

Abstract

A series of 2-pyrazolyl quinolones has been designed and synthesized in 5–7 steps to optimize for both in vitro antimalarial potency and various in vitro drug metabolism and pharmacokinetics (DMPK) features. The most potent compounds display no cross-resistance with multidrug resistant parasite strains (W2) compared to drug sensitive strains (3D7), with IC50 (concentration of drug required to achieve half maximal growth suppression) values in the range of 15–33 nM. Furthermore, members of the series retain moderate activity against the atovaquone-resistant parasite isolate (TM90C2B). The described 2-pyrazoyl series displays improved DMPK properties, including improved aqueous solubility compared to previously reported quinolone series and acceptable safety margin through in vitro cytotoxicity assessment. The 2-pyrazolyl quinolones are believed to bind to the ubiquinone-reducing Qi site of the parasite bc1 complex, which is supported by crystallographic studies of bovine cytochrome bc1 complex.
Keywords: Quinolone, antimalarial, Plasmodium falciparum, cytochrome bc1, atovaquone, drug resistance
Malaria was responsible for nearly 216 million cases and an estimated 445,000 deaths in 2016.1 Approximately half of the global population is at risk of infection, particularly in the tropical and subtropical regions where malaria is widespread.
Malaria is a disease caused by the parasite of the genus Plasmodium and is transmitted to people through the bites of infected female Anopheles mosquitoes. Plasmodium falciparum is the most prevalent and lethal species of the parasite to human and has developed resistance to most of the classical antimalarials.2,3
The quinolone scaffold is present in several antibiotics, and this chemotype possesses a wide range of biological activities including anticancer, anti-HIV, and antiviral.4−7 The antimalarial activity of Endochin was first identified in the 1940s,8 and recent publications have highlighted the promising potential antimalarial properties of aryl and alkyl substituted quinolones.9−12 Studies by Nilsen and co-workers discovered the quinolone-3-diaryl ethers ELQ-300 and P4Q-391, which have excellent profiles and selectively inhibit Plasmodium cytochrome bc1 complex.13 Our group14 and others15 have focused on 2-aryl quinolones, and we have shown that representative 2-aryl quinolones eg. (1) can inhibit two mitochondrial enzymes in the electron transport chain, the cytochrome bc1 complex and the recently identified PfNDH2 (Type II NADH:ubiquinone oxidoreductase).16,17 This inhibition results in the collapse of the mitochondrial membrane potential, the inhibition of de novo pyrimidine biosynthesis, and ultimately the death of the parasite.18
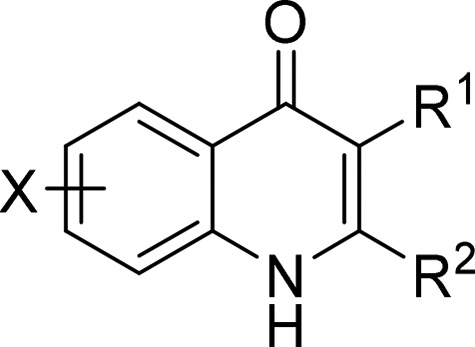
Previously compound 1 was identified by us as one of the lead compounds with good antimalarial activity in a drug discovery program14 (Figure 1). While compound 1 demonstrated good antimalarial activities against various strains of P. falciparum, it required further optimization of its physiochemical properties, especially lipophilicity (ClogP) and aqueous solubility. In this Letter we describe the further optimization and the synthesis of a series of 2-pyrazolyl quinolone with the aim of reducing ClogP and improving the aqueous solubility while maintaining/improving the antimalarial activity. It has been well documented that pyrazole is a bioisostere for benzene ring and can improve physiochemical properties (i.e., aqueous solubility) by reducing CLogP.19 This strategy was applied to compound 1 by replacing the C-ring with a pyrazolyl ring. Different substituents on other parts of the molecule such as A-ring, B-ring, and D-ring were also explored. In addition to medicinal chemistry optimization, we were also interested in probing the effect of chemical substitution on bc1 (Qi) site binding by comparing our previously published bc1 enzyme–inhibitor complexes with lead pyrazoles prepared in this work.
Figure 1.

Initial lead 1 and its antimalarial activities and physiochemical properties.
