

Abstract
Peptides represent a promising source of new medicines, but improved technologies are needed to facilitate discovery and optimization campaigns. In particular, longer peptides with multiple disulfide bridges are challenging to produce, and producing large numbers of structurally related variants is dissuasively costly and time-consuming. The principal cost and time drivers are the multiple column chromatography purification steps that are used during the multistep chemical synthesis procedure, which involves both ligation and oxidative refolding steps. In this study, we developed a method for multiplex parallel synthesis of complex peptide analogs in which the structurally variant region of the molecule is produced as a small peptide on a 384-well synthesizer with subsequent ligation to the longer, structurally invariant region and oxidative refolding carried out in-well without any column purification steps. To test the method, we used a panel of 96 analogs of the chemokine RANTES (regulated on activation normal T cell expressed and secreted)/CCL5 (69 residues, two disulfide bridges), which had been synthesized using standard approaches and characterized pharmacologically in an earlier study. Although, as expected, the multiplex method generated chemokine analogs of lower purity than those produced in the original study, it was nonetheless possible to closely match the pharmacological attributes (anti-HIV potency, capacity to elicit G protein signaling, and capacity to elicit intracellular receptor sequestration) of each chemokine analog to reference data from the earlier study. This rapid, low-cost approach has the potential to support discovery and optimization campaigns based on analogs of other chemokines as well as those of other complex peptide and small protein targets of a similar size.
Keywords: peptide chemical synthesis, chemokine, protein engineering, C-C chemokine receptor type 5 (CCR5), HIV, drug discovery, high-throughput screening (HTS)
Introduction
Peptides and small proteins have the potential to become an important source of new medicines, but progress would be facilitated by technological advances related to procedures for the discovery and optimization of promising compounds (1–3). For smaller peptides (up to 15 residues in length), small-molecule medicinal chemistry principles can be readily applied. Production and screening of large synthetic libraries is now a mature technology, and identified hits can be optimized by synthesizing and screening families of structurally related molecules (2, 4, 5). Similar approaches are not currently possible for longer peptides and small proteins, however. Although the introduction of the native chemical ligation approach (6) has made chemical synthesis of longer peptides and small proteins feasible (7, 8), the multistep synthesis strategies involved, which feature several intermediate column chromatography purifications, are not amenable to parallel synthesis.
Libraries of longer peptides and small proteins can be produced recombinantly either as soluble molecules (9) or in surface display systems (10–12), but it is challenging to explore beyond the set of 20 coded amino acids using recombinant approaches, and, despite advances (9), the subcloning, expression, and purification steps required to validate and characterize candidate hits remain onerous and difficult to perform in parallel.
Chemokines are small disulfide-bridged proteins typically 70–130 residues in length (13). The structure–activity relationships governing their interaction with chemokine receptors have been studied using chemical synthesis (14, 15), recombinant approaches (16, 17), and semisynthesis (18). Although many different studies have been performed (19, 20), making a major contribution to the delineation of a set of common rules for chemokine–chemokine receptor engagement (21, 22), only those carried out on one chemokine receptor, CCR5, yielded analogs with improvements sufficient to encourage follow-up work.
CCR5 is the major HIV coreceptor (23), and its native chemokine ligands have modest but detectable anti-HIV activity (24). One of the native ligands, RANTES/CCL5, has been the focus of intense discovery and optimization activity. Initially, AOP-RANTES, an analog identified previously as having enhanced anti-HIV potency (18), was optimized by a medicinal chemistry approach in which a total of 45 rationally designed variants were chemically synthesized and evaluated, with beneficial modifications at different parts of the pharmacophore combined. The most potent analog, PSC-RANTES, has been shown to have anti-HIV potency 50-fold higher than the starting analog (25) and full efficacy in a stringent animal model of HIV transmission (26). Subsequently, a chemokine phage display approach (27, 28) involving iterative rounds of library design, selection, chemical synthesis, and pharmacological evaluation of 120 hits led to the identification of several fully recombinant chemokine analogs with potency comparable with that of PSC-RANTES together with an improved pharmacological profile (29). One of these analogs, 5P12-RANTES, has been taken into clinical development as an HIV prevention agent (30–32).
This body of work provided has valuable insights into the structural dynamics of CCR5 modulation by its ligands, indicating that sites in the N-terminal region of RANTES/CCL5 can be modified to independently affect three different pharmacological parameters: anti-HIV potency, CCR5 agonist activity, and the capacity to elicit intracellular sequestration activity, with the latter two parameters particularly sensitive to modifications at positions corresponding to Tyr3, Ser5, Asp6, and Thr7 of the parent protein (25). Crystallographic studies of one of the analogs from the panel in complex with CCR5 provided a structural explanation for these observations, indicating that these residues make contact with parts of the transmembrane core of CCR5 that are associated with conformational changes leading to receptor activation (33).
It is enticing to envisage attempting to replicate the success with RANTES/CCL5 analogs on CCR5 on another chemokine/chemokine receptor system, but because synthesis of hundreds of chemokine analogs is dissuasively costly and time-consuming using current methods, we set out in this study to significantly streamline the workflow, eliminating the major cost-driving and time-consuming steps by setting up a multiplex parallel synthesis approach without column chromatography purification (Fig. 1). We evaluated the approach by using a set of 96 previously characterized RANTES/CCL5 analogs (29) and comparing their pharmacological properties with those obtained using individually synthesized and purified analogs in the original study.
Figure 1.

Design and evaluation of a streamlined process for rapid and inexpensive production of large panels of chemokine analogs. Until now (left panel), work exploring chemokine structure–activity relationships has been based on chemical synthesis of individual molecules. In this study (right panel), we designed and evaluated a process for the multiplex production of chemokine analogs. Black boxes, steps performed in series; red boxes, multiplex steps; blue boxes, column chromatography steps; green boxes, multiplex procedures used to replace column chromatography steps.
Results
In-well synthesis and folding of chemokine analogs
The streamlined synthesis strategy involves producing large batches of the C-terminal fragment, RANTES(11–68) (core fragment, unchanged across the panel of analogs), and using it for ligation reactions performed in parallel with multiplex-synthesized fragments corresponding to the N-terminal regions of each of the target analogs in the panel.
