficient to abolish its interaction with PCNA, even in the presence of MG132 (Fig. 5A). In addition, we found that the levels of CDT2 YF/AA mutant protein were consistently higher than that of the WT CDT2 protein (Fig. 5A). These results indicate that the PIP box–like motif of CDT2 indeed mediates its interaction with PCNA in vivo. These studies also suggest that the PIP box–like motif destabilizes the CDT2 protein, likely through its interaction with PCNA.
Figure 5.

The PIP box–like mutant of CDT2 is more stable and functionally defective to interact with PCNA and mediate CDT1 degradation. A, the PIP box–like mutant of CDT2 is more stable and fails to interact with PCNA in vivo. 293 cells were transfected with expression constructs of the FLAG-CDT2 and YF/AA mutant, for 48 h. The cells were treated with MG132 (10 μg/ml) for 3 h as indicated. The CDT2 protein complexes were isolated by anti-FLAG antibodies and Western blotted with anti-CDT2 and PCNA antibodies. PCNA in the lysate serves as a source for total PCNA. B, the PIP box–like mutants of CDT2 are defective in CDT1 interaction. 293 cells were transfected with expression constructs of the FLAG-CDT2, and the YF/AA and ΔPIP mutants of CDT2, in the presence of absence of PCNA expression construct, for 48 h. The cells were treated with MG132 in the presence of MG132 (10 μg/ml) for 3 h and then UV irradiation (50 J/cm2) in the presence of MG132 (10 μg/ml) for 1 h. The interaction between CDT1 and CDT2 was analyzed by anti-CDT1 immunoprecipitation and Western blotting with anti-FLAG–CDT2 and CDT1 antibodies. C, both CDT2 and PCNA are required for CDT1 degradation in response to UV irradiation. HeLa cells were transfected with 50 nm siRNAs for luciferase (control), CDT2, or PCNA in the presence or absence of UV irradiation, as indicated. The protein levels of CDT1, CDT2, and PCNA were analyzed by Western blotting with indicated antibodies. Actin is used as a protein loading control. D, the PIP box–like mutant of CDT2 failed to rescue CDT1 degradation. The 3×FLAG–3×HA–tagged CDT2 (coding region). The YF/AA and C-terminal ΔPIP deletion mutants were cloned in pMCSV-Puro and expressed in HeLa cells by transfection. The endogenous CDT2 was knocked down by the siRNA that targets the 3′-UTR of CDT2 after 12 h of FLAG-CDT2 transfection. The effects of ectopically expressed CDT2, the YF/AA, and C-terminal ΔPIP deletion mutants on CDT1 degradation were analyzed after UV irradiation.
Mutation of the PIP box–like region of CDT2 is defective in CDT1 interaction
The function of CDT2 is to serve as a substrate-specific subunit of the CRL4CDT2 ubiquitin E3 ligase complex to target the critical replication-licensing protein, CDT1, for degradation in S phase to prevent re-replication of the genomic DNA (4, 8). We have previously shown that CDT1 is degraded after UV- or γ-irradiation by CRL4CDT2 and the interaction between CDT1 and CDT2 can be detected in vivo (6, 7, 9). To examine whether CDT2 mutants affect its interaction with CDT1, the expression constructs of the WT CDT2 and the PIP mutants were transfected into 293 cells in the presence or absence of PCNA. The interaction between the transfected CDT2 and endogenous CDT1 was analyzed after UV-irradiation by co-immunoprecipitation (Fig. 5B). To prevent CDT1 protein degradation, cells were treated with MG132 prior to UV-irradiation. We found that in the absence of co-transfected PCNA, CDT2 interacted with CDT1 poorly (Fig. 5B). Co-expression of PCNA significantly promoted the interaction between CDT1 and CDT2 (Fig. 5B). However, both ΔPIP and YF/AA mutants of CDT2 reduced their interaction with CDT1, even in the presence of PCNA (Fig. 5B). These results suggest that the PIP box–like motif in CDT2 is required for CDT1 and CDT2 interaction in vivo.
