

Abstract
On January 21, 2017, I received an E-mail from Herb Tabor that I had been simultaneously hoping for and dreading for several years: an invitation to write a “Reflections” article for the Journal of Biological Chemistry. On the one hand, I was honored to receive an invitation from Herb, a man I have admired for over 40 years, known for 24 years, and worked with as a member of the Editorial Board and Associate Editor of the Journal of Biological Chemistry for 17 years. On the other hand, the invitation marked the waning of my career as an academic scientist. With these conflicting emotions, I wrote this article with the goals of recording my career history and recognizing the many mentors, trainees, and colleagues who have contributed to it and, perhaps with pretension, with the desire that students who are beginning a career in research will find inspiration in the path I have taken and appreciate the importance of luck.
Keywords: protein synthesis, cholesterol, bile acid, bile acids, androgen, androgens, vitamin D, lipid, lipids, genetic disease, genetic disorders
A beginning
I was born in Dallas in 1954 to British emigrant parents who after processing through Ellis Island rode a tandem bicycle from New York to Paris, Texas, during the summer of 1952. My father, Gordon A. Russell, and mother, Celia M. Russell (nee Hyde), were born within days of each other in Oxford, England, met at a dance in that city as young adults and, after a traditional courtship and marriage, made the fortunate decision to emigrate to the United States to escape the class repression indigenous to British society at the time and to take advantage of the opportunities available here in the post-war economy. Neither of my parents went to university, but my father did attend a technical school in Oxford at which he learned drafting skills. These skills, together with the publicity they received from their arduous bicycle trip to Texas and the pro-British atmosphere in the States after World War II, led to my father being hired by the Mobil Oil Company, whose research laboratories were then located in Dallas.
I was home-schooled through the age of six by my mother, who taught me to read, write, and calculate. As a lucky consequence of this early preparation, a late September birthdate, and the then rather low standards of the Dallas public schools, I began my formal education in the third grade at the James S. Hogg Elementary School. I had one traumatic experience at that institution that affected my subsequent professional career. All pupils in Mrs. Pace's third-grade class had to deliver an oral book report, and I remember vividly the absolute fear that gripped me as I walked to the front of the classroom and the paralysis that set in once there. I failed the assignment, and after this experience, which occurred over 50 years ago, I have spent an inordinate amount of time organizing and practicing every lecture, seminar, and presentation delivered since!
The Mobil Oil research laboratories at which my father worked moved from downtown Dallas to Duncanville, Texas, in the early 1960s and, with this relocation, the Russell family, which had since grown to include my sister, Anne, and two brothers, Stephen and Greg, purchased a house in southwest Dallas. There, I finished my elementary, junior high, and high school educations, graduating in 1971 at the age of 16 from Justin F. Kimball High School. I was not a diligent student, and despite having some talent in mathematics, I had no special interest in science nor any defined career goals. During this time, my mother obtained an undergraduate degree in language from the University of Texas at Arlington, then largely a commuter school and component of the now 16-institution University of Texas System. Thus, when asked within the first few seconds of my one meeting (exit interview?) with the high school career counselor what I was going to do after graduation, I replied that I too wanted to attend UT Arlington. To which the counselor responded, “OK” and showed me the door. The total elapsed time of this meeting, which was at most 3 minutes, contrasts with the extensive preparation, career counseling, and dozen or more college campus visits that high school graduates undergo today. If my subsequent occupation as a university professor is a measure of the importance of high school career counseling, then one might conclude much of today's extra effort is wasted and that many students would be better off finding their own ways to professional fulfillment.
Undergraduate directions
I applied to and was accepted to UT Arlington, despite having a low score on the standardized entrance examination and at best a mediocre grade point average. My major was biology and my minor was chemistry. Two highlights among the courses I took as a freshman and sophomore were introductory biology and ecology, which were taught by Robert L. Neill, a talented naturalist whose knowledge of birds impressed me. I did not do well in any other courses, including chemistry, in large part due to a lack of effort. Then still a teenager and living at home, I was more interested in working for what at the time seemed like good money (now known as minimum wage) and in ice skating than I was in my future. I would have benefited from two gap years, which, if done in a structured and goal-oriented environment, would have allowed me to grow up prior to entering college. My time at UT Arlington did strengthen my interests in biology, and in 1972, I did manage to meet my future wife while ice skating at the Fair Park Coliseum in Dallas.
Aging is fortunately inevitable, and by 18 I had finally begun to mature and to realize that my future depended on a more serious educational effort. My grades had continuously improved, and my parents were now willing to allow me to move out of the house provided that I remained in college. To these ends, I applied as a transfer student to the University of Texas at Austin, a much larger and more prestigious university that offered many educational opportunities. In retrospect, applying to UT Austin was the first manifestation of ambition on my part and the first of several life-altering decisions I have made. Transferring between institutions within the UT System in the early 1970s required only a C average, and thus my application to do so was accepted. I changed my major to pre-dental with the goal of becoming a small-town dentist in Texas. I took organic chemistry (Chemistry 810) during my first semester in Austin, a course I had failed at UT Arlington and, much to my surprise, got a good grade on the first test. This outcome, in what remains one of the toughest courses on college campuses today, made me realize that I might have a future in science.
Three other courses at UT Austin set my career path. The first was Biochemistry 334, which was taught by G. Barrie Kitto, an accomplished enzymologist with a pronounced New Zealand accent and a knack for making intricate metabolic pathways interesting. The second was Microbiology 366, a difficult course taken by both undergraduate and graduate students and taught by Charles E. Lankford. I developed a strong foundation in and a great appreciation for bacterial genetics from this experience, and these in turn were crucial to my subsequent efforts in molecular cloning. The third and most important course I took was Chemistry 394, an advanced biochemistry course taught by Joanne M. Ravel, which focused on metabolic pathways and mechanisms of protein synthesis, an area in which Joanne carried out NIH-funded research. I found the latter subject fascinating, and based on this interest asked Joanne whether I could do research in her laboratory. Luckily, I had done well on the first two tests in her course and, based on this performance, she offered me a position.
After a month or two of working in Joanne's laboratory, my ambition to become a dentist disappeared and that to become a professor blossomed. Although my “experiments” were limited to a few protein measurements via the Lowry method and tRNA purifications on benzoylated DEAE-cellulose columns, they kindled a passion for research and, of equal importance, introduced me to the career of the academic scientist. Additionally, this undergraduate research experience led to my introduction to Linda L. Spremulli, then a postdoctoral fellow in Joanne's laboratory (Fig. 1), and soon to be an assistant professor in the Department of Chemistry at the University of North Carolina in Chapel Hill. Despite my major in biology, Linda suggested in the summer of 1976 that I apply to graduate school at UNC. I was lucky in that the entering class that year in Chapel Hill was exceptionally small (∼13 students versus the normal 30), and so UNC accepted both my late application and their first biology major into their nationally ranked chemistry graduate program.
Figure 1.

Joanne M. Ravel, second from right, and Linda L. Spremulli, third from right. Ravel was the author's research mentor as an undergraduate student at the University of Texas at Austin. Spremulli, a postdoctoral fellow in the Ravel lab when this photograph was taken in the mid-1970's, was the author's mentor and dissertation advisor when he was a graduate student in the Department of Chemistry at the University of North Carolina. Photograph provided by Margaret Elman.
East to North Carolina
At the age of 21, I packed up a Ford Pinto to drive to North Carolina with $200 in my pocket and a Mobil credit card for backup (which I subsequently found had expired two months before I left and was thus worthless). I camped a night each in Arkansas and Tennessee and on the third day drove into Chapel Hill with a blown radiator that had required stopping every 40 or so miles to allow the engine to cool down and thereafter the addition of water. Upon arrival, I immediately set out to find a roommate (I could not afford to live by myself), locate, rent, and furnish an apartment, register for classes, learn the campus and Chapel Hill, take multiple chemistry prelim exams, and prepare to be a teaching assistant for two freshman chemistry lab courses (25 students each) and a freshman chemistry lecture course (200+ students).
Once classes started, I stayed busy studying, teaching, and grading. The classes I took were all advanced: inorganic, organic, and physical chemistry, math, and physics, none of which had a major in biology prepared me for. Add to that teaching for the first time, and you have an 80–90-hour workweek and little else. Many a time during that first year of graduate school I thought about quitting, returning to Dallas, and getting a job on a loading dock, which would have paid ∼$10/hour (again considered good money in the mid-1970s). When I mentioned this desire to the first-year graduate student advisor, John H. Harrison, a former Marine, professor, and biological chemist in the Department of Chemistry, he “advised” me that I was not going to quit but I was going to work harder. This counsel was dispensed in rather more colorful terms than I indicate here, which today would be considered politically incorrect if not grounds for termination of his tenured position!
Looking back, the trial by fire that chemistry graduate school put me through was transformational. Ambition, self-discipline, and organization got me through it. I thought my ambition to become a professor would ultimately bring respect, job security, and a lifetime in research, all of which were motivational. Self-discipline, which likely came from my mother, allowed me to maintain the above harried schedule in single-minded fashion and to increase my effort in response to Harrison's beratement. Multi-tasking as both student and teacher required considerable organization and the honing of time-management skills. Together, these behaviors were essential to success then and in my subsequent professional years.
Graduate school was not all hardship. A physical chemistry course in quantum mechanics taught by Richard Jarnagin, was arguably the best course I ever took; and one in advanced differential equations taught by Martin Karel in the Department of Mathematics was similarly enlightening. And then there was my dissertation research, which involved a project conceived in Joanne Ravel's laboratory at UT Austin and brought to Chapel Hill. How the ribosome cycle, the process by which free ribosomal subunits are made available for the initiation of protein synthesis, was effected in bacteria was known in some detail through the research of Ravel and others. Two initiation factors, IF-1 and IF-3, bind the small ribosomal subunit and prevent its interaction with the large ribosomal subunit, thus allowing the eventual assembly of an initiation complex composed of the small ribosomal subunit, an mRNA, and an initiator fMet–tRNA. Subsequent release of the initiation factors allowed the large ribosomal subunit to join and translation of the mRNA to commence. In this scenario, IF-1 and IF-3 opposed the ionically favored association of the ribosomal subunits and thus acted as ribosome anti-reassociation factors rather than as ribosome dissociation factors.
The details of this process were less clear in eukaryotes. Others had shown that a large, multi-subunit protein, termed eIF-3, bound to the small ribosomal subunit and prevented association with the large ribosomal subunit. It was not known whether this mechanism involved the protein acting as an anti-reassociation factor or a dissociation factor, or whether other initiation factors were involved in the ribosome cycle. To gain insight into these steps, I developed a rapid assay for ribosome anti-reassociation factors based on an idea that Joanne Ravel and Linda Spremulli had conceived. They reasoned that it should be possible to incubate ribosomes maintained as subunits in a buffer containing a low (1 mm) concentration of magnesium ions with an extract containing potential ribosome anti-reassociation factors, and then, in a second step, raise the concentration of magnesium to a somewhat higher level (3–5 mm) to favor association of free ribosomal subunits and thereafter to measure the amount of free small ribosomal subunit remaining. Free small subunits and hence anti-reassociation activity were quantified by their ability to bind a ternary complex composed of radiolabeled methionine–tRNA, the trinucleotide initiation codon AUG, and another initiation factor termed eIF-2. If the amounts of small and large ribosomal subunits were equal in the initial incubation and if either subunit was bound by an anti-reassociation factor, then some fraction of the small subunits would be available for binding by the ternary complex containing the radiolabeled initiator tRNA. This binding could be detected at the end of the incubation by scintillation counting after passing the mixture through a Millipore filter under conditions that would retain the ribosomal subunits but allow unbound ternary complexes to pass through.
With the filter-binding assay (1), dozens of samples could be assayed for ribosomal anti-reassociation activity in an hour or two, whereas to do the same with the traditional sucrose density gradient assay would have required days (assuming the availability of a dedicated ultracentrifuge, which I did not have access to). I used the assay to identify a ribosome anti-reassociation factor activity from wheat germ that was distinct from the previously identified eIF-3 (2), and then to purify the new activity to near homogeneity, which we named eIF-6 (3). Purification of eIF-6 required eight steps, beginning with ammonium sulfate fractionation of a post-ribosomal supernatant derived from a wheat germ extract followed by chromatography on columns containing anionic, cationic, and gel filtration resins as well as absorption chromatography on hydroxyapatite. With highly purified protein in hand, we showed that eIF-6 bound to the large subunit to effect ribosome anti-reassociation, and not to the small subunit as did eIF-3, and thus that the ribosome cycle in eukaryotic cells was more complex than that in prokaryotic cells (4).
These studies revealed functions for eIF-6 in protein synthesis and the ribosome cycle of plants. Similar roles were subsequently confirmed by others in mammalian cells (5) and, over the ensuing decades, eIF-6 was shown to be required for the assembly of ribosomal subunits in the nucleolus (6) and to associate with the RNA-induced silencing complex (RISC) to regulate the processing of microRNAs (7). The X-ray crystal structure of eIF-6 was determined in 2000 (8), and a structure of eIF-6 bound to the large ribosomal subunit was reported in 2011 (9). Watching the roles of eIF-6 expand since our initial identification of the protein in the late 1970s has been gratifying.
The purification of eIF-6 formed the basis of my thesis dissertation. I also collaborated with fellow graduate students in the lab, including Caroline Breitenberger, Marsha Moore, and Joan Sperrazza, on several related projects studying ribosomes and other factors involved in protein synthesis (10, 11). Looking back, my research in protein synthesis provided an excellent background in assay development, protein purification, and reconstitution of complex biochemical systems. In turn, this bench biochemistry combined with the rigorous coursework of the UNC chemistry graduate program gave me the confidence in my subsequent professional career to pursue a research problem wherever it led.
There were several other aspects of graduate school training that profoundly influenced my future. One was the emphasis that both Joanne Ravel and Linda Spremulli placed on reading the literature. They convened journal clubs every week in which one or two papers were examined in detail down to the concentration of salt used to elute a protein of interest from an ion-exchange column. The publication of other papers in a variety of fields was noted. In Chapel Hill, all graduate students in the Spremulli lab were responsible for reading the Journal of Biological Chemistry, and each was assigned a list of journals for which they were responsible for reporting back to the group any and all papers that might be of relevance to projects in the laboratory. My list included the Proceedings of the National Academy of Sciences, Archives of Biophysics and Biochemistry, and the European Journal of Biochemistry. The newest member of the lab was assigned the onerous task of covering Biochimica et Biophysica Acta, a voluminous journal with many different subjournals, each with a different focus.
As a consequence of these weekly journal clubs, I was aware of the great strides being made in molecular biology in the 1970s. These advances arose from discoveries in bacteriology and made possible the cloning, sequencing, and manipulation of genes. They were dramatically changing biomedical research, and it was clear that the application of molecular biology to biochemistry and other fields would be similarly enlightening. Based on this awareness, in the summer of 1979, I wrote to academic scientists in the U.S., Europe, and the U.K. who were using molecular biology in fields ranging from virology to plant biology to inquire about postdoctoral training opportunities. One such individual was Michael Smith at the University of British Columbia in Vancouver, Canada. Michael and colleagues had published a paper in the Journal of Biological Chemistry in 1978 describing site-directed mutagenesis (12), a technique that allowed the alteration of any nucleotide in a gene and thus the ability to change the amino acid sequence of an encoded protein. The potential of this technology was obvious even to a third-year graduate student in chemistry, and thus I chose to work with Michael.
In response to my letter, Michael had written back to describe four ongoing projects in his laboratory, including further applications of site-directed mutagenesis, and to indicate that he would occupy new laboratory space in the fall in which to accommodate me as a postdoctoral fellow. He also noted that I would need to bring my own funding and included a list of possible sources of postdoctoral fellowships. To this end and being new to the funding game, I submitted seven different applications to as many agencies over the next several months and ultimately received six fellowship offers. In retrospect, submitting seven applications was excessive given the obvious power of site-directed mutagenesis, but I was naïve and as giddy with the prospects of the technology as the fellowship review boards must have been.
Diagonally to British Columbia
During my first year of graduate school, I had proposed to Karen Baty, the woman I met as a college sophomore while ice skating in Dallas. Luckily for me, Karen accepted my over-the-telephone proposal from Chapel Hill, and we were married in May of 1977. Karen finished her undergraduate degree at UNC in May of 1980 at the same time that I finished my Ph.D. Shortly thereafter, we packed up the Ford Pinto for the diagonal cross-country drive from Chapel Hill to Vancouver. Karen had been accepted to the University of British Columbia's MBA program and was to begin classes in the fall, and I was to begin postdoctoral studies in July with Michael Smith supported by a fellowship from the Damon Runyon Cancer Fund. Upon arrival, we rented a basement apartment and set about our new life in Canada.
Bacteriophage M13 had just been developed by Joachim Messing as a useful DNA sequencing and cloning system, and thus Michael suggested as a first project that I establish M13 in the laboratory. The facts that the M13 genome is packaged as ssDNA and that unlike other bacteriophages there is no strict limit on the size of the DNA that can be packaged also suggested that M13 would be a useful vector for site-directed mutagenesis. We received M13 protocols and reagents from the Messing laboratory and, leaning heavily on my undergraduate bacteriology courses and advice from Shirley Gillam, a research associate in Michael's laboratory, I began learning the M13 system. The M13mp7 vector then in use relied on α-complementation of β-gal to identify recombinant viruses containing cloned DNA. Although I experienced many failed attempts at cloning into M13, I distinctly remember taking the Petri dishes out of the incubator one morning and seeing a mixture of white (recombinant) and blue (nonrecombinant) bacteriophages on the plate. The successful experiment utilized yeast DNA cleaved with the Sau3AI restriction enzyme and ligated into the BamHI site of the polylinker in the M13mp7 vector. To this day, I still remember this experiment and the DNA sequences recognized and cleaved by these two enzymes!
Valerie Williamson, a postdoctoral fellow working with Elton (Ted) Young in the Department of Genetics at the University of Washington in Seattle, Washington, provided the cloned yeast DNA for these experiments. Prior to my joining the laboratory, Michael Smith had entered into a collaboration with Ted to determine the sequence of several mutant alleles of the alcohol dehydrogenase 2 gene (ADR2), which had been identified by Michael Ciriacy in Germany. These mutations, which rendered expression of the normally tightly regulated ADR2 gene constitutive, were of interest for their potential to reveal mechanisms of eukaryotic gene regulation, about which little was known at the time. Valerie cloned several different constitutive ADR2 alleles and then sent plasmids containing these DNAs and the WT ADR2 gene to Vancouver for sequencing. I then used a combination of bacteriophage M13 sequencing, which utilized the enzymatic dideoxy terminator (Sanger) method and a chemical sequencing (Maxam–Gilbert) method to sequence the ADR2 DNAs. Shirley Gillam taught me enzymatic sequencing, while Caroline Astell, who was then also a research associate in Michael's laboratory, taught me chemical sequencing.
