

Abstract
KRAS is the most frequently mutated driver oncogene in human cancer, and KRAS mutations are commonly associated with poor prognosis and resistance to standard treatment. The ability to effectively target and block the function of mutated KRAS has remained elusive despite decades of research. Recent findings have demonstrated that directly targeting KRAS-G12C with electrophilic small molecules that covalently modify the mutated codon 12 cysteine is feasible. We have discovered a series of tetrahydropyridopyrimidines as irreversible covalent inhibitors of KRAS-G12C with in vivo activity. The PK/PD and efficacy of compound 13 will be highlighted.
Keywords: Cancer, KRAS, G12C, covalent
KRAS is the single most frequently mutated oncogene and the first of more than 700 genes to be causally implicated in human cancer (COSMIC).1 Mutations in KRAS are prevalent among the top three most deadly cancer types in the United States: pancreatic (95%), colorectal (45%), and lung (35%).2 The frequent mutation of KRAS across a spectrum of aggressive cancers has stimulated an intensive drug discovery effort to develop therapeutic strategies that block KRAS function for cancer treatment. Despite nearly four decades of research, a clinically viable KRAS cancer therapy has remained elusive. However, recent findings have stimulated a new wave of activities to develop KRAS-targeted therapies.
Capping off an era marred by drug development failures and punctuated by waning interest and presumed intractability toward direct targeting of KRAS, new technologies and strategies are aiding in the target’s resurgence.3,4 Central to this renewal is a single mutation: KRAS-G12C, a well-validated driver mutation and the most frequent individual KRAS mutation in lung cancer.5 Associated with poor prognosis and resistance to treatment, KRAS-G12C represents both an extraordinary unmet clinical need and opportunity. This mutation has a causal role in 14% of lung adenocarcinomas (∼14 000 new U.S. cases annually), 5% of colorectal adenocarcinomas (∼5000 new U.S. cases annually), and smaller fractions of other cancers. Collectively, KRAS-G12C mutations comprise a patient population with a worldwide annual incidence of >100 000 individuals. The scientific basis for targeting KRAS-G12C was originally described in a breakthrough article by Shokat and colleagues.6 In their paper, the authors reported the identification of compounds that bound to a previously unappreciated pocket near the KRAS Switch-II effector region. The compounds form an irreversibly covalent bond to the mutant cysteine 12, locking the protein in its inactive GDP-bound state.7 Targeting the switch-II binding site was a clear advance in the field. This Letter describes the discovery of covalent small-molecule KRAS-G12C inhibitors with in vivo activity.
The Array BioPharma covalent fragment collection was screened against KRAS-G12C in a protein modification assay similar to that described by Ostrem.6 Subsequent hit elaboration was performed in an iterative library format. Primary observations from this exercise were incorporated into a structure-based drug design effort, which led to the discovery of compound 4. Our methods were validated by the publication of papers containing other KRAS-G12C inhibitors.8,9
The synthesis of compound 4 is shown in Scheme 1. Commercially available 4-Cl N-Boc tetrahydropyridopyrimidine 1 was treated with Cbz-piperazine and base in DMA under microwave conditions, followed by TFA, to give the amine compound 2. The naphthalene was installed using Buchwald conditions with racemic-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl/tris(dibenzylideneacetone)dipalladium(0) (BINAP/Pd2DBA3) catalyst. Deprotection of the Cbz group by hydrogenation and acetylation using acryloyl chloride gave the covalent inhibitor 4.
Scheme 1. Reactants.

(a) Cbz-piperazine, Cs2CO3, DMA, 150 °C, 95%. (b) TFA, DCM, 95%. (c) NaOtBu, BINAP/Pd2DBA3, toluene, 100 °C, 24 h, 33%. (d) Pd/C, H2, MeOH, 69%. (e) Acryloyl chloride, Hunig’s base, DCM, 19%.
Initial SAR was developed using the protein modification assay. In brief, compounds were incubated at a given time and concentration with KRAS-G12C lite,10 and the protein was assayed by mass spectrometry (MS). The percentage of protein + adduct was compared with control to give a percent of control (POC) value. As the program progressed and compounds became more potent, the incubation time was shortened and the concentration of compound was reduced. In the POC assay under the 3 h/5 μM conditions, compound 4 showed 13% modification of KRAS-G12C. Compound 4 was also assayed in an H358 KRAS-G12C-driven cell line and showed no suppression of ERK phosphorylation at the top 16 μM drug concentration. The mitogen-activated protein kinase (MAPK) pathway plays an important role in gene expression, cellular growth, and survival.11 Signaling through this pathway begins with RAS activation, followed by RAF recruitment to the membrane. RAS phosphorylates and activates MEK, which, in turn, phosphorylates ERK. Suppression of ERK phosphorylation is a mechanistic hallmark of pathway inhibition and was the readout of the cell assays.