The PIP box–like motif of CDT2 is required for CDT1 degradation in response to DNA damage
We have previously shown that CDT2 is required for CDT1 proteolysis in response to DNA damage (Fig. 5C, left panel) (6, 7, 9). Similarly, loss of PCNA also impaired the CDT2-dependent CDT1 degradation in the presence of DNA damage (Fig. 5C, right panel). Because the PIP box mutant of CDT2 appeared defective in its interaction with PCNA and CDT1 (Fig. 5, A and B), we wondered whether the PIP box–like motif in CDT2 is required for CDT1 degradation in response to DNA damage. To test this possibility, we tried to replace the endogenous CDT2 with the ectopically expressed CDT2 or its mutants (Fig. 5D). We first eliminated the endogenous CDT2 with a siRNA for the 3′-UTR region of the CDT2 mRNA so it would not interfere with the ectopically expressed CDT2 that contains only the coding region (Fig. 5D). We then expressed either the WT FLAG–CDT2 or the PIP box–like CDT2 mutants to see whether ectopically expressed CDT2 or its PIP mutants can replace the endogenous CDT2 function on CDT1 degradation in these cells. Our studies revealed that the 3′-UTR siRNA effectively reduced CDT2 to stabilize the CDT1 protein in response to UV irradiation (Fig. 5D). Expression of the CDT2 cDNA, which contains only the coding region so it is insensitive to the 3′-UTR siRNA, was sufficient to restore the degradation of CDT1 in cells treated with the 3′-UTR siRNA (Fig. 5D). However, expression of either the deletion of the PIP box–like motif (ΔPIP) or the YF/AA mutant of CDT2 failed to restore the ability of cells to degrade CDT1 in response to DNA damage (Fig. 5D). These results indicate that the PIP box–like motif of CDT2 is required for CDT1 degradation in response to DNA damage.
The PIP box mutant of CDT2 is defective to degrade CDT1 during S phase in the cell cycle
It is well established that CDT1 is degraded by the CRL4CDT2 ubiquitin E3 ligase in the S phase to prevent DNA re-replication (2). Our previous studies have shown that loss of CDT2 increased the stability of CDT1 protein in the S phase, resulting in both the accumulation of enlarged nuclei and the activation of phosphorylated H2AX staining in the nuclei due to partial re-replication of genomic DNA that activates the DNA damage checkpoint (23). To further test the function of the PIP box–like motif of CDT2, we conducted a replacement experiment by expressing the WT CDT2 or its PIP box mutant to determine whether their expression suppresses enlarged nucleus and phosphorylated H2AX staining in CDT2 knockdown cells. Knockdown of CDT2 using 3′-UTR–based siRNA caused a small fraction of cells to exhibit enlarged nuclei that are also positively stained with anti-phospho-H2AX (Fig. 6A), which are consistent with our previous finding that cells with enlarged nucleus represent cells undergoing partial DNA re-replication (23). We found that expression of the WT CDT2 cDNA construct is sufficient to reduce enlarged nucleus, decrease CDT1 protein level, and down-regulate the phospho-H2AX staining in these cells (Fig. 6A), demonstrating that the exogenously expressed CDT2 can functionally replace endogenous CDT2 that was removed by our CDT2 3′-UTR siRNA (Fig. 6A). However, our studies revealed that expression of the ΔPIP mutant of CDT2 failed to rescue the defects of CDT2 deficiency, so that the expression of the ΔPIP mutant of CDT2 did not significantly affect the accumulation of cells with enlarged nucleus positively stained with the phosphorylated H2AX (Fig. 6A).
Figure 6.

The PIP box–like region in CDT2 is required for CDT1 degradation in the cell cycle. A, the PIP box–like mutant of CDT2 failed to rescue the activation of DNA damage checkpoint caused by knockdown of endogenous CDT2. H1299 were transfected with the WT FLAG–CDT2 and ΔPIP mutant for 12 h. The cells were then transfected with luciferase siRNA or the CDT2 siRNA that targets the 3′-UTR of CDT2 for an additional 12 h. Nuclei of cells were stained with DAPI and anti-H2AX antibodies. The expression of CDT2 and PIP mutant proteins, CDT1 and Actin (loading control) were analyzed by Western blotting. Scale bar, 20 μm. B, failure of the PIP box–like mutant of CDT2 to reduce the accumulation of cells with enlarged nucleus in CDT2 knockdown cells. HCT116 cells with p53 deletion are transfected as in A and treated with 10 μm EdU for 2 h before performing the Click-iT reaction for EdU incorporation analyses. The cells were then stained with DAPI. About 200 cells from each sample were counted and the fractions of cells with enlarged nucleus were quantified and compared. The quantifications are represented by bar graph with mean ± S.D. for error bars from three technical replicates. The p value of vector, WT, and ΔPIP CDT2 to control (Luc) siRNAs was calculated by independent t test and the p values of single knockdown to WT and ΔPIP CDT2 were calculated by paired Student's t test (***, p < 0.001). Experiments were repeated three independent times with the same conclusion and one example is shown. Scale bar, 20 μm. C, the PIP box–like mutant of CDT2 failed to abolish the re-replication caused by knockdown of endogenous CDT2. Preparation is the same as in B except the cells are harvested by trypsinization and stained with phosphatidylinositol (PI). Two-dimensional flow cytometry analysis of DNA synthesis (EdU) and DNA content (PI) is shown.