The 2-pyrazolyl quinolone analogues were prepared by three different synthetic routes. The synthesis of quinolones 4a–h is depicted in Scheme 1. 2-Bromo-4-chloroquinoline 2, synthesized from oxidation of corresponding 4-chloroquinoline followed by bromination, was coupled with readily available pyrazole boronic acid pinacol ester, giving the quinoline 3 in 38–93% yields. Upon hydrolysis using acetic acid or formic acid, quinoline 3 provided quinolones 4a–h in excellent yields. Some selected 3H-quinolones were further chlorinated by sodium dichloroisocyanurate to give the 3-Cl analogue 5a–c in 56–72% yields.
Scheme 1. General Route 1 for Synthesis of Pyrazole Quinolones.

Conditions and reagents: (a) pyrazole boronic acid pinacol ester, 10 mol % PdCl2(dppf), K2CO3·1.5H2O, dioxane, reflux, 24 h; (b) AcOH, H2O, 120 °C, 24–48 h or HCl(aq), dioxane, reflux, 48 h or HCOOH/H2O, DMF, 140 °C, 4 h; (c) sodium dichloroisocyanurate, MeOH, NaOH(aq), r.t., o/n.
The synthesis of quinolones 11a–j was accomplished in 3–6 steps from commercially available starting materials according to the synthetic methodology showed in Scheme 2. Oxazoline 7 was synthesized from the corresponding isatoic anhydride 6 in 60–75% yields. Substituted pyrazole 9, synthesized from corresponding iodopyrazole 8 and benzyl bromide in excellent yield (see Supporting Information), was converted to ketone2010 in 26–55% yields. Cyclization of oxazoline 7 with ketone 10 in the presence of catalytic trifluoromethanesulfonic acid afforded the desired quinolones 11a–j in 42–84% yields.
Scheme 2. General Route 2 for Synthesis of Pyrazole Quinolones.

Conditions and reagents: (a) 2-amino-2-methyl-propanol, ZnCl2, PhCl, 135 °C, 24 h; (b) corresponding benzyl bromide, K2CO3, acetone, reflux, 3 h; (c) Pd2(dba)3, dppp, pyrrolidine, 4 Å M.S., DMF, 110 °C, 6 h; (d) CF3SO3H, n-BuOH, N2, 130 °C, 24 h.
Investigations also focused on the possibility of formulating the series as salts and improving the solubility by extending the side chain and introducing the morpholine group at the terminal as illustrated by 13a and 13b. The synthesis of the extended side chain quinolones 13a and 13b was shown in Scheme 3. Quinolone 12 (see Supporting Information) was coupled with the corresponding boronic acid pinacol ester to provide quinolones 13a and 13b.
Scheme 3. General Route 3 for Synthesis of Pyrzaole Quinolones with Extended Side-Chains.

Conditions and reagents: (a) 5 mol % PdCl2(dppf), K2CO3, H2O/dioxane, 100 °C, 5 h.
In vitro antimalarial activity of the quinolone analogues was assessed against the 3D7 strain (chloroquine sensitive) of Plasmodium falciparum (Table 1). Several analogues exhibit improved antimalarial activity compared with the original lead 1. As observed from previous work, a p-OCF3 substituent on the D-ring in the 2-pyrazolyl series provides better antimalarial activity than p-F. The terminal phenyl ring is more favorable than a pyridinyl or morpholine ring. Longer side chains, as seen in 11j, 13a, and 13b, results in a significant loss in antimalarial activity. A clear trend is seen in the nature of the A-ring substituent X. In general, the presence of F, Cl, and OMe on the A-ring is well tolerated and often improves the activity as shown when comparing 4e (100 nM), 11c (33 nM), 11g (80 nM), and 11h (50 nM). A small electron withdrawing substituent on the 6-position of quinolone is more favorable (see 11d and 11e). While F and Cl at the 7-position of quinolone exhibit potent activity, 7-CF3 is less tolerated and a 8-fold drop in activity is observed. Among the substituents on the A-ring, 7-OMe enhances the activity greatly. The position of the pyrazolyl ring that links to the quinolone core also effects the activity. When the 3-position of pyrazolyl ring is linked to the quinolone core (4h, 5c, and 11i) (instead of the 4-position), there is a reduction in potency. Looking into the substituents at the 3-position of the quinolone, most of substituents, except isopropyl group, are well-tolerated. In contrast to previous SAR studies, 3-chloro analogues are less potent than the 3-Me analogue (as seen in 5b and 11c), which is the most active in this series.