We learned that constitutive expression of ADR2 arose from two classes of mutations. In the first class, a transposable element referred to as Ty1 was inserted in the immediate 5′-flanking region of the gene (13) and, in the second class, a tract of 20 adenine residues located ∼200 nucleotides upstream of the coding region of the normal gene (14) was expanded to 54 or 55 residues (15). We speculated that insertion of the roughly 5-kb Ty1 transposable element into the 5′-flanking region of the gene caused constitutive expression by displacing an essential regulatory element, a prediction that was later confirmed by David Beier in Ted's laboratory (16). For the poly(dA) tract mutations, we doubted that the ∼30-bp displacement caused by the expansion was large enough to disrupt the postulated regulatory sequence and instead guessed that the long tract of adenine residues disrupted the chromatin structure of the 5′-flanking region. Evidence to support this idea was published a decade later by others (17).
The collaboration between Michael's lab and Ted's lab on the ADR2 gene was characteristic of many that Michael participated in during my time in the laboratory. In most of these collaborations, trainees in Michael's lab provided a technology such as DNA sequencing or site-directed mutagenesis that was not widely available at the time, and the other laboratory provided the biology or genes. Although this situation was not ideal if one wanted to learn biology, it was at least conducive to reading about different areas of biology and to collaborating with other laboratories. To these ends, my collaboration with James Haber and colleagues was informative regarding yeast mating type mutations (18), as was a project with Ira Herskowitz and colleagues characterizing the homothallism (HO) locus of yeast (19). Interactions with others in Michael's lab were also enlightening, including those with postdocs Mark Zoller (a fellow American who perfected the use of site-directed mutagenesis using bacteriophage M13 vectors) and Tom Atkinson (a nucleotide chemist). During this time, I worked with several graduate students, including Susan Porter, Andrew Spence (whose collaboration with Steve McKnight at the Fred Hutchinson Institute in Seattle led to my life-long friendship with Steve), and Johnny Ngsee; as well as with technicians Patricia Jahnke, Janice Long, and Lena Ahlstrom.
My research in Michael's laboratory went sufficiently well that after a year or so, I began scanning journal want-ads advertising faculty positions. This behavior, which both Mark Zoller and I manifested with some regularity, used to rankle Michael, who would huff that we had only just arrived in his laboratory. Nevertheless, he began notifying us of unadvertised job opportunities at various universities and in the then-burgeoning biotechnology industry. At about this time, Michael, together with Earl Davie and Benjamin Hall at the University of Washington, formed a company in Seattle named Zymos. The Zymos business plan proposed to use yeast to express recombinant clotting factors, which were just being cloned in Earl's laboratory. The company was in need of a molecular biologist, and thus after an interview with Ben Hall during one of his visits to Vancouver, I traveled to Seattle to meet several other postdocs, including Glenn Kawasaki, who also were being courted by Zymos. I was subsequently offered a position at the company with a starting salary of $40,000 (huge!), plus options that amounted to 1% of the company's stock.
Coincident with this nascent job hunting effort, Michael's status as a scientist was increasing exponentially due to the widespread application of site-directed mutagenesis in both biology and industry. He was in demand as a seminar speaker and as a member of prize juries, one of which judged the Gairdner Foundation Award competition in Toronto. In this capacity, he met Mike Brown and Joe Goldstein, who received the award in the fall of 1981 for their discovery of the low density lipoprotein (LDL)2 receptor pathway that mammalian cells use to meet their cholesterol requirements. At the award ceremony held on a Saturday night, Joe asked Michael Smith whether he had any molecular biologists in his laboratory who were looking for a job, to which Michael is said to have replied “Why yes I do, and in fact, he's from Texas.” At 8 a.m. the following Monday, I received a telephone call from Joe inviting me to give a seminar in the Department of Molecular Genetics at what was then called the University of Texas Health Science Center at Dallas. As the proposed dates of the visit coincided with an already planned trip by Karen and me to visit our relatives in Dallas over the Christmas holidays, I accepted Joe's offer. My ready acceptance was hastened by Joe's promise to pay half of my air fare, which, being naïve to the world of recruiting, I thought was remarkably generous!
I had little or no knowledge of the LDL receptor, cholesterol, or lipid metabolism in general, but reading several reviews that Mike and Joe had written was enough to convince me that there were many interesting questions in these areas that could be answered with molecular biology. Dennis Vance, who at the time was the resident lipid expert in the Department of Biochemistry at the University of British Columbia (see Dennis' Journal of Biological Chemistry Reflections article (20)), confirmed this thinking and further indicated that Mike and Joe were outstanding scientists. Dennis also noted that one of his former postdoctoral fellows, Wolfgang Schneider, had accepted a position in Dallas with Mike and Joe and was purifying the LDL receptor protein.
While waiting outside Joe's office on the morning of my visit, I realized that I knew his research prior to the LDL receptor. When he was a postdoctoral fellow in Marshall Nirenberg's laboratory, Joe, along with Tom Caskey, had published papers on a factor they termed “protein S” that facilitated the binding of release factors to the prokaryotic ribosome. I had read these papers as an undergraduate student and, as a class assignment for Chemistry 394, had written a paper that summarized the work. I mentioned this coincidence to Joe in our initial conversation, and he seemed pleasantly surprised that I knew the work. The rest of my visit to Dallas went equally well and, at dinner that evening, Mike and Joe offered me a position as an assistant professor in the Department of Molecular Genetics to begin on July 1, 1982, with the goal of collaborating with them to clone the LDL receptor cDNA and gene.
Thus, in the course of two years of postdoctoral training with Michael Smith, I had learned how to sequence, mutagenize, and otherwise manipulate DNA. This molecular biology training, together with that in biochemistry and protein synthesis as a graduate student, gave me the background necessary to tackle the difficult problem of cloning the LDL receptor gene. Knowledge in these diverse disciplines was instilled in me by a series of talented mentors, four of whom, Joanne Ravel, Linda Spremulli, Shirley Gillam, and Caroline Astell, were women, and the fifth, Michael Smith, would go on to win the 1993 Nobel Prize in Chemistry for his discovery of site-directed mutagenesis. I had received two job offers, one at a biotechnology company (Zymos) in Seattle and the other an academic position at the University of Texas Health Science Center at Dallas (now named the University of Texas Southwestern Medical Center). Of these, I chose the academic position because I thought it offered the best opportunities for discovery and biology. In retrospect, it would have been more financially rewarding to have accepted the Zymos offer as the company was subsequently bought by Novo Nordisk, making its founders and initial hires instant millionaires!
Return to Texas
By June of 1982, Karen had finished her MBA degree, and so we packed up the Ford Pinto again for the drive back to Dallas. We took a circuitous route, camping in British Columbia and visiting friends in Boise, Idaho, and Berkeley, California, prior to our arrival in Texas. We purchased and began renovating a house, and Karen accepted a position in banking. In the months preceding my start at UT Southwestern, I had sent dozens of purchase orders to Joe's secretary (Dorothy Lund) for laboratory equipment and supplies, and these had been received and unpacked, which in turn allowed me to begin doing experiments on almost my first day of employment. The total cost of these purchases, which amounted to my start-up package, was approximately $180,000, a sum that pales compared with the multi-million dollar packages we routinely offer assistant professors today.
Cholesterol supply
By the time I joined the faculty in Dallas, Wolfgang Schneider had succeeded in purifying the LDL receptor protein from bovine adrenal glands and, together with Clive Slaughter, in obtaining the sequence of several peptides from the purified protein. In addition, a former postdoctoral fellow with Mike and Joe, Ulrike Beisiegel, had raised polyclonal and monoclonal antibodies against the receptor. Thus, with these tools in hand, I began the cloning project, which we predicted would be difficult given the rather primitive cloning tools of the day and the rarity of the LDL receptor, which was estimated to represent less than 0.01% of total membrane protein in an expressing cell.
As a first step, we modified a magnesium precipitation protocol, originally developed by Richard Palmiter (21), to prepare large quantities of polyribosomes from hundreds of bovine adrenal glands that Richard Gibson, a technician in the department, and I obtained from a local slaughterhouse. Adrenals were used because they were known to contain relatively large amounts of the LDL receptor, which was required to supply cholesterol for steroid hormone synthesis by the gland. Gloria Brunschede, one of Mike and Joe's first technicians, assisted in stockpiling dozens of polyribosome preparations, and I began using the polyclonal antibody that recognized the LDL receptor protein in attempts to immunoprecipitate the rare polyribosomes that were translating the receptor mRNA. In preparing the polyribosomes, we included the antibiotic trichodermin, which preferentially blocks the termination step of protein synthesis. This was done to trap the longest possible nascent LDL receptor peptides on the polyribosomes, which would presumably increase the chance that epitope(s) recognized by the precipitating antibody would be present on the translating polyribosome. We isolated RNA from the precipitated polyribosomes by phenol–chloroform extraction and ethanol precipitation and then translated it using in vitro protein synthesis extracts prepared from rabbit reticulocytes. After multiple attempts, I succeeded in purifying the LDL receptor mRNA to the point where a large protein of the estimated mass of the intact receptor (∼120,000 Da) could be detected by SDS-PAGE after translation.
The next step was to construct a cDNA library from the partially purified receptor mRNA that could be screened with oligonucleotide probes made from the peptide sequences generated by Wolfgang and Clive. I had no experience in making cDNA libraries, and of course there were no kits available to do so in 1982. Luckily, I was joined in the cloning effort at this point by Tokuo Yamamoto, a new postdoctoral fellow in Mike and Joe's laboratory. Tokuo had obtained his Ph.D. with Shosaku Numa at Kyoto University in Japan and was one of the few people in the world who was skilled in the art of constructing cDNA libraries by the Okayama–Berg method (22). This method, which used an oligo(dT)-tailed plasmid vector to prime cDNA synthesis, was designed to allow the cloning of long cDNAs like that encoding the LDL receptor, which we estimated would be ∼3.0 kb in length given the size of the protein after SDS-PAGE. Tokuo used the RNA extracted from the immunoprecipitated polyribosomes to construct a cDNA library, which after transformation into Escherichia coli was estimated to contain over 500,000 independent cDNAs.
I next asked Michael Smith if he would synthesize oligonucleotide probes based on the sequence of a single polypeptide from the purified LDL receptor protein. He agreed to do so, and several weeks later, we received from Mark Zoller and Tom Atkinson several families of oligonucleotides that were 14 nucleotides in length and represented all possible coding sequences of two adjacent five-amino acid sequences. We first screened the cDNA library made from the enriched polyribosomes with oligonucleotides representing one five-amino acid sequence to identify ∼30 putative LDL receptor cDNAs, and then we rescreened these 30 clones with oligonucleotide probes representing the second five-amino acid sequence. Two cDNAs hybridized with all oligonucleotide probes, and the ∼2.7-kb inserts in these plasmids, were dutifully mapped using a dozen or so restriction endonucleases, including PvuII.
I subcloned a 432-bp PstI fragment from the cDNA insert that hybridized with the oligonucleotide probes into bacteriophage M13, and I then determined the sequence of this insert using the enzymatic dideoxy terminator method. When I developed the autoradiographic film from the sequencing experiment the next day and then held it up to the dim red light in the darkroom, I knew instantaneously that we had isolated the LDL receptor cDNA based on the presence of sequences specifying a PvuII site that were outside those used to construct the oligonucleotide primers. This news quickly made its way by excited word of mouth to Mike Brown, who, after reading the sequence and comparing it with that of the polypeptide, agreed that we had in fact isolated a cDNA for the bovine LDL receptor. Joe was then at a Gordon Conference in New Hampshire, which at the time had a no-telephone policy for attendees (this was long before cell phones), and thus we were at a loss as to how to transmit the good news to him. Later that afternoon, Mike came up with the idea of asking the conference organizers to inform Joe that his wife had had a baby and that he should call home as quickly as possible. Joe is a lifelong bachelor and thus I would have liked to have seen the look on his face when he received this message!
It took Tokuo and me one year to isolate the two bovine LDL receptor cDNA clones. We spent another several months characterizing and sequencing their cDNA inserts and using these in Southern and RNA blotting experiments to gain preliminary insight into the cow LDL receptor gene and its expression, and then published these findings in the fall of 1983 (23) and summer of 1984 (24). By November of 1984, a full-length (5.3-kb) cDNA for the human LDL receptor had been isolated, sequenced, and expressed in transfected mammalian cells (25).
We worked at a feverish pace during this time because other laboratories with more skill and bigger reputations were also trying to isolate LDL receptor cDNAs and genes. These included the lab of Barbara Wold at the California Institute of Technology, who had trained with Richard Axel at Columbia and who was attempting to isolate the LDL receptor gene by transfection of genomic DNA, selection, and use of a red blood cell rosetting assay. Additionally, a postdoctoral fellow in Paul Berg's laboratory at Stanford, Michael McPhaul, a former summer student of Mike and Joe's, was trying to isolate LDL receptor cDNAs using transfection and cell sorting with the mAb raised in Dallas by Ulrike Beisiegel against the receptor protein. Knowing that disciples of Richard Axel and Paul Berg were the competition was highly motivating!
While Tokuo and I were cloning the LDL receptor cDNA, I began to work with other postdoctoral fellows in Mike and Joe's laboratory who were using molecular biology to study other genes. Dan Chin, Ken Luskey, and Gregorio Gil were collaborating with Hiroto Okayama and Paul Berg at Stanford in the isolation and characterization of cDNAs encoding 3-hydroxy-3-methyl-glutaryl CoA reductase (HMG CoA-reductase), the rate-limiting enzyme in cholesterol biosynthesis and the target of statin drugs, which were under development by Merck and Co. Working in collaboration with Ray MacDonald (Department of Biochemistry, UT Southwestern), Dan isolated a partial cDNA for the hamster HMG CoA-reductase using a clever differential hybridization method, which was used to screen a large library made by Hiroto. The four of us in Dallas (Gregorio, Ken, Dan, and I) then spent a year sequencing the full-length cDNA using a combination of enzymatic and chemical methods to reveal the 887-amino acid sequence of the hamster HMG CoA-reductase. Analysis of the predicted sequence on a desktop computer, which had a then-whopping 128 KB of data storage (KB, not today's multi GB or even PB), indicated that the protein contained multiple transmembrane domains linked to a more hydrophilic region containing the active site of the enzyme. These findings were published in April of 1984 (26).
Thus, by 1984, two years after I had joined the faculty at UT Southwestern, we had isolated and characterized cDNA clones encoding the LDL receptor and HMG CoA-reductase. Earlier work by Mike and Joe had revealed the central role of the LDL receptor in mammalian cholesterol supply and the pathological consequences of inherited deficiencies in the encoding gene (27). They had shown that cholesterol synthesis was regulated by a feedback mechanism in which excess cholesterol suppressed expression of HMG CoA-reductase as well as other enzymes in the biosynthetic pathway, and that the LDL receptor pathway was also regulated in this manner (28). Their work with Richard Anderson had defined the cell biology of the LDL receptor pathway, including the intracellular itinerary taken by the nascent LDL receptor from its site of synthesis in the endoplasmic reticulum through the Golgi apparatus and to the coated pits of the plasma membrane from which LDL particles were delivered to the interior of the cell by receptor-mediated endocytosis (29). With the cloned receptor and reductase cDNAs in hand, it was possible to begin defining the molecular details of the LDL receptor and cholesterol biosynthetic pathways.
Over the ensuing several years, an exceptionally talented group of postdoctoral fellows, students, and staff began doing exactly this (Fig. 2). Those with whom I worked and their projects included Geoff Davis, who expressed the LDL receptor in cultured cells and defined many functional domains in the protein (30–33); Mark Lehrman, who worked out the molecular bases of numerous LDL receptor gene mutations in subjects with familial hypercholesterolemia (34–39); Thomas Südhof, who cloned and elucidated the exon–intron structure of the LDL receptor gene and found evidence that the gene arose through exon shuffling (41) and also identified cholesterol-regulated DNA sequences in the promoter of the gene (42); Helen Hobbs, who showed how repetitive sequences (Alu elements) within the LDL receptor gene were a source of both inter-individual variation and mutation in the gene (43–45); Richard Bishop, who together with Tokuo Yamamoto elucidated the molecular defect in the Watanabe heritable hyperlipidemic rabbit, an animal model of familial hypercholesterolemia (46); Sandra Hofmann, who used in situ mRNA hybridization to reveal cell type–selective expression of the LDL receptor in the brain (47) and, together with Robert Hammer, constructed a line of transgenic mice that overexpressed the LDL receptor (48); Susan Peacock, who by expressing the human LDL receptor in Xenopus laevis oocytes showed that signals for O-linked glycosylation and receptor-mediated endocytosis were conserved between species (49); Victoria Esser, who characterized mutations leading to defective intracellular trafficking of the LDL receptor (50), and together with Lee Limbird, who worked with us while on sabbatical leave from Vanderbilt University, mapped the domains of the receptor to which LDL and very-low-density lipoprotein (VLDL) ligands bind (51); and Jennifer Cuthbert, who characterized expression of the LDL receptor in circulating human monocytes while on sabbatical leave from the Department of Internal Medicine at UT Southwestern (52).
Figure 2.

Faculty, fellows, and staff of the Department of Molecular Genetics, December 1983. Mentioned in the text are: 1. Daphne Davis; 2. Gloria Brunschede; 3. the author; 4. Mark Lehrman; 5. Ken Luskey; 6. Gregorio Gil; 7. Helen Hobbs; 8. Geoff Davis; 9. Wolfgang Schneider; 10. Tokuo Yamamoto; 11. Thomas Südhof; and 12. Dan Chin.
In many of these projects, Daphne Davis (now Rye) provided invaluable technical assistance. Daphne was the first person I ever hired, and we ended up working together for over 30 years. She continues to work with my colleague Arun Radhakrishnan in the Department of Molecular Genetics at UT Southwestern.