To further verify that the cells with the enlarged nuclei are caused by re-replication of genomic DNA after loss of endogenous CDT2, we examined DNA replication by labeling the cells with 5-ethynyl-2′-deoxyuridine (EdU), a nucleoside analogue of thymidine that can be incorporated into replicating DNA, to directly monitor active DNA synthesis in the cell cycle in HCT116 cells with p53 deficiency that facilitates the detection of DNA re-replication (Fig. 6, B and C). We found that knockdown of CDT2 led to about 25–30% of HCT116 cells to exhibit enlarged nucleus (Fig. 6B). These cells also showed prominent EdU incorporation, suggesting they were actively replicating their DNA (Fig. 6B). Although ectopic expression of the WT CDT2 cDNA construct suppressed the enlarged nucleus in CDT2-deficient cells, expression of the CDT2 ΔPIP mutant showed the same enlarged nucleus defects as endogenous CDT2 knockdown alone (Fig. 6B). Because our previous studies indicate that loss of CDT2 promoted re-replication of the genome due to the failure to degrade CDT1 (23), we employed flow cytometry analyses to monitor DNA replication in these experiments. Our studies revealed that knockdown of endogenous CDT2 significantly increased the fraction of cells with genomic DNA greater than 4N, indicating DNA re-replication in the absence of CDT2 activity (Fig. 6C). Our analysis using two-dimensional flow cytometry further showed that a significant number of cells with more than 4 n DNA content incorporated EdU, indicating that they were undergoing active DNA re-replication (Fig. 6C). Expression of the WT CDT2 is sufficient to reduce the percentage of cells with genomic DNA greater than 4 n, whereas the PIP mutant of CDT2 could not rescue the defects induced by CDT2 deficiency (Fig. 6C). These studies indicate that knockdown of CDT2 causes DNA re-replication due to the stabilization of CDT1 protein (Fig. 6B), and the CDT2 mutant lacking the PIP box–like motif is defective to target CDT1 for degradation during DNA replication in the cell cycle.
Discussion
We have previously shown that the CDT2 complex also contains PCNA (9). In this report, we found that the PCNA–protein complexes also contained CDT2 (Fig. 1). Our studies revealed that CDT2 contains a highly conserved PIP box–like motif at its C terminus and this motif directly interacts with PCNA both in vitro and in vivo (Fig. 2–5). Our studies further showed that the PIP box–like motif in CDT2 is required for CDT1 degradation in response to DNA damage and DNA replication in the cell cycle (Figs. 5 and 6). Although we and others have established that the CRL4CDT2 ubiquitin E3 ligase complex targets CDT1 degradation via a PCNA-dependent process (9, 10), how the CRL4CDT2 ubiquitin E3 ligase is recruited to interact with the PCNA–CDT1 complex target CDT1 only during DNA replication or repair synthesis remains unclear (2). Our studies are consistent with a previous report that the C-terminal half of CDT2 is required for the PCNA-dependent degradation of Xenopus CDK inhibitor