Table 1. In Vitro Antimalarial Activities of Quinolones versus the 3D7 Strain+ of Plasmodium falciparuma.

50% inhibitory concentration in vitro against P. falciparum chloroquine-sensitive (3D7) lines.
A selection of compounds was tested against the chloroquine resistant strain of P. falciparum, W2, and atovaquone resistant TM90C2B containing the Y268S mutation in the quinol oxidation Qo site of the parasite mitochondrial cytochrome bc1 complex21−23 (Table 2). The SAR trends observed from the 3D7 data are similar to the W2 data with the presence of a 7-methoxy (11c) enhancing activity when compared to unsubstituted analogue (4e). Interestingly, unlike the activity data against 3D7 strain, the presence of 3-Cl in the quinolone core enhances activity compared with 3-methylation. In a confirmatory study that assessed antimalarial potency against the transgenic P. falciparum TX13 strain,24 expressing yeast dihydroorotate dehydrogenase,255b showed no inhibition at >1000 nM, further supporting that the series is targeting the respiratory chain of the parasite mitochondrion. To determine if the antimalarial activity is a result of on-target plasmodial bc1 inhibition, the enzymatic activity was determined by monitoring cytochrome c reduction using decylubiquinol as electron donor as previously reported.26 This enzymatic study confirmed 11c as a potent Pf bc1 inhibitor with an IC50 of 0.75 nM (Figure S1). However, it is noteworthy that, although relative to atovaquone, some of the selected compounds in this series are active against the TM90C2B strain, and reduced potency is seen compared to 3D7 and W2. A possible explanation for this observation could be that for this series, there is a contributing element of Qo site inhibition; it has been noted by Riscoe and co-workers that minor modifications to the quinolone core of a series of related endochin quinolone analogues can subtly affect both Pf bc1 Qo/Qi sites binding.27 This observation may well explain in part the reduced potency of 11c versus the Qo site mutated TM90C2B strain.
Table 2. In Vitro Antimalarial Activities of Selected Quinolones versus W2 and TM90C2B and PfNDH2 Enzyme Inhibition Dataa.
| compound | IC50 W2 (nM) | IC50 TM90C2B (nM) | IC50PfNDH2 (nM) |
|---|---|---|---|
| chloroquine | 12.3 | 14.5 | NDb |
| atovaquone | 0.3 | 9908 | 10,000 |
| 1 | 26 | 122 | 20 |
| 4e | 33 | ND | 837 |
| 5a | 14 | ND | ND |
| 5b | 11 | 110 | ND |
| 11c | 15 | 500 | 1,000 |
| 11i | 49 | 300 | 68 |
50% inhibitory concentration in vitro against P. falciparum chloroquine-resistant W2 strain (Indochina), Atovaquone resistant TM90C2B strain, and PfNDH2 enzyme inhibition data.24
ND, not determined.
One of the major aims in this lead optimization process was to improve the physiochemical properties of compound 1, especially its aqueous solubility. Aqueous solubility of molecules is related to lipophilicity (CLogP) and crystal packing via π-stacking of aromatic ring systems (as reflected in the melting point).28 Replacement of the benzene C-ring to a pyrazole ring and incorporating various substitutions at the 3-position of the quinolone can dramatically change both CLogP and melting point of the analogues in this series, and thus improve the aqueous solubility profile (Table 3). Replacement of the benzene C-ring with pyrazole reduced CLogP by between 1.5 to 2 units. Incorporation of a substituent, such as Me or Et, at the 3-position of the quinolone ring likely reduces the planarity of the side-chain, reducing packing, and this reduces the melting point. The most significant reduction in melting point came as the result of modification of the linkage of the pyrazole heterocycle from a 1,4 to 1,3 arrangement. The combination of reduction in both lipophilicity and aggregation via π-stacking of aromatic ring systems resulted in over 10-fold improvement in aqueous solubility for some selected analogues in this series (11c and 11i).