Synthesis of the core fragment batch
Two batches of the core chemokine fragment RANTES(11–68) were prepared, one as a ligation product consisting of a RANTES(11–34)-SEA2 thioester fragment synthesized by Fmoc chemistry to the RANTES(35–68) fragment synthesized by Boc chemistry, using a thiazolidine protection approach to block Cys11 during ligation (34) (core fragment batch 1), the other a single long fragment synthesized by Fmoc chemistry (core fragment batch 2).
MALDI MS analysis of core fragment batch 1 revealed a mass consistent with that of the target product (expected mass, 6812 Da; observed mass, 6806 Da). For core fragment batch 2, MALDI MS analysis revealed a mass (6832 Da) consistent with the target product carrying an oxidized methionine residue (expected mass, 6828 Da). Complete oxidation of Met67 to Met(O)67 in this fragment was confirmed by MALDI MS analysis of tryptic peptides (data not shown). Oxidation of methionine residues is a phenomenon that has been observed during Fmoc synthesis of longer peptide fragments (35).
Multiplex synthesis of the N-terminal fragments
96 N-terminal-SEA peptides, corresponding to residues 0–10 of a group of 96 previously described RANTES/CCL5 analogs, were synthesized in parallel in individual wells on a multiplex synthesis plate. 87 of the 96 reactions yielded products corresponding to peptides with the expected masses, with nine syntheses yielding products corresponding to capped truncated peptides (Table S1).
Multiplex in-well ligation and folding
87 chemokine analogs were synthesized in parallel by performing in-well native chemical ligation between the core fragment with each of the synthesized N-terminal SEA-thioester peptides, 51 using core fragment batch 1 and 36 using core fragment batch 2. Following a membrane filtration step to remove excess unreacted N-terminal peptide and thiol scavengers, reaction products were subjected to an in-well folding step and a final in-well desalting step. For the 87 reactions, all but two (those for 7P14-RANTES and M7-RANTES) yielded exploitable material. These samples were analyzed by MALDI MS (Table S2, A and B) and RP-HPLC (Fig. S1, A and B).
Despite the absence of any chromatography purification steps, the multiplex in-well ligation and folding reactions all yielded products with a single observed mass corresponding to that of the folded target product (reactions using core fragment batch 1, Table S1A), a single observed mass corresponding to that of the folded target product incorporating Met(O)67 (a subset of the reactions using core fragment batch 2, Table S1B), or two observed masses, one corresponding to that of the folded target product and the other corresponding to that of the folded target product incorporating Met(O)67 (the remainder of reactions using core fragment batch 2, Table S1B).
Similarly, analysis by RP-HPLC revealed either a single major peak (reactions using core fragment batch 1, Fig. S1A) or two major peaks (reactions using core fragment batch 2, e.g. 2P14-RANTES or 8P2-RANTES, Fig. S1B). Further analysis of the double major peaks in two representative wells (2P14-RANTES and 8P2-RANTES) showed that, in both cases, the peak with the longer retention time had a mass consistent with that of the target protein, and the peak with the shorter retention time had a mass consistent with that of the target protein incorporating a single oxidized methionine, Met(O)67 (Fig. S2). The product with nonoxidized Met67 derived from full Met(O)67 core fragment batch 2 in these syntheses is presumably the consequence of a partial reduction of the oxidized Met(O)67 residue under the reducing conditions used for the ligation reaction.
Retention time analysis with reference standards
Six selected wells, representing syntheses of differing quality in the preliminary analysis (5P12-RANTES, 7P1-RANTES, 5P6-RANTES 5P7-RANTES, 5P2-RANTES, and 6P9-RANTES), were subjected to further analysis by RP-HPLC. Retention times of the major peaks were compared with those of the corresponding reference standard chemokines (Fig. 2).
Figure 2.

Comparison of RP-HPLC retention times of multiplex synthesis products with reference standard chemokine analogs. The RP-HPLC profiles of representative multiplex synthesis products (in-well mixture) were compared with those of reference standard chemokine analogs (ref. std) (29) using the same analytical column and under identical conditions. Peak retention times for major peaks are noted for each sample. A, representative samples produced using core fragment batch 1. B, representative samples produced using core fragment batch 2.
For the syntheses using core fragment batch 1, the single major peak in each case had an elution profile consistent with that of the reference standard sample. For the syntheses using core fragment batch 2, in the cases where double major peaks were apparent (5P12-RANTES and 7P1-RANTES), the peaks with the longer retention times had elution profiles consistent with those of the reference standard samples. In the case where only a single major peak was apparent (5P6-RANTES), its retention time was not consistent with that of the reference standard sample but had the reduced retention time characteristic of a Met67(O) variant. A shoulder peak showed a retention time consistent with that of the reference standard sample, indicating that the nonoxidized Met67 variant may have been present as a minority product.
Estimation of yields
To estimate the range of yields in this multiplex synthesis, we made use of the same six wells, which included syntheses providing both high yields (e.g. 7P1-RANTES and 5P7-RANTES) and lower yields (e.g. 5P6-RANTES and 5P2-RANTES) and used HPLC analysis software to estimate percentage purity, based on peak area, of the peaks corresponding to the target product. Because modifications at the C terminus of RANTES/CCL5 do not affect pharmacological activity (36), we opted to consider peaks corresponding to the Met67(O) congener as part of the total target product yield when estimating final yields per well. For the same six wells, we also estimated total protein content by dissolving the contents of the well in 250 μl of water and measuring absorbance at 280 nm, making use of the predicted extinction coefficients of the analogs.
The in-well ligation and folding procedure purities in this group of six wells provided target protein purities spanning a range of 17–56%, corresponding to yields of ∼7–14% with respect to the C-terminal target fragment. For well contents dissolved in 250 μl of water, the estimated concentration of target protein ranged from 26–56 μm (Table S3). We opted to define a nominal concentration of 50 μm for each target protein, noting that this concentration was likely to be an overestimate for certain well mixtures whose analytical RP-HPLC traces (Fig. S1) indicated the lowest levels of purity and yield (e.g. M44-RANTES, 7P19-RANTES, and M23-RANTES).