Xic1, which is a substrate of Xenopus CRL4CDT2 (12). Based on our observation, we propose that both CDT1 and CDT2 are PIP box–containing proteins that are capable to bind to PCNA. The initiation of DNA replication or DNA damage-induced DNA repair synthesis leads to the recruitment of PCNA to the ongoing replication forks to form the trimeric clamp by the clamp loader, RFC. The formation of the trimeric PCNA clamp also leads to the recruitment of both CDT1 and CDT2 to the same trimeric PCNA clamp so their proximity to each other facilitate the interaction between CDT1 and CDT2, promoting the ubiquitin-dependent proteolysis of CDT1 by the CRL4CDT2 ubiquitin E3 ligase complex (Fig. 7). Our studies indicate that the PIP boxes in both CDT1 and CDT2 are required to be recruited onto the replicating DNA strands through their binding to the trimeric PCNA clamp, promoting the ubiquitin-dependent degradation of CDT1 by further recruitment of the CRL4 core E3 ligase complex. Because CDT1 is only targeted for degradation by the CRL4CDT2 ubiquitin E3 ligase complex during DNA replication or repair synthesis, our finding provides a model that the trimerization of PCNA encircling the replicating DNA acts as a molecular bridge to bring the substrates and the CRL4CDT2 ubiquitin E3 ligase complex together. Our studies thus reveal for the first time a novel mechanism by which the CRL4CDT2 ubiquitin E3 ligase is recruited to the replicating DNA by its binding to PCNA to target PCNA-bound CDT1 and other substrates for degradation. Previous studies suggest that certain specific amino acid residues, such as Asp-122 in PCNA, may be involved in recruitment of CDT2 to interact with the CDT1–PCNA complex (17). It is possible that after CDT2 is recruited to the trimeric PCNA complex and the replicating DNA, CDT2 may further interact with the interface of the PCNA–CDT1 binary complex on the same PCNA ring and this interaction may require the presence of Asp-122 in PCNA. Nevertheless, our studies indicate that PCNA is required for recruiting both CDT1 and CDT2 to the replicating DNA to target CDT1 and other PCNA-binding protein substrates for degradation during S phase or in response to DNA damage. Thus, our studies revealed for the first time a novel mechanism by which the replicating DNA strands serve as a recruitment mechanism for the binding of both substrates and ubiquitin E3 ligase to trigger the degradation of the substrates only during DNA replication or repair synthesis in the cell cycle.
Figure 7.
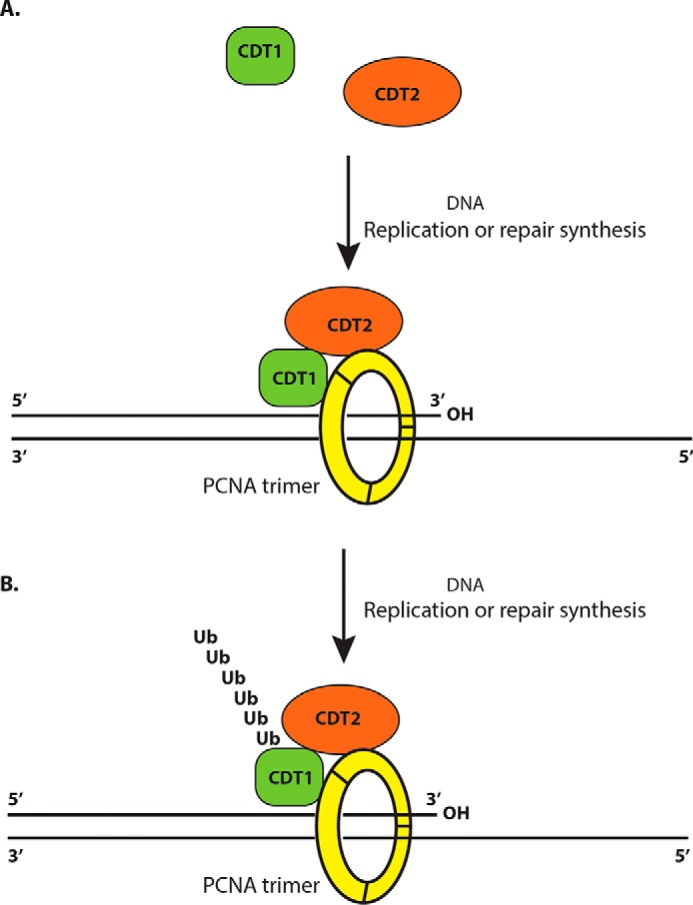
A model for DNA replication-dependent degradation of CDT1 by trimeric PCNA and CDT2. A, both CDT2 and CDT1 interact with PCNA through their respective PIP box motifs. During DNA replication or DNA repair synthesis, the trimeric form of PCNA brings CDT1 and CDT2 together to the same PCNA clamp. B, the proximity of CDT1 and CDT2 leads to the polyubiquitination and eventual proteolysis of CDT1 by the CRL4DCAT2 ubiquitin E3 ligase complex in response to DNA replication and DNA damage.