Table 3. CLogP Value, Melting Point, and Aqueous Solubility at pH 7.4 for Selected 2-Pyrazolyl Quinolones.
| compound | CLogP | melting point (°C) | solubilitya (μM) |
|---|---|---|---|
| 1 | 5.67 | 213 | 0.03 |
| 4e | 3.71 | 194 | 0.1 |
| 5a | 4.04 | 256 | 0.01 |
| 11a | 4.24 | 143 | 0.2 |
| 11c | 3.70 | 172 | 0.3 |
| 11i | 3.92 | 64 | 0.4 |
Solubility in pH 7.4 PBS buffer.
To further examine the DMPK properties, selected compounds in the series have also been screened for metabolic stability and plasma protein binding in vitro (Table S1). From the human microsomal stability and rat hepatocyte stability data, all selected representatives in the 2-pyrazolyl quinolone series had very low clearance and good metabolic stability. Most of the tested compounds, except 5a, had high human plasma protein binding level, but below 99.9% bound, which is comparable with other antimalarial quinolones.
To gain insight into the key protein/ligand binding interactions of 2-pyrazole quinolones within a bc1 complex, we cocrystallized bovine heart-derived cytochrome bc1(29) with compound 11c. Clear and defined omit Fo–Fc electron density within the Qi pocket near heme bH (Figure 2A,B) showed unambiguous binding of the quinolones to the Qi site. The carbonyl of quinolone core forms H-bonds with His201 side chain, and the aromatic tail is positioned within the hydrophobic region. The planar quinolone ring of 11c makes an aromatic stacking with the phenyl ring of Phe220, and its amine points to the side chain of Ser35. The aromatic tail is packed in the hydrophobic cavity conferred by Ile39 and Ile42.
Figure 2.
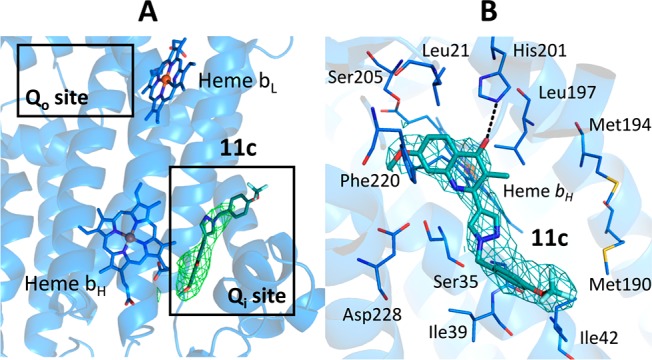
Cytochrome bc1 Qi site (bovine heart derived) bound inhibitor 11c. (A) The omit Fo–Fc map (green) contoured at 3σ level around 11c (teal) compounds shown as sticks. The cartoon representation of cytochrome b subunit is shown in blue. The Qi and Qo sites are marked by black boxes. (B) The 2Fo–Fc electron density map (cyan) contoured at 1σ level around the inhibitors. Surrounding residues are drawn as blue lines and hydrogen bonds as black dashed lines.
As there is no structure of P. falciparum cytochrome bc1, its homology model was generated by SWISS-MODEL online tool30 based on the primary sequence (Q02768) and the bovine cytochrome b (PDB: 5OKD) template. The Pf model was aligned to the bovine crystal structure to visualize inhibitor interactions within the Pf Qi site (Figure S2). The parasite’s Qi binding pocket appears to be smaller than bovine, and there could be a steric contact of Phe30 (Ser35 in bovine) side chain with the pyrazole ring of 11c. The inhibitors had to adopt different poses in the Pf Qi site because of steric clashes with the calculated protein model. To predict possible binding poses in the parasite enzyme, in silico docking was performed by SwissDock31 with defined interest region of Qi site. The final solution for 11c was determined based on the compound pose in bovine crystal structure with the highest FullFitness scoring of −858.01 kcal/mol. As the absence of 7-methoxy group on the A-ring often reduces antimalarial activities, compound 4e, which is the unsubstituted analogue of 11c, was docked into the Pf Qi site with FullFitness score of −854.04 kcal/mol. The molecular docking results are shown in Figure 3. Both compounds can form a hydrogen bond with His192, but the presence of 7-methoxy group in 11c causes a shift in binding location away from the 4e position with stronger binding explained by π-stacking interaction of D-ring with Phe30 and Phe37 side chains. This observation provides insight as to how 7-methoxy quinolone analogues have improved potency over other derivatives. Future work will utilize the homology Pf bc1 model with the mammalian bovine structures described here to guide chemical substitution that enhances parasite potency and selectivity further.
