

Abstract
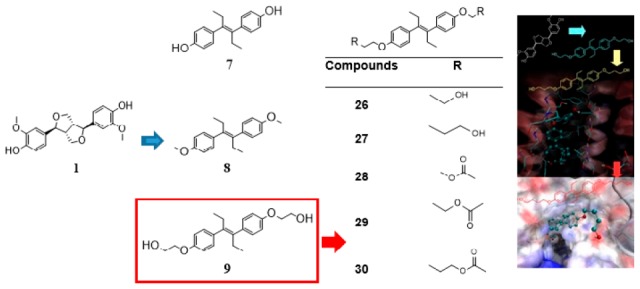
To find novel P-gp-inhibitors, a library of pregnane X receptor (PXR) ligands and the ZINC DrugsNow library were superimposed on the P-gp inhibitor (+)-pinoresinol (1) used as a query for a three-dimensional similarity search. After determining the TanimotoCombo index of similarity with 1, eight compounds from the PXR library and two ZINC compounds were selected for biological evaluation. The P-gp inhibition study showed that compounds 7, 8, and 9 successfully increased intracellular doxorubicin (DOX) accumulation in the P-gp overexpressed Lucena 1 cells from 25, 12.5, and 6.25 μM, respectively. Among a series of analogues of 9, compounds 26–30 were shown to be active, with 26 and 27 causing a significant increase in DOX accumulation from 1.56 μM and rendering Lucena 1 sensitive to DOX from 1.56 and 0.78 μM, respectively. Molecular modeling studies showed that both compounds bind to the P-gp at transmembrane helices (TMH) 4, 5, and 6, with 27 also showing contacts with TMH 3.
Keywords: MDR reversing agents, P-glycoprotein, pinoresinol, diethylstilbestrol derivatives, structure−activity relationship, molecular docking
The development of drug resistance in cancer cells is a major barrier to successful chemotherapy. Among the various mechanisms behind multidrug resistance (MDR),1 the increased energy-dependent efflux of a broad set of structurally unrelated cytostatic drugs is one of the most commonly encountered.2 This phenomenon is mediated by transport proteins,3 the best characterized of which is P-glycoprotein (P-gp) encoded by the mdr-1 gene, one of the 49 identified ATP-binding cassette (ABC) transporters.4 P-gp contains two transmembrane domains, each consisting of six transmembrane α-helices (TMHs) and a nucleotide-binding domain (NBD).5 The latter hydrolyzes ATP to drive the transport of the binding substrate. The importance of P-gp is that it is expressed in almost half of human tumors,2 being observed in many types of leukemia,2 including chronic myelogenous leukemia.6
Despite some encouraging results,7 most of the P-gp inhibitors submitted to clinical trials were disappointing, mainly due to their side effects, interactions with coadministered drugs, or defects in the experimental design.8,9 The development of drugs able to counteract the resistance mediated by P-gp is thus imperative. Natural compounds have attracted great attention and are considered fourth generation P-gp inhibitors.9 A preceding paper reported that the lignan (±)-pinoresinol, isolated from Melia azedarach,10 showed inhibitory activity on the transport of doxorubicin (DOX) out of P-gp overexpressed chronic myelogenous leukemia cells, Lucena 1 from 56 μM, as demonstrated by an accumulation assay.2 By multidrug resistance reversal assay, (±)-pinoresinol was able to sensitize resistant cells to DOX from 7 μM.2 Further studies revealed that (±)-pinoresinol bonds to P-gp through residues of the TMH 4, 5, and 6, some of which are also involved in the binding of the reference P-gp inhibitors, verapamil and tariquidar.2
This scenario and the success of plant-derived products as starting points for drug development11 led us to search for suitable candidates with improved activity compared to (±)-pinoresinol to specifically counteract the mechanism of drug resistance in cells with enhanced P-gp expression. Since the chemical structure of (+)-pinoresinol (1) (Figure 1) includes two hydroxyl groups at a distance of 1.3 nm bound to a central scaffold, we virtually screened our own previously synthesized pregnane X receptor (PXR) library (Figure 1; compounds 2–9), which has a similar structural feature,12,13 for similarity with 1 using ROCS (ROCS 3.2.2.2: OpenEye Scientific Software, Santa Fe, NM. http://www.eyesopen.com).14 TanimotoCombo similarity index of shape (shape Tanimoto index) and pharmacophore features (color Tanimoto index) of the query and of compounds 2–9 were calculated. Additionally, the ZINC DrugsNow library, containing more than 10 million compounds, was also screened for three-dimensional similarity to 1 using the TanimotoCombo similarity index (sum of shape and color Tanimoto index).
