

Abstract
Polypharmacology is often a key contributor to the efficacy of a drug, but is also a potential risk. We investigated two hits discovered via a cell-based phenotypic screen, the CDK9 inhibitor CCT250006 (1) and the pirin ligand CCT245232 (2), to establish methodology to elucidate their secondary protein targets. Using computational pocket-based analysis, we discovered intrafamily polypharmacology for our kinase inhibitor, despite little overall sequence identity. The interfamily polypharmacology of 2 with B-Raf was used to discover a novel pirin ligand from a very small but privileged compound library despite no apparent ligand or binding site similarity. Our data demonstrates that in areas of drug discovery where intrafamily polypharmacology is often an issue, ligand dissimilarity cannot necessarily be used to assume different off-target profiles and that understanding interfamily polypharmacology will be important in the future to reduce the risk of idiopathic toxicity and in the design of screening libraries.
Keywords: Polypharmacology, Privileged structure, Binding site comparison, Pirin, B-Raf
Polypharmacology, where small molecules bind to more than one protein target at concentrations relevant to their therapeutic free exposure,1,2 is a key element in drug discovery and development. It can be beneficial as an essential part of the efficacy and phenotype of a drug, particularly in complex diseases such as cancer,3 but can also be detrimental, leading to toxicity by hitting undesirable protein targets.4 This balance in therapeutic index can represent a significant challenge in lead optimization.5
Privileged structures are defined as small molecule scaffolds that are able to bind more than one receptor,6 a concept which has been useful in compound library design and in understanding polypharmacology.7
We recognized that there are four types of polypharmacology that are directly relevant to drug discovery.
(i) The first is often encountered in small molecule probes used in chemical biology, for example, PROTACs,8 and in the rational design of dual inhibitors, such as the combination of histone deacetylase (HDAC) inhibitors with Janus kinase9 and IDO1-targeting compounds.10 In this approach, two distinct compounds, usually with different functions and selectivities, are attached to each other via a flexible linker, with the aim that despite being attached, each motif of the chemical probe will retain its activity.
(ii) The second is described by weak and often nonspecific interactions with proteins outside of the target protein’s family.11 These antitargets are broadly characterized in in vitro safety pharmacology screens,12 where binding to promiscuous targets can cause adverse drug reactions at high dose, for example, adenosine A2a receptor and hERG.
(iii) Owing to high sequence similarity in the active sites within a protein family, small molecule ligands often bind with high affinity to multiple members of that family.13,14 This type of polypharmacology is particularly well established in kinase drug discovery,15 where privileged structures of certain hinge-binding motifs display low selectivity within the family,16 an effect that can lead to a poor therapeutic index (staurosporin)17 or be crucial for efficacy (aurora/FLT3).18
(iv) The final type of polypharmacology that can impact drug discovery has only recently been recognized,19 largely due to broad proteome screening platforms becoming readily available. High affinity specific interactions can occur for ligands binding to different protein families despite no apparent binding site or sequence similarity.20,21 This concept was recently highlighted by the discovery that the well characterized potent kinase inhibitor, BI-2536 (PLK1, IC50 = 0.83 nM),22 binds with high affinity to the bromodomain family protein, BRD4 (IC50 = 25–37 nM). However, it is unclear whether the interfamily activity encountered represents an inherent conservation in protein structure, expressed through an underlying similarity between protein binding sites, or is simply an anomaly.
We recently carried out a cell-based high-throughput phenotypic screen to discover inhibitors of the heat shock transcription factor 1 (HSF1) stress pathway.23 From the screen, we identified two novel series and exploited several molecular target identification strategies to discover direct protein targets for each chemotype. The first series, exemplified by aminopyrimidine CCT250006 (1) (Figure 1C), proved to bind with high affinity to the RNA polymerase II regulator, cyclin-dependent kinase 9 (CDK9, IC50 ≤ 3.0 nM).24 The second series, exemplified by bisamide CCT245232 (2) (Table 2, entry 1), bound with high affinity to the putative transcription factor regulator, pirin (SPR KD = 38 nM).23 As these compounds were discovered via a phenotypic screen, which did not discriminate against mechanisms of action (MOA) derived from the inhibition of multiple protein targets,25,26 we were interested in determining what role polypharmacology could play in both the efficacy and potential toxicity of these chemotypes.
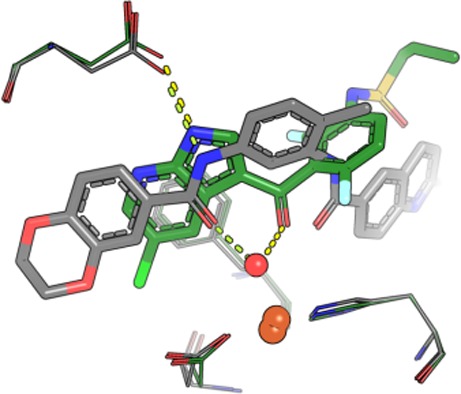
Figure 1.

Rational polypharmacology within the kinase family. (A) Close analogue of aminopyrimidine 1 bound to CDK2 (PDB 4BZD, green) and overlaid with CDK9 (PDB 4EC8, turquoise) and homology models of TAOK1 (brown) and HIPK2 (yellow); despite little overall sequence identity, the proteins show clear pocket shape similarity, which was detected by the pocket analysis tool. C, gray; N, blue; O, red; H and solvent omitted for clarity. (B) Key interactions of the close analogue of 1 with CDK2 (PDB 4BZD, green), compared to the equivalent residues in CDK9 (blue), TAOK1 (brown), and HIPK2 (yellow), showing that these key residues are retained. (C) Chemical structure of aminopyrimidine 1 and the well validated CDK9 inhibitor dinaciclib 3.
Table 2. Inter-Family Polypharmacology Screen of Pirin and B-Rafe.

ND = Not determined
All compounds were purchased from Selleckchem (www.selleckchem.com).
Compared to the dual pirin/B-Raf ligand bisamide 2, calculated using ECFC_4 molecular fingerprint Tanimoto coefficients as implemented in Pipeline Pilot v9.5.
For references of the described B-Raf activities, see SI.
Measured using the pirin FP-assay and represents the geometric mean of at least n = 2 repeats.
We began our investigation into polypharmacology by exploring the well-established promiscuity of ATP-competitive kinase inhibitors. This was accomplished through binding site analysis to assess the pocket similarity of CDK9 with various other kinases using the pocket comparison tool, SiteHopper, to rank and select proteins for further analysis.27,28 The SiteHopper tool represents pockets as 3D patches encoded with spatial information concerning the local molecular surface (shape) and chemical properties (color) of residues lining protein binding sites. Binding site patches are aligned and their overlap scored, yielding a continuous PatchScore between 0 and 4, conveying dissimilarity and perfect similarity, respectively. Binding sites were identified using the fpocket cavity detection tool,29 enabling the use of homology models and unbound protein structures in the analysis30 (see pocket analysis section in the Supporting Information (SI)). To narrow the search space, we retrieved the off-target kinase profile of the well-validated CDK9 inhibitor dinaciclib 3 (Figure 1C).31 The 1,5-pyrazolopyrimidine structure of dinaciclib 3 is a distinct chemotype from our aminopyrimidine inhibitor 1 when compared using their ECFC_4 molecular fingerprint Tanimoto coefficients (TC = 0.41).32 We hypothesized that as there was limited structural similarity between the potent