Looking back, the insight this team generated over a short period of time into cell biology, human genetics, evolution, gene regulation, lipid metabolism, and biochemistry is remarkable. It is perhaps not surprising that they went on as individuals to win the Nobel Prize (Thomas Südhof, in 2013), win the Breakthrough Prize (Helen Hobbs, in 2016), become full professors (Mark, Thomas, Tokuo, Helen, Sandra, Robert, Lee, and Jennifer), start successful biotechnology companies (Geoff, Richard), and succeed in law (Susan). Also, three of us (Thomas, Helen, and I) were elected to the U.S. National Academy of Sciences. The camaraderie of the group was high and discord was low for many reasons. One, there was so much to do and so many interesting projects that personal jealousies were nonexistent; two, this was a uniquely talented, secure, and mature group of individuals; three, we had access to outstanding resources and funding; and four, and most importantly, Mike and Joe mentored each of us with an exceptionally equitable hand, and they set a golden standard of inquiry, experimentation, and presentation that were ours to emulate. Another important factor contributing to the harmony and success of the group was the recognition that poured in at the same exponential rate that findings were being made and that culminated in Mike and Joe winning the Nobel Prize in Physiology or Medicine in 1985.
The downside of the rapid progress made during the period between 1982 and 1987 was that the body of work I thought would occupy my entire professional career was more or less completed. I had accepted the job in Dallas with the belief that a lifetime would be spent cloning the LDL receptor gene, understanding its function, and working out the molecular genetics of familial hypercholesterolemia. Such was not to be the case, and thus around 1987 I began thinking of other research projects in which molecular biology might be impactful.
Cholesterol breakdown: Bile acids
A lucky hallway conversation with Mike Brown around this time introduced me to a field that would be the center point of my research for the next 25 years. He knew that I was trying to come up with new cloning projects and suggested that I read up on cholesterol 7α-hydroxylase, which was thought to be the rate-limiting enzyme in bile acid synthesis, the major pathway by which cholesterol is metabolized and thereafter secreted from the body. A trip to the library revealed that bile acids play many important roles in lipid metabolism and physiology and that the pathway by which they were made had not yet been fully elucidated—thank you, Mike!
Two talented postdoctoral fellows joined my laboratory and agreed to work on new projects involving bile acid metabolism. Stefan Andersson arrived from Uppsala University in Sweden after graduate training with Kjell Wikvall and Henry Danielsson, who had both made major contributions to bile acid research. Stefan wanted to learn molecular biology and brought with him the N-terminal sequence of an enzyme in the rabbit bile acid synthesis pathway, sterol 27-hydroxylase, which he and Kjell had purified. Coincidentally, Hans Jörnvall had determined this sequence, and I was familiar with his work on horse alcohol dehydrogenase from my postdoctoral studies on yeast alcohol dehydrogenases. The second postdoc to join the effort was Diane Jelinek, who had done her graduate training in immunology with Peter Lipsky at UT Southwestern, and who also wanted to learn molecular biology. Diane, a fearless scientist, agreed to take on the difficult project of purifying cholesterol 7α-hydroxylase with the goal of obtaining peptide sequences that could be used to clone the encoding cDNA.
By this time, the synthesis of oligonucleotide probes had been automated, and we had purchased a desktop instrument for this purpose. Stefan used the sequence of sterol 27-hydroxylase to design and synthesize a family of oligonucleotides that were 35 bases in length and contained inosine at wobble positions to reduce the number of oligonucleotides required to represent all possible codons of the protein sequence. As this sequence was derived from the N terminus of the protein, which was estimated to contain about 500 amino acids, he knew that the cDNA library to be screened would have to be enriched for longer cDNAs. Luckily, Tokuo Yamamoto had made such a library from rabbit liver using the Okayama–Berg cloning method for his studies on the Watanabe heritable hyperlipidemic rabbit mentioned above (46). Stefan screened this library, isolated and sequenced full-length cDNAs encoding sterol 27-hydroxylase, and expressed the enzyme via transfection in cultured COS-M6 cells (53).
These experiments revealed several interesting features regarding the enzyme that also provided new insight into bile acid biosynthesis. First, the enzyme was a mitochondrial member of the cytochrome P450 gene family. Second, in addition to hydroxylating cholesterol and bile acid intermediates at carbon 27 of the sterol side chain, the expressed enzyme appeared to catalyze the further oxidation of this hydroxyl group to an aldehyde and then to a carboxylic acid. Earlier work had suggested that the latter oxidation steps were catalyzed by two separate enzymes in the bile acid synthesis pathway, an alcohol dehydrogenase and an aldehyde dehydrogenase, but our results suggested that one enzyme, sterol 27-hydroxylase, catalyzed all three reactions. We could not rule out the possibility that the sequential oxidation activity was a consequence of transfection and overexpression of the enzyme in cultured cells, but subsequent and careful kinetic studies with purified recombinant enzyme by Irina Pikuleva and colleagues confirmed that these activities were intrinsic to sterol 27-hydroxylase (54).
The paper reporting the cDNA cloning of sterol 27-hydroxylase (54) is highly cited. One might think that this citation was because the study reported an early example of the cDNA cloning of a bile acid biosynthetic enzyme, but in reality the paper is recognized because we had constructed a new plasmid vector named pCMV4 to express sterol 27-hydroxylase in transfected cells. Up until this time, most mammalian expression vectors used a late-region promoter from the simian virus 40 (SV40) to drive expression of a cloned cDNA. In contrast, pCMV4 utilized the immediate early gene 4 promoter of the human cytomegalovirus together with a translational enhancer derived from the alfalfa mosaic virus to drive expression; this combination was stronger and showed none of the cell-specific tropism of the SV40 promoter plasmids. The pCMV4 vector was one of several plasmids (pCMV1–pCMV8) that we assembled and sent to hundreds of laboratories around the world, which in turn repeatedly referenced Stefan's paper as a source of this versatile vector series.
The rabbit sterol 27-hydroxylase cDNA was used by a graduate student in the lab, James Cali, to isolate the human cDNA by cross-hybridization (55), which he subsequently used to elucidate the molecular basis of a human autosomal-recessive genetic disease named cerebrotendinous xanthomatosis (CTX), (56). CTX is a progressive neuropathy marked by reduced bile acid synthesis and the gradual accumulation of cholesterol and cholestanol in the central nervous system; this accumulation in turn disrupts the myelin sheaths surrounding neuronal axons, leading to a loss of mobility and eventually death (57). At the time the human sterol 27-hydroxylase cDNA was isolated, it was unclear whether the disease arose from loss of sterol 27-hydroxylase activity or loss of sterol 24-hydroxylase activity, both of which are required for the oxidation of the sterol side chain in bile acid biosynthesis. With the cloned cDNA in hand, and the use of the then newly developed PCR method, James was quickly able to show that in fact loss-of-function mutations in the sterol 27-hydroxylase gene caused CTX. Since then, many different mutations in the gene (symbol CYP27A1) have been identified in CTX subjects. This work was particularly gratifying because the development of neurological symptoms in affected individuals can be prevented by oral bile acids if the disease is diagnosed early in its course.
At the same time Stefan and James were working on sterol 27-hydroxylase, Diane Jelinek was spending hours in the cold room purifying the rat cholesterol 7α-hydroxylase enzyme (protein symbol CYP7A1). Kjell Wikvall and Stefan had reported a partial purification of the enzyme, and Diane began her studies by scaling up their method to obtain more starting material. She placed 1,200 adult male rats on a diet containing cholestyramine, which increases CYP7A1 activity in the liver, for 7 days. She then harvested the livers at the mid-point of the dark circadian cycle (the peak of CYP7A1 expression) and prepared twice-washed microsomal membrane pellets from the ∼1,200 organs. She commandeered every centrifuge on the building floor for weeks on end to prepare what amounted to buckets of starting microsomes. With this material, she developed a six-step purification protocol that enriched CYP7A1 activity over 600-fold. SDS-PAGE analysis of the final fraction revealed four proteins. Further purification proved futile so, to determine which of the four proteins was CYP7A1, Diane prepared hepatic microsomes from 200 rats fed a diet containing a bile acid, which suppresses enzyme activity in the liver. She then subjected these microsomes to her purification protocol. She found that the protein with the largest molecular weight was decreased in an amount relative to that in preparations from the cholestyramine-fed rat livers and thus was likely CYP7A1. Clive Slaughter, who together with Wolfgang Schneider had sequenced the purified LDL receptor protein, next obtained sequences of seven peptides from the putative CYP7A1 protein, which were used by Diane to synthesize oligonucleotide probes and clone the encoding cDNA. She went on to express the cDNA in transfected COS-M6 cells and to show that regulation of enzyme activity by dietary cholestyramine and bile acid occurred at the transcriptional level (58).
At this point, which was in 1991, our initial forays into bile acids had produced cDNAs and genes (59, 60) encoding two enzymes in the bile acid biosynthetic pathway, sterol 27-hydroxylase and cholesterol 7α-hydroxylase, and had elucidated the molecular basis of cerebrotendinous xanthomatosis, an inherited deficiency in bile acid synthesis. By the mid-1990s, Robert Hammer and Joachim Herz had established the technology to produce knockout mice on campus. Margrit Schwarz joined my lab as a postdoctoral fellow in 1994 after graduate training with Wilhelm Stoffel in Cologne, Germany, and together with Shun Ishibashi, a postdoc with Mike and Joe who was working with Joachim Herz, began a project to construct and characterize mice deficient in cholesterol 7α-hydroxylase (mouse gene symbol Cyp7a1). Margrit's and Shun's analysis of the deficient animals confirmed the requirement for cholesterol 7α-hydroxylase in bile acid synthesis and many of the important physiological functions of bile acids including their role as detergents in the solubilization of vitamins A, E, D, and K from the diet (61).
One experiment documenting the need for bile acids to solubilize dietary fat-soluble vitamins was particularly memorable. Cyp7a1−/− animals appeared to move around less in their cages, and to quantify this loss of mobility, Margrit held the feet of WT mice to a blue ink pad and then placed them in an enclosure for 1 min that had a sheet of Whatman 3M filter paper on the floor. The movement of the animals during this time was marked by blue footprints, which were located along the walls and across the middle of the enclosure. She held the feet of Cyp7a1−/− mice to a red ink pad and repeated the experiment on the same pad of filter paper. In contrast to the WT mice, the red footprints of the Cyp7a1−/− animals were located exclusively along the wall that the mice had been placed next to at the start of the experiment. The resulting red–blue pattern of footprints was striking (Fig. 3, reproduced from Ref. 61), but we had no idea how to explain the obvious difference in behaviors until the data were shown to Helen Hobbs, who within seconds of seeing the pattern said, “They're blind!” She further explained that there was a long hallway in her house linking the master bedroom and kitchen and that she would often get up in the middle of the night for a drink of water. Not wanting to wake her family by turning on the lights, and thus being effectively blind, she would navigate the hallway by running her hand along the wall. This movement based on tactile cues exactly mirrored that of the Cyp7a1−/− mice as revealed by their red footprints along the wall of the enclosure, thus her diagnosis of blindness. We subsequently tested this hypothesis by feeding the knockout mice supraphysiological levels of vitamin A, which restored their vision and movement to normal. This anecdote involving insights on the part of Helen is characteristic of many I have experienced over the years with her as well as numerous other UT Southwestern colleagues.
Figure 3.
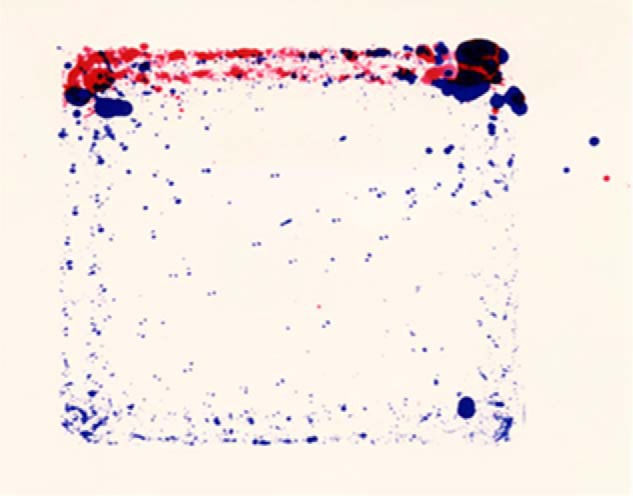
Paw prints of normal and cholesterol 7α-hydroxylase–deficient (Cyp7a1−/−) mice. The paws of normal mice were dipped in blue ink and those of Cyp7a1−/− mice were dipped in red ink. The animals were allowed to run around on a sheet of Whatman 3M paper, generating the composite shown. Reproduced with permission from Ref. 61. This research was originally published in the Journal of Biological Chemistry. S. Ishibashi, M. Schwarz, P. K. Frykman, J. Herz, and D. W. Russell. Disruption of cholesterol 7α-hydroxylase gene in mice. I. Postnatal lethality reversed by bile acid and vitamin supplementation. J. Biol. Chem.1996; 271:18017–18023. © the American Society for Biochemistry and Molecular Biology.
A second puzzling observation that Margrit made concerning the Cyp7a1−/− mice related to the time course of their death. In the absence of dietary supplementation with bile acids and vitamins, 90% of the knockout mice died before the age of 21 days; however, the 10% that made it to day 21 thereafter experienced a normal lifespan (61). Analysis of the surviving mice showed that they synthesized bile acids by an alternate pathway that did not involve cholesterol 7α-hydroxylase but rather an enzyme termed oxysterol 7α-hydroxylase, the substrates of which were the so-called side-chain oxysterols, 25-hydroxycholesterol and 27-hydroxycholesterol (62). The paper describing these findings was published in 1996 and was the first of many studies in which we collaborated with Ken Setchell at the University of Cincinnati Children's Hospital. Ken developed numerous analytical methods to quantify bile acids and sterol intermediates in the bile acid biosynthetic pathway and has used these methods to identify a majority of currently known inherited defects in the pathway. Of equal importance for the direction of my research, the study provided unequivocal genetic evidence for an alternate pathway of bile acid biosynthesis that at least in mice could compensate for the classic pathway.
We spent the next twenty-plus years annotating many of the 16 enzymes and 17 reactions they catalyze in what are now referred to as the classic and alternate pathways of bile acid synthesis (Fig. 4). As the figure illustrates, cholesterol is the starting substrate for both pathways, and the difference between the two is whether synthesis is initiated by hydroxylation of the ring structure of the sterol by cholesterol 7α-hydroxylase (the classic pathway) or by hydroxylation of the side chain of the sterol by one of either cholesterol 24-hydroxylase, cholesterol 25-hydroxylase, or sterol 27-hydroxylase (the alternate pathway). Thereafter, the sterol products of these initiating enzymes are acted upon by 11–13 different enzymes to produce the end products of the pathway, which are conjugated bile acids. The linear pathways depicted in Fig. 4 are an explanatory convenience only, as with the possible exception of the requirement for 7α-hydroxylated substrates by the enzyme 3β hydroxysteroid-Δ5-C27-steroid oxidoreductase (HSD3B7), there is little evidence in vitro or in vivo to suggest an exact order of the many steps in these pathways.
Figure 4.
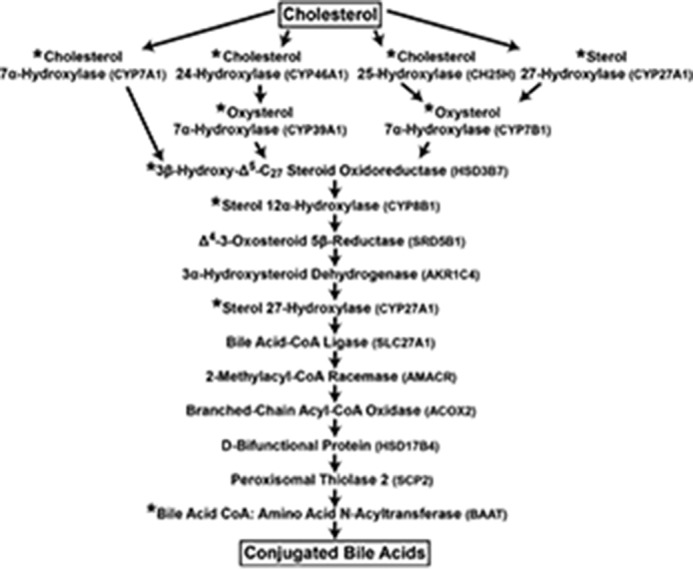
The enzymes of bile acid synthesis. The biosynthetic pathway from cholesterol to conjugated bile acids is shown. Individual enzymes are noted together with their symbols. Those marked with an asterisk were studied in the author's laboratory. The pathway is an explanatory convenience; with few exceptions, the order of reactions catalyzed and the flow of metabolites are not known.
Much of this research was carried out in the 1990s, and during this decade, improvements in in vitro RNA synthesis, mammalian cell expression vectors, cDNA cloning, and bacterial transformation allowed us to develop efficient expression cloning strategies utilizing cultured cells and Xenopus oocytes to isolate cDNAs specifying numerous enzymes in the pathways shown in Fig. 4 as well those in steroid hormone metabolism (see below). In a typical project, a library containing hundreds of thousands of independent cDNAs of average length >1.0 kb was constructed and transformed into E. coli, and then plasmid pools containing 500–10,000 cDNAs were prepared. These pools were either used to produce synthetic mRNA for subsequent injection into Xenopus oocytes or transfected directly into cultured cells. Thereafter, a radioactive sterol or steroid substrate was added to the medium, and after an incubation period, lipids were extracted from the cells and medium and separated by TLC. Once a pool expressing the desired enzyme activity was identified, it was progressively subdivided and assayed to identify the cDNA encoding the enzyme.
Over the years, trainees in the laboratory used these and other cloning methods to isolate cDNAs specifying multiple enzymes in the two pathways shown in Fig. 4. For example, Erik Lund, a talented postdoctoral fellow who obtained his graduate degree with Ingemar Björkhem at the Karolinska Institute, isolated cDNAs encoding the cholesterol 24-hydroxylase (63) and the cholesterol 25-hydroxylase (64). Margrit Schwarz, in a collaboration with Richard Lathe in Edinburgh, Scotland, identified cDNAs and genes encoding the CYP7B1 oxysterol 7α-hydroxylase (65), and Julie Li-Hawkins, a postdoctoral fellow who joined us after graduate training with Savio Woo at the Baylor College of Medicine, identified the CYP39A1 oxysterol 7α-hydroxylase (66). In some of their projects, it was possible to generate in situ novel substrates for enzymes whose cDNAs were being cloned by transfecting cells with expression plasmids specifying one or more known enzymes. For example, Margrit added commercially available radioactive 25-hydroxycholesterol to the medium of cells transfected with an expression vector encoding the CYP7B1 oxysterol 7α-hydroxylase to generate in situ 3β,7α,25-trihydroxy-cholest-5-en, which is a substrate for the HSD3B7 enzyme. Co-transfecting these cells with pools of cDNAs derived from mouse liver allowed her to identify cDNAs encoding HSD3B7 (67).