Figure 3.

In silico docking of 4e (pink) and 11c (teal) into the Plasmodium falciparum Qi site. The protein structure and residues shown in magenta. The binding surface shown in gray. Hydrogen bonds are indicated by black dashed lines.
Finally, given that members of this series have the propensity to bind to mammalian bc1, we examined the cytotoxicity profiles in the Hep G2 cell line (Table 4). From this in vitro toxicity assessment, the tested 2-pyrazolyl quinolone analogues showed similar or higher IC50 values than the negative control, Tamoxifen, which indicate low cytotoxicity for the analogues tested. Based on the 3D7 IC50 data, there is a sufficient safety windows for the tested analogues with 11c expressing the highest therapeutic index ratio of 333.
Table 4. In Vitro Cytotoxicity Assessment Using Hep G2 Cells for Selected 2-Pyrazolyl Quinolones.
| compound | Hep G2 toxicity IC50 (μM) ± SEM | therapeutic indexa |
|---|---|---|
| 4e | 13.0 ± 1.7 | 130 |
| 5a | 28.4 ± 8.3 | 171 |
| 11a | 19.3 ± 3.3 | 219 |
| 11c | 11.0 ± 0.7 | 333 |
| 11i | 21.2 ± 0.8 | 79 |
| rotenone | 1.52 ± 0.24 | |
| tamoxifen | 12.0 ± 0.5 |
Therapeutic index is determine by comparing the HepG2 IC50 values with the corresponding 3D7 IC50 values.
To conclude, a series of 2-pyrazolyl quinolones with potent antimalarial activity against the 3D7 strain and W2 strain of P. falciparum have been identified. Representative analogue 11c has improved antimalarial activity, physiochemical, and DMPK properties in comparison to previously reported lead molecules in addition to low cytotoxicity. While the series on a whole have improved solubility compared with previous quinolone derivatives, further work is required to find quinolone derivatives with solubility in a more desired range (>50 μM). Crystallography and homology based modeling of mammalian and parasite bc1 complexes have now been produced that may allow rational drug design approaches to be initiated for more selective Pf bc1 Qi inhibitors. It is noteworthy that, despite the enzymatic and crystallographic data described above, we cannot rule out that this series of 2-pyrazolyl quinolones may potentially target other components of the electron transport chain of the parasite mitochondrion.
Further work also is in progress to investigate the in vivo PK profiles and efficacy of this series and to profile the lead compounds for their activity against liver and sexual stage of the parasites.
Acknowledgments
We thank Professor Dennis Kyle (College of Public Health, University of South Florida; present address Centre for Tropical and Emerging Global Diseases, University of Georgia, USA) for supplying the atovaquone resistant isolate TM90C2B (Thailand) and Dr. Jiri Gut and Professor Phil Rosenthal for the W2 data in Table 2 (Department of Medicine, University of California, San Francisco, USA). We also thank the staff and patients of Ward 7Y and the Gastroenterology Unit, Royal Liverpool Hospital, for their generous donation of blood. We also want to thank the DMPK group (led by Peter Webborn) in AstraZeneca U.K. for providing the in vitro measurement of DMPK properties, including aqueous solubility, human plasma protein binding, mouse microsome clearance, and rat hepatocyte clearance described in Table 3 and Table S1 (Supporting Information). We thank Diamond Light Source for access to beamline I04 (proposal number 11740) that contributed to the results presented here.
Glossary
ABBREVIATIONS
- CCR2
CC chemokine receptor ;
- CCL2
CC chemokine ligand 2
- CCR5
CC chemokine receptor 5
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00371.