Figure 1.

Chemical structures of (+)-pinoresinol (1), the PXR ligands 2–9, and the ZINC compounds 10–25 showing high similarity with 1 by ROCS analysis.
ROCS demonstrated that the identified ZINC molecules 10–25 (Figure 1) overlay better with 1 (TanimotoCombo between 1.413 and 1.606) than the bazedoxifene scaffold-based PXR antagonists 2–6 (Figure 1, TanimotoCombo between 0.717 and 0.909) and the diethylstilbestrol scaffold-based PXR ligands 7–9 (Figure 1, TanimotoCombo between 0.823 and 0.851) (see Supporting Information for representative overlays, Figure S1). On the basis of these results, the optimized structures of these compounds were studied in silico by molecular docking, using the human P-gp model previously built based on the homologous P-gp from mouse (Mus musculus) as template.15 In relevant cases, both stereoisomers (±) were investigated, and both OH and NH tautomers (keto and enol forms) for lactam compounds 20–25. As observed in Table S1, the compounds evaluated showed binding energies ranging from −7.07 to −9.23 kcal/mol. To have a first estimation of the ability to reverse the P-gp-resistant phenotype of cancer cells, compounds 2–9, the most strongly binding nontetrahydrofuran based ZINC compound 23 (binding energy of NH tautomer, −9.23 kcal/mol) and its close structural analogue 21 (binding energy of OH tautomer, −8.56 kcal/mol) were evaluated by a DOX accumulation assay using flow cytometry2 (see Supporting Information). For this purpose, the chronic myelogenous leukemia cells, Lucena 1,16 with 58% of the cells expressing P-gp at the surface, were used as a model. These cells derived from the sensitive K562 cell line with less P-gp in the outer membrane (2% of cells). The retention of the cytotoxic clinical drug DOX, a known P-gp fluorescent substrate, inside the cells is commonly used to measure the capacity of a substance to inhibit P-gp-mediated outward transport. Efflux inhibition positively correlates with the intracellular concentration of the probe.17 The same assay was carried out in the K562 cell line with the aim of discarding any other synergism between the compounds and the fluorescent drug different from the effect on P-gp.
Notably, diethylstilbestrol-based PXR ligands 7, 8, and 9 effectively increased intracellular DOX (p < 0.05) in Lucena 1 cells with fluorescence intensity ratio (FIR) values corresponding to 1.06, 1.19, and 1.16, respectively, at 25 μM, while the bazedoxifene PXR antagonists 2–6 and the ZINC compounds 21 and 23 did not enhance DOX-associated fluorescence (p > 0.05) (Table 1). The activity of 7–9 was not significantly different with respect to the activity of 25 μM of the well-known reference P-gp modulators verapamil, tamoxifen, and cyclosporine A (CsA) (p > 0.05). It is worth noting that 7–9 did not cause a significant increase in DOX accumulation in the K562 cell line (p > 0.05) (Table S2) indicating that these compounds specifically inhibited P-gp transport. Taken together, the better reversal activity displayed by 7–9 compared to that of 2–6 and 21 and 23 would indicate that the diethylstilbestrol scaffold is more beneficial for achieving P-gp inhibitory activity than the bazedoxifene- and 1,4,5,7-tetrahydro-6H-pyrazolo[3,4-b]pyridin-6-one scaffold.
Table 1. Effect of PXR and Selected ZINC Compounds on P-gp Function by Doxorubicin Accumulation and Resistance Reversal Assays in Lucena 1 Cell Linea.