In general, once a cDNA for a given enzyme was isolated, we took two approaches to understand the physiological roles of these and other enzymes in the bile acid synthesis pathways. As with cholesterol 7α-hydroxylase (61, 62), we made and characterized the phenotypes of mice deficient in various enzymes, including cholesterol 24-hydroxylase (68), cholesterol 25-hydroxylase (69), sterol 27-hydroxylase (70), CYP7B1 oxysterol 7α-hydroxylase (71), HSD3B7 (72), and sterol 12α-hydroxylase (73). The mutant mice presented with strikingly diverse phenotypes, which included liver failure (cholesterol 7α-hydroxylase and HSD3B7), immune system dysfunction (cholesterol 25-hydroxylase, see below), learning difficulties (cholesterol 24-hydroxylase) (74, 75), hepatomegaly and hypertriglyceridemia (sterol 27-hydroxylase), and minor (CYP7B1 oxysterol 7α-hydroxylase) and major (sterol 12α-hydroxylase) alterations in cholesterol and bile acid metabolism. Working through these phenotypes required talented trainees in the lab, including Erik Lund, Tina Kotti, Denise Ramirez, Rebekkah Halford, David Bauman, Julie Li-Hawkins, Ashlee Stiles, and Heidi Shea, and collaborations with many colleagues at UT Southwestern and other universities, including John Dietschy, Steve Turley, Brad Pfeiffer, Kim Huber, Andrew Bitmansour, Jeff McDonald, Joyce Repa, Jay Horton, Eran Leitersdorf, Gösta Eggertsen, Mats Gåfvels, and Ingemar Björkhem.
In a second approach, we obtained cells, tissues, or DNA from subjects with suspected inborn errors in bile acid synthesis and characterized mutations in genes encoding the affected enzymes. To these ends, mutations in the sterol 27-hydroxylase gene (noted above), the CYP7B1 oxysterol 7α-hydroxylase gene (76), HSD3B7 (67), and the bile acid CoA:amino acid N-acyltransferase (77) were identified. As I am not a physician, these studies required collaborations with sharp-eyed clinicians who recognized the prismatic value of unusual phenotypes in their patients. For example, Ron Sokol at the Children's Hospital Colorado treated an infant with liver failure who Ken Setchell subsequently showed had abnormal levels of oxysterols in the blood. This was the predicted presentation of an inherited defect in the CYP7B1 oxysterol 7α-hydroxylase, which our molecular analyses with Ron and Ken documented (76). As noted above, Margrit Schwarz used a clever expression cloning approach to isolate the mouse HSD3B7 cDNA, which she then used to identify the human cDNA and HSD3B7 gene. Working with Ingemar Björkhem and with Hisham Nazer, a pediatrician at the King Faisal Hospital in Riyadh, Saudi Arabia, she showed that inherited mutations in this gene underlie a different form of liver failure in newborns. In so doing, she confirmed at the molecular level a large body of chemical precedent set by Ken Setchell, Peter Clayton, and other mass spectrometrists.
Our work, and that of others, has largely confirmed at the biochemical, physiological, and genetic levels the pathways of bile acid synthesis shown in Fig. 4. As of this writing, inherited defects in nine genes in these pathways have been identified (Fig. 5), and at least as many mice with induced mutations in these DNAs have been characterized. The availability of cloned genes also allowed the regulation of bile acid synthesis to be worked out at the molecular level, as well as the demonstration of the central roles played by numerous nuclear receptors in this regulation. I collaborated with others in some of this research, including projects completed by Tom Kerr and Wenling Chen with my UT Southwestern colleague David Mangelsdorf (78, 79), and a study done by Tom Kerr with Bei Shan and Margrit Schwarz, who were then at Tularik Corporation (80), but for the most part I was but a witness to these insightful regulatory studies.
Figure 5.
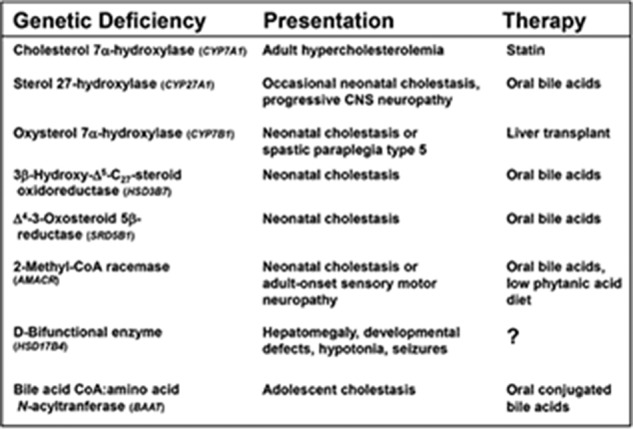
Inborn errors of bile acid synthesis. The nine known inherited disorders in which bile acid synthesis is disrupted are tabulated together with their associated presentations (symptoms) and therapies.
Cholesterol metabolism: Androgens
When Diane Jelinek began purifying cholesterol 7α-hydroxylase in 1987 with the goal of obtaining the encoding cDNA, I thought it best to have a back-up approach in case the enzyme proved unstable or otherwise difficult to isolate. Susan Peacock was using Xenopus oocytes to express the LDL receptor, so it seemed reasonable to co-opt this system to establish an expression cloning assay for the cholesterol 7α-hydroxylase mRNA. To this end, Stefan Andersson purified hepatic poly(A)+ mRNA from female rats, size-fractioned it on methylmercury-containing sucrose gradients, and learned from Susan how to farm frogs. He injected oocytes with aliquots of size-fractionated mRNAs from the gradients and allowed the mRNAs to express overnight. The next day he added radioactive cholesterol to the oocyte medium, and after a second incubation, extracted lipids and resolved sterols by TLC. The thinking was that if an injected mRNA fraction contained the cholesterol 7α-hydroxylase mRNA, then this mRNA would be translated into the enzyme, which in turn would convert the exogenously added radioactive cholesterol substrate to 7α-hydroxycholesterol product. Once an active fraction was identified in this manner, the mRNA could be converted into cDNA and cloned into an RNA expression vector, and then the resulting library could be screened and subdivided to identify cDNAs encoding cholesterol 7α-hydroxylase. A similar expression cloning approach had been used by others to isolate ion channel cDNAs, and David Julius in Richard Axel's laboratory had used it to identify serotonin receptor cDNAs. David kindly provided his oocyte preparation protocols to us, and their use greatly improved our efficiency.
Once the Xenopus project started, Stefan disappeared into the frog room, and I did not see him for a period of several weeks. When I finally ran into him in the hallway and enquired about his progress, he indicated that nothing was working. He had been unable to detect cholesterol 7α-hydroxylase enzyme activity despite numerous attempts. Given the large number of steps in the approach and the fickleness of Xenopus oocytes, any number of reasons could explain the negative outcome of his studies. I thus asked him whether he had a positive control in the experiment that would indicate an injected mRNA was being translated into an active enzyme. He replied “yes” and that he had remembered from his graduate studies that expression of the enzyme steroid 5α-reductase (abbreviated henceforth as 5α-reductase) was sexually dimorphic in the rat, being 20 times more abundant in female rat liver than in male rat liver. As he had purified mRNA for the experiment from female rats, he added radioactive testosterone to the mRNA-injected oocytes and assayed its conversion to dihydrotestosterone, the reaction catalyzed by 5α-reductase (Fig. 6). He detected this activity and thus knew that most steps in the expression cloning protocol were working.
Figure 6.
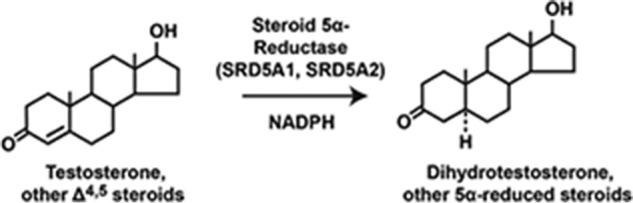
The conversion of testosterone to dihydrotestosterone by steroid 5α-reductase type 1 (SRD5A1) and steroid 5α-reductase type 2 (SRD5A2).
Upon hearing this report, I replied that his results indicated he could clone 5α-reductase cDNAs, to which Stefan replied “Well, sure, but why would you want to do that?” From a series of coincidences, I knew exactly why. Luckily, I had purchased Merck stock several years earlier based on their development of the first statin and as a consequence received the company's quarterly reports describing new drug candidates, one of which was an inhibitor of 5α-reductase. Merck was developing this compound, then known as MK-906, as a treatment for benign prostatic hyperplasia, a common affliction of men over the age of 50 that arises in part from excess dihydrotestosterone production. I knew that if Merck was developing inhibitors of 5α-reductase, then it must be an important enzyme. I thought it might be possible to identify 5α-reductase cDNAs by cross-hybridization at low stringency with HMG-CoA reductase cDNA probes. Thus, the idea of cDNA cloning 5α-reductase was not a new one.
From this point forward, Stefan began using the Xenopus expression system to identify 5α-reductase cDNAs, and over the next several months he made excellent progress. At about this time, Mike Brown asked me how the Xenopus project was going, and I replied that it looked like we were going to be able to clone the rat 5α-reductase cDNA. To which he replied, “You mean the cholesterol 7α-hydroxylase cDNA?” I summarized the above series of events to him, and in response he told me I had to go speak with our colleague Jean Wilson in the Department of Internal Medicine. I knew Jean to be an accomplished endocrinologist, but I did not know that he had discovered dihydrotestosterone, 5α-reductase, and human subjects with an inherited deficiency in the encoding gene! In defense of my ignorance, because the decision to clone 5α-reductase cDNAs had been made, I had read the 5α-reductase literature in reverse chronological order (i.e. from the current to the past) and had not gotten to the decade between 1968 and 1978 when Jean made many of his discoveries in the field. Regardless, a subsequent conversation with Jean led to what would ultimately be a 29-year collaboration with him in which molecular biology was applied to the study of 5α-reductase.
Stefan was successful in cloning the rat 5α-reductase cDNA from liver, and he used this cDNA as a probe to document that the same gene was expressed in the prostate and therein regulated by androgens (81). He was assisted in the project by Richard Bishop, who was the first graduate student to complete his Ph.D. dissertation research under my direction. Stefan used the rat cDNA as a cross-hybridization probe to isolate the orthologous human 5α-reductase cDNA from a prostate library, which he then expressed in cultured cells (82). Biochemical and pharmacological characterization of the activity expressed in the transfected cells revealed two puzzling features: the enzyme had a pH optimum centered around 7.0, and the activity was poorly inhibited (Ki ≥300 nm) by the Merck MK-906 compound. These results contrasted with those of others who had studied 5α-reductase activity in human prostate extracts, which showed an acidic pH optimum (∼4.5) and inhibition by low nanomolar concentrations on MK-903.
We considered several explanations for these contrasting results, including that the recombinant enzyme lacked an essential subunit or post-translational modification versus the prostatic enzyme or that there were two distinct 5α-reductase enzymes. At this time (1990), Jean Wilson steered us to a paper he had published in the Journal of Biological Chemistry in 1976, which provided biochemical evidence for the existence of two 5α-reductase enzymes, one exhibiting an acidic pH optimum and the other a more basic pH optimum, and genetic evidence for the same in that fibroblasts from subjects with inherited 5α-reductase deficiency contained only the basic pH optimum activity (83). With Jean's guidance, Stefan, together with David Berman, an M.D./Ph.D. student, began a project to clone cDNAs encoding the second enzyme. Stefan took an expression cloning approach in mammalian cells, while David used oligonucleotides derived from the first 5α-reductase cDNA (then referred to as 5α-reductase type 1) in reduced stringency PCRs. They were both successful in identifying a related but different 5α-reductase cDNA, which was termed 5α-reductase type 2, and which encoded an enzyme that exhibited an acidic pH optimum and was exquisitely sensitive to inhibition by MK-906. The final evidence for the existence of two 5α-reductases was provided by Elizabeth Jenkins, an endocrine fellow in the laboratory, who extracted genomic DNA from fibroblasts of subjects with inherited 5α-reductase deficiency and showed via Southern blotting that these subjects had a large deletion in their 5α-reductase type 2 gene (84).
With the cloned cDNAs and genes for 5α-reductase 1 and 2 in hand, a host of trainees in the laboratory and collaborators all over the world spent the next 25 years elucidating the molecular basis of 5α-reductase type 2 deficiency (Fig. 7), the pharmacology of the two isozymes, and their overlapping and distinct physiological roles. Jean Wilson and Julianne Imperato-McGinley had shown that the absence of 5α-reductase 2 in men gives rise to an intersex phenotype in which the internal male genitalia (epididymis, seminal vesicles, and vas deferens) develop normally whereas the external genitalia (penis and scrotum) and the internal prostate gland fail to develop. Affected individuals are often raised as females until the time of puberty when a surge of dihydrotestosterone produced by the intact 5α-reductase 1 enzyme leads to their virilization, and in more cases than not, a change to a male identity. Thus, research on the genetic basis of 5α-reductase 2 deficiency, done in collaboration with Jean and Julianne and many other physicians, led us into human genetics, sexual development, and the complexities of gender identity.
Figure 7.

Mutations in the human gene encoding steroid 5-reductase type 2 (SRD5A2) that cause an intersex phenotype. The SRD5A2 gene is shown as a five-exon schematic with intervening introns. Point mutations that give rise to substitutions and premature truncations in the protein are shown in panel A. Insertions, deletions, and splicing mutations that disrupt the gene are shown in panel B. Reproduced with permission from Ref. 40. This research was originally published in Genetic Steroid Disorders. D. W. Russell and J. D. Wilson. Steroid 5α-reductase 2 deficiency. Genetic Steroid Disorders. (New, M. I., Lekarev, L., Parsa, A., Yuen, T., O'Malley, B., and Hammer, G., eds), pp. 199–214. © 2013 Elsevier.
Similarly, pharmacological studies involving collaborations with scientists at Merck, GlaxoSmithKline, and Eli Lilly ultimately led to FDA-approved drugs (Proscar®, Propecia®, and Avodart®) to treat benign prostatic hyperplasia, male- and female-pattern baldness, and prostate cancer. Anice Thigpen, a postdoctoral fellow in the laboratory, carried out structure–function studies that provided insight into where and how the various 5α-reductase inhibitors acted (85, 86). This research introduced me to the inherent difficulties of drug development and later to appearances in the British High Court and others as an expert witness on 5α-reductase inhibitors for Merck, which was trying to stave off the efforts of multiple generic drug companies to circumvent the company's patents on 5α-reductase inhibitors!
Multiple trainees in the laboratory characterized the physiological roles of the two 5α-reductase enzymes in animal models. David Berman, mentioned above, and Hui Tian, a graduate student, determined the cell-type–specific expression patterns of 5α-reductase 1 and 2 in the rat and mouse urogenital tracts (87, 88), while Karl Normington, a postdoctoral fellow, showed how the kinetic characteristics and tissue distributions of the two 5α-reductase enzymes influenced their biological roles in the rat (89). Mala Mahendroo constructed and characterized mice deficient in 5α-reductase 1 and 5α-reductase 2, and bred the two strains to produce doubly deficient animals (90). Her studies revealed that 5α-reductase 1 plays an essential catabolic role in the pregnant female mouse and is required to metabolize progesterone in the cervix to allow ripening and subsequent delivery of pups. In the absence of this breakdown, the cervix fails to ripen and pups are not delivered despite normal uterine contractions at term (91). Mala found that male mice lacking 5α-reductase 2 virilized normally, which at the outset was a disappointing finding as the prediction was that these mice would be an animal model of human 5α-reductase 2 deficiency. We initially attributed virilization to compensation by 5α-reductase 1 but then found that the doubly deficient mice also underwent normal male sexual development (90). This conundrum was solved when Mala measured steroid hormone levels in the male urogenital tract and found that testosterone accumulates to levels 10-fold higher in tissues from mice lacking 5α-reductase 1 and 2 than in normal mice. Jean Wilson pointed out that these results supported a long-standing idea in andrology, namely that the conversion of testosterone to dihydrotestosterone by 5α-reductase serves to amplify the androgenic signal during virilization of certain tissues such as the penis, scrotum, and prostate, and not that dihydrotestosterone has any unique hormonal properties (90). This notion has been largely confirmed in molecular studies by others, which have failed to identify genes that respond to dihydrotestosterone but not testosterone. That loss of 5α-reductase 2 in the human male is sufficient to generate an intersex phenotype likely means that testosterone does not accumulate to high enough levels to drive differentiation of the human male urogenital tract during development.
As noted above, our initial entry into androgen research was afforded by Stefan Andersson's application of expression cloning to isolate the rat 5α-reductases 1 cDNA. This technique was also used to identify cDNAs encoding 17β-hydroxysteroid dehydrogenase type 3 (HSD17B3), which catalyzes the last step in the testosterone biosynthetic pathway, the conversion of androstenedione to testosterone (92). Together with Jean Wilson, Stefan showed that inherited deficiencies in the HSD17B3 gene give rise to a disorder of male sexual differentiation that differs slightly from 5α-reductase 2 deficiency (93). In a separate project, Michael Biswas, an M.D./Ph.D. student in the laboratory, used expression cloning to isolate another 17β-hydroxysteroid dehydrogenase cDNA, which he termed 17β-hydroxysteroid dehydrogenase type 6 (HSD17B6). Michael showed that this member of the 17β-hydroxysteroid dehydrogenase family oxidized 5α-androstane-3α,17β-diol to 5α-androstane-3α-diol,17β-one (androsterone) and thus was likely involved in the inactivation of androgens (94). Unexpectedly, he found that HSD17B6 shared 65% sequence identity with a class of enzymes referred to as retinol dehydrogenases, which interconvert retinols, retinaldehydes, and retinoic acids. His careful kinetic analyses revealed that retinol dehydrogenase type 1 (RoDH1) was a far better androgen-metabolizing enzyme than retinol-metabolizing enzyme, and furthermore, that RoDH1 converted the inactive androgen 5α-androstane-3α,17β-diol to dihydrotestosterone, the most potent androgen yet described (94). Subsequent independent work by Jean Wilson, Marilyn Renfree, Richard Auchus, and Nima Sharifi showed that this reaction was part of an alternate pathway leading to dihydrotestosterone synthesis in the tammar wallaby (95) and in human castration–resistant prostate cancer (96).