Synthetic methods, procedures, and chemical analysis data of all final compounds (except compound 1) and the intermediates; biological testing methods and procedures; and cytochrome bc1 preparation and crystallography (PDF)
Author Present Address
∥ ARUK Oxford Drug Discovery Institute, University of Oxford, Oxford, OX3 7FZ, U.K.
Author Contributions
W.D.H., S.C.L., K.A., S.V.A., N.B., G.A.B., and P.M.O. contributed to writing of the manuscript; P.M.O., S.A.W., S.V.A., S.S.H., and G.A.B. conceived this work; W.D.H. and G.N. designed, synthesized, and characterized chemical compounds; J.D. and R.S.P. conducted biological studies; K.A. and S.V.A. performed crystallographic studies. All authors have given approval to the final version of the manuscript.
This work was supported by grants from the Leverhulme Trust, the Wellcome Trust (Seeding Drug Discovery Initiative), the National Institute of Health Research (NIHR, BRC Liverpool), and Mahidol-Liverpool Stang Mongkolsuk PhD scholarship.
The authors declare no competing financial interest.
This paper published ASAP on November 1, 2018 with errors in Table S2 in the Supporting Information file. The corrected paper reposted to the Web on November 7, 2018.
Supplementary Material
References
- WHO . World Malaria Report 2017. https://www.who.int/malaria/publications/world-malaria-report-2017/en/.
- Burrows J. N.; Chibale K.; Wells T. N. C. The state of the art in anti-malarial drug discovery and development. Curr. Top. Med. Chem. 2011, 11 (10), 1226–1254. 10.2174/156802611795429194. [DOI] [PubMed] [Google Scholar]
- Kaur K.; Jain M.; Reddy R. P.; Jain R. Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem. 2010, 45 (8), 3245–3264. 10.1016/j.ejmech.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Wube A.; Hufner A.; Seebacher W.; Kaiser M.; Brun R.; Bauer R.; Bucar F. 1,2-Substituted 4-(1H)-Quinolones: Synthesis, Antimalarial and Antitrypanosomal Activities in Vitro. Molecules 2014, 19 (9), 14204–14220. 10.3390/molecules190914204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarveswari S.; Vijayakumar V.; Siva R.; Priya R. Synthesis of 4-Hydroxy-2(1H)-Quinolone Derived Chalcones, Pyrazolines and Their Antimicrobial, In Silico Antimalarial Evaluations. Appl. Biochem. Biotechnol. 2015, 175 (1), 43–64. 10.1007/s12010-014-1256-9. [DOI] [PubMed] [Google Scholar]
- Rajabalian S.; Foroumadi A.; Shafiee A.; Emami S. Functionalized N-(2-oxyiminoethyl) piperazinyl quinolones as new cytotoxic agents. Journal of Pharmacy and Pharmaceutical Sciences 2007, 10 (2), 153–158. [PubMed] [Google Scholar]
- Sancineto L.; Iraci N.; Barreca M. L.; Massari S.; Manfroni G.; Corazza G.; Cecchetti V.; Marcello A.; Daelemans D.; Pannecouque C.; Tabarrini O. Exploiting the anti-HIV 6-desfluoroquinolones to design multiple ligands. Bioorg. Med. Chem. 2014, 22 (17), 4658–4666. 10.1016/j.bmc.2014.07.018. [DOI] [PubMed] [Google Scholar]
- Stephen J. M. L.; Tonkin I. M.; Walker J. 192. Tetrahydroacridones and related compounds as antimalarials. J. Chem. Soc. 1947, (0), 1034–1039. 10.1039/jr9470001034. [DOI] [PubMed] [Google Scholar]