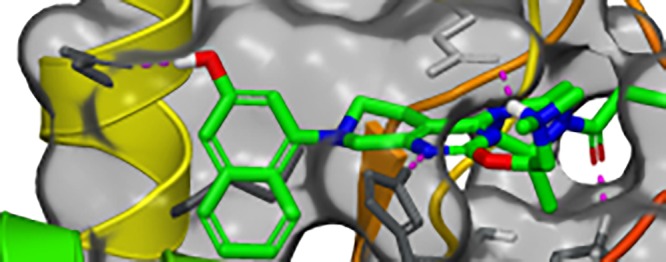
Figure 1 illustrates the X-ray crystal structure of 4 bound in the switch II pocket of KRAS-G12C. The covalent bond between the Cys12 side chain and the acrylamide of 4 is clearly visible. Key noncovalent interactions include hydrogen bonds between the amine nitrogen of Lys16 and the acrylamide carbonyl (2.91 Å) and the NE2 nitrogen of His95 and N-1 of the pyrimidine (2.77 Å). The naphthyl ring of 4 binds in a hydrophobic pocket formed by Val9, Met72, Phe78, Tyr96, Ile100, and Val103. The side-chain carboxylate of Asp69 is positioned ca. 3.3 Å from the naphthyl ring of 4. Whereas Asp69 forms a salt bridge with Arg102, it may be possible to form an additional H-bond to this carboxylate.
Figure 1.
X-ray crystal structure of 4 in complex with KRAS-G12C at 1.8 Å resolution. Hydrogens are added for clarity (PDB ID 6N2J).
Table 1 describes a series of analogs that were synthesized in an attempt to make an H-bond to Asp69; the POC data at 3 h/5 μM as well as the H358 cell IC50 data are presented. Compound 4 with no H-bond donor is included for reference. Indazole 5 was equipotent to naphthyl in the POC assay and had no activity in the cellular assay. As noted above, the naphthyl region of the binding pocket is highly lipophilic, suggesting that loss of contacts in this region would lead to significant potency decreases (i.e., naphthyl to phenyl substitution). This result was borne out experimentally; the two phenol compounds 6 and 7 show 2% protein modification in the 3 h/5 μM conditions. Naphthol compound 8 was the most potent compound in this series with 99% protein modification and measurable cell activity with an IC50 = 7.6 μM. Finally, compound 9 showed 0% modification under these assay conditions, presumably due to poor positioning of the hydroxyl group.
Table 1. Comparison of Naphthyl Replacement SARa.

In vitro activity assays. Percent of control (POC) protein modification assay measured at the 3 h time point with 5 μM compound. H358 cell assay IC50 showing concentration at which 50% suppression of ERK phosphorylation is observed. See the Supporting Information for the standard deviation and the number of tests.
We hypothesized that substitution at the C-2 position of the pyrimidine ring would afford access to the carboxylate of Glu62. Shown in Table 2, potential ionic interactions with Glu62 were tested with compounds 10 and 11 by varying the chain length to place a basic amine near the carboxylate. Both of these compounds were more active in the protein modification assay, under the 15 min/3 μM conditions, and cell assay compared with compound 8 (included in Table 2 for comparison). The rigidified α-methyl analog 12 showed a further increase in cell potency.
Table 2. Exploration of the Basic C-2 Side Chaina.


In vitro activity assays. Percent of control (POC) protein modification assay measured at the 15 min time point with 3 μM compound. H358 cell assay IC50 showing concentration at which 50% suppression of ERK phosphorylation is observed. See the Supporting Information for the standard deviation and the number of tests.