Michael Biswas worked on a second androgen project that led to a brief incursion into plant biology. Jianming Li and Joanne Chory at the Salk Institute had shown that mutations in the DET2 gene encoding an enzyme in the brassinolide biosynthetic pathway give rise to a pleiotropic phenotype in Arabidopsis that includes defects in light-regulated gene expression, development, and male sexual differentiation (97). Brassinolide is one of many steroids found in plants and has a 28-carbon structure with a 5α-reduced configuration in the A-ring of the steroid backbone. Joanne and Jianming found that DET2 shared substantial sequence identity with mammalian 5α-reductases, and thus Michael and Jianming did a series of experiments to determine whether a mammalian 5α-reductase could substitute for the plant ortholog, DET2. Surprisingly, they found that human 5α-reductase 1 and 2 could complement DET2 deficiency in Arabidopsis and that all three enzymes shared steroid substrates and were blocked by mammalian enzyme inhibitors developed by the pharmaceutical industry (98). We concluded from this study that steroid hormone biosynthesis and signaling were conserved over great evolutionary distances, as was the need for 5α-reduction in male sexual development.
Looking back on our work with androgens, I am tempted to write that it was all part of a grand design to study the various pathways by which cholesterol is actively metabolized to products such as bile acids and steroid hormones. The truth as noted above was that my entry into the field of andrology hinged on a lucky financial decision to purchase Merck stock, and the fortuitous happenstance of having as a colleague Jean Wilson, a world expert in androgens and sexual differentiation. This path illustrates yet again the role of chance in basic research and suggests that the myriad attempts by funding agencies to influence its course through targeted requests for applications and other mechanisms are frivolous.
Sterol metabolism: Vitamin D
Jeff Cheng joined the lab as an M.D./Ph.D. student in 1999 and, after completing a study in which he characterized a series of HSD3B7 gene mutations in subjects with bile acid synthesis defects and liver failure (99), he asked whether he could tackle a completely new project in the laboratory. After considering several possibilities, Jeff chose to go after the gene encoding the vitamin D 25-hydroxylase (Fig. 8). This was a brave choice on his part as talented investigators in the vitamin D field had been searching for this enzyme for close to 30 years. Undaunted, Jeff established an expression cloning strategy together with another M.D./Ph.D student (Dan Motola) in David Mangelsdorf's laboratory in which they co-transfected cultured cells with a vitamin D receptor–responsive luciferase gene, expression vectors encoding a modified vitamin D receptor, mouse adrenodoxin, E. coli β-gal, and the adenoviral VA-1 RNA, and pools of ∼100 cDNAs made from the livers of mice lacking sterol 27-hydroxylase. Their thinking was that if a given cDNA pool expressed a vitamin D 25-hydroxylase, then the enzyme would 25-hydroxylate 1α-hydroxyvitamin D added to the cellular medium. The resulting 1α,25-dihydroxyvitamin D would bind to the expressed vitamin D receptor, resulting in activation of the vitamin D–responsive luciferase construct, which in turn could be detected by measuring luciferase enzyme activity in a luminometer. The expressed E. coli β-gal served as a control for transfection efficiency; the adenoviral VA-1 gene boosted expression from transfected plasmids; and the adrenodoxin cDNA was added in case the vitamin D 25-hydroxylase was a mitochondrial cytochrome P450 enzyme and as such would need this protein co-factor. Lastly, the choice of the cDNA library from sterol 27-hydroxylase–deficient mouse liver mRNA was important because there were reports in the literature that in at least some species, this enzyme had vitamin D 25-hydroxylase activity and Jeff did not want to reclone the sterol 27-hydroxylase cDNA.
Figure 8.
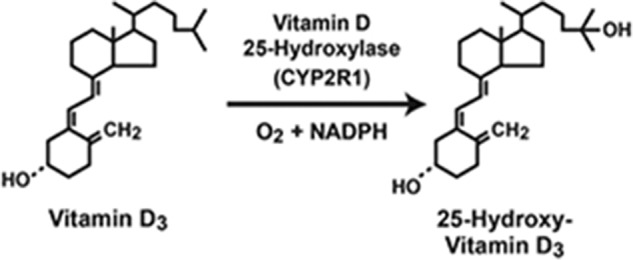
Reaction catalyzed by the vitamin D 25-hydroxylase (CYP2R1).
As cockamamie as this strategy reads, it worked! Success was realized because Jeff and Dan optimized each step of the protocol with the result being that they identified a vitamin D 25-hydroxylase activity in one cDNA pool out of 1,056 screened (100). Their ultimate reward was the identification of a cDNA specifying a bona fide vitamin D 25-hydroxylase, which turned out to be a microsomal cytochrome P450, CYP2R1, with all of the properties and tissue distribution predicted for this long-lost enzyme. In a subsequent study, Jeff collaborated with two physicians, Mike Levine and Norman Bell, to identify a mutation in the human CYP2R1 gene as a cause of vitamin D deficiency (101). Two additional mutations in the CYP2R1 gene in a family from Saudi Arabia with rickets were identified later in a collaboration with Angham Al Mutair and Ghada Nasrat (102). Together, these genetic studies suggested that CYP2R1 is the physiologically relevant vitamin D 25-hydroxylase in humans. This conclusion may not be true in other species as Hector DeLuca has shown that mice deficient in CYP2R1 have half-normal levels of circulating 25-hydroxyvitamin D and no obvious vitamin D deficiencies (103).
To reiterate a theme, one might conclude that because vitamin D is produced from the sterol 7-dehydrocholesterol in the skin by the actions of UV light and then activated by 25-hydroxylation in the liver and 1α-hydroxylation in target tissues, our studies in this field were part of a grand design to study cholesterol metabolism in all of its guises. But again, no: My research in this field was driven by a desire to apply molecular biology to gain insight into a metabolic pathway and by a lucky association with Jeff Cheng, a talented student with aspirations to be a dermatologist (he is now on the dermatology faculty at UCSF).
Brief forays
The above research in cholesterol, bile acid, androgen, and vitamin D metabolism represented the lion's share of my scientific efforts. There were other minor excursions into different areas of research through the years. I collaborated with Janet Robishaw, Bruce Harris, and Al Gilman in the cloning of cDNAs encoding the Gsα subunit of G proteins (104); with Ming-Shi Chang, Ellen Vitetta, and Jonathan Uhr in the cloning of ricin B chain cDNA (105); with Wen-Ji Chen, Mike, and Joe in the cloning of farnesyltransferase β-subunit cDNAs (106); with Joe Naglich and Leon Eidels in the isolation of cDNAs specifying the diphtheria toxin receptor (107); and with Hui Tian and Steve McKnight in identification and characterization of HIF2α (EPAS1) and several other PAS domain–containing proteins (108–110). In other studies, a collaboration with Yildiz Yildiz, Heidrun Matern, and Siegfried Matern led to the identification of β-glucosidase 2 (GBA2), an enzyme that metabolizes both bile acid–glucose conjugates and glucocerebrosides, and when knocked out in the mouse causes a lipid storage disease and sperm abnormalities (111). Two other groups later showed that mutations in human GBA2 cause an inherited form of spastic paraplegia (112, 113). Lastly, Jeff Cheng identified, cDNA-cloned, and characterized three enzymes that compose the mammalian wax biosynthetic pathway (114, 115).
Swansong: 25-Hydroxycholesterol
From 1987 to the present, we exerted considerable effort in studying a class of cholesterol metabolites referred to as side-chain oxysterols, which as noted above differ from cholesterol by the presence of an additional hydroxyl group on carbons 24, 25, or 27 of the side chain (Fig. 9). To these ends, cDNAs encoding sterol 27-hydroxylase (CYP27A1), cholesterol 24-hydroxylase (CYP46A1), and cholesterol 25-hydroxylase (CH25H) were isolated by various members of the laboratory. Studies by us and others showed that the oxysterols produced by these enzymes are substrates for bile acid synthesis via the alternate pathway (Fig. 4), ligands for the liver X receptor (LXR) nuclear receptor (116), and intermediates in the turnover of cholesterol by the brain, lung, and other tissues (117). Of the three enzymes and their respective oxysterol products, determining the physiological roles of cholesterol 25-hydroxylase and 25-hydroxycholesterol proved to be both the most challenging and the most rewarding. This project also was the last of my scientific career.
Figure 9.
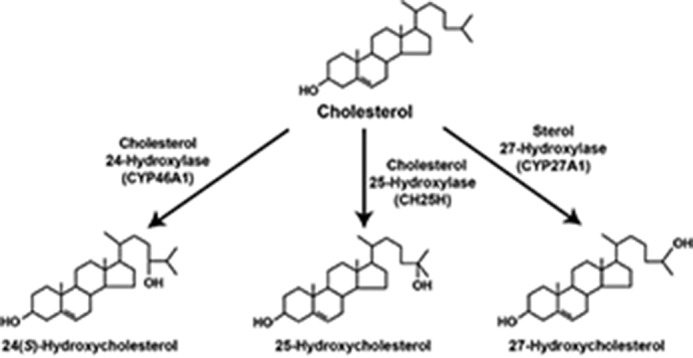
Biosynthesis of side-chain oxysterols. The conversion of cholesterol into three side-chain oxysterols is shown together with the enzymes that synthesize each.
Papers published in 1974 by Mike Brown and Joe Goldstein (118) and Herbert Chen and Andrew Kandutsch (119) reported that 25-hydroxycholesterol was more potent than cholesterol in mediating feedback repression of cholesterol synthesis in cultured cells. Thus, when we began working on oxysterols, we did so with the assumption that 25-hydroxycholesterol would play a central role in regulating cholesterol metabolism in vivo. As Mike and Joe and their colleagues progressively defined the machinery underlying sterol-mediated feedback regulation through the 1990s (reviewed in Ref. 120), including the identification of sterol regulatory elements in responsive genes, transcription factors that bind these sequences (SREBP-1 and SREBP-2), and proteins that regulate the intracellular movement and activation of SREBPs (SCAP, INSIG-1, INSIG-2, S1P, and S2P), they consistently found that both cholesterol and 25-hydroxycholesterol were potent mediators of feedback inhibition. Their biochemical studies with purified proteins showed that cholesterol mediates feedback repression by binding to SCAP (121) while 25-hydroxycholesterol does so by binding to INSIG proteins (122).
Our work on 25-hydroxycholesterol began in 1997 when Erik Lund set out to isolate cDNAs and genes encoding the putative cholesterol 25-hydroxylase enzyme. He made a cDNA library from the livers of transgenic mice expressing SREBP-1, which we knew secreted measurable amounts of 25-hydroxycholesterol in the stool, and then screened pools of this library in transfected mammalian cells for their ability to 25-hydroxylate radiolabeled sterol substrates. After screening 255 pools of ∼3,800 cDNAs each, he succeeded in identifying a single pool that catalyzed the desired reaction. Purification of the cDNA expressing this activity followed by DNA sequencing indicated that the mouse cholesterol 25-hydroxylase (hereafter abbreviated as CH25H) was a polytopic membrane protein of 298 amino acids and a member of a small family of enzymes that utilize di-iron co-factors to catalyze hydroxylation and other reactions on hydrophobic substrates (64). As expected from Mike and Joe's studies, expression of CH25H in cultured cells and the subsequent synthesis of 25-hydroxycholesterol reduced the synthesis of cholesterol and blocked the activation of the SREBP pathway; however, RNA blotting studies indicated that expression of the CH25H gene did not correlate with cholesterol synthesis rates in different tissues of the mouse. This was an unexpected result and one that did not support an in vivo role for 25-hydroxycholesterol as a regulator of cholesterol metabolism.
Guosheng Liang next knocked out the mouse cholesterol 25-hydroxylase gene (gene symbol Ch25h), which turned out to be a rare example of a mammalian gene lacking exons, and in 1998, he and others in my laboratory began characterizing the mutant mice. Over roughly the next ten years, every aspect of lipid metabolism in these mice was studied in detail, sometimes in isolated cells (e.g. embryonic stem cells and fibroblasts) and other times in the whole animal, and not a single difference was found between WT and Ch25h−/− mice. The lack of a phenotype was demoralizing to various trainees who cycled through the lab during this decade, and the cholesterol 25-hydroxylase project became known as the black hole of the Russell laboratory. Several fortuitous events broke this logjam.
First, David Bauman joined us as a postdoctoral fellow after graduate training with Trevor Penning in androgen metabolism at the University of Pennsylvania. Second, Jeffrey McDonald joined the Department of Molecular Genetics faculty after graduate training in MS with Ronald Hites at Indiana University and postdoctoral training in explosives analysis at the FBI. Third, I joined the LIPID MAPS Consortium at Ed Dennis' invitation. LIPID MAPS was a large-scale, glue-grant effort underwritten by the National Institute of General Medical Sciences to identify all lipids in a mammalian cell using MS (reviewed in Ed's Journal of Biological Chemistry Reflections article (123)). In the experimental design chosen by LIPID MAPS, the lipids present in resting and activated macrophages were to be compared. To this end, RAW 264.7 cells, an immortalized line of mouse macrophages, were incubated in the absence or presence of a Toll 4 receptor (TLR4) agonist, and after a period of time, lipids were extracted and then analyzed by MS.
The LIPID MAPS charge to the Russell laboratory was to develop methods to quantify sterols, a challenge that Jeff took on. He developed chromatography methods to resolve a majority of known sterols and, using authentic standards synthesized by Walt Shaw at Avanti Polar Lipids, Inc., established MS methods to identify and quantify this class of lipids (124). With these analytical methods in hand and with superb technical assistance from Bonne Thompson, another long-term technician whom I hired, Jeff found that activation of RAW macrophages via TLR4 led to a large increase (30–50–fold) in the levels of 25-hydroxycholesterol both within cells and in the medium in which they were grown. Additional experiments in RAW cells by David Bauman, who being new to the lab and thus unfamiliar with the dark history of the cholesterol 25-hydroxylase project had agreed to take a stab at elucidating the phenotype of the Ch25h knockout mice, revealed a concomitant induction of the CH25H mRNA and gene. They next showed that the CH25H gene was induced in different tissues of WT mice treated with a TLR4 agonist and that the level of 25-hydroxycholesterol in the serum reached low nanomolar concentrations in these animals.
The fact that RAW cells were derived originally from primary macrophages, an innate immune cell type, suggested that perhaps the Ch25h−/− mice might have an immunological phenotype versus an alteration in the regulation of cholesterol metabolism as originally supposed. To test this idea, David carried out a series of microarray studies, which revealed that mRNAs encoding the heavy and J chains of IgA were elevated in immune system tissues of the knockout mice. Measurement of IgA levels in the sera, lungs, and mucosa by ELISA confirmed the microarray results, and indicated that Ch25h−/− animals had more circulating IgA than IgG, which is normally the predominant immunoglobulin subtype in the bloodstream.
From our earlier work annotating the alternate pathway of bile acid synthesis, we knew that 25-hydroxycholesterol is metabolized by the CYP7B1 oxysterol 7α-hydroxylase (Fig. 4). In agreement with this pathway, Julie Li-Hawkins and Erik Lund had shown that mice lacking the encoding Cyp7b1 gene had elevated levels of 25-hydroxycholesterol in their sera (71). When David measured IgA levels in the Cyp7b1−/− mice, he found that they were suppressed relative to those in WT mice. Thus, mice that could not synthesize 25-hydroxycholesterol (Ch25h−/− animals) had elevated levels of IgA and those with excessive 25-hydroxycholesterol (Cyp7b1−/− animals) had suppressed levels of IgA. With help from Andrew Bitmansour, a postdoctoral fellow in Ellen Vitetta's laboratory in the Department of Immunology at UT Southwestern, David went on to show that addition of nanomolar concentrations of 25-hydroxycholesterol to naïve B cells (splenic B220+ cells) suppressed class switch recombination to an IgA secreting phenotype but had no effect on class switching to IgG or IgE phenotypes. Although we were unable to determine the exact mechanism by which 25-hydroxycholesterol affected class switch recombination, the oxysterol was shown to reduce the proliferation of B cells that is required prior to rearrangement of the immunoglobulin heavy chain locus, and it suppressed expression of the gene encoding the activation-induced cytosine deaminase, which catalyzes class switching in naïve B cells.
The paper reporting our mouse results was published in September of 2009 with David Bauman as first author (69). In November of 2009, Ulf Diczfalusy and colleagues published similar findings with respect to the induction of CH25H in bone marrow–derived macrophages after TLR4 activation and importantly, showed that serum levels of 25-hydroxycholesterol rose in human subjects treated with a TLR4 agonist (125). Together, the two papers provided initial evidence that 25-hydroxycholesterol had a function in the immune system.
A tsunami of papers on the role of 25-hydroxycholesterol and its metabolites in the immune system arrived in the following years! First, dendritic cells were shown to induce CH25H in response to TLR4 agonists, just as we found macrophages to do, and interferons, which are secreted by TLR4-stimulated innate immune cells, were shown to be the inducer of the CH25H gene (126). Second, 7α, 25-dihydroxycholesterol, the product of the combined actions of CH25H and the CYP7B1 oxysterol 7α-hydroxylase on cholesterol (Fig. 10), was shown by Jason Cyster, Andreas Sailer, and colleagues (127) and by Changlu Liu and colleagues (128) to be the ligand of the Epstein–Barr virus–induced receptor 2 (EBI2; also known as GPR183), a G protein–coupled receptor required for B cell and dendritic cell migration within lymphoid tissues such as the spleen and lymph nodes, and in non-lymphoid tissues (reviewed in Refs. 129 and 130). Third, 25-hydroxycholesterol was shown by two different groups to have potent anti-viral activities against many different DNA and RNA viruses (131, 132). Fourth, the Cyster laboratory showed that interferon-induced 25-hydroxycholesterol synthesis in activated macrophages serves to suppress the inflammatory response once initiated (133). They traced the mechanism by which the production of pro-inflammatory cytokines like interleukin-1β is reduced to the suppression of the SREBP pathway by 25-hydroxycholesterol. A later paper by this group revealed that 25-hydroxycholesterol–mediated suppression of SREBP-2,in particular, blocked activation of AIM2-containing inflammasomes, leading to the subsequent reduction in interleukin-1β production (134). Further research has shown that 25-hydrocycholesterol–mediated suppression of SREBP is a general mechanism by which the innate immune response is shut off.