| FIR/FR |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| concentration
(μM) |
|||||||||||
| compound | 25 | 12.50 | 6.25 | 3.12 | 1.56 | 0.78 | 0.39 | 0.19 | 0.098 | 0.048 | 0.024 |
| 2 | 0.80 ± 0.02 | ||||||||||
| 3 | 0.72 ± 0.03 | ||||||||||
| 4 | 0.75 ± 0.08 | ||||||||||
| 5 | 1.01 ± 0.03 | ||||||||||
| 6 | 0.90 ± 0.15 | ||||||||||
| 7 | 1.06 ± 0.01* | 1.10 ± 0.08 | 1.04 ± 0.03 | ||||||||
| 8 | 1.19 ± 0.03* | 1.09 ± 0.02** | 1.09 ± 0.07 | ||||||||
| 9 | 1.16 ± 0.08* | 1.20 ± 0.04* | 1.08 ± 0.007*** | 1.01 ± 0.06 | |||||||
| 21 | 1.11 ± 0.06 | ||||||||||
| 23 | 0.96 ± 0.01 | ||||||||||
| 26 | 1.23 ± 0.08* | 1.06 ± 0.02* | 1.09 ± 0.01* | 1.04 ± 0.009*/1.55 ± 0.09** | 1.05 ± 0.003*/1.30 ± 0.12* | 0.98 ± 0.02/1.15 ± 0.11 | |||||
| 27 | 1.32 ± 0.12* | 1.18 ± 0.07* | 1.08 ± 0.02* | 1.06 ± 0.01*/1.61 ± 0.14* | 1.10 ± 0.02*/1.34 ± 0.07* | 0.99 ± 0.02/1.16 ± 0.08* | 1.00 ± 0.02/1.03 ± 0.03 | ||||
| 28 | 1.21 ± 0.07* | 1.15 ± 0.05* | 1.12 ± 0.02** | 1.07 ± 0.009** | 1.03 ± 0.03 | ||||||
| 29 | 1.21 ± 0.03*** | 1.10 ± 0.02* | 1.06 ± 0.02* | 1.04 ± 0.01* | 1.02 ± 0.01 | ||||||
| 30 | 1.18 ± 0.07* | 1.10 ± 0.02* | 1.09 ± 0.03** | 1.08 ± 0.02* | 1.01 ± 0.02 | ||||||
| 31 | 0.99 ± 0.04 | ||||||||||
| 32 | 1.04 ± 0.05 | ||||||||||
| 33 | 1.03 ± 0.02 | ||||||||||
| 34 | 0.95 ± 0.06 | ||||||||||
| Ver | 1.35 ± 0.009*** | 1.26 ± 0.03** | 1.20 ± 0.06* | 1.16 ± 0.03* | 1.07 ± 0.03* | 1.07 ± 0.07 | |||||
| Tam | 1.13 ± 1.03* | 1.07 ± 0.02* | 1.05 ± 0.01* | 1.01 ± 0.02 | |||||||
| CsA | 1.40 ± 0.01* | 1.47 ± 0.12* | 1.44 ± 0.06** | 1.47 ± 0.07** | 1.46 ± 0.12* | 1.43 ± 0.09* | 1.30 ± 0.04** | 1.25 ± 0.06* | 1.16 ± 0.04* | 1.15 ± 0.01** | 1.01 ± 0.04 |
Ver: verapamil. Tam: tamoxifen. CsA: cyclosporine A. Fluorescence intensity ratio (FIR); analysis of differences between the intracellular accumulations of DOX in cells treated with the tested compounds compared to control cells were performed by one-tailed paired t-test. Fold reversal (FR) values were calculated as IC50 of DOX alone/IC50 of DOX in the presence of the tested compounds. Statistical comparisons between IC50 of DOX alone or with compound in each treatment were performed by one-tailed paired t-test. Results represent the mean ± SE. ***p < 0.001, **p < 0.01 and *p < 0.05.
Following primary screening, and with the aim of determining the minimum effective concentrations (MEC), 7–9 were further tested at serial dilutions. As seen in Table 1, compound 7 did not restore DOX accumulation in Lucena 1 cells at concentrations below 25 μM (p > 0.05), while 8 and 9 turned out to be effective from 12.5 (p < 0.01) and 6.25 μM (p < 0.001), respectively, with no differences in activity with respect to verapamil, tamoxifen, and CsA (p > 0.05). A dose-dependent response was observed for 8 (b = 0.0097; p = 0.0076; CI 95% = 0.0043 to 0.0151) and 9 (b = 0.0090; p = 0.0496; CI 95% = 0.00002 to 0.01797). To the best of our knowledge, this is the first report of the inhibition of diethylstilbestrol 7 on P-gp-mediated transport, although inhibitory properties have been previously reported against the breast cancer resistance protein (BCRP) at 30 μM.18 In another work, 7 showed a weak activation (45%) of P-gp ATPase activity at 10 μM.19 Although mild stimulation of ATP hydrolysis may be associated with modulatory activity on P-gp function, as observed for the compound (±)-pinoresinol2 or cyclosporine A,20 no studies have previously been performed about the inhibitory effect of 7 on the P-gp-mediated efflux of drugs. Such investigations must be carried out based on the assumption that some molecules able to activate P-gp-ATPase, for instance, the powerful modulator tariquidar,21 behave as inhibitors, while others, such as DOX, vinblastine, progesterone, or propranolol, act only as substrates.20,22
The limited clinical efficacy of multiple drugs as a result of resistance is a complex mechanism not only attributed to the efflux mediated by an ABC transporter.1 It is therefore highly beneficial to develop multitargeted agents for overcoming the resistance to pharmacotherapies.1 As well as P-gp, PXR also plays a key role in the MDR of cancer cells, acting as a xenobiotic sensor that regulates the transcription of genes that ultimately increase the activity of transporters and drug-metabolizing enzymes.12,13,23 The dual activity of the PXR antagonists 7 and 9, negatively modulating PXR activity13 and, at the same time, the outward transport mediated by P-gp, is encouraging for these antagonists to be agents for overcoming MDR.