Experimental procedures
Cells and tissue cultures
HeLa cells and 293 cells were purchased from ATCC and cultured in Dulbecco's modified Eagle's medium, supplemented with 10% fetal bovine serum and penicillin/streptomycin (Invitrogen) (24). The cells have been recently authenticated and tested for contamination based on protein markers such as p53, p16, and p21 for both HeLa and 293 cells (23). UV irradiation (UV-C) of cells was conducted using a UV Stratalinker 1800 (Stratagene, La Jolla, CA), as described previously (6).
Antibodies and immunological methods
The anti-PCNA (ab29 and ab18197) and anti-GST (ab19256) antibodies were purchased from Abcam. Phospho-histone H2AX (Ser-139) antibody was purchased from Cell Signaling. Anti-CDT2 (A300–948A) and CDT1 (A300–786A) were from Bethyl Laboratories. The anti-FLAG antibody (Phe-1804) and MG132 (474790) were from Sigma. The anti-HA (12CA5) antibody was from Roche Applied Science. The immunoprecipitation and Western blotting analyses were performed as described previously (6, 7, 9).
Immunoaffinity purification and MS analysis
Cell lysates from 20 dishes (150 × 25 mm) of 293 cells expressing 3×FLAG–3×HA–PCNA or equal dishes of control 293 cells were employed for immunoaffinity purification using the anti-FLAG M2 affinity gel (Sigma) (19). The bound protein complexes were eluted with 3×FLAG peptide, as described previously (7, 19). The eluted protein complexes were further purified by anti-HA affinity antibody Sepharose (Roche Applied Science) (19). The isolated proteins were separated on an SDS-PAGE gel, excised, trypsinized, fractionated on an Easy nanoliter LC, and identified using a LTQ Orbitrap XL mass-spectrometer system (Thermo Scientific) (19).
Recombinant proteins and binding assays
The human p21, CDT1, CDT2 and various deletion mutant constructs of CDT2 were fused in-frame with GST in pGEXKG and expressed in Escherichia coli BL21 strain (6, 25). These cDNAs, as well as that of human p27 and CUL4A, were also cloned as GST fusion in baculoviral pVL1392-GST vector and expressed in SF9 cells as described previously (7, 26). GST proteins were purified by GSH-Sepharose (GE Healthcare). Human PCNA cDNA was cloned into the pET vector and expressed in E. coli BL21 strain. The site-directed mutagenesis was conducted as previously described (22). For binding assays, 1 μg of GST fusion proteins were mixed with bacterial lysates containing the expressed PCNA and incubated at 4 °C for 2 h. The protein complexes were isolated by 30 μl of GSH-Sepharose and detected by Western blotting with anti-GST or anti-PCNA antibodies.
Transfection and siRNAs
Lipofectamine 2000 was used for transient expression constructs into HeLa or 293 cells. Oligofectamine (Life Technologies) was used for transfection of siRNAs for knockdown analysis (7, 24). Typically 50 nm of each siRNA or their combinations were transfected into target cells for 48–60 h. The siRNAs for human CDT2 in the coding region was AAUCCCUUCUGACUGGUUAUC and in the 3′-UTR regions were GAGGAUGAAUGCUGUGUUU, PCNA siRNA, GGAGGAAGCUGUUACCAUA, and Luciferase, CGUACGCGGAAUACUUCGA. All siRNAs were synthesized from GE Dharmacon.
Immunostaining
Cells were cultured on coverslips in 35-mm dishes and were fixed with 3.7% paraformaldehyde and then permeabilized with 0.3% Triton X-100. Cells were incubated with the primary antibodies overnight at 4 °C, washed, and stained for 1 h with fluorescence-labeled secondary antibodies at room temperature as described previously (6). Coverslips were mounted with Mowiol containing 1 μg/ml of 4′,6-diamidino-2-phenylindole (DAPI) for DNA staining. Images were captured on an Olympus fluorescence microscope (Olympus CKX41, Japan) coupled to a cooled charge-coupled device camera (QICAM, Japan) and processed by using the QCapture Pro 6.0 program.