Pharmacological evaluation of chemokine analogs
In the initial study of this panel of chemokine analogs (29), pharmacological activity was determined according to three key criteria: anti-HIV potency, determined in an R5-dependent envelope-mediate cell fusion assay; CCR5 agonist activity, an undesirable characteristic from a safety perspective, determined using a calcium flux assay; and CCR5 sequestration activity, which, for certain chemokine analogs, is crucial for anti-HIV activity (25), using a cell surface antibody binding assay. In this study, to reduce the time and cost burden, we opted to replace the full-range nine-point dose–response curves in the cell fusion assay with a four point dose–response procedure, adapt the calcium flux assay from a 96-well plate format to a 384-well format, and replace the costly mAb-based CCR5 sequestration assay with a technique based on bystander BRET (37).
Anti-HIV potency
Each chemokine analog was tested for anti-HIV potency using a cell fusion assay, scoring each compound for its capacity to block cell fusion at each of four estimated concentrations: 1 nm, 4.6 nm, 21.5 nm, and 100 nm. We then divided the compounds into five groups: 1, complete inhibition not achieved at any concentration; 2, complete inhibition only achieved at the highest concentration (100 nm); 3, complete inhibition achieved at the two highest concentrations (21.5 nm and 100 nm); 4, complete inhibition achieved at three concentrations (4.6 nm, 21.5 nm, and 100 nm); and 5, complete inhibition achieved at all four concentrations (Fig. 3A). When the multiplex-synthesized analogs were divided into anti-HIV potency groups in this way and compared with the pIC50 values obtained using the corresponding reference standard chemokine analogs in the earlier study (29), a good correlation was obtained (Fig. 3B), with analogs in groups 1 to 5 corresponding to pIC50 values from the original study spanning the ranges 7.2–8.2, 7.8–9.6, 8.6–10.0, 9.4–10.7, and 10.0–11.0, respectively. This indicates that screening multiplex-synthesized chemokine analogs with this method is sufficient to identify the most potent anti-HIV chemokines from a panel as well as to stratify the less potent analogs with a reasonably high resolution.
Figure 3.
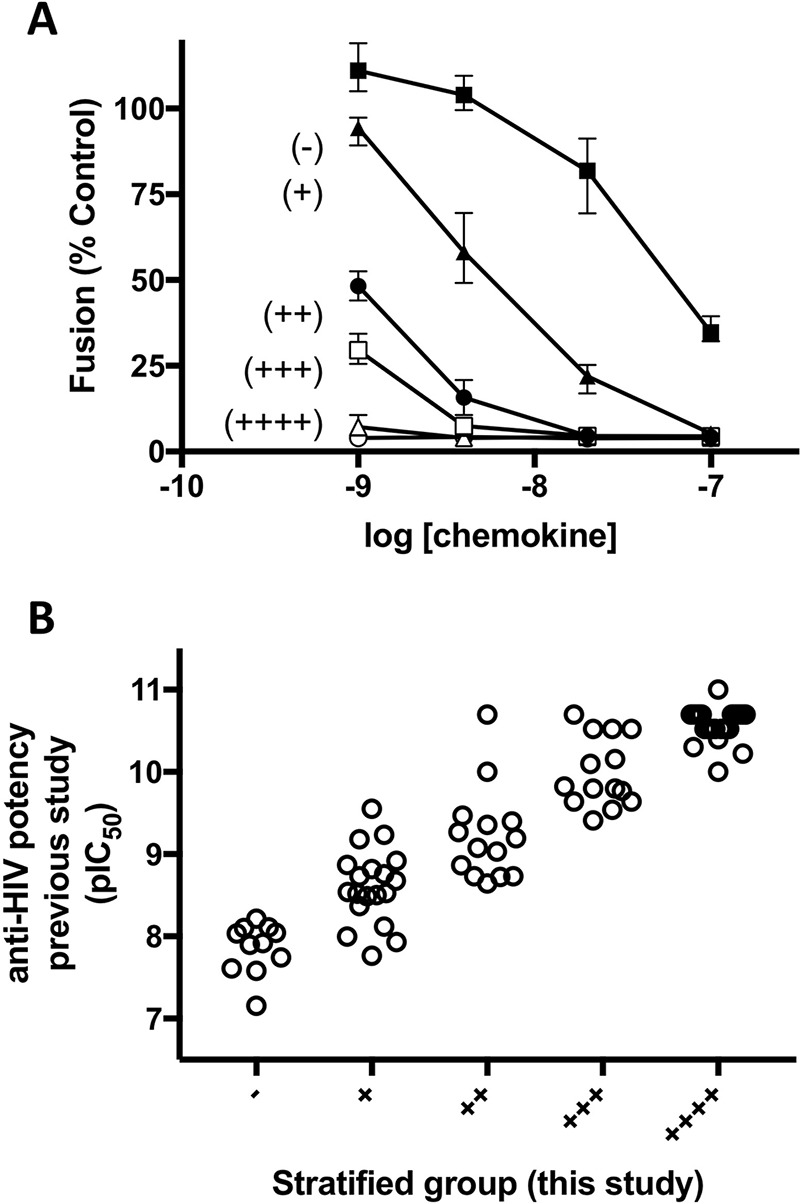
Comparison of the anti-HIV potencies of multiplex-synthesized chemokine analogs with those previously obtained for the corresponding reference standard samples. A, examples of data used for stratification; R5-tropic envelope-dependent cell fusion assays were performed at four different nominal concentrations, with samples ranked from (−) to (++++) according to the number of concentrations at which complete inhibition of cell fusion was achieved. Symbols indicate mean cell fusion activity ± range (n = 3). Black squares, M9-RANTES; black triangles, M19-RANTES; black circles, 8P5-RANTES; white squares, 8P6-RANTES; white triangles, M21-RANTES; white circles, 5P12-RANTES reference standard. B, the potency of each compound produced and stratified in this study compared with the potency (pIC50) of the corresponding molecule produced and tested in the original study (29).