- Beteck R. M.; Smit F. J.; Haynes R. K.; N’Da D. D. Recent progress in the development of anti-malarial quinolones. Malar. J. 2014, 13, 10. 10.1186/1475-2875-13-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáenz F. E.; LaCrue A. N.; Cross R. M.; Maignan J. R.; Udenze K. O.; Manetsch R.; Kyle D. E. 4-(1H)-Quinolones and 1,2,3,4-Tetrahydroacridin-9(10H)-Ones Prevent the Transmission of Plasmodium falciparum to Anopheles freeborni. Antimicrob. Agents Chemother. 2013, 57 (12), 6187–6195. 10.1128/AAC.00492-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter R. W.; Kelly J. X.; Smilkstein M. J.; Dodean R.; Hinrichs D.; Riscoe M. K. Antimalarial quinolones: Synthesis, potency, and mechanistic studies. Exp. Parasitol. 2008, 118 (4), 487–497. 10.1016/j.exppara.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Clark J. A.; Connelly M. C.; Zhu F.; Min J.; Guiguemde W. A.; Pradhan A.; Iyer L.; Furimsky A.; Gow J.; Parman T.; El Mazouni F.; Phillips M. A.; Kyle D. E.; Mirsalis J.; Guy R. K. Lead Optimization of 3-Carboxyl-4(1H)-Quinolones to Deliver Orally Bioavailable Antimalarials. J. Med. Chem. 2012, 55 (9), 4205–4219. 10.1021/jm201642z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen A.; LaCrue A. N.; White K. L.; Forquer I. P.; Cross R. M.; Marfurt J.; Mather M. W.; Delves M. J.; Shackleford D. M.; Saenz F. E.; Morrisey J. M.; Steuten J.; Mutka T.; Li Y.; Wirjanata G.; Ryan E.; Duffy S.; Kelly J. X.; Sebayang B. F.; Zeeman A.-M.; Noviyanti R.; Sinden R. E.; Kocken C. H. M.; Price R. N.; Avery V. M.; Angulo-Barturen I.; Jiménez-Díaz M. B.; Ferrer S.; Herreros E.; Sanz L. M.; Gamo F.-J.; Bathurst I.; Burrows J. N.; Siegl P.; Guy R. K.; Winter R. W.; Vaidya A. B.; Charman S. A.; Kyle D. E.; Manetsch R.; Riscoe M. K. Quinolone-3-Diarylethers: A New Class of Antimalarial Drug. Sci. Transl. Med. 2013, 5 (177), 177ra37–177ra37. 10.1126/scitranslmed.3005029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidathala C.; Amewu R.; Pacorel B.; Nixon G. L.; Gibbons P.; Hong W. D.; Leung S. C.; Berry N. G.; Sharma R.; Stocks P. A.; Srivastava A.; Shone A. E.; Charoensutthivarakul S.; Taylor L.; Berger O.; Mbekeani A.; Hill A.; Fisher N. E.; Warman A. J.; Biagini G. A.; Ward S. A.; O’Neill P. M. Identification, Design and Biological Evaluation of Bisaryl Quinolones Targeting Plasmodium falciparum Type II NADH:Quinone Oxidoreductase (PfNDH2). J. Med. Chem. 2012, 55 (5), 1831–1843. 10.1021/jm201179h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Yu Y.; Li X.; Li J.; Wu Y.; Yu J.; Ge J.; Huang Z.; Jiang L.; Rao Y.; Yang M. Target Elucidation by Cocrystal Structures of NADH-Ubiquinone Oxidoreductase of Plasmodium falciparum (PfNDH2) with Small Molecule To Eliminate Drug-Resistant Malaria. J. Med. Chem. 2017, 60 (5), 1994–2005. 10.1021/acs.jmedchem.6b01733. [DOI] [PubMed] [Google Scholar]
- Biagini G. A.; Viriyavejakul P.; O’Neill P. M.; Bray P. G.; Ward S. A. Functional characterization and target validation of alternative complex I of Plasmodium falciparum mitochondria. Antimicrob. Agents Chemother. 2006, 50 (5), 1841–1851. 10.1128/AAC.50.5.1841-1851.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher N.; Bray P. G.; Ward S. A.; Biagini G. A. The malaria parasite type II NADH:quinone oxidoreductase: an alternative enzyme for an alternative lifestyle. Trends Parasitol. 2007, 23 (7), 305–310. 10.1016/j.pt.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Srivastava I. K.; Rottenberg H.; Vaidya A. B. Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J. Biol. Chem. 1997, 272 (7), 3961–3966. 10.1074/jbc.272.7.3961. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011, 54 (8), 2529–2591. 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- Ruan J.; Saidi O.; Iggo J. A.; Xiao J. Direct Acylation of Aryl Bromides with Aldehydes by Palladium Catalysis. J. Am. Chem. Soc. 2008, 130 (32), 10510–10511. 10.1021/ja804351z. [DOI] [PubMed] [Google Scholar]