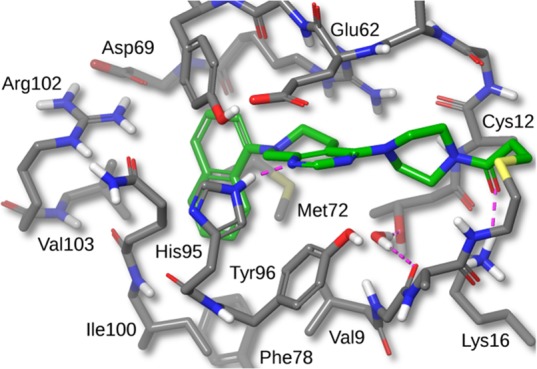
A high-resolution crystal structure of compound 12 bound to KRAS-G12C is shown in Figure 2. The binding mode for 12 is similar to that of compound 4. Interestingly, even though the KRAS protein was reacted with racemic compound 12 in solution, only the (R)-enantiomer was seen in the crystal structure. Docking studies suggested that the (S)-enantiomer would display more strain energy in the bound conformation.
Figure 2.
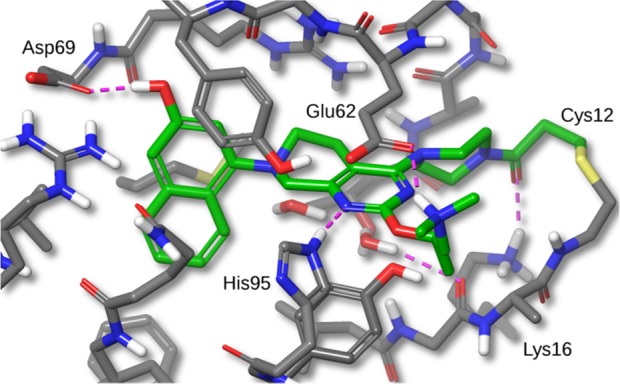
X-ray crystal structure of compound 12 bound to KRAS-G12C with 1.7 Å resolution. Hydrogens added for clarity (PDB ID 6N2K).
In addition to the previously mentioned interactions, there is a 2.65 Å H-bond from the naphthyl OH to the carboxylate of Asp69. The C-2 amino substituent forms a salt-bridge with the carboxylate of Glu62, with an N–O distance of 2.80 Å, and a cation−π interaction with His95 (His95 ring centroid to amine nitrogen distance is 5.21 Å). On the basis of this X-ray structure and the observed amine interactions with Glu62 and His95, we designed compound 13 with the aim of eliminating one rotatable bond and the introduction of more hydrophobic contacts with His95. Docking studies of compound 13 suggested that the pyrrolidine ring amine would make the same salt-bridge interaction with Glu62 and cation−π interaction with His95. The N-methyl pyrrolidine C-2 amine substituent of compound 13 displayed a dramatic boost in potency, protein modification POC = 84%, and cell IC50 = 0.070 μM. We attributed this potency increase to the aforementioned interactions and the removal of one rotatable bond.
The importance of the RAS-MAPK pathway was previously mentioned; PI3K is another effector pathway of RAS, regulating cell growth, cell-cycle entry, cell survival, and metabolism.12 Both of these pathways play key roles in the physiology of healthy cells such that it is of critical importance for a KRAS-G12C inhibitor to spare wild-type (WT) KRAS. To determine selectivity for the KRAS-G12C mutation, the activity of compound 13 against three non-G12C cell lines was determined. The cell lines studied included the KRAS-WT RKO and SNU-C5 lines as well as the KRAS-G12D AGS line. In all three lines, compound 13 was found to be inactive with IC50 values exceeding 16 μM, the highest concentration tested.
To determine if compound 13 was a suitable candidate for in vivo antitumor efficacy studies, the ADME and PK properties were evaluated. Compound 13 displayed low permeability with P-gp efflux in the MDR1-transfected LLC-PK1 cell permeability assay, and mouse plasma protein binding (PPB) was 95% (Table 3). The predicted hepatic clearance (CLh) in mouse was calculated to be 53 and 76 mL/min/kg from compound 13 half-life data in mouse liver microsome and mouse hepatocyte incubations, respectively. The lower stability in hepatocytes indicated the involvement of Phase II metabolism. Following a 3 mg/kg IV dose of compound 13 to CD-1 mice, the clearance was 46 mL/min/kg (Table 4). Oral administration of 100 mg/kg compound 13 to mice resulted in a Cmax of 0.59 μg/mL (Table 4), which was below the calculated free-fraction-adjusted cell IC50 of 1.4 μM (0.74 μg/mL). Although the PK parameters required to drive antitumor efficacy were not known, PO dosing did not appear to warrant further evaluation in an efficacy study. However, the moderate clearance of compound 13 supported intraperitoneal (IP) dosing as an opportunity to demonstrate antitumor activity. IP dosing resulted in appreciably higher concentrations of compound 13 in mouse plasma compared with oral dosing. Following the 15 or 50 mg/kg doses, drug plasma concentrations were above the free-fraction-adjusted IC50 for 1–3 h. However, the 100 mg/kg dose provided >6 h coverage of the free-fraction-adjusted cellular IC50 (Table 5). On the basis of these results, 30 and 100 mg/kg IP doses were selected for tumor growth inhibition experiments.