CCR5 agonist assay
We next tested the panel of multiplex-synthesized chemokine analogs on a plate-based G protein signaling assay similar to that used in the earlier study (29), except that CCR5 was expressed in a human embryonic kidney (HEK) cell background rather than a HeLa cell background. Compounds were tested a single Emax concentration (300 nm), and the signal obtained was expressed as a percentage of the signal obtained in the same experiment using a reference standard samples of the CCR5 superagonist PSC-RANTES (100% signaling) and the nonsignaling ligand 5P12-RANTES (0% signaling). Expressed on this scale (Fig. S3), compounds ranged in activity between −5 and 200%. Compounds were divided into three groups: absent or low signaling (0–25% signal), medium signaling (25–100% signal) and high signaling (over 100% signal). Divided in this way and compared with the Emax values obtained using the corresponding reference standard chemokine analogs in the earlier study (29), a good correlation was obtained (Fig. 4), indicating that screening multiplex-synthesized chemokine analogs for G protein signaling is sufficient to stratify multiplex-synthesized chemokine analogs nonsignaling, medium-signaling, and high-signaling groups with reasonable accuracy, although we note that the medium-signaling group in this study contains a number of analogs considered nonsignaling molecules and three compounds belonging to the group of high-signaling molecules in the earlier study (29). It has been noted that G protein–coupled receptor signaling responses to agonists can vary to some extent according to the cellular background used (38), and this is the most likely explanation for the discrepancy between the results of this study and from the original work.
Figure 4.

Comparison of the calcium signaling activity of multiplex-synthesized chemokine analogs with those previously obtained for the corresponding reference standard samples. The calcium signaling assay of each multiplex-synthesized chemokine analog was determined at an Emax concentration (300 nm) in a 384-well plate–based assay (Fig. S3). According to the results, analogs were stratified into three groups (low, medium, and high signaling). This figure shows the distribution of signaling efficacies determined using molecules produced and tested in the original study (29) for the analogs in each group.
Cell surface downmodulation assay
Our final analysis involved measuring the capacity of the multiplex-synthesized chemokine analogs to elicit steady-state downmodulation of CCR5. In this assay, we made use of a BRET biosensor cell line in which CCR5 C-terminally tagged with a derivative of Renilla luciferase (Rluc8) is coexpressed with YFP fused to the prenylation CAAX box of KRas to direct plasma membrane expression (37). Proximity between CCR5-Rluc and cell surface YFP generates a BRET signal that is lost upon receptor internalization. BRET signals in individual wells were recorded after 25-min incubation with multiplex-synthesized chemokine analogs at a single Emax concentration (300 nm), and the level of receptor internalization was expressed as a percentage of the internalization signal obtained by reference standard samples of the CCR5 superagonist PSC-RANTES (100% internalization) and the noninternalizing ligand 5P12-RANTES (0% signaling). Expressed on this scale (Fig. S4), compounds ranged in activity between −10% and 115%. Compounds were divided into three groups: absent or low downmodulation (0–25%), medium downmodulation (25–80%), and high downmodulation (over 80%). Divided in this way and compared with the values obtained using the corresponding reference standard chemokine analogs in the earlier study (29), a good correlation was obtained (Fig. 5), indicating that screening multiplex-synthesized chemokine analogs for CCR5 downmodulation using this method is suitable for rapidly and inexpensively stratifying multiplex-synthesized chemokine analogs into nonsignaling, medium-signaling, and high-signaling groups.
Figure 5.

Comparison of the CCR5 downmodulation activity of multiplex-synthesized chemokine analogs with those previously obtained for the corresponding reference standard samples. The CCR5 downmodulation activity of each multiplex-synthesized chemokine analog was determined at an Emax concentration (300 nm) in a BRET bystander assay (Fig. S4). According to the results, analogs were stratified into three groups (low, medium and high downmodulation). This figure shows the distribution of downmodulation efficacies determined using molecules produced and tested in the original study (29) for the analogs in each group.
Discussion
Previous success using both chemical synthesis-based (25) and recombinant (29) protein engineering approaches to enhance the pharmacological properties of RANTES/CCL5 highlights the value of exploring shape space in the key pharmacophore region located at the N terminus of the protein. The chemokine analogs that were identified have been valuable not only in elucidation of the pharmacology and cell biology of CCR5 (36, 39, 40) and the structural biology of chemokine receptors (33) but also in understanding the HIV transmission mechanism and its prevention (26) and the identification of a candidate drug for clinical development as a new anti-HIV medicine (30, 31).
The relative lack of success in pharmacological optimization achieved using other chemokine/chemokine receptor systems (20, 28) could, in theory, be due to unknown unique properties of CCR5 compared with other chemokine receptors. However, because chemokines have a broadly shared structure–activity relationship for engaging their receptors (13, 22), a more likely explanation is that insufficient shape space was explored. Although well over 100 RANES/CCL5 analogs differing only in the key N-terminal pharmacophore region were synthesized and evaluated during the discovery campaigns on CCR5, work on other chemokine receptors has typically involved either individual analogs or small groups of less than ten structurally related variants. Researchers involved in these studies were likely to have been dissuaded from conducting a wider shape space exploration because of the cost and time required to produce each analog.
In this study, we set out to develop and validate a platform for multiplex synthesis and evaluation of panels of chemokine analogs to minimize the time and expense required (Fig. 1). As expected, the multiplex approach has some drawbacks compared with the standard procedures. Eleven of 96 analogs were lost during synthesis, and the variable final purities and yields meant target product concentrations were approximated rather than determined (Table S3). It was also necessary to work on the assumption that the contaminants present in the unpurified samples would not detectably enhance or reduce pharmacological activity. Nonetheless, it was possible to fully recapitulate the pharmacological profiles described previously for analogs synthesized and purified individually using three independent criteria: anti-HIV potency (Fig. 3), CCR5 agonist activity (Fig. 4), and CCR5 sequestration activity (Fig. 5). Importantly, this represents a level of structure–activity information that would be sufficient to guide the discovery analogs such as PSC-RANTES and 5P12-RANTES. Our approach could be used to discover new anti-HIV chemokines with pharmacological profiles better than these benchmark compounds, but, perhaps more importantly, it could be used to provide a rapid and inexpensive route to optimization of analogs based on other chemokine/chemokine receptor systems.
Beyond chemokines, this approach could, in principle, be used to discover and optimize analogs of other peptides and small proteins that are too large to be compatible with current peptide library medicinal chemistry approaches (2, 4, 5). Although, in its current form, the approach is limited to introducing diversity into the N-terminal region of target molecules, it would be possible to adapt the technique to enable multiplex production of analogs of peptides and small protein analogs such as anaphylatoxin C5a (41), for which the key pharmacophore region is located at the C terminus rather than the N terminus. This could be achieved by producing the larger core fragment incorporating a C-terminal thioester by either chemical synthesis or biosynthesis using expressed protein ligation technology (42) prior to in-well ligation with multiplex-synthesized C-terminal peptides carrying an N-terminal cysteine residue.