Figure 10.
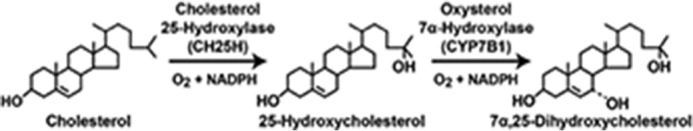
Biosynthetic pathway of immune modulatory oxysterols. The conversion of cholesterol into two sterols that regulate different aspects of the innate and adaptive immune systems is shown.
Jeff, Guosheng, and I provided oxysterol measurements and various mouse strains in some of these studies, but for the most part, we were but voyeurs to the incursion of oxysterol metabolism into immunology and virology, and later bone biology (135). Nevertheless, it was particularly gratifying to participate in elucidating the in vivo roles of 25-hydroxycholesterols as interferon-responsive antiviral and immunomodulatory sterols. That this body of work ultimately confirmed the initial findings of Mike and Joe and others from 1974 and represented my publishing swansong in 2017 (134) made the icing even sweeter!
On books and editorial boards
I have always felt that an academic should publish at least one book in their lifetime, and thus in 1990 when Joe Sambrook offered me a chance to co-author with him the third edition of the classic Cold Spring Harbor Laboratory Press cloning manual entitled Molecular Cloning, I jumped at the chance. Little did I know then that we would not finish the book until 2000 or that the tome would expand to three separate volumes encompassing over 2,000 pages! The style of the book morphed several times during this ten-year effort and eventually involved extended descriptions of the background and theory behind each experimental protocol described. I have fond memories of writing the various chapters and updates in my office on Saturday mornings when interruptions were few and access to the UT Southwestern library unfettered. The effort allowed me to reconnect with and extend my knowledge of bacterial genetics and to gain newfound appreciation for the history of molecular biology, and greatly benefited our application of molecular biology in general to problems in lipid metabolism. It was a distinct pleasure to work with Joe, who is one of the most erudite individuals I know and whose knowledge of the English language and writing prowess are unique. The staff at Cold Spring Harbor Laboratory Press was equally delightful to work with, including Jan Argentine, Nancy Ford, Nina Irwin, Kaaren Janssen, Maryliz Dickerson, and Inez Sialiano; and John Inglis as the director of the press kept us all in line. The book sold well after it was finally published in December of 2000, and royalties from the sales went a long way toward covering the college tuitions of my children!
Serving as an editor of a book is another quintessential academic task, and I was able to carve this notch by co-editing a volume of the Methods in Enzymology series with David Mangelsdorf. Volume 364 is entitled “Nuclear Receptors” and contains 27 chapters by leading lights in the field. David and I planned the volume, solicited authors for the various chapters, and edited each one. The hardest part was lobbying contributors to turn in their chapters in a timely fashion. Having spent many hours poring over earlier volumes of Methods in Enzymology at all stages of my career, it was an honor to contribute to this venerable series.
Editorial board service is another must in academia. I reviewed my first manuscript for the journal Nucleic Acids Research in 1983, and with typical rookie enthusiasm, wrote an overly detailed critique that almost certainly upset the authors of the paper. Since then, having reviewed, edited, or otherwise handled thousands of manuscripts, I have learned how to write more succinct reviews that judge the overall contribution of the study, point out needed improvements, and assess whether the paper is an appropriate match for the journal's stated mission. This education came from service on the editorial boards of many journals, including Biochemistry (1991–1995), the Journal of Lipid Research (1987–1995; 2005–2017), Annual Review of Biochemistry (1996–2000), Molecular Endocrinology (1997–2001), Trends in Biochemical Sciences (2004–2010), Cell Metabolism (2004–2016), and the Proceedings of the National Academy of Sciences (2010–2018), as well as from completing hundreds of ad hoc reviews for various publications.
The periodical that I had the longest and most enriching experiences with was the Journal of Biological Chemistry. From day one of my reading the scientific literature under the tutelage of Joanne Ravel, I was made aware of the prestige of the Journal and the high quality of papers published in it. Thus, when I received a letter from Herb Tabor, then Editor-in-Chief of the Journal, to join the Editorial Board in January of 1992, I accepted it immediately. At this time, everything was hard copy, and roughly 7,800 manuscripts were handled by the board, with each paper being reviewed by two board members. This system led to tens of thousands of green-striped first-class, envelopes flowing through the U.S. Mail between Herb's office, those of the Associate Editors, and we board members. My desk remained awash in these distinctive envelopes, as did the mailbox outside the building where my office and lab were located. The promise made upon joining the board was that a member would be asked to review approximately one manuscript per week, or about 50 manuscripts per year. I was slow to learn that the faster one reviewed and returned a manuscript the more were sent to you, and thus, on my first year on the board, I reviewed considerably more than 50 papers. Thereafter, I wised up and began requesting of Herb that no more paper be sent to me once I had reviewed 52 in a given calendar year. He and the Associate Editors honored these requests, even when I sent them, as I did in one year, in March!
The early 1990s were a time of great change in scientific publishing when all steps in manuscript submission, review, and publication were going electronic. Herb Tabor and the Journal, together with Bob Simoni and HighWire Press, and Associate Editor Jim Stull and his first-generation Osiris manuscript tracking software, led the way in this publishing revolution. In turn, the early adoption and increased efficiency of digital publishing caused a doubling of the number of manuscripts submitted to the Journal and corresponding increases in the numbers of Associate Editors and board members to handle the bigger load. During my time on the Editorial Board, first as a member from 1993–1997, and then as an Associate Editor from 1999–2010, the number of submitted manuscripts increased from roughly 7,800 in 1992 to a peak of >15,000 in 2004, and then leveled off to ∼9,600 by 2010 as more journals went electronic. The acceptance rate of submitted manuscripts varied between 32 and 45% during this period, and to accommodate the larger number of accepted papers, the frequency with which the Journal was published increased to once per week. A reasonably fit scientist could pick up a year's worth (12 editions) of the Journal when I first began reading it in the mid-1970s, but by 2010, perhaps only a handful of Olympic weight lifters could heft the 52 editions that came out that year!
A particularly enjoyable aspect of my time with the Journal involved attending the quarterly meetings of my fellow Associate Editors together with Herb (Fig. 11). The real work of planning the Journal took place at these meetings, which were held on alternating coasts and in conjunction with the annual meeting of the American Society of Biochemistry and Molecular Biology. Looking back, it is surprising that we were able to make any forward progress given the no small amounts of alcohol that were consumed at the nightly dinners! But we were blessed with an exceptionally competent ASBMB staff, including Barbara Gordon, Jeanne Gladfelter, Nancy Rodnan, and others, who both organized the meetings and kept us on track.
Figure 11.

Members of the Editorial Board of the Journal of Biological Chemistry in 2010. A, Associate Editors and Editor-in-Chief. From left to right: Norma Allewell, Vince Hascall, Bill Smith, Jerry Lingrel, Jim Stull, Linda Spremulli, Ken Neet, Tom Vanaman, the author, Chuck Samuels, Herb Tabor, Fred Guengerich, Dale Benos, Jim Siedow, Judy Bond, John Exton, Joel Gottesfeld, Bob Lehman, Martha Fedor (Editor-in-Chief), Bob Simoni, Xiao-Fan Wang, and George Carman. B, the author with Herb Tabor, then co-editor of the Journal. Photographs provided by Bill Smith.
Dean, one letter away from dead
Between 1982, when I joined the UT Southwestern faculty, and today, the institution expanded in size to occupy four separate campuses centered on a cloverleaf intersection between Inwood Road and Harry Hines Boulevard, and dramatically expanded clinical operations by hiring thousands of faculty physicians, building numerous hospitals and clinics in North Texas, and increasing the sizes of our graduate and postgraduate medical training programs. This maturation into a full-fledged medical center was begun by President Kern Wildenthal and greatly accelerated when Dan Podolsky became President in September of 2008. The rapid growth of clinical programs was of some concern to the chairs and directors of the institution's basic science departments and centers, who were afraid that UT Southwestern's traditional emphasis on research might be shifting. To ensure their continued representation in the administration, they proposed the creation of a new position, the Dean of Basic Research. This individual would be responsible for championing basic science across the campus and ensuring that the institution remained at the forefront of modern biomedical research.
President Dan Podolsky and Provost Greg Fitz accepted this proposal and in 2010 charged a search committee chaired by Steve McKnight, then chairman of the Department of Biochemistry, with identifying qualified candidates. Joe Goldstein brought me up to date on this situation, and Steve later came by my office to describe the position and its responsibilities. Perhaps because I was suffering from late-onset “50-year–old man syndrome” and thus in need of new professional challenges (other remedies to the disorder such as buying a sports car or bagging a trophy wife were unpalatable), I indicated to Steve an interest in the job. The fact that I had no formal administrative experience and had done little more than run a laboratory at that point in my career did not faze the search committee, which subsequently recommended me to Dan and Greg as the top (and only) candidate. In retrospect, someone savvier than me would have recognized that the committee was not so much enthusiastic regarding my candidacy but rather surprised that any self-respecting scientist would want to become a dean. Regardless, in December of 2010 I joined the UT Southwestern administration as the inaugural Dean of Basic Science, and because the Dean of the Graduate School of Biomedical Sciences was to report to me, I was also made a Vice Provost.
These two positions were supposed to require a 50% time commitment, thus allowing me to continue running my laboratory, but within a few months I understood that they would be full-time occupations. I also quickly realized that my interpersonal skills were severely underdeveloped, that my budgetary skills were nonexistent, and that the range of human personalities and behaviors in a major medical center was large. Fortunately, a talented group of administrators (Fig. 12) in the Provost's Office, including Dwain Thiele, Greg Fitz, and Charles Ginsburg, mentored me and provided invaluable advice, support, and friendship as I learned the decanal ropes.
Figure 12.

Administrative mentors, colleagues, and friends in the UT Southwestern Provost's Office. Left to right: the author; Dwain Thiele, Vice Provost and Senior Associate Dean of Clinical and Faculty Initiatives; Greg Fitz, Executive Vice President for Academic Affairs, Provost, and Dean of Southwestern Medical School; and Charles Ginsburg, Vice Provost and Senior Associate Dean of Education. Photograph provided by David Gresham.
As my administrative duties increased, I gradually reduced the size of my laboratory and closed it in December of 2016. I continue today as a full-time administrator in the position of Vice Provost and Dean of Research, with responsibility for research, broadly defined, across the medical center.
Looking back
I have few regrets concerning my 40-plus year research career. Overall, I could have been a better scientist had I not been distracted by the many financially lucrative opportunities available to academics such as seminar and meeting honorariums, consulting contracts, book and patent royalties, and service on paid editorial, scientific advisory, and review boards. These addictions, which arose from a combination of hubris and a childhood of modest means, led to my violating a fundamental tenant of Mike Brown's and Joe Goldstein's mentorship advice, namely to focus on research, not rewards.
From a research point of view, I regret that we were never able to elucidate the mechanism by which the compound LY295427 antagonizes the cellular actions of 25-hydroxycholesterol. LY295427, a steroid, was identified by scientists at the Eli Lilly and Company as a cholesterol-lowering agent that acted by inducing LDL receptor gene expression (136, 137). Bethany Janowski, a postdoctoral fellow in the laboratory, showed that LY295427 antagonized the ability of 25-hydroxycholesterol to suppress SREBP processing (138). As LDL receptor gene expression depends on SREBP processing, her findings provided a rational mechanism of action for the compound's hypocholesterolemic effect. Similarly, in unpublished studies, David Bauman showed that LY295427 antagonized the actions of 25-hydroxycholesterol in B cells, and Chris Glass showed that the compound blocked the activation of the integrated stress response by 25-hydroxycholesterol in macrophages (see Ref. 139). Whether the latter actions were due to inhibition of SREBP processing was never determined, nor has the presumed protein target of LY295427 been identified.
I am similarly remorseful that we never identified inherited mutations in the human cholesterol 24-hydroxylase (CYP46A1) and cholesterol 25-hydroxylase (CH25H) genes, loss of which is likely to give rise to learning and memory difficulties (CYP46A1 mutations) and immune system deficiencies (CH25H mutations) based on our work with these genes in mice (69, 74).
These few regrets aside, I have been exceptionally fortunate in my career as an academic scientist. I have been blessed with generous and wise mentors, hard-working trainees, great colleagues, and many golden opportunities at UT Southwestern. These, combined with a supporting and forgiving wife, understanding children, and dumb luck, have allowed me to accomplish far more than I ever should have.
Acknowledgments
I thank Mike Brown, Joe Goldstein, Helen Hobbs, and Linda Spremulli for critical reading of the manuscript, and Nancy Heard for the figures appearing in this manuscript as well as all of those I have published over the last several decades. The research described in this article was supported by National Institutes of Health Grants HL01287, HL20948, HL31346, HD38127, DK47657, AR51943, and DK81182; and by grants from the Robert A. Welch Foundation, the Eugene McDermott Foundation, the Perot Family Foundation, and the Clayton Foundation for Research.
The author declares that he has no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- LDL
- low density lipoprotein
- CTX
- cerebrotendinous xanthomatosis
- SREBP
- sterol regulatory element–binding protein
- TLR4
- Toll 4 receptor
- HMG
- 3-hydroxy-3-methylglutaryl.