- Hutchinson D. B.; Viravan C.; Webster H. K.; Kyle D. E.; Canfield C. J.; Looareesuwan S. Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am. J. Trop. Med. Hyg. 1996, 54 (1), 62–66. 10.4269/ajtmh.1996.54.62. [DOI] [PubMed] [Google Scholar]
- Fisher N.; Majid R. A.; Antoine T.; Al-Helal M.; Warman A. J.; Johnson D. J.; Lawrenson A. S.; Ranson H.; O’Neill P. M.; Ward S. A.; Biagini G. A. Cytochrome b Mutation Y268S Conferring Atovaquone Resistance Phenotype in Malaria Parasite Results in Reduced Parasite bc(1) Catalytic Turnover and Protein Expression. J. Biol. Chem. 2012, 287 (13), 9731–9741. 10.1074/jbc.M111.324319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon G. L.; Moss D. M.; Shone A. E.; Lalloo D. G.; Fisher N.; O’Neill P. M.; Ward S. A.; Biagini G. A. Antimalarial pharmacology and therapeutics of atovaquone. J. Antimicrob. Chemother. 2013, 68 (5), 977–985. 10.1093/jac/dks504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagini G. A.; Fisher N.; Shone A. E.; Mubaraki M. A.; Srivastava A.; Hill A.; Antoine T.; Warman A. J.; Davies J.; Pidathala C.; Amewu R. K.; Leung S. C.; Sharma R.; Gibbons P.; Hong D. W.; Pacorel B.; Lawrenson A. S.; Charoensutthivarakul S.; Taylor L.; Berger O.; Mbekeani A.; Stocks P. A.; Nixon G. L.; Chadwick J.; Hemingway J.; Delves M. J.; Sinden R. E.; Zeeman A. M.; Kocken C. H. M.; Berry N. G.; O’Neill P. M.; Ward S. A. Generation of quinolone antimalarials targeting the Plasmodium falciparum mitochondrial respiratory chain for the treatment and prophylaxis of malaria. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (21), 8298–8303. 10.1073/pnas.1205651109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter H. J.; Morrisey J. M.; Mather M. W.; Vaidya A. B. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 2007, 446 (7131), 88–91. 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- Biagini G. A.; Fisher N.; Berry N.; Stocks P. A.; Meunier B.; Williams D. P.; Bonar-Law R.; Bray P. G.; Owen A.; O’Neill P. M.; Ward S. A. Acridinediones: Selective and potent inhibitors of the malaria parasite mitochondrial bc(1) complex. Mol. Pharmacol. 2008, 73 (5), 1347–1355. 10.1124/mol.108.045120. [DOI] [PubMed] [Google Scholar]
- Stickles A. M.; de Almeida M. J.; Morrisey J. M.; Sheridan K. A.; Forquer I. P.; Nilsen A.; Winter R. W.; Burrows J. N.; Fidock D. A.; Vaidya A. B.; Riscoe M. K. Subtle Changes in Endochin-Like Quinolone Structure Alter the Site of Inhibition within the Cytochrome bc(1) Complex of Plasmodium falciparum. Antimicrob. Agents Chemother. 2015, 59 (4), 1977–1982. 10.1128/AAC.04149-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa M.; Hashimoto Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem. 2011, 54 (6), 1539–54. 10.1021/jm101356p. [DOI] [PubMed] [Google Scholar]
- Capper M. J.; O’Neill P. M.; Fisher N.; Strange R. W.; Moss D.; Ward S. A.; Berry N. G.; Lawrenson A. S.; Hasnain S. S.; Biagini G. A.; Antonyuk S. V. Antimalarial 4(1H)-pyridones bind to the Qi site of cytochrome bc1. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (3), 755–760. 10.1073/pnas.1416611112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini M.; Bienert S.; Waterhouse A.; Arnold K.; Studer G.; Schmidt T.; Kiefer F.; Gallo Cassarino T.; Bertoni M.; Bordoli L.; Schwede T. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–8. 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosdidier A.; Zoete V.; Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–7. 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.