Other strategies for reducing the time and cost of production of peptides and small proteins can be envisaged. Loibl et al. (43) recently published an approach with a similar aim to ours, i.e. streamlining chemical synthesis of longer peptides and small proteins by eliminating column chromatography steps. Although it was only exemplified using nine different target molecules that do not require oxidative refolding, this resin capture–based approach is compatible with multi-fragment ligation assembly and provided higher levels of final purity than our method. It is hampered, however, by the requirement to retain an N-terminal affinity tag and a hydrazide-modified C terminus on the target molecule, the relatively slow reaction rate of on-resin ligation, and the losses inherent in multiple resin capture–release steps (43). In conclusion, the approach we present in this study has the potential to rapidly and inexpensively guide programs for the discovery and optimization of pharmacologically active longer peptides and small proteins, helping to enable this emerging drug class to reach its full potential (1–3).
Experimental procedures
Synthesis of core fragment RANTES CCL5(11–68)
Two different synthesis batches were used for this study. One was prepared by native chemical ligation of the RANTES/CCL5(34–68) peptide, synthesized by Boc chemistry on an adapted ABI 433 peptide synthesizer, to a thiazolidine (Thz)11-RANTES/CCL5(12–33) C-terminal thioester peptide, synthesized using Fmoc chemistry on the Prelude synthesizer, followed by deprotection of Thz11 to generate the N-terminal Cys11 residue (34) (core fragment batch 1) and the other as a full-length fragment using Fmoc chemistry on a Prelude (Protein Technologies Inc.) synthesizer (core fragment batch 2). Following synthesis, core fragments were purified by reverse-phase HPLC (RP-HPLC) using the Waters 1525 system with a Vydac 250 × 22 mm C8 column and subjected to MALDI MS analysis on an AB Sciex 4800 MALDI TOF/TOF mass spectrometer (linear positive mode using 2,5-dihydroxybenzoic acid as a matrix).
Plate-based synthesis of N-terminal fragments
Fragments corresponding to the first 11 N-terminal residues of a set of previously identified RANTES/CCL5 analogs (29) were synthesized at 2 μmol scale on a MultiPep RSi 384-well peptide synthesizer (Intavis) synthesizer using bis(2-sulfanylethyl)amino (SEA) resin prepared according to the methods of Ollivier et al. (44) so that cleavage would yield fragments in the C-terminal thioester format required for the in-well native chemical ligation step. Following resin cleavage, the crude product in each well was dissolved in 500 μl of water/acetonitrile (1:1) containing 1% TFA. A volume of this solution corresponding to an estimated 0.6-μmol peptide (150 μl) was transferred to a 2-ml deep-well 96-well polypropylene plate and lyophilized.
In-well native chemical ligation
In-well native chemical ligation was carried out using an estimated 6-fold excess (0.6 μmol) of the N-terminal SEA fragment over the core fragment (0.1 μmol). A 1 mm solution of core fragment was prepared in ligation buffer (0.2 m sodium phosphate buffer (pH 7.2) containing 6 m guanidine hydrochloride, 50 mm methionine, 0.1 m 4-mercaptophenylacetic acid, and 0.1 m tris(2-carboxyethyl)phosphine). 100 μl of this solution was added to each well containing lyophilized crude N-terminal SEA fragment synthesis product; plates were sealed, and the reaction mixtures were stirred overnight at 37 °C.
Multiplex size exclusion step
Following ligation, excess unreacted N-terminal peptide and the other constituents of the ligation buffer were removed using a multiplex size exclusion step. Ligation mixtures from wells were applied to Microcon tubes (Millipore) with 10-kDa cutoff membranes that had been prewet with a solution of 6 m guanidine hydrochloride. The tubes were then centrifuged for 10 min at 14,000 × g, and the flow-through was discarded. Three washing steps were carried out by applying 150 μl of 6 m guanidine hydrochloride solution and centrifuging for 10 min at 14,000 × g, and then retentates were supplemented with 50 μl of a solution of 0.28 m tris(2-carboxyethyl)phosphine dissolved in 6 m guanidine hydrochloride (pH 5.3) and left at ambient temperature for 30 min without agitation. Eight further 6 m guanidine hydrochloride solution washing steps were carried out, and then the retentate (100 μl) was recovered by inverting the Microcon tube insert and placing it above a receiving tube provided by the manufacturer and centrifuging at 1000 × g for 4 min.
Multiplex folding step
Folding of the ligated material was performed by adding 1.2 ml of folding buffer (2 m guanidine hydrochloride, 0.1 m Tris base, 0.5 mm reduced GSH, 0.3 mm oxidized GSH, and 10 mm methionine (pH 8.0) directly to the recovered retentate and leaving the mixture at ambient temperature for 3 days without agitation.
In-well desalting step
The folding reactions were acidified by adding 50 μl of acetic acid (33% v/v) with each reaction, divided into three 45-μl aliquots, and placed in wells of a 2-ml deep-well 96-well polypropylene plate. 900 μl of 2 m guanidine hydrochloride was added to each well, and the well contents were transferred into wells of fritted 96-well plates that had been filled with C18 Chromabond resin (Macherey-Nagel, 130 mg/well), pretreated with 500 μl of acetonitrile, and equilibrated with two washes (500 μl) of 5% solvent B (0.1% TFA in 90% acetonitrile and 10% water) and 95% solvent A (0.1% TFA in water).
Resin in wells was washed four times using 500 μl of 5% solvent B and eluted into recovery deep-well plates using two 200-μl volumes of a mixture of 50% solvent B followed by one 200-μl volume of 90% solvent B. Eluates were lyophilized.
Characterization by HPLC and MS
Lyophilized eluates were dissolved in 250 μl of water, with 0.5-μl samples taken for MALDI MS analysis on an AB Sciex 4800 MALDI TOF/TOF mass spectrometer (linear positive mode using 2,5-dihydroxybenzoic acid as a matrix) and 2.5-μl samples taken for RP-HPLC using an Alliance 2695 system (Waters) and a nucleosil C8-300-5 column (Macherey-Nagel), with a gradient of 10% to 70% solvent B/solvent A at 1%/min.