References
- 1. Russell D. W., and Spremulli L. L. (1979) A rapid and sensitive assay for the detection of eukaryotic ribosome dissociation factors. Anal. Biochem. 93, 238–243 10.1016/S0003-2697(79)80144-X [DOI] [PubMed] [Google Scholar]
- 2. Russell D. W., and Spremulli L. L. (1978) Identification of a wheat germ ribosome dissociation factor distinct from initiation factor eIF-3. J. Biol. Chem. 253, 6647–6649 [PubMed] [Google Scholar]
- 3. Russell D. W., and Spremulli L. L. (1979) Purification and characterization of a ribosome dissociation factor (eukaryotic initiation factor 6) from wheat germ. J. Biol. Chem. 254, 8796–8800 [PubMed] [Google Scholar]
- 4. Russell D. W., and Spremulli L. L. (1980) Mechanism of the wheat germ ribosome dissociation factor: interaction with the 60S ribosomal subunit. Arch. Biochem. Biophys. 201, 518–526 10.1016/0003-9861(80)90540-8 [DOI] [PubMed] [Google Scholar]
- 5. Valenzuela D. M., Chaudhuri A., and Maitra U. (1982) Eukaryotic ribosomal subunit anti-association activity of calf liver is contained in a single polypeptide chain protein of Mr = 25,500 (eukaryotic initiation factor 6). J. Biol. Chem. 257, 7712–7719 [PubMed] [Google Scholar]
- 6. Basu U., Si K., Warner J. R., and Maitra U. (2001) The Saccharomyces cerevisiae TIF6 gene encoding translation initiation factor 6 is required for 60S ribosomal subunit biogenesis. Mol. Cell. Biol. 21, 1453–1462 10.1128/MCB.21.5.1453-1462.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chendrimada T. P., Finn K. J., Ji X., Baillat D., Gregory R. I., Liebhaber S. A., Pasquinelli A. E., and Shiekhattar R. (2007) MicroRNA silencing through RISC recruitment of eIF6. Nature 447, 823–828 10.1038/nature05841 [DOI] [PubMed] [Google Scholar]
- 8. Groft C. M., Beckmann R., Sali A., and Burley S. K. (2000) Crystal structures of ribosome anti-association factor IF6. Nat. Struct. Biol. 7, 1156–1164 10.1038/82017 [DOI] [PubMed] [Google Scholar]
- 9. Klinge S., Voigts-Hoffmann F., Leibundgut M., Arpagaus S., and Ban N. (2011) Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor 6. Science 334, 941–948 10.1126/science.1211204 [DOI] [PubMed] [Google Scholar]
- 10. Breitenberger C. A., Moore M. N., Russell D. W., and Spremulli L. L. (1979) Purification of eukaryotic cytoplasmic elongation factor 2 and organellar elongation factor G by an affinity binding procedure. Anal. Biochem. 99, 434–440 10.1016/S0003-2697(79)80029-9 [DOI] [PubMed] [Google Scholar]
- 11. Sperrazza J. M., Russell D. W., and Spremulli L. L. (1980) Reversible dissociation of wheat germ ribosomal subunits: cation-dependent equilibria and thermodynamic parameters. Biochemistry 19, 1053–1058 10.1021/bi00547a001 [DOI] [PubMed] [Google Scholar]
- 12. Hutchison C. A. 3rd, Phillips S., Edgell M. H., Gillam S., Jahnke P., and Smith M. (1978) Mutagenesis at a specific position in a DNA sequence. J. Biol. Chem. 253, 6551–6560 [PubMed] [Google Scholar]
- 13. Williamson V. M., Cox D., Young E. T., Russell D. W., and Smith M. (1983) Characterization of transposable element-associated mutations that alter yeast alcohol dehydrogenase II expression. Mol. Cell. Biol. 3, 20–31 10.1128/MCB.3.1.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Russell D. W., Smith M., Williamson V. M., and Young E. T. (1983) Nucleotide sequence of the yeast alcohol dehydrogenase II gene. J. Biol. Chem. 258, 2674–2682 [PubMed] [Google Scholar]
- 15. Russell D. W., Smith M., Cox D., Williamson V. M., and Young E. T. (1983) DNA sequences of two yeast promoter-up mutants. Nature 304, 652–654 10.1038/304652a0 [DOI] [PubMed] [Google Scholar]
- 16. Beier D. R., Sledziewski A., and Young E. T. (1985) Deletion analysis identifies a region, upstream of the ADH2 gene of Saccharomyces cerevisiae, which is required for ADR1-mediated derepression. Mol. Cell. Biol. 5, 1743–1749 10.1128/MCB.5.7.1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iyer V., and Struhl K. (1995) Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J. 14, 2570–2579 10.1002/j.1460-2075.1995.tb07255.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weiffenbach B., Rogers D. T., Haber J. E., Zoller M., Russell D. W., and Smith M. (1983) Deletions and single base pair changes in the yeast mating type locus that prevent homothallic mating type conversions. Proc. Natl. Acad. Sci. U.S.A. 80, 3401–3405 10.1073/pnas.80.11.3401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Russell D. W., Jensen R., Zoller M. J., Burke J., Errede B., Smith M., and Herskowitz I. (1986) Structure of the yeast HO gene and analysis of its upstream regulatory region. Mol. Cell. Biol. 6, 4281–4294 10.1128/MCB.6.12.4281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vance D. E. (2017) From masochistic enzymology to mechanistic physiology and disease. J. Biol. Chem. 292, 17169–17177 10.1074/jbc.X117.815100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Palmiter R. D. (1974) Magnesium precipitation of ribonucleoprotein complexes. expedient techniques for the isolation of undergraded polysomes and messenger ribonucleic acid. Biochemistry 13, 3606–3615 10.1021/bi00714a032 [DOI] [PubMed] [Google Scholar]
- 22. Okayama H., and Berg P. (1983) A cDNA cloning vector that permits expression of cDNA inserts in mammalian cells. Mol. Cell. Biol. 3, 280–289 10.1128/MCB.3.2.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Russell D. W., Yamamoto T., Schneider W. J., Slaughter C. J., Brown M. S., and Goldstein J. L. (1983) cDNA cloning of the bovine LDL receptor: feedback regulation of a receptor mRNA. Proc. Natl. Acad. Sci. U.S.A. 80, 7501–7505 10.1073/pnas.80.24.7501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Russell D. W., Schneider W. J., Yamamoto T., Luskey K. L., Brown M. S., and Goldstein J. L. (1984) Domain map of the LDL receptor: sequence homology with the epidermal growth factor precursor. Cell 37, 577–585 10.1016/0092-8674(84)90388-X [DOI] [PubMed] [Google Scholar]
- 25. Yamamoto T., Davis C. G., Brown M. S., Schneider W. J., Casey M. L., Goldstein J. L., and Russell D. W. (1984) The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell 39, 27–38 10.1016/0092-8674(84)90188-0 [DOI] [PubMed] [Google Scholar]
- 26. Chin D. J., Gil G., Russell D. W., Liscum L., Luskey K. L., Basu S. K., Okayama H., Berg P., Goldstein J. L., and Brown M. S. (1984) Nucleotide sequence of HMG CoA reductase, a glycoprotein of the endoplasmic reticulum. Nature 308, 613–617 10.1038/308613a0 [DOI] [PubMed] [Google Scholar]
- 27. Brown M. S., and Goldstein J. L. (1974) Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. Proc. Natl. Acad. Sci. U.S.A. 71, 788–792 10.1073/pnas.71.3.788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brown M. S., and Goldstein J. L. (1980) Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J. Lipid Res. 21, 505–517 [PubMed] [Google Scholar]
- 29. Goldstein J. L., Anderson R. G., and Brown M. S. (1979) Coated pits, coated vesicles, and receptor-mediated endocytosis. Nature 279, 679–685 10.1038/279679a0 [DOI] [PubMed] [Google Scholar]
- 30. Davis C. G., Elhammer A., Russell D. W., Schneider W. J., Kornfeld S., Brown M. S., and Goldstein J. L. (1986) Deletion of clustered O-linked carbohydrates does not impair function of low density lipoprotein receptor in transfected fibroblasts. J. Biol. Chem. 261, 2828–2838 [PubMed] [Google Scholar]
- 31. Davis C. G., Goldstein J. L., Südhof T. C., Anderson R. G. W., Russell D. W., and Brown M. S. (1987) Growth factor homology region in LDL receptor mediates acid-dependent ligand dissociation and receptor recycling. Nature 326, 760–765 10.1038/326760a0 [DOI] [PubMed] [Google Scholar]
- 32. Davis C. G., Lehrman M. A., Russell D. W., Anderson R. G. W., Brown M. S., and Goldstein J. L. (1986) The J. D. mutation in familial hypercholesterolemia: substitution of cysteine for tyrosine in cytoplasmic tail impedes internalization of LDL receptors. Cell 45, 15–24 10.1016/0092-8674(86)90533-7 [DOI] [PubMed] [Google Scholar]
- 33. Davis C. G., van Driel I. R., Russell D. W., Brown M. S., and Goldstein J. L. (1987) The LDL receptor: identification of amino acids in cytoplasmic domain required for rapid endocytosis. J. Biol. Chem. 262, 4075–4082 [PubMed] [Google Scholar]
- 34. Lehrman M. A., Goldstein J. L., Brown M. S., Russell D. W., and Schneider W. J. (1985) Internalization-defective LDL receptors produced by genes with nonsense and frameshift mutations that truncate the cytoplasmic domain. Cell 41, 735–743 10.1016/S0092-8674(85)80054-4 [DOI] [PubMed] [Google Scholar]
- 35. Lehrman M. A., Goldstein J. L., Russell D. W., and Brown M. S. (1987) Duplication of seven exons in LDL receptor gene caused by Alu-Alu recombination in a subject with familial hypercholesterolemia. Cell 48, 827–835 10.1016/0092-8674(87)90079-1 [DOI] [PubMed] [Google Scholar]
- 36. Lehrman M. A., Russell D. W., Goldstein J. L., and Brown M. S. (1986) Exon-Alu recombination deletes 5 kilobases from low density lipoprotein receptor gene, producing null phenotype in familial hypercholesterolemia. Proc. Natl. Acad. Sci. U.S.A. 83, 3679–3683 10.1073/pnas.83.11.3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lehrman M. A., Russell D. W., Goldstein J. L., and Brown M. S. (1987) Alu-Alu recombination deletes splice acceptor sites and produces secreted LDL receptor in a subject with familial hypercholesterolemia. J. Biol. Chem. 262, 3354–3361 [PubMed] [Google Scholar]
- 38. Lehrman M. A., Schneider W. J., Brown M. S., Davis C. G., Elhammer A., Russell D. W., and Goldstein J. L. (1987) The Lebanese allele at the LDL receptor locus: nonsense mutation produces truncated receptor that is retained in endoplasmic reticulum. J. Biol. Chem. 262, 401–410 [PubMed] [Google Scholar]
- 39. Lehrman M. A., Schneider W. J., Südhof T. C., Brown M. S., Goldstein J. L., and Russell D. W. (1985) Mutation in LDL receptor: Alu-Alu recombination deletes exons encoding transmembrane and cytoplasmic domains. Science 227, 140–146 10.1126/science.3155573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Russell D. W., and Wilson J. D. (2013) Steroid 5α-reductase 2 deficiency. in Genetic Steroid Disorders (New M. I., Lekarev L., Parsa A., Yuen T., O'Malley B., and Hammer G., eds), pp. 199–214, Elsevier, San Diego, CA [Google Scholar]
- 41. Südhof T. C., Goldstein J. L., Brown M. S., and Russell D. W. (1985) The LDL receptor gene: a mosaic of exons shared with different proteins. Science 228, 815–822 10.1126/science.2988123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Südhof T. C., Russell D. W., Brown M. S., and Goldstein J. L. (1987) 42-bp element from LDL receptor gene confers end-product repression by sterols when inserted into viral TK promoter. Cell 48, 1061–1069 10.1016/0092-8674(87)90713-6 [DOI] [PubMed] [Google Scholar]
- 43. Hobbs H. H., Lehrman M. A., Yamamoto T., and Russell D. W. (1985) Polymorphism and evolution of Alu sequences in the human low density lipoprotein receptor gene. Proc. Natl. Acad. Sci. U.S.A. 82, 7651–7655 10.1073/pnas.82.22.7651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hobbs H. H., Brown M. S., Goldstein J. L., and Russell D. W. (1986) Deletion of exon encoding cysteine-rich repeat of LDL receptor alters its binding specificity in a subject with familial hypercholesterolemia. J. Biol. Chem. 261, 13114–13120 [PubMed] [Google Scholar]
- 45. Hobbs H. H., Brown M. S., Russell D. W., Davignon J., and Goldstein J. L. (1987) Deletion in LDL receptor gene occurs in majority of French Canadians with familial hypercholesterolemia. N. Engl. J. Med. 317, 734–737 10.1056/NEJM198709173171204 [DOI] [PubMed] [Google Scholar]
- 46. Yamamoto T., Bishop R. W., Brown M. S., Goldstein J. L., and Russell D. W. (1986) Deletion in cysteine-rich region of LDL receptor impedes transport to cell surface in WHHL rabbit. Science 232, 1230–1237 10.1126/science.3010466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hofmann S. L., Russell D. W., Goldstein J. L., and Brown M. S. (1987) Detection of mRNA for LDL receptor in brain and spinal cord of immature and mature rabbits. Proc. Natl. Acad. Sci. U.S.A. 84, 6312–6316 10.1073/pnas.84.17.6312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hofmann S. L., Russell D. W., Brown M. S., Goldstein J. L., and Hammer R. E. (1988) Overexpression of human LDL receptor eliminates LDL from plasma of transgenic mice. Science 239, 1277–1281 10.1126/science.3344433 [DOI] [PubMed] [Google Scholar]
- 49. Peacock S. L., Bates M. P., Russell D. W., Brown M. S., and Goldstein J. L. (1988) The human LDL receptor expressed in Xenopus oocytes: conserved signals for O-linked glycosylation and receptor-mediated endocytosis. J. Biol. Chem. 263, 7838–7845 [PubMed] [Google Scholar]
- 50. Esser V., and Russell D. W. (1988) Transport-deficient mutations in the low density lipoprotein receptor: alterations in the cysteine-rich and cysteine-poor regions of the protein block intracellular transport. J. Biol. Chem. 263, 13276–13281 [PubMed] [Google Scholar]
- 51. Esser V., Limbird L. E., Brown M. S., Goldstein J. L., and Russell D. W. (1988) Mutational analysis of the ligand binding domain of the low density lipoprotein receptor. J. Biol. Chem. 263, 13282–13290 [PubMed] [Google Scholar]
- 52. Cuthbert J. A., Russell D. W., and Lipsky P. E. (1989) Regulation of low density lipoprotein receptor gene expression in human lymphocytes. J. Biol. Chem. 264, 1298–1304 [PubMed] [Google Scholar]
- 53. Andersson S., Davis D. L., Dahlbäck H., Jörnvall H., and Russell D. W. (1989) Cloning, structure, and expression of the mitochondrial cytochrome P-450 sterol 26-hydroxylase, a bile acid biosynthetic enzyme. J. Biol. Chem. 264, 8222–8229 [PubMed] [Google Scholar]
- 54. Pikuleva I. A., Babiker A., Waterman M. R., and Björkhem I. (1998) Activities of recombinant human cytochrome P450c27 (CYP27) which produces intermediates of alternate bile acid biosynthetic pathways. J. Biol. Chem. 273, 18153–18160 10.1074/jbc.273.29.18153 [DOI] [PubMed] [Google Scholar]
- 55. Cali J. J., and Russell D. W. (1991) Characterization of human sterol 27-hydroxylase: a mitochondrial cytochrome P-450 that catalyzes multiple oxidation reaction in bile acid biosynthesis. J. Biol. Chem. 266, 7774–7778 [PubMed] [Google Scholar]
- 56. Cali J. J., Hsieh C. L., Francke U., and Russell D. W. (1991) Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J. Biol. Chem. 266, 7779–7783 [PMC free article] [PubMed] [Google Scholar]
- 57. Björkhem I., Boberg K. M., and Leitersdorf E. (2001) Inborn errors in bile acid biosynthesis and storage of sterols other than cholesterol. in The Metabolic and Molecular Bases of Inherited Disease. (Scriver C. R., Beaudet A. L., Sly W. S., Valle D., Childs B., Kinzler K. W., and Vogelstein B., eds), Eighth Ed., pp. 2961–2988, McGraw-Hill, Inc., New York [Google Scholar]
- 58. Jelinek D. F., Andersson S., Slaughter C. A., and Russell D. W. (1990) Cloning and regulation of cholesterol 7α-hydroxylase, the rate limiting enzyme in bile acid biosynthesis. J. Biol. Chem. 265, 8190–8197 [PMC free article] [PubMed] [Google Scholar]
- 59. Jelinek D. F., and Russell D. W. (1990) Structure of the rat gene encoding cholesterol 7α-hydroxylase. Biochemistry 29, 7781–7785 10.1021/bi00486a001 [DOI] [PubMed] [Google Scholar]
- 60. Cohen J. C., Cali J. J., Jelinek D. F., Mehrabian M., Sparkes R. S., Lusis A. J., Russell D. W., and Hobbs H. H. (1992) Cloning of the human cholesterol 7 α-hydroxylase gene (CYP7) and localization to chromosome 8q11-q12. Genomics 14, 153–161 10.1016/S0888-7543(05)80298-8 [DOI] [PubMed] [Google Scholar]
- 61. Ishibashi S., Schwarz M., Frykman P. K., Herz J., and Russell D. W. (1996) Disruption of cholesterol 7α-hydroxylase gene in mice. I. Postnatal lethality reversed by bile acid and vitamin supplementation. J. Biol. Chem. 271, 18017–18023 10.1074/jbc.271.30.18017 [DOI] [PubMed] [Google Scholar]
- 62. Schwarz M., Lund E. G., Setchell K. D. R., Kayden H. J., Zerwekh J. E., Björkhem I., Herz J., and Russell D. W. (1996) Disruption of cholesterol 7α-hydroxylase gene in mice. II. Bile acid deficiency is overcome by induction of oxysterol 7α-hydroxylase. J. Biol. Chem. 271, 18024–18031 10.1074/jbc.271.30.18024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lund E. G., Guileyardo J. M., and Russell D. W. (1999) cDNA cloning of cholesterol 24-hydroxylase, a regulator of cholesterol homeostasis in the brain. Proc. Natl. Acad. Sci. U.S.A. 96, 7238–7243 10.1073/pnas.96.13.7238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lund E. G., Kerr T. A., Sakai J., Li W.-P., and Russell D. W. (1998) cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J. Biol. Chem. 273, 34316–34348 10.1074/jbc.273.51.34316 [DOI] [PubMed] [Google Scholar]
- 65. Schwarz M., Lund E. G., Lathe R., Björkhem I., and Russell D. W. (1997) Identification and characterization of a mouse oxysterol 7α-hydroxylase cDNA. J. Biol. Chem. 272, 23995–24001 10.1074/jbc.272.38.23995 [DOI] [PubMed] [Google Scholar]
- 66. Li-Hawkins J., Lund E. G., Bronson A. D., and Russell D. W. (2000) Expression cloning of an oxysterol 7α-hydroxylase selective for 24-hydroxycholesterol. J. Biol. Chem. 275, 16543–16549 10.1074/jbc.M001810200 [DOI] [PubMed] [Google Scholar]
- 67. Schwarz M., Wright A. C., Davis D. L., Nazer H., Björkhem I., and Russell D. W. (2000) Expression cloning of 3β-hydroxy-Δ5-C27-steroid oxidoreductase gene of bile acid synthesis and its mutation in progressive intrahepatic cholestasis. J. Clin. Invest. 106, 1175–1184 10.1172/JCI10902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lund E. G., Xie C., Kotti T., Turley S. D., Dietschy J. M., and Russell D. W. (2003) Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J. Biol. Chem. 278, 22980–22988 10.1074/jbc.M303415200 [DOI] [PubMed] [Google Scholar]
- 69. Bauman D. R., Bitmansour A. D., McDonald J. G., Thompson B. M., Liang G., and Russell D. W. (2009) 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl. Acad. Sci. U.S.A. 106, 16764–16769 10.1073/pnas.0909142106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Repa J. J., Lund E. G., Horton J. D., Leitersdorf E., Russell D. W., Dietschy J. M., and Turley S. D. (2000) Disruption of the sterol 27-hydroxylase gene in mice results in hepatomegaly and hypertriglyceridemia: reversal by cholic acid feeding. J. Biol. Chem. 275, 39685–39692 10.1074/jbc.M007653200 [DOI] [PubMed] [Google Scholar]
- 71. Li-Hawkins J., Lund E. G., Turley S. D., and Russell D. W. (2000) Disruption of the oxysterol 7α-hydroxylase gene in mice. J. Biol. Chem. 275, 16536–16542 10.1074/jbc.M001811200 [DOI] [PubMed] [Google Scholar]