As previously reported, compound 7 was able to decrease P-gp expression levels in MCF-7/MDR cells.24 Therefore, the effectiveness of this compound could be a sum of effects, possibly inhibiting both P-gp transport and expression.
Given that compounds 7–9 displayed higher P-gp modulating activity than (±)-pinoresinol,2 and aiming to optimize the P-gp-mediated MDR reversal activity of the identified compounds, an additional series of O-functionalized diethylstilbestrol derivatives 26–34, originally prepared as potential PXR modulators (Figure 2), were assayed for P-gp inhibition at progressive 2-fold dilutions from 25 μM. Cotreatment of Lucena 1 with 26–30 resulted in enhanced DOX accumulation (p < 0.001–0.05), with FIR values ranging from 1.04 to 1.32. While 28–30 increased DOX-associated MFI from 3.12 μM (p < 0.01–0.05), 26 and 27 were active from 1.56 μM (p < 0.05), with no differences with respect to the same concentrations of verapamil and CsA (p > 0.05) (Table 1 and Figure S2A). Dose-dependency was observed for 26 (b = 0.0082; p = 0.0106; CI 95% = 0.0032 to 0.0133), 27 (b = 0.0120; p = 0.0025; CI 95% = 0.0071 to 0.0169), 28 (b = 0.0071; p = 0.0109; CI 95% = 0.0031 to 0.0110), 29 (b = 0.0077; p = 0.0001; CI 95% = 0.0068 to 0.0087), and 30 (b = 0.0056; p = 0.0009; CI 95% = 0.0009 to 0.0103). The lack of activity on K562 was confirmed indicating that the effect was due to P-gp inhibition (Table S2).
Figure 2.

Chemical structures of the diethylstilbestrol derivatives: 26–34.
In addition to the DOX accumulation study, a similar assay using the dye rhodamine 123 (Rho123), a classic P-gp substrate, was further carried out by flow cytometry. As observed in Table S3 and Figure S2B, compounds 26 and 27 increased the intracellular Rho123 from 50 μM and 12.5 μM, respectively, and were significantly different from verapamil and CsA, both at 25 and 12.5 μM, respectively (p < 0.05), but were comparable with tamoxifen at the same concentrations (p > 0.05).
To evaluate the capabilities of the most effective compounds, 26 and 27, more thoroughly, a multidrug resistance reversal assay was carried out (see Supporting Information). The different parameters measured by both types of technique allowed us to address different aspects related to their pharmacological profile to determine the effectiveness of the compounds.25 The accumulation assay enables assessment of the quantity of substrate accumulated inside the cells resulting from inhibition of the efflux, while the multidrug resistant reversal assay determines the intracellular activity of a cytotoxic drug applied in combination with P-gp modulators compared with its individual activity.2 It is worth mentioning that Lucena 1 cells displayed 34-fold more resistance to DOX toxicity than parental K562 cells, with IC50 values of 48.49 ± 3.8 and 1.42 ± 0.18 μM, respectively. As expected, 26 and 27 showed their ability to reverse the P-gp-mediated DOX resistance from 1.56 and 0.78 μM, respectively (Table 1 and Figure 3), displaying fold reversal (FR) values of 1.30 and 1.16 (p < 0.05), respectively (Table 1). These results match those found in the accumulation assay. To determine whether the DOX cytotoxicity restoring effect was specific to P-gp, K562 cells were treated with 3.12–0.78 μM of 26 or 27. No sensitization to DOX on K562 was observed with compound 26 at 3.12 μM, while 27 was not able to increase DOX toxicity in these cells at 1.56 (Table S2). These results showed that both molecules appear as chemosensitizers specific to P-gp modulation.
Figure 3.

Dose–response curves for cytotoxicity of doxorubicin (DOX) in Lucena 1 and K562 cells with and without compounds 26 (A) and 27 (B) as determined by the multidrug resistance reversal assay. Values are expressed as mean ± SE of at least three independent experiments.