Cell lines
HEK-CCR5 cells
A stably transduced clonal HEK293 cell line was obtained by transduction with a lentiviral vector (25) followed by clonal selection by FACS. Cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FBS.
CHO-CCR5-Rluc8 cells
An ORF encoding CCR5 fused via its C terminus to RLuc8 was generated by PCR assembly and inserted into the pCDNA 3.1(−) expression vector using the XbaI and NotI sites. CHO-K1 cells were transfected with the pCDNA3.1(−)-CCR5-RLuc8 plasmid using X-tremeGENE HP DNA transfection reagent (Roche), and a clone of stably transfected CHO-CCR5-Rluc8 cells was isolated. Cells were maintained in RPMI medium supplemented with 10% FBS and 1% Geneticin.
CHO-CCR5-RLuc8/YFP-CAAX cells
An ORF encoding YFP appended with the prenylation CAAX box sequence (KKKKKKSKTKCVIM) from KRas (37) was inserted by Gibson assembly (New England Biolabs) into the FUGW lentiviral vector (45) that had been digested at the BamHI and EcoRI sites to generate the FUGW-YFP-CAAX vector. CHO-CCR5-RLuc8 cells were transduced with FUGW-YFP-KRas, and a YFP-positive population was isolated by flow cytometry. The resulting CHO-CCR5-RLuc8/YFP-CAAX clone was maintained in RPMI medium supplemented with 10% FBS and 1% Geneticin at 37 °C and 5% CO2.
Cell fusion assay
R5-tropic envelope-dependent cell fusion assays were carried out as described previously (25, 29, 46) using HeLa-P5L (18) and HeLa-Env-ADA (47) cell lines.
CCR5 calcium flux assay
HEK-CCR5 cells were seeded (20,000 cells/well) overnight in 384-well plates that had been pretreated with 10 μg/ml of polyornithine (37 °C, 1 h). Cells were then loaded with Fluo-4/AM (Invitrogen) according to the manufacturer's recommendations and incubated for 1 h at 37 °C. The culture medium was removed, and cells were washed with PBS and incubated in assay buffer (143 mm NaCl, 6 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 0.1% glucose, and 20 mm HEPES (pH 7.4)). Ca2+-dependent fluorescence measurements were carried out on an FDSS 384-well plate reader (Hamamatsu). Molecules were screened (n = 4) at a single concentration (300 nm) at which PSC RANTES gives a maximal signal (48). Signaling activity was expressed as a percentage of the value obtained for the 300 nm PSC-RANTES reference standard (maximum signaling) after subtraction of the value obtained for the 300 nm 5P12-RANTES reference standard (background signaling).
CCR5 internalization assay
CHO-CCR5-RLuc8/YFP-CAAX cells were seeded overnight in 96-well-plates (20,000 cells/well), and then the medium was removed and replaced with chemokine analogs (300 nm) diluted in BRET buffer (5 m NaCl, 1 m KCl, 100 mm MgSO4, 1 m HEPES, 20% glucose, 1% BSA, and 5 μm coelenterazine H).
BRET measurements were performed on a Polarstar (BMG Labtech) plate reader with a filter set (center wavelength/bandwidth) of 475/30 nm (donor) and 535/30 nm (acceptor). Luminescence was recorded immediately after 25 min of incubation at 37 °C, and BRET ratios, defined as emission from the acceptor YFP (535 nm) divided by emission from the donor RLuc8 (475 nm), were calculated. Molecules were screened (n = 4) at a single Emax concentration (300 nm) (48). Internalization activity was expressed as a percentage of the value obtained for the 300 nm PSC-RANTES reference standard (maximum internalization) after subtraction of the value obtained for the 300 nm 5P12-RANTES reference standard (background internalization).
Author contributions
M. P.-B. and O. H. conceptualization; M. P.-B., E. M., and I. S. methodology; M. P.-B., F. C., E. M., I. S., and O. H. writing-review and editing; F. C., E. M., and I. S. investigation; O. H. supervision; O. H. writing-original draft.
Supplementary Material
We acknowledge support from the Swiss National Science Foundation (grant 310030_163085 to O. H). O. H. is the inventor of the chemokine analogs described in this study and a shareholder of Orion Biotechnology Switzerland, to whom the rights to the invention have been assigned.
This article contains Figs. S1–S4 and Tables S1–S3.
- SEA
- bis(2-sulfanylethyl)amino
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- BRET
- bioluminescence resonance energy transfer
- RP-HPLC
- reverse-phase HPLC
- HEK
- human embryonic kidney
- YFP
- yellow fluorescent protein
- FBS
- fetal bovine serum
- CHO
- Chinese hamster ovary.