- 72. Shea H. C., Head D. D., Setchell K. D., and Russell D. W. (2007) Analysis of HSD3B7 knockout mice reveals that a 3α-hydroxyl stereochemistry is required for bile acid function. Proc. Natl. Acad. Sci. U.S.A. 104, 11526–11533 10.1073/pnas.0705089104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Li-Hawkins J., Gåfvels M., Olin M., Lund E. G., Andersson U., Schuster G., Björkhem I., Russell D. W., and Eggertsen G. (2002) Cholic acid mediates negative feedback regulation of bile acid synthesis. J. Clin. Invest. 110, 1191–1200 10.1172/JCI0216309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kotti T. J., Ramirez D. M., Pfeiffer B. E., Huber K. M., and Russell D. W. (2006) Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc. Natl. Acad. Sci. U.S.A. 103, 3869–3874 10.1073/pnas.0600316103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kotti T., Head D. D., McKenna C. E., and Russell D. W. (2008) Biphasic requirement for geranylgeraniol in hippocampal long-term potentiation. Proc. Natl. Acad. Sci. U.S.A. 105, 11394–11399 10.1073/pnas.0805556105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Setchell K. D. R., Schwarz M., O'Connell N. C., Lund E. G., Davis D. L., Lathe R., Thompson H. R., Weslie Tyson R., Sokol R. J., and Russell D. W. (1998) Identification of a new inborn error in bile acid synthesis: mutation of the oxysterol 7α-hydroxylase gene causes severe neonatal liver disease. J. Clin. Invest. 102, 1690–1703 10.1172/JCI2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Setchell K. D., Heubi J. E., Shah S., Lavine J. E., Suskind D., Al-Edreesi M., Potter C., Russell D. W., O'Connell N. C., Wolfe B., Jha P., Zhang W., Bove K. E., Knisely A. S., Hofmann A. F., Rosenthal P., and Bull L. N. (2013) Genetic defects in bile acid conjugation cause fat-soluble vitamin deficiency. Gastroenterology 144, 945–955 10.1053/j.gastro.2013.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lu T. T., Makishima M., Repa J. J., Schoonjans K., Kerr T. A., Auwerx J., and Mangelsdorf D. J. (2000) Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 6, 507–515 10.1016/S1097-2765(00)00050-2 [DOI] [PubMed] [Google Scholar]
- 79. Chen W., Chen G., Head D. L., Mangelsdorf D. J., and Russell D. W. (2007) Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice. Cell Metab. 5, 73–79 10.1016/j.cmet.2006.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kerr T. A., Saeki S., Schneider M., Schaefer K., Berdy S., Redder T., Shan B., Russell D. W., and Schwarz M. (2002) Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Developmental Cell 2, 713–720 10.1016/S1534-5807(02)00154-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Andersson S., Bishop R. W., and Russell D. W. (1989) Expression cloning and regulation of steroid 5α-reductase, an enzyme essential for male sexual differentiation. J. Biol. Chem. 264, 16249–16255 [PMC free article] [PubMed] [Google Scholar]
- 82. Andersson S., and Russell D. W. (1990) Structural and biochemical properties of cloned and expressed human and rat steroid 5α-reductases. Proc. Natl. Acad. Sci. U.S.A. 87, 3640–3644 10.1073/pnas.87.10.3640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Moore R. J., and Wilson J. D. (1976) Steroid 5α-reductase in cultured human fibroblasts. Biochemical and genetic evidence for two distinct enzyme activities. J. Biol. Chem. 251, 5895–5900 [PubMed] [Google Scholar]
- 84. Andersson S., Berman D. M., Jenkins E. P., and Russell D. W. (1991) Deletion of steroid 5α-reductase 2 gene in male pseudohermaphroditism. Nature 354, 159–161 10.1038/354159a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Thigpen A. E., and Russell D. W. (1992) Four-amino acid segment in steroid 5α-reductase 1 confers sensitivity to finasteride, a competitive inhibitor. J. Biol. Chem. 267, 8577–8583 [PubMed] [Google Scholar]
- 86. Thigpen A. E., Cala K. M., and Russell D. W. (1993) Characterization of Chinese hamster ovary cell lines expressing human steroid 5α-reductase isozymes. J. Biol. Chem. 268, 17404–17412 [PubMed] [Google Scholar]
- 87. Tian H., and Russell D. W. (1997) Expression and regulation of steroid 5α-reductase in the genital tubercle of the fetal rat. Dev. Dyn. 209, 117–126 10.1002/(SICI)1097-0177(199705)209:1%3C117::AID-AJA11%3E3.0.CO%3B2-Z [DOI] [PubMed] [Google Scholar]
- 88. Berman D. M., Tian H., and Russell D. W. (1995) Expression and regulation of steroid 5α-reductase in the urogenital tract of the fetal rat. Mol. Endocrinol. 9, 1561–1570 10.1210/mend.9.11.8584033 [DOI] [PubMed] [Google Scholar]
- 89. Normington K., and Russell D. W. (1992) Tissue distribution and kinetic characteristics of rat steroid 5α-reductase isozymes: evidence for distinct physiological functions. J. Biol. Chem. 267, 19548–19554 [PubMed] [Google Scholar]
- 90. Mahendroo M. S., Cala K. M., Hess D. L., and Russell D. W. (2001) Unexpected virilization in male mice lacking steroid 5α-reductase enzymes. Endocrinology 142, 4652–4662 10.1210/endo.142.11.8510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mahendroo M. S., Porter A., Russell D. W., and Word R. A. (1999) The parturition defect in steroid 5α-reductase type 1 deficient mice is due to impaired cervical ripening. Mol. Endocrinol. 13, 981–992 10.1210/mend.13.6.0307 [DOI] [PubMed] [Google Scholar]
- 92. Geissler W. M., Davis D. L., Wu L., Bradshaw K. D., Patel S., Mendonca B. B., Elliston K. O., Wilson J. D., Russell D. W., and Andersson S. (1994) Male pseudohermaphroditism caused by mutations of testicular 17-hydroxysteroid dehydrogenase 3. Nat. Genet. 7, 34–39 10.1038/ng0594-34 [DOI] [PubMed] [Google Scholar]
- 93. Andersson S., Russell D. W., and Wilson J. D. (1996) 17β-Hydroxysteroid dehydrogenase 3 deficiency. Trends Endocrinol. Metab. 7, 121–126 10.1016/1043-2760(96)00034-3 [DOI] [PubMed] [Google Scholar]
- 94. Biswas M. G., and Russell D. W. (1997) Expression cloning and characterization of oxidative 17β- and 3α-hydroxysteroid dehydrogenases from rat and human prostate. J. Biol. Chem. 272, 15959–15966 10.1074/jbc.272.25.15959 [DOI] [PubMed] [Google Scholar]
- 95. Shaw G., Fenelon J., Sichlau M., Auchus R. J., Wilson J. D., and Renfree M. B. (2006) Role of the alternate pathway of dihydrotestosterone formation in virilization of the Wolffian ducts of the tammar wallaby, Macropus eugenii. Endocrinology 147, 2368–2373 10.1210/en.2005-1251 [DOI] [PubMed] [Google Scholar]
- 96. Chang K. H., Li R., Papari-Zareei M., Watumull L., Zhao Y. D., Auchus R. J., and Sharifi N. (2011) Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 108, 13728–13733 10.1073/pnas.1107898108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Li J., Nagpal P., Vitart V., McMorris T. C., and Chory J. (1996) A role for brassinosteroids in light-dependent development of Arabidopsis. Science 272, 398–401 10.1126/science.272.5260.398 [DOI] [PubMed] [Google Scholar]
- 98. Li J., Biswas M. G., Chao A., Russell D. W., and Chory J. (1997) Conservation of function between mammalian and plant steroid 5α-reductases. Proc. Natl. Acad. Sci. U.S.A. 94, 3554–3559 10.1073/pnas.94.8.3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Cheng J. B., Jacquemin E., Gerhardt M., Nazer H., Cresteil D., Heubi J. E., Setchell K. D. R., and Russell D. W. (2003) Molecular genetics of 3β-hydroxy-Δ5-C27-steroid oxidoreductase deficiency in 16 patients with loss of bile acid synthesis and liver disease. J. Clin. Endocrinol. Metab. 88, 1833–1841 10.1210/jc.2002-021580 [DOI] [PubMed] [Google Scholar]
- 100. Cheng J. B., Motola D. L., Mangelsdorf D. J., and Russell D. W. (2003) De-orphanization of cytochrome P450 2R1, a microsomal vitamin D 25-hydroxylase. J. Biol. Chem. 278, 38084–38093 10.1074/jbc.M307028200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cheng J. B., Levine M. A., Bell N. H., Mangelsdorf D. J., and Russell D. W. (2004) Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc. Natl. Acad. Sci. U.S.A. 101, 7711–7715 10.1073/pnas.0402490101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Al Mutair A. N., Nasrat G. H., and Russell D. W. (2012) Mutation of the CYP2R1 vitamin D 25-hydroxylase in a Saudi Arabian family with severe vitamin D deficiency. J. Clin. Endocrinol. Metab. 97, E2022–2025 10.1210/jc.2012-1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zhu J. G., Ochalek J. T., Kaufmann M., Jones G., and Deluca H. F. (2013) CYP2R1 is a major, but not exclusive, contributor to 25-hydroxyvitamin D production in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, 15650–15655 10.1073/pnas.1315006110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Robishaw J. D., Russell D. W., Harris B. A., Smigel M. D., and Gilman A. G. (1986) Primary structure of the a-subunit of the GTP-binding stimulatory protein of adenylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 83, 1251–1255 10.1073/pnas.83.5.1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chang M.-S., Russell D. W., Uhr J. W., and Vitetta E. S. (1987) Cloning and expression of recombinant, functional, ricin B chain. Proc. Natl. Acad. Sci. U.S.A. 84, 5640–5644 10.1073/pnas.84.16.5640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Chen W.-J., Andres D. A., Goldstein J. L., Russell D. W., and Brown M. S. (1991) cDNA cloning and expression of the peptide-binding β-subunit of rat p21ras farnesyltransferase, the counterpart of yeast DPR1/RAM1. Cell 66, 327–334 10.1016/0092-8674(91)90622-6 [DOI] [PubMed] [Google Scholar]
- 107. Naglich J. G., Metherall J. E., Russell D. W., and Eidels L. (1992) Expression cloning of a diphtheria toxin receptor: identity with a heparin-binding EGF-like growth factor precursor. Cell 69, 1051–1061 10.1016/0092-8674(92)90623-K [DOI] [PubMed] [Google Scholar]
- 108. Tian H., McKnight S. L., and Russell D. W. (1997) Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 11, 72–82 10.1101/gad.11.1.72 [DOI] [PubMed] [Google Scholar]
- 109. Tian H., Hammer R. E., Matsumoto A. M., Russell D. W., and McKnight S. L. (1998) The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev. 12, 3320–3324 10.1101/gad.12.21.3320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zhou Y. D., Barnard M., Tian H., Li X., Ring H. Z., Francke U., Shelton J., Richardson J., Russell D. W., and McKnight S. L. (1997) Molecular characterization of two mammalian bHLH-PAS domain proteins selectively expressed in the central nervous system. Proc. Natl. Acad. Sci. 94, 713–718 10.1073/pnas.94.2.713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yildiz Y., Matern H., Thompson B., Allegood J. C., Warren R. L., Ramirez D. M., Hammer R. E., Hamra F. K., Matern S., and Russell D. W. (2006) Mutation of β-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J. Clin. Invest. 116, 2985–2994 10.1172/JCI29224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Martin E., Schüle R., Smets K., Rastetter A., Boukhris A., Loureiro J. L., Gonzalez M. A., Mundwiller E., Deconinck T., Wessner M., Jornea L., Oteyza A. C., Durr A., Martin J. J., Schöls L., Mhiri C., Lamari F., Züchner S., De Jonghe P., Kabashi E., Brice A., and Stevanin G. (2013) Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am. J. Hum. Genet. 92, 238–244 10.1016/j.ajhg.2012.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Hammer M. B., Eleuch-Fayache G., Schottlaender L. V., Nehdi H., Gibbs J. R., Arepalli S. K., Chong S. B., Hernandez D. G., Sailer A., Liu G., Mistry P. K., Cai H., Shrader G., Sassi C., Bouhlal Y., Houlden H., Hentati F., Amouri R., and Singleton A. B. (2013) Mutations in GBA2 cause autosomal-recessive cerebellar ataxia with spasticity. Am. J. Hum. Genet. 92, 245–251 10.1016/j.ajhg.2012.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Cheng J. B., and Russell D. W. (2004) Mammalian wax biosynthesis. I. Identification of two mammalian fatty acyl-coenzyme A reductases with different substrate specificities and tissue distributions. J. Biol. Chem. 279, 37789–37797 10.1074/jbc.M406225200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Cheng J. B., and Russell D. W. (2004) Mammalian wax biosynthesis. II. Expression cloning of mammalian wax synthase cDNAs encoding a member of the diacyl glycerol acyl transferase family. J. Biol. Chem. 279, 37798–37807 10.1074/jbc.M406226200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Janowski B. A., Willy P. J., Devi T. R., Falck J. R., and Mangelsdorf D. J. (1996) An oxysterol signalling pathway mediated by the nuclear receptor LXRa. Nature 383, 728–731 10.1038/383728a0 [DOI] [PubMed] [Google Scholar]
- 117. Björkhem I., Diczfalusy U., and Lütjohann D. (1999) Removal of cholesterol from extrahepatic sources by oxidative mechanisms. Curr. Opin. Lipidol. 10, 161–165 10.1097/00041433-199904000-00010 [DOI] [PubMed] [Google Scholar]
- 118. Brown M. S., and Goldstein J. L. (1974) Suppression of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity and inhibition of growth of human fibroblasts by 7-ketocholesterol. J. Biol. Chem. 249, 7306–7314 [PubMed] [Google Scholar]
- 119. Kandutsch A. A., and Chen H. W. (1974) Inhibition of sterol synthesis in cultured mouse cells by cholesterol derivatives oxygenated in the side chain. J. Biol. Chem. 249, 6057–6061 [PubMed] [Google Scholar]
- 120. Brown M. S., and Goldstein J. L. (2009) Cholesterol feedback: from Schoenheimer's bottle to Scap's MELADL. J. Lipid Res. 50, S15–S27 10.1194/jlr.R800054-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Radhakrishnan A., Sun L.-P., Kwon H. J., Brown M. S., and Goldstein J. L. (2004) Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol. Cell 15, 259–268 10.1016/j.molcel.2004.06.019 [DOI] [PubMed] [Google Scholar]
- 122. Radhakrishnan A., Ikeda Y., Kwon H. J., Brown M. S., and Goldstein J. L. (2007) Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to INSIG. Proc. Natl. Acad. Sci. U.S.A. 104, 6511–6518 10.1073/pnas.0700899104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Dennis E. A. (2016) Liberating chiral lipid mediators, inflammatory enzymes, and LIPID MAPS from biological grease. J. Biol. Chem. 291, 24431–24448 10.1074/jbc.X116.723791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. McDonald J. G., Smith D. D., Stiles A. R., and Russell D. W. (2012) A comprehensive method for extraction and quantitative analysis of sterols and secosteroids from human plasma. J. Lipid Res. 53, 1399–1409 10.1194/jlr.D022285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Diczfalusy U., Olofsson K. E., Carlsson A. M., Gong M., Golenbock D. T., Rooyackers O., Fläring U., and Björkbacka H. (2009) Marked up-regulation of cholesterol 25-hydroxylase expression by lipopolysaccharide. J. Lipid Res. 50, 2258–2264 10.1194/jlr.M900107-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Park K., and Scott A. L. (2010) Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J. Leukoc. Biol. 88, 1081–1087 10.1189/jlb.0610318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hannedouche S., Zhang J., Yi T., Shen W., Nguyen D., Pereira J. P., Guerini D., Baumgarten B. U., Roggo S., Wen B., Knochenmuss R., Noël S., Gessier F., Kelly L. M., Vanek M., Laurent S., Preuss I., Miault C., Christen I., Karuna R., Li W., Koo D. I., Suply T., Schmedt C., Peters E. C., Falchetto R., Katopodis A., Spanka C., Roy M. O., Detheux M., Chen Y. A., Schultz P. G., Cho C. Y., Seuwen K., Cyster J. G., and Sailer A. W. (2011) Oxysterols direct immune cell migration via EBI2. Nature 475, 524–527 10.1038/nature10280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Liu C., Yang X. V., Wu J., Kuei C., Mani N. S., Zhang L., Yu J., Sutton S. W., Qin N., Banie H., Karlsson L., Sun S., and Lovenberg T. W. (2011) Oxysterols direct B-cell migration through EBI2. Nature 475, 519–523 10.1038/nature10226 [DOI] [PubMed] [Google Scholar]
- 129. Pereira J. P., Kelly L. M., and Cyster J. G. (2010) Finding the right niche: B-cell migration in the early phases of T-dependent antibody responses. Int. Immunol. 22, 413–419 10.1093/intimm/dxq047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Chan T. D., Gardam S., Gatto D., Turner V. M., Silke J., and Brink R. (2010) In vivo control of B-cell survival and antigen-specific B-cell responses. Immunol. Rev. 237, 90–103 10.1111/j.1600-065X.2010.00942.x [DOI] [PubMed] [Google Scholar]
- 131. Blanc M., Hsieh W. Y., Robertson K. A., Kropp K. A., Forster T., Shui G., Lacaze P., Watterson S., Griffiths S. J., Spann N. J., Meljon A., Talbot S., Krishnan K., Covey D. F., Wenk M. R., Craigon M., Ruzsics Z., Haas J., Angulo A., Griffiths W. J., Glass C. K., Wang Y., and Ghazal P. (2013) The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38, 106–118 10.1016/j.immuni.2012.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Liu S. Y., Aliyari R., Chikere K., Li G., Marsden M. D., Smith J. K., Pernet O., Guo H., Nusbaum R., Zack J. A., Freiberg A. N., Su L., Lee B., and Cheng G. (2013) Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38, 92–105 10.1016/j.immuni.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Reboldi A., Dang E. V., McDonald J. G., Liang G., Russell D. W., and Cyster J. G. (2014) Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 345, 679–684 10.1126/science.1254790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Dang E. V., McDonald J. G., Russell D. W., and Cyster J. G. (2017) Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell 171, 1057–1071 10.1016/j.cell.2017.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Nevius E., Pinho F., Dhodapkar M., Jin H., Nadrah K., Horowitz M. C., Kikuta J., Ishii M., and Pereira J. P. (2015) Oxysterols and EBI2 promote osteoclast precursor migration to bone surfaces and regulate bone mass homeostasis. J. Exp. Med. 212, 1931–1946 10.1084/jem.20150088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Bowling N., Matter W. F., Gadski R. A., McClure D. B., Schreyer T., Dawson P. A., and Vlahos C. J. (1996) LY295427, a novel hypocholesterolemic agent, enhances [3H]25-hydroxycholesterol binding to liver cytosolic proteins. J. Lipid Res. 37, 2586–2598 [PubMed] [Google Scholar]
- 137. Bensch W. R., Gadski R. A., Bean J. S., Beavers L. S., Schmidt R. J., Perry D. N., Murphy A. T., McClure D. B., Eacho P. I., Breau A. P., Archer R. A., and Kauffman R. F. (1999) Effects of LY295427, a low-density lipoprotein (LDL) receptor up-regulator, on LDL receptor gene transcription and cholesterol metabolism in normal and hypercholesterolemic hamsters. J. Pharmacol. Exp. Ther. 289, 85–92 [PubMed] [Google Scholar]
- 138. Janowski B. A., Shan B., and Russell D. W. (2001) The hypercholesterolemic agent LY295427 reverses suppression of sterol regulatory element-binding protein processing mediated by oxysterols. J. Biol. Chem. 276, 45408–45416 10.1074/jbc.M108348200 [DOI] [PubMed] [Google Scholar]
- 139. Shibata N., Carlin A. F., Spann N. J., Saijo K., Morello C. S., McDonald J. G., Romanoski C. E., Maurya M. R., Kaikkonen M. U., Lam M. T., Crotti A., Reichart D., Fox J. N., Quehenberger O., Raetz C. R., Sullards M. C., Murphy R. C., Merrill A. H. Jr., Brown H. A., Dennis E. A., Fahy E., Subramaniam S., Cavener D. R., Spector D. H., Russell D. W., and Glass C. K. (2013) 25-Hydroxycholesterol activates the integrated stress response to reprogram transcription and translation in macrophages. J. Biol. Chem. 288, 35812–35823 10.1074/jbc.M113.519637 [DOI] [PMC free article] [PubMed] [Google Scholar]