As previously mentioned by Syed et al.,26 P-gp modulators submitted to clinical trials present octanol/water partition coefficients ranging from 3.64 to 6.81. The partition coefficients (ClogP, calculated with ChemDraw Ultra software) found for compounds 7–9 and 26–30 are mostly in agreement with these values (see Table S4) suggesting that the assayed compounds show a similar partition into the lipid bilayer to that observed with the anti-P-gp candidates. In addition, the ClogP values obtained are higher than 2.92/3.25, the minimum ClogP values established for a compound to be considered as an effective P-gp inhibitor.27,28 In the present work, the most potent compounds 26–30 showed the highest calculated logP values (5.17 to 7.22, Table S4) in agreement with many authors20,29,30 who proposed that lipophilicity is a key factor for a better inhibition of P-gp.
The binding mode of 1 was previously described,2 with the main contacts occurring with residues S222, A223, K234, F303, Y307, Y310, L339, A342, and F343, most of which are amino acids from TMH 4, 5, and 6 at the top of the inverted “V” shaped by the transmembrane α-helices. All these residues were proposed as essential for binding on theoretical and experimental bases, as previously stated.2,15 The inhibitors 7–9 and 26–30, which were found active by DOX accumulation assay and by docking, overlap their binding regions with that of the parent compound 1, which in turn overlaps with the reference inhibitor tariquidar.2 In particular, compounds 26, 27, and 30 showed the maximum superposition with tariquidar, with 27 being the only compound of the 26–30 series, which also showed contacts with TMH 3 (T199, F200, and G203). The superimposition of 1, 26, and 27 with tariquidar is shown in Figure 4. Further details on the binding mode of 26 and 27 are available in Figures S3 and S4. One of the most plausible reasons behind the difference in activity of 26 and 27 with respect to DOX and Rho123 accumulation techniques, which were also observed in treatments with (±) pinoresinol,2 concerns the affinity of each substrate to the binding sites of P-gp. This issue may be explained based on the results obtained by performing docking studies. The lowest energy docked poses of both 26 and 27 bind to the two main sites of DOX, the latter with −8.23 and −8.16 kcal/mol binding energies (Figure S5). As shown in Figure S5, also other higher energy conformations of 26 and 27 (up to 1.2 kcal/mol above the lowest one) overlapped these sites as well. However, none of these conformations are able to overlap the primary and one of the secondary sites of Rho123 (which have similar affinities within 0.3 kcal/mol), as illustrated in Figures S6 and S7. Clearly, neither 26 nor 27 would be able to inhibit the Rho123 efflux (at least not in the same competitive way) as it does with DOX. In sharp contrast, the much more flexible and bigger molecule CsA binds in a region that occlude the three main Rho123 binding sites (binding energies −10.0 to −9.0 kcal/mol), as shown in Figure S8. Although smaller than CsA, verapamil still showed a similar pattern (Figure S9, conformations from −9.1 to −8.1 kcal/mol shown) These differences in the binding preferences of both target compounds, of the two model substrates, and of the reference inhibitors are consistent with the experimental observations regarding the preference of 26 and 27, and also of tamoxifen, for DOX, whereas CsA and verapamil showed a similar behavior with Rho123 or DOX as substrates.
Figure 4.
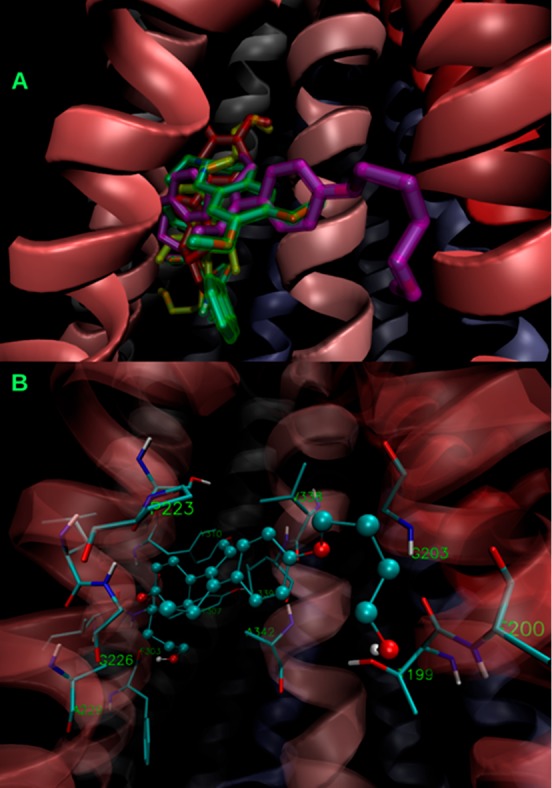
(A) Superimposition of the lowest energy poses of the inhibitors 1 (highlighted in red), 26 (in yellow), 27 (in violet), and the reference inhibitor tariquidar (in green). (B) Binding of the most active compound 27 (balls and sticks) showing its main contacts (licorice). The cartoon representation of the α-helices is colored according to the sequence, from TMH 1 (red) to TMH 12 (blue).