References
- 1. Fosgerau K., and Hoffmann T. (2015) Peptide therapeutics: current status and future directions. Drug Discov. Today 20, 122–128 10.1016/j.drudis.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 2. Henninot A., Collins J. C., and Nuss J. M. (2018) The current state of peptide drug discovery: back to the future? J. Med. Chem. 61, 1382–1414 10.1021/acs.jmedchem.7b00318 [DOI] [PubMed] [Google Scholar]
- 3. Lau J. L., and Dunn M. K. (2018) Therapeutic peptides: historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 26, 2700–2707 10.1016/j.bmc.2017.06.052 [DOI] [PubMed] [Google Scholar]
- 4. Vlieghe P., Lisowski V., Martinez J., and Khrestchatisky M. (2010) Synthetic therapeutic peptides: science and market. Drug Discov. Today 15, 40–56 10.1016/j.drudis.2009.10.009 [DOI] [PubMed] [Google Scholar]
- 5. Breitling F., Nesterov A., Stadler V., Felgenhauer T., and Bischoff F. R. (2009) High-density peptide arrays. Mol. Biosyst. 5, 224–234 10.1039/b819850k [DOI] [PubMed] [Google Scholar]
- 6. Dawson P. E., Muir T. W., Clark-Lewis I., and Kent S. B. (1994) Synthesis of proteins by native chemical ligation. Science 266, 776–779 10.1126/science.7973629 [DOI] [PubMed] [Google Scholar]
- 7. Raibaut L., El Mahdi O., and Melnyk O. (2015) Solid phase protein chemical synthesis. Top. Curr. Chem. 363, 103–154 [DOI] [PubMed] [Google Scholar]
- 8. Engelhard M. (2016) Quest for the chemical synthesis of proteins. J. Pept. Sci. 22, 246–251 10.1002/psc.2880 [DOI] [PubMed] [Google Scholar]
- 9. Koehn J., and Hunt I. (2009) High-throughput protein production (HTPP): a review of enabling technologies to expedite protein production. Methods Mol. Biol. 498, 1–18 10.1007/978-1-59745-196-3_1 [DOI] [PubMed] [Google Scholar]
- 10. Pande J., Szewczyk M. M., and Grover A. K. (2010) Phage display: concept, innovations, applications and future. Biotechnol. Adv. 28, 849–858 10.1016/j.biotechadv.2010.07.004 [DOI] [PubMed] [Google Scholar]
- 11. Plückthun A. (2012) Ribosome display: a perspective. Methods Mol. Biol. 805, 3–28 10.1007/978-1-61779-379-0_1 [DOI] [PubMed] [Google Scholar]
- 12. Angelini A., Chen T. F., de Picciotto S., Yang N. J., Tzeng A., Santos M. S., Van Deventer J. A., Traxlmayr M. W., and Wittrup K. D. (2015) Protein engineering and selection using yeast surface display. Methods Mol. Biol. 1319, 3–36 10.1007/978-1-4939-2748-7_1 [DOI] [PubMed] [Google Scholar]
- 13. Allen S. J., Crown S. E., and Handel T. M. (2007) Chemokine: receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 25, 787–820 10.1146/annurev.immunol.24.021605.090529 [DOI] [PubMed] [Google Scholar]
- 14. Clark-Lewis I., Vo L., Owen P., and Anderson J. (1997) Chemical synthesis, purification, and folding of C-X-C and C-C chemokines. Methods Enzymol. 287, 233–250 10.1016/S0076-6879(97)87018-8 [DOI] [PubMed] [Google Scholar]
- 15. Dawson P. E. (1997) Synthesis of chemokines by native chemical ligation. Methods Enzymol. 287, 34–45 10.1016/S0076-6879(97)87005-X [DOI] [PubMed] [Google Scholar]
- 16. Hesselgesser J., and Horuk R. (1997) Alanine scan mutagenesis of chemokines. Methods Enzymol. 287, 59–69 10.1016/S0076-6879(97)87007-3 [DOI] [PubMed] [Google Scholar]
- 17. Horuk R., Reilly D., and Yansura D. (1997) Expression, purification, and characterization of Escherichia coli-derived recombinant human melanoma growth stimulating activity. Methods Enzymol. 287, 3–12 10.1016/S0076-6879(97)87003-6 [DOI] [PubMed] [Google Scholar]
- 18. Simmons G., Clapham P. R., Picard L., Offord R. E., Rosenkilde M. M., Schwartz T. W., Buser R., Wells T. N., and Proudfoot A. E. (1997) Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science 276, 276–279 10.1126/science.276.5310.276 [DOI] [PubMed] [Google Scholar]
- 19. Chevigné A., Fievez V., Schmit J. C., and Deroo S. (2011) Engineering and screening the N-terminus of chemokines for drug discovery. Biochem. Pharmacol. 82, 1438–1456 10.1016/j.bcp.2011.07.091 [DOI] [PubMed] [Google Scholar]
- 20. Hartley O., and Offord R. E. (2005) Engineering chemokines to develop optimized HIV inhibitors. Curr. Protein Pept. Sci. 6, 207–219 10.2174/1389203054065400 [DOI] [PubMed] [Google Scholar]
- 21. Wells T. N., Power C. A., Lusti-Narasimhan M., Hoogewerf A. J., Cooke R. M., Chung C. W., Peitsch M. C., and Proudfoot A. E. (1996) Selectivity and antagonism of chemokine receptors. J. Leukoc. Biol. 59, 53–60 10.1002/jlb.59.1.53 [DOI] [PubMed] [Google Scholar]
- 22. Kufareva I., Salanga C. L., and Handel T. M. (2015) Chemokine and chemokine receptor structure and interactions: implications for therapeutic strategies. Immunol. Cell Biol. 93, 372–383 10.1038/icb.2015.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Berger E. A., Murphy P. M., and Farber J. M. (1999) Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 17, 657–700 10.1146/annurev.immunol.17.1.657 [DOI] [PubMed] [Google Scholar]
- 24. Cocchi F., DeVico A. L., Garzino-Demo A., Arya S. K., Gallo R. C., and Lusso P. (1995) Identification of RANTES, MIP-1α, and MIP-1β as the major HIV-suppressive factors produced by CD8+ T cells [see comments]. Science 270, 1811–1815 10.1126/science.270.5243.1811 [DOI] [PubMed] [Google Scholar]
- 25. Hartley O., Gaertner H., Wilken J., Thompson D., Fish R., Ramos A., Pastore C., Dufour B., Cerini F., Melotti A., Heveker N., Picard L., Alizon M., Mosier D., Kent S., and Offord R. (2004) Medicinal chemistry applied to a synthetic protein: development of highly potent HIV entry inhibitors. Proc. Natl. Acad. Sci. U.S.A. 101, 16460–16465 10.1073/pnas.0404802101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lederman M. M., Veazey R. S., Offord R., Mosier D. E., Dufour J., Mefford M., Piatak M. Jr, Lifson J. D., Salkowitz J. R., Rodriguez B., Blauvelt A., and Hartley O. (2004) Prevention of vaginal SHIV transmission in rhesus macaques through inhibition of CCR5. Science 306, 485–487 10.1126/science.1099288 [DOI] [PubMed] [Google Scholar]