Table 3. ADME Properties of Compound 13a.
| ADME assay | result |
|---|---|
| MDR1-transfected LLC-PK1 permeability | Papp(A to B) = 1.4 × 10–6 cm/sec |
| Papp(B to A) = 37 × 10–6 cm/sec | |
| BA/AB ratio = 27 | |
| mouse PPB | 95% |
| mouse predicted CLh (microsomes) | 53 mL/min/kg |
| mouse predicted CLh (hepatocytes) | 76 mL/min/kg |
Bidirectional transport of 1 μM compound 13 was measured in MDR1-transfected LLC-PK1 cell monolayers; A = apical side; B = basolateral side. PPB was determined by equilibrium dialysis in CD-1 mouse plasma with 1 μM compound 13. Hepatic CL was predicted based on the t1/2 of 1 μM compound 13 in CD-1 mouse microsomes (1 mg/mL) or hepatocytes (1 × 106 cells/mL).
Table 4. PK of Compound 13 in CD-1 Micea.
| dose | parameter | value |
|---|---|---|
| 3 mg/kg IV | CL | 46 mL/min/kg |
| t1/2 | 0.96 h | |
| VSS | 0.53 L/kg | |
| 100 mg/kg PO | Cmax | 0.59 μg/mL |
| Tmax | 1.00 h | |
| AUCinf | 0.88 h·μg/mL | |
| F | 2.4% |
CD-1 mice (n = 3/time-point) were dosed IV with 3 mg/kg or PO with 100 mg/kg compound 13 in 10% captisol and 50 mM sodium citrate, pH 5. Plasma was collected at 1, 5 (IV only), 15, and 30 min and 1, 2, 4, 8, 12, and 24 h, and PK parameters were calculated on the mean concentration data using noncompartmental analysis.
Table 5. Compound 13 Concentrations after IP Dose in Micea.
| compound 13 concentration (μg/mL) |
|||
|---|---|---|---|
| time (h) | 15 mg/kg | 50 mg/kg | 100 mg/kg |
| 0.5 | 1.5 ± 1.3 | 8.3 ± 1.8 | 26 ± 16 |
| 1 | 0.97 ± 0.24 | 2.6 ± 0.69 | 17 ± 2 |
| 3 | BLQ | 0.25 ± 0.04 | 1.4 ± 1.2 |
| 6 | ND | ND | 1.3 ± 1.2 |
CD-1 mice (n = 3/time-point) were dosed IP with 15, 50, or 100 mg/kg compound 13 in 10% captisol and 50 mM sodium citrate, pH 5. Concentrations are reported as the arithmetic mean ± standard deviation. BLQ = below limit of quantitation. ND = not determined.
The KRAS-G12C mutant H358 cell line was used for in vitro screening of pERK modulation because the conditions for the assay were well developed. However, the MIA PaCa-2 line outperformed the H358 cell line in terms of tumor growth in nude mice and reproducibility of tumor doubling times. For those reasons, tumor growth inhibition studies were conducted in the MIA PaCa-2 line. Similar to what was observed in H358 cells, the IC50 of 13 was 48 nM in a pERK inhibition assay in MIA PaCa-2 cells. A tumor growth inhibition study was run with compound 13 administered IP QD in MIA PaCa-2 tumor-bearing mice. The animals were randomized with six animals per group, and dosing began on day 15 when the tumor volume had reached a size of 150 mm3. Tumor growth curves for vehicle control and the 30 and 100 mg/kg doses are illustrated in Figure 3. The vehicle control tumors grew unabated, tripling in size by day 25. Both of the treated groups showed rapid tumor regressions after 5 days of dosing and then a leveling off of tumor volume with continued dosing. It should be noted that on day 25 there were two animals in the 30 mg/kg group and four animals in the 100 mg/kg group that were found to be tumor-free. Compound 13 was well tolerated throughout this experiment, and animals did not show weight loss or adverse symptoms.