While compounds 9, 26, and 27, differing only in the length of the hydroxyalkoxy side chains (2 to 4 carbon atoms), appear as encouraging lead compounds for P-gp inhibition, the sulfate ester derivatives 32–34 had no effect. As observed in Table 1, the increase in the length of the side chain in 26–27 with respect to 9 enhanced effectiveness. However, the addition of an O-acetyl group in the side chain (compounds 29–30) decreased the activity compared to the hydroxy compounds 26–27. The presence of O–CH3 groups at positions 4 and 4′ of compound 8 slightly increased P-gp modulation activity with respect to bis-hydroxy compound 7, while the addition of the 2-hydroxyethyl group in the lateral chain of 7 to give 9 markedly increased the MDR-modulating efficiency.
To further determine the suitability of compounds 26 and 27 as potential inhibitors of P-gp function, their toxicity on nontumoral peripheral blood mononuclear cells (PBMC) was evaluated. With an MTT assay,31,3226 and 27 showed 34.67 ± 0.88 and 41.00 ± 0.58% cytotoxicity at 25 μM, while a complete absence of toxicity was observed at 6.25 μM. A compound can be considered as cytotoxic when the IC50 values are lower than 10 μM.33
With these results, the newly identified compounds 26–27 appear as promising starting points for the development of inhibitors of the P-gp-mediated transport of chemotherapeutic drugs such as DOX. The information obtained about the structural requirements for activity sheds light on the design of further synthetic pinoresinol-related agents able to reverse the MDR/P-gp phenotype.
Acknowledgments
We thank Joss Heywood for revising the English language. M.C.C., D.M.A.V., and S.M.P. are staff members of the National Research Council of Argentina (CONICET). J.L. acknowledges receipt of a Scholarship from CONICET. We thank OpenEye Scientific Software, Santa Fe, NM, for free academic licenses for the use of their software. This research project was conducted with the support of the Catholic University of Córdoba, CONICET (PIP 11220130100437), FONCyT (PICT 2014-1594), MINCyT-MHEST AR/1408, and of the Slovenian Research Agency (Grant No. P1-0208; Bilateral project Slovenia–Argentina ARRS-BI-AR/15-17-008).
Glossary
ABBREVIATIONS
- BCRP
breast cancer resistance protein
- ClogP calculated octanol
calculated octanol/water partition coefficient
- DOX
doxorubicin
- FR
fold reversal
- FIR
fluorescence intensity ratio
- MEC
minimum effective concentration
- MFI
medium fluorescence intensity
- PBMC
peripheral blood mononuclear cells
- P-gp
P-glycoprotein
- PXR
pregnane X receptor
- Rho123
rhodamine 123
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00324.
Detailed experimental procedures and biological assays, summary of docking results for PXR ligands and ZINC compounds (Table S1), effect of PXR and ZINC compounds on P-gp function in the K562 cell line by DOX accumulation assay (Table S2), effect of 26 and 27 on P-gp function by rhodamine 123 accumulation assay (Table S3), ClogP values (Table S4), overlays of PXR-ligands and ZINC compounds with 1 according to ROCS (Figure S1), representative histograms of DOX and Rho 123 accumulation assays (Figure S2), and poses of binding to P-gp of the most active compounds determined by docking (Figures S3–S9) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Chen Y.; Tang Y.; Guo C.; Wang J.; Boral D.; Nie D. Nuclear receptors in the multidrug resistance through the regulation of drug-metabolizing enzymes and drug transporters. Biochem. Pharmacol. 2012, 83, 1112–1126. 10.1016/j.bcp.2012.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González M. L.; Vera D. M. A.; Laiolo J.; Joray M. B.; Maccioni M.; Palacios S. M.; Molina G.; Lanza P. A.; Gancedo S.; Rumjanek V.; Carpinella M. C. Mechanism Underlying the Reversal of Drug Resistance in P-Glycoprotein-Expressing Leukemia Cells by Pinoresinol and the Study of a Derivative. Front. Pharmacol. 2017, 8, x. 10.3389/fphar.2017.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Shi T.; Zhang L.; Zhu P.; Deng M.; Huang C.; Hu T.; Jiang L.; Li J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. 10.1016/j.canlet.2015.10.010. [DOI] [PubMed] [Google Scholar]