Flow cytometry (FACS) analysis
Cells were labeled with 10 μm EdU for 2 h, trypsinized, and processed according to the protocol of Clik-iTTM Plus EdU Flow Cytometry Assay Kit (Invitrogen, catalog number C10632) and stained by FxCycleTM PI/RNase Staining Solution (Invitrogen, catalog number F10797) at room temperature for 30 min. They were analyzed by BD FACSCalibur Flow Cytometry and evaluated with FlowJo version 7.6.5.
Author contributions
F. Leng, L. S., and C. Z. data curation; F. Leng, L. S., N. H., C. Z., L. L., X. G., and H. S. investigation; F. Leng, N. H., W. L., and F. Lu methodology; L. S., N. H., and C. Z. validation; H. Z. conceptualization; H. Z. supervision; H. Z. funding acquisition; H. Z. writing-original draft; H. Z. project administration.
This work was supported by National Institutes of Health Grants R15NS096694 (to H. S.), R15GM116087 and R15GM131255 (to H. Z.) and the Cancer Research Gift Fund to the College of Sciences at University of Nevada, Las Vegas (to H. S. and H. Z.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PCNA
- proliferating cell nuclear antigen
- PIP box
- PCNA-interacting peptide box
- CRL
- cullin-RING ubiquitin ligase
- EDU
- 5-ethynyl-2′-deoxyuridine
- HA
- hemagglutinin
- GST
- glutathione S-transferase
- DAPI
- 4′,6-diamidino-2-phenylindole.
References
- 1. Mailand N., Gibbs-Seymour I., and Bekker-Jensen S. (2013) Regulation of PCNA-protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 14, 269–282 10.1038/nrm3562 [DOI] [PubMed] [Google Scholar]
- 2. Arias E. E., and Walter J. C. (2007) Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev. 21, 497–518 10.1101/gad.1508907 [DOI] [PubMed] [Google Scholar]
- 3. Gilljam K. M., Feyzi E., Aas P. A., Sousa M. M., Muller R., Vågbo C. B., Catterall T. C., Liabakk N. B., Slupphaug G., Drabløs F., Krokan H. E., and Otterlei M. (2009) Identification of a novel, widespread, and functionally important PCNA-binding motif. J. Cell Biol. 186, 645–654 10.1083/jcb.200903138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Higa L. A., and Zhang H. (2007) Stealing the spotlight: CUL4-DDB1 ubiquitin ligase docks WD40-repeat proteins to destroy. Cell Div. 2, 5 10.1186/1747-1028-2-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mihaylov I. S., Kondo T., Jones L., Ryzhikov S., Tanaka J., Zheng J., Higa L. A., Minamino N., Cooley L., and Zhang H. (2002) Control of DNA replication and chromosome ploidy by geminin and cyclin A. Mol. Cell Biol. 22, 1868–1880 10.1128/MCB.22.6.1868-1880.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Higa L. A., Mihaylov I. S., Banks D. P., Zheng J., and Zhang H. (2003) Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat. Cell Biol. 5, 1008–1015 10.1038/ncb1061 [DOI] [PubMed] [Google Scholar]
- 7. Higa L. A., Wu M., Ye T., Kobayashi R., Sun H., and Zhang H. (2006) CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol. 8, 1277–1283 10.1038/ncb1490 [DOI] [PubMed] [Google Scholar]
- 8. Jin J., Arias E. E., Chen J., Harper J. W., and Walter J. C. (2006) A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 23, 709–721 10.1016/j.molcel.2006.08.010 [DOI] [PubMed] [Google Scholar]
- 9. Higa L. A., Banks D., Wu M., Kobayashi R., Sun H., and Zhang H. (2006) L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell Cycle 5, 1675–1680 10.4161/cc.5.15.3149 [DOI] [PubMed] [Google Scholar]
- 10. Arias E. E., and Walter J. C. (2006) PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat. Cell Biol. 8, 84–90 10.1038/ncb1346 [DOI] [PubMed] [Google Scholar]
- 11. Senga T., Sivaprasad U., Zhu W., Park J. H., Arias E. E., Walter J. C., and Dutta A. (2006) PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J. Biol. Chem. 281, 6246–6252 10.1074/jbc.M512705200 [DOI] [PubMed] [Google Scholar]