- 27. Hartley O., Dorgham K., Perez-Bercoff D., Cerini F., Heimann A., Gaertner H., Offord R. E., Pancino G., Debré P., and Gorochov G. (2003) Human immunodeficiency virus type 1 entry inhibitors selected on living cells from a library of phage chemokines. J. Virol. 77, 6637–6644 10.1128/JVI.77.12.6637-6644.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dorgham K., Cerini F., Gaertner H., Melotti A., Rossitto-Borlat I., Gorochov G., and Hartley O. (2016) Generating chemokine analogs with enhanced pharmacological properties using phage display. Methods Enzymol. 570, 47–72 10.1016/bs.mie.2015.09.014 [DOI] [PubMed] [Google Scholar]
- 29. Gaertner H., Cerini F., Escola J. M., Kuenzi G., Melotti A., Offord R., Rossitto-Borlat I., Nedellec R., Salkowitz J., Gorochov G., Mosier D., and Hartley O. (2008) Highly potent, fully recombinant anti-HIV chemokines: reengineering a low-cost microbicide. Proc. Natl. Acad. Sci. U.S.A. 105, 17706–17711 10.1073/pnas.0805098105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Veazey R. S., Ling B., Green L. C., Ribka E. P., Lifson J. D., Piatak M. Jr., Lederman M. M., Mosier D., Offord R., and Hartley O. (2009) Topically applied recombinant chemokine analogues fully protect macaques from vaginal simian-human immunodeficiency virus challenge. J. Infect. Dis. 199, 1525–1527 10.1086/598685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cerini F., Gaertner H., Madden K., Tolstorukov I., Brown S., Laukens B., Callewaert N., Harner J. C., Oommen A. M., Harms J. T., Sump A. R., Sealock R. C., Peterson D. J., Johnson S. K., Abramson S. B., et al. (2016) A scalable low-cost cGMP process for clinical grade production of the HIV inhibitor 5P12-RANTES in Pichia pastoris. Protein Expr. Purif. 119, 1–10 10.1016/j.pep.2015.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cerini F., Offord R., McGowan I., and Hartley O. (2017) Stability of 5P12-RANTES, a candidate rectal microbicide, in human rectal lavage. AIDS Res. Hum. Retroviruses 33, 768–777 10.1089/aid.2016.0199 [DOI] [PubMed] [Google Scholar]
- 33. Zheng Y., Han G. W., Abagyan R., Wu B., Stevens R. C., Cherezov V., Kufareva I., and Handel T. M. (2017) Structure of CC chemokine receptor 5 with a potent chemokine antagonist reveals mechanisms of chemokine recognition and molecular mimicry by HIV. Immunity 46, 1005–1017.e5 10.1016/j.immuni.2017.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bang D., and Kent S. B. (2004) A one-pot total synthesis of crambin. Angew. Chem. Int. Ed. Engl. 43, 2534–2538 10.1002/anie.200353540 [DOI] [PubMed] [Google Scholar]
- 35. Ramli S., Gentle I. R., and Ross B. P. (2009) Efficient manual Fmoc solid-phase synthesis of the N-terminal segment of surfactant protein B (SP-B(1–25)). Protein Peptide Letters 16, 810–814 10.2174/092986609788681706 [DOI] [PubMed] [Google Scholar]
- 36. Escola J. M., Kuenzi G., Gaertner H., Foti M., and Hartley O. (2010) CC chemokine receptor 5 (CCR5) desensitization: cycling receptors accumulate in the trans-Golgi network. J. Biol. Chem. 285, 41772–41780 10.1074/jbc.M110.153460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Namkung Y., Le Gouill C., Lukashova V., Kobayashi H., Hogue M., Khoury E., Song M., Bouvier M., and Laporte S. A. (2016) Monitoring G protein-coupled receptor and β-arrestin trafficking in live cells using enhanced bystander BRET. Nat. Commun. 7, 12178 10.1038/ncomms12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kenakin T. (2002) Efficacy at G-protein-coupled receptors. Nat. Rev. Drug Discov. 1, 103–110 10.1038/nrd722 [DOI] [PubMed] [Google Scholar]
- 39. Colin P., Bénureau Y., Staropoli I., Wang Y., Gonzalez N., Alcami J., Hartley O., Brelot A., Arenzana-Seisdedos F., and Lagane B. (2013) HIV-1 exploits CCR5 conformational heterogeneity to escape inhibition by chemokines. Proc. Natl. Acad. Sci. U.S.A. 110, 9475–9480 10.1073/pnas.1222205110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bönsch C., Munteanu M., Rossitto-Borlat I., Fürstenberg A., and Hartley O. (2015) Potent anti-HIV chemokine analogs direct post-endocytic sorting of CCR5. PLoS ONE 10, e0125396 10.1371/journal.pone.0125396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Monk P. N., Scola A. M., Madala P., and Fairlie D. P. (2007) Function, structure and therapeutic potential of complement C5a receptors. Br. J. Pharmacol. 152, 429–448 10.1038/sj.bjp.0707332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Muir T. W. (2003) Semisynthesis of proteins by expressed protein ligation. Annu. Rev. Biochem. 72, 249–289 10.1146/annurev.biochem.72.121801.161900 [DOI] [PubMed] [Google Scholar]
- 43. Loibl S. F., Harpaz Z., Zitterbart R., and Seitz O. (2016) Total chemical synthesis of proteins without HPLC purification. Chem. Sci. 7, 6753–6759 10.1039/C6SC01883A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ollivier N., Dheur J., Mhidia R., Blanpain A., and Melnyk O. (2010) Bis(2-sulfanylethyl)amino native peptide ligation. Organic Letters 12, 5238–5241 10.1021/ol102273u [DOI] [PubMed] [Google Scholar]
- 45. Lois C., Hong E. J., Pease S., Brown E. J., and Baltimore D. (2002) Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295, 868–872 10.1126/science.1067081 [DOI] [PubMed] [Google Scholar]
- 46. Cerini F., Landay A., Gichinga C., Lederman M. M., Flyckt R., Starks D., Offord R. E., Le Gal F., and Hartley O. (2008) Chemokine analogues show suitable stability for development as microbicides. J. Acquir. Immune Defic. Syndr. 49, 472–476 10.1097/QAI.0b013e31818c953f [DOI] [PubMed] [Google Scholar]
- 47. Pleskoff O., Tréboute C., Brelot A., Heveker N., Seman M., and Alizon M. (1997) Identification of a chemokine receptor encoded by human cytomegalovirus as a cofactor for HIV-1 entry [see comments]. Science 276, 1874–1878 10.1126/science.276.5320.1874 [DOI] [PubMed] [Google Scholar]
- 48. Gaertner H., Lebeau O., Borlat I., Cerini F., Dufour B., Kuenzi G., Melotti A., Fish R. J., Offord R., Springael J. Y., Parmentier M., and Hartley O. (2008) Highly potent HIV inhibition: engineering a key anti-HIV structure from PSC-RANTES into MIP-1β/CCL4. Protein Eng. Des. Sel. 21, 65–72 10.1093/protein/gzm079 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.