- Palmeira A.; Sousa E.; H Vasconcelos M.; M Pinto M. Three decades of P-gp inhibitors: skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. 10.2174/092986712800167392. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. Thiol-reactive drug substrates of human P-glycoprotein label the same sites to activate ATPase activity in membranes or dodecyl maltoside detergent micelles. Biochem. Biophys. Res. Commun. 2017, 488, 573–577. 10.1016/j.bbrc.2017.05.106. [DOI] [PubMed] [Google Scholar]
- Rumjanek V. M.; Vidal R. S.; Maia R. C. Multidrug resistance in chronic myeloid leukaemia: how much can we learn from MDR–CML cell lines?. Biosci. Rep. 2013, 33, 875 10.1042/BSR20130067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- List A. F.; Kopecky K. J.; Willman C. L.; Head D. R.; Persons D. L.; Slovak M. L.; Dorr R.; Karanes C.; Hynes H. E.; Doroshow J. H.; Shurafa M.; Appelbaum F. R. Benefit of cyclosporine modulation of drug resistance in patients with poor-risk acute myeloid leukemia: a Southwest Oncology Group study. Blood 2001, 98, 3212–3220. 10.1182/blood.V98.12.3212. [DOI] [PubMed] [Google Scholar]
- Szakács G.; Hall M. D.; Gottesman M. M.; Boumendjel A.; Kachadourian R.; Day B. J.; Baubichon-Cortay H.; Di Pietro A. Targeting the Achilles Heel of Multidrug-Resistant Cancer by Exploiting the Fitness Cost of Resistance. Chem. Rev. 2014, 114, 5753–5774. 10.1021/cr4006236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva R.; Vilas-Boas V.; Carmo H.; Dinis-Oliveira R. J.; Carvalho F.; de Lourdes Bastos M.; Remião F. Modulation of P-glycoprotein efflux pump: induction and activation as a therapeutic strategy. Pharmacol. Ther. 2015, 149, 1–123. 10.1016/j.pharmthera.2014.11.013. [DOI] [PubMed] [Google Scholar]
- Carpinella M. C.; Giorda L. M.; Ferrayoli C. G.; Palacios S. M. Antifungal Effects of Different Organic Extracts from Melia azedarach L. on Phytopathogenic Fungi and Their Isolated Active Components. J. Agric. Food Chem. 2003, 51, 2506–2511. 10.1021/jf026083f. [DOI] [PubMed] [Google Scholar]
- Carpinella M. C.; Rai M.. Novel Therapeutic Agents from Plants; Science Publishers, 2009. [Google Scholar]
- Hodnik Ž.; Peterlin Mašič L.; Tomašić T.; Smodiš D.; D’Amore C.; Fiorucci S.; Kikelj D. Bazedoxifene-Scaffold-Based Mimetics of Solomonsterols A and B as Novel Pregnane X Receptor Antagonists. J. Med. Chem. 2014, 57, 4819–4833. 10.1021/jm500351m. [DOI] [PubMed] [Google Scholar]
- Hodnik Ž.; Tomašič T.; Smodiš D.; D’Amore C.; Mašič L. P.; Fiorucci S.; Kikelj D. Diethylstilbestrol-scaffold-based pregnane X receptor modulators. Eur. J. Med. Chem. 2015, 103, 551–562. 10.1016/j.ejmech.2015.09.005. [DOI] [PubMed] [Google Scholar]
- Hawkins P. C. D.; Skillman A. G.; Nicholls A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- Jara G. E.; Vera D. M. A.; Pierini A. B. Binding of modulators to mouse and human multidrug resistance P-glycoprotein. A computational study. J. Mol. Graphics Modell. 2013, 46, 10–21. 10.1016/j.jmgm.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Moreira M. A. M.; Bagni C.; de Pinho M. B.; Mac-Cormick T. M.; dos Santos Mota M.; Pinto-Silva F. E.; Daflon-Yunes N.; Rumjanek V. M. Changes in gene expression profile in two multidrug resistant cell lines derived from a same drug sensitive cell line. Leuk. Res. 2014, 38, 983–987. 10.1016/j.leukres.2014.06.001. [DOI] [PubMed] [Google Scholar]
- Castagnolo D.; Contemori L.; Maccari G.; Avramova S. I.; Neri A.; Sgaragli G.; Botta M. From Taxuspine X to Structurally Simplified Taxanes with Remarkable P-Glycoprotein Inhibitory Activity. ACS Med. Chem. Lett. 2010, 1, 416–421. 10.1021/ml100118k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y.; Tsukahara S.; Imai Y.; Sugimoto Y.; Ueda K.; Tsuruo T. Reversal of Breast Cancer Resistance Protein-mediated Drug Resistance by Estrogen Antagonists and Agonists. Mol. Cancer Ther. 2003, 2, 105–112. [PubMed] [Google Scholar]