- 12. Kim D. H., Budhavarapu V. N., Herrera C. R., Nam H. W., Kim Y. S., and Yew P. R. (2010) The CRL4Cdt2 ubiquitin ligase mediates the proteolysis of cyclin-dependent kinase inhibitor Xic1 through a direct association with PCNA. Mol. Cell Biol. 30, 4120–4133 10.1128/MCB.01135-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chuang L. C., and Yew P. R. (2005) Proliferating cell nuclear antigen recruits cyclin-dependent kinase inhibitor Xic1 to DNA and couples its proteolysis to DNA polymerase switching. J. Biol. Chem. 280, 35299–35309 10.1074/jbc.M506429200 [DOI] [PubMed] [Google Scholar]
- 14. Petroski M. D., and Deshaies R. J. (2005) Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20 10.1038/nrm1547 [DOI] [PubMed] [Google Scholar]
- 15. He Y. J., McCall C. M., Hu J., Zeng Y., and Xiong Y. (2006) DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 20, 2949–2954 10.1101/gad.1483206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Angers S., Li T., Yi X., MacCoss M. J., Moon R. T., and Zheng N. (2006) Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443, 590–593 [DOI] [PubMed] [Google Scholar]
- 17. Havens C. G., Shobnam N., Guarino E., Centore R. C., Zou L., Kearsey S. E., and Walter J. C. (2012) Direct role for proliferating cell nuclear antigen in substrate recognition by the E3 ubiquitin ligase CRL4Cdt2. J. Biol. Chem. 287, 11410–11421 10.1074/jbc.M111.337683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jackson S., and Xiong Y. (2009) CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem. Sci. 34, 562–570 10.1016/j.tibs.2009.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yin F., Lan R., Zhang X., Zhu L., Chen F., Xu Z., Liu Y., Ye T., Sun H., Lu F., and Zhang H. (2014) LSD1 regulates pluripotency of embryonic stem/carcinoma cells through histone deacetylase 1-mediated deacetylation of histone H4 at lysine 16. Mol. Cell Biol. 34, 158–179 10.1128/MCB.00631-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qin W., Leonhardt H., and Pichler G. (2011) Regulation of DNA methyltransferase 1 by interactions and modifications. Nucleus 2, 392–402 10.4161/nucl.2.5.17928 [DOI] [PubMed] [Google Scholar]
- 21. Pacaud R., Brocard E., Lalier L., Hervouet E., Vallette F. M., and Cartron P. F. (2014) The DNMT1/PCNA/UHRF1 disruption induces tumorigenesis characterized by similar genetic and epigenetic signatures. Sci. Rep. 4, 4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang C., Hoang N., Leng F., Saxena L., Lee L., Alejo S., Qi D., Khal A., Sun H., Lu F., and Zhang H. (2018) LSD1 demethylase and the methyl-binding protein PHF20L1 prevent SET7 methyltransferase-dependent proteolysis of the stem-cell protein SOX2. J. Biol. Chem. 293, 3663–3674 10.1074/jbc.RA117.000342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu F., Wu X., Yin F., Chia-Fang Lee C., Yu M., Mihaylov I. S., Yu J., Sun H., and Zhang H. (2016) Regulation of DNA replication and chromosomal polyploidy by the MLL-WDR5-RBBP5 methyltransferases. Biol. Open 5, 1449–1460 10.1242/bio.019729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang X., Lu F., Wang J., Yin F., Xu Z., Qi D., Wu X., Cao Y., Liang W., Liu Y., Sun H., Ye T., and Zhang H. (2013) Pluripotent stem cell protein Sox2 confers sensitivity to LSD1 inhibition in cancer cells. Cell Rep. 5, 445–457 10.1016/j.celrep.2013.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Higa L. A., Yang X., Zheng J., Banks D., Wu M., Ghosh P., Sun H., and Zhang H. (2006) Involvement of CUL4 ubiquitin E3 ligases in regulating CDK inhibitors Dacapo/p27Kip1 and cyclin E degradation. Cell Cycle 5, 71–77 10.4161/cc.5.1.2266 [DOI] [PubMed] [Google Scholar]
- 26. Tsvetkov L. M., Yeh K. H., Lee S. J., Sun H., and Zhang H. (1999) p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr. Biol. 9, 661–664 10.1016/S0960-9822(99)80290-5 [DOI] [PubMed] [Google Scholar]