Figure 3.
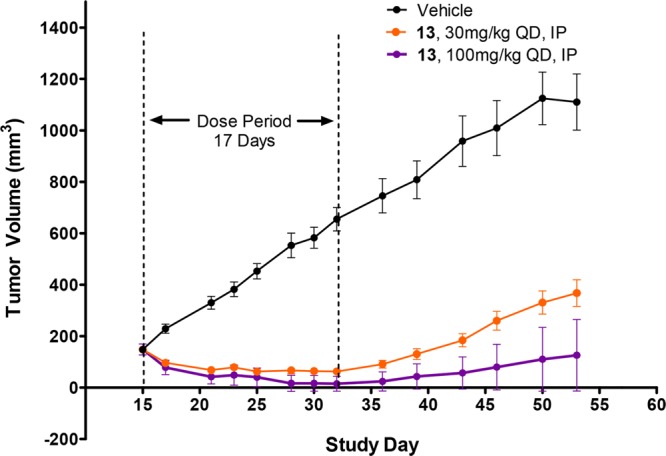
Antitumor efficacy of compound-13-dosed IP in the MIA PaCa-2 tumor xenograft model. The mean tumor volume for 12 tumors is plotted, and the error bars represent the standard error of the mean.
On the basis of a published report by Patricelli,13 compound 13 target engagement was evaluated in MIA PaCa-2 tumor tissue from a mouse subcutaneous xenograft model. Mice were dosed IP QD for 5 days, and tumors were harvested at 3 and 24 h post-dose on days 1 and 5. An LC–MS-based KRAS-G12C engagement assay was developed to quantitatively measure the interaction of compound 13 with its intended protein target. The decrease in the cysteine-12-containing peptide from tryptic digests of KRAS-G12C mutant tumors following compound treatment was quantified relative to a control peptide, representing total KRAS. Following multiple doses of compound 13, KRAS-G12C engagement was maintained at >65% in tumors from both dose groups (Figure 4). Target engagement only slightly decreased between 3 and 24 h, despite a large decrease in concentration of compound 13 in the plasma (Table 5), supporting a sustained target residence time and indicating a relatively slow rate of KRAS-G12C protein synthesis. Coupled to the tumor growth inhibition results, these data show on-target efficacy for the tetrahydropyridopyrimidine KRAS-G12C inhibitor 13.
Figure 4.
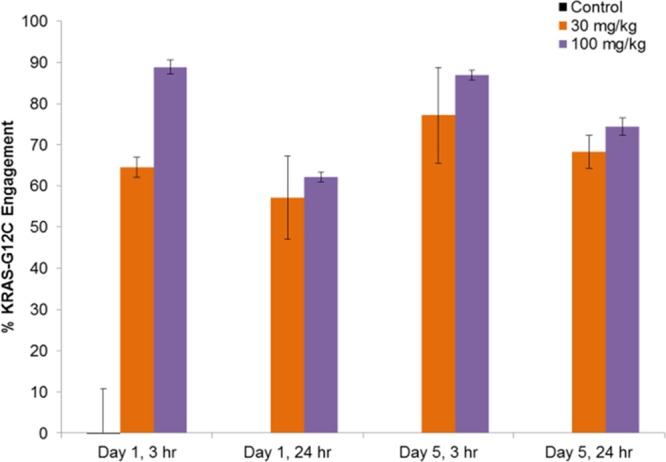
KRAS-G12C target engagement in MIA PaCa-2 tumors. The mean % KRAS-G12C engagement (see the Supporting Information) for three tumors is plotted, and the error bars represent the standard deviation.
In summary, we have identified a series of tetrahydropyridopyrimidines as irreversible covalent inhibitors of KRAS-G12C. A crystal structure of prototype compound 4 was used to optimize the interactions with the protein for enhanced potency. The replacement of the naphthyl with a naphthol and the substitution at C-2 of the pyrimidine ring resulted in compound 13 possessing a cell IC50 = 70 nM. Tumor regressions and cures were observed in a TGI study with compound-13-dosed IP QD at 30 and 100 mg/kg. In addition, an on-target mechanism of action was confirmed via KRAS-G12C target engagement experiments. Compound 13 is an advanced lead; future publications will detail our findings toward potent, orally bioavailable compounds with enhanced PK.