- Rao U. S.; Fine R. L.; Scarborough G. A. Antiestrogens and steroid hormones: substrates of the human P-glycoprotein. Biochem. Pharmacol. 1994, 48, 287–292. 10.1016/0006-2952(94)90099-X. [DOI] [PubMed] [Google Scholar]
- Seelig A.; Landwojtowicz E. Structure–activity relationship of P-glycoprotein substrates and modifiers. Eur. J. Pharm. Sci. 2000, 12, 31–40. 10.1016/S0928-0987(00)00177-9. [DOI] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. Tariquidar inhibits P-glycoprotein drug efflux but activates ATPase activity by blocking transition to an open conformation. Biochem. Pharmacol. 2014, 92, 558–566. 10.1016/j.bcp.2014.10.006. [DOI] [PubMed] [Google Scholar]
- Watanabe T.; Kokubu N.; Charnick S. B.; Naito M.; Tsuruo T.; Cohen D. Interaction of cyclosporin derivatives with the ATPase activity of human P-glycoprotein. Br. J. Pharmacol. 1997, 122, 241–248. 10.1038/sj.bjp.0701377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Q.; Han Z.; Zuo X.; Wei H.; Huang W. Co-expression of pregnane X receptor and ATP-binding cassette sub-family B member 1 in peripheral blood: A prospective indicator for drug resistance prediction in non-small cell lung cancer. Oncol. Lett. 2016, 11, 3033–3039. 10.3892/ol.2016.4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutoh K.; Tsukahara S.; Mitsuhashi J.; Katayama K.; Sugimoto Y. Estrogen-mediated post transcriptional down-regulation of P-glycoprotein in MDR1-transduced human breast cancer cells. Cancer Sci. 2006, 97, 1198–1204. 10.1111/j.1349-7006.2006.00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferté J.; Kühnel J.-M.; Chapuis G.; Rolland Y.; Lewin G.; Schwaller M. A. Flavonoid-Related Modulators of Multidrug Resistance: Synthesis, Pharmacological Activity, and Structure–Activity Relationships. J. Med. Chem. 1999, 42, 478–489. 10.1021/jm981064b. [DOI] [PubMed] [Google Scholar]
- Syed S. B.; Arya H.; Fu I.-H.; Yeh T.-K.; Periyasamy L.; Hsieh H.-P.; Coumar M. S. Targeting P-glycoprotein: Investigation of piperine analogs for overcoming drug resistance in cancer. Sci. Rep. 2017, 7, 7972. 10.1038/s41598-017-08062-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. B.; Kuo C. L.; Lien L. L.; Lien E. J. Structure–activity relationship: analyses of p-glycoprotein substrates and inhibitors. J. Clin. Pharm. Ther. 2003, 28, 203–228. 10.1046/j.1365-2710.2003.00487.x. [DOI] [PubMed] [Google Scholar]
- Li D.; Chen L.; Li Y.; Tian S.; Sun H.; Hou T. ADMET Evaluation in Drug Discovery. 13. Development of in Silico Prediction Models for P-Glycoprotein Substrates. Mol. Pharmaceutics 2014, 11, 716–726. 10.1021/mp400450m. [DOI] [PubMed] [Google Scholar]
- Zamora J. M.; Pearce H.; Beck W. T. Physical-chemical properties shared by compounds that modulate multidrug resistance in human leukemic cells. Mol. Pharmacol. 1988, 33, 454–462. [PubMed] [Google Scholar]
- Pearce H.; Winter M.; Beck W. Structural characteristics of compounds that modulate P-glycoprotein-associated multidrug resistance. Adv. Enzyme Regul. 1990, 30, 357–373. 10.1016/0065-2571(90)90026-X. [DOI] [PubMed] [Google Scholar]
- González M. L.; Joray M. B.; Laiolo J.; Crespo M. I.; Palacios S. M.; Ruiz G. M.; Carpinella M. C. Cytotoxic Activity of Extracts from Plants of Central Argentina on Sensitive and Multidrug-Resistant Leukemia Cells: Isolation of an Active Principle from Gaillardia megapotamica. Evid. Based Complement. Alternat. Med. 2018, 2018, 1. 10.1155/2018/9185935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joray M. B.; Trucco L. D.; González M. L.; Diaz Napal G. N.; Palacios S. M.; Bocco J. L.; Carpinella M. C. Antibacterial and Cytotoxic Activity of Compounds Isolated from Flourensia oolepis. Evid. Based Complement. Alternat. Med. 2015, 2015, 11. 10.1155/2015/912484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C. D.; Bracher F. Cytotoxic ring A-modified steroid analogues derived from Grundmann’s ketone. Eur. J. Med. Chem. 2011, 46, 3227–3236. 10.1016/j.ejmech.2011.04.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.