Acknowledgments
We thank the in vitro ADME group at Array BioPharma, Inc.
Glossary
Abbreviations
- ADME
absorption, distribution, metabolism, and excretion
- AUCinf
area under the curve extrapolated to infinity
- BLQ
below the lower limit of quantitation
- CBZ
carboxybenzyl
- CL
clearance
- Cmax
maximum concentration
- DCM
dichloromethane,
- DMA
dimethylacetamide
- F
bioavailability
- LC–MS
liquid chromatography–mass spectrometry
- POC
percent of control
- ND
not detected
- PD
pharmacodynamics
- P-gp
P-glycoprotein
- PK
pharmacokinetics
- PPB
plasma protein binding
- Tmax
time at Cmax
- TFA
trifluoroacetic acid
- TGI
tumor growth inhibition
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00382.
1. Experimental section. 2. KRAS LC–MS modification assay procedure (POC assay). 3. G12C cell assay. 4. Liver microsomal incubation. 5. Hepatocyte incubations. 6. Analytical quantitation of hepacyte and microsomal incubations. 7. Institutional animal care and use committee statement. 8. Antitumor efficacy study. 9. K-Ras G12C engagement (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Notes
Coordinates for the X-ray structures shown in Figures 1 and 2 have been deposited with the Protein Data Bank.
Supplementary Material
References
- Bos J. Ras oncogenes in human cancer: a review. Cancer Res. 1989, 49, 4682–4689. [PubMed] [Google Scholar]
- Jarvis L. Have drug hunters finally cracked KRAS?. C&EN 2016, 94, 28–33. [Google Scholar]
- Ledford H. Cancer: The Ras renaissance. Nature 2015, 520, 278–280. 10.1038/520278a. [DOI] [PubMed] [Google Scholar]
- Papke B.; Der C. Drugging RAS: Know the enemy. Science 2017, 355, 1158–1163. 10.1126/science.aam7622. [DOI] [PubMed] [Google Scholar]
- Matikas A.; Mistriotis D.; Georgoulias V.; Kotsakis A. Targeting KRAS mutated non-small cell lung cancer: A history of failures and a future of hope for a diverse entity. Crit. Rev. Oncol. Hematol. 2017, 110, 1–12. 10.1016/j.critrevonc.2016.12.005. [DOI] [PubMed] [Google Scholar]
- Ostrem J. M.; Peters U.; Sos M. L.; Wells J. A.; Shokat K. M. KRAS (-G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem J. M.; Shokat K. M. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat. Rev. Drug Discovery 2016, 15, 771–785. 10.1038/nrd.2016.139. [DOI] [PubMed] [Google Scholar]
- Zeng M.; Lu J.; Li Ls.; Feru F.; Quan C.; Gero T. W.; Ficarro S. B.; Xiong Y.; Ambrogio C.; Paranal R. M.; Catalano M.; Shao J.; Wong K.-K.; Marto J. A.; Fischer E. S.; Janne P. A.; Scott D. A.; Westover K. D.; Gray N. S. Potent and Selective Covalent Quinazoline Inhibitors of K-Ras G12C. Cell Chem. Bio. 2017, 24, 1005–1016. 10.1016/j.chembiol.2017.06.017. [DOI] [PubMed] [Google Scholar]
- Janes M. R.; Zhang J.; Li L.-S.; Hansen R.; Peters U.; Guo X.; Chen Y.; Babbar A.; Firdaus S. J.; Darjania L.; et al. Targeting KRAS Mutant Cancers with a covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589. 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- KRAS-G12C lite has all cysteines except G12C mutated to other amino acids (C51S/C80L/C118S), as in Ostrem et al.6
- Knight T.; Irving J. A. Ras/Raf/Mek/Erk pathway activation in childhood acute lymphoblastic leukemia and its therapeutic targeting. Front. Oncol. 2014, 4, 1–12. 10.3389/fonc.2014.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano E.; Downward J. RAS Interactions with PI3K. Genes Cancer 2011, 2, 261–274. 10.1177/1947601911408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli M. P.; Janes M. R.; Li L. S.; Hansen R.; Peters U.; Kessler L. V.; Chen Y.; Kucharski L. M.; Feng J.; Ely T.; Chen J. H.; Firdaus S. J.; Babbar A.; Ren P.; Liu Y. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discovery 2016, 6, 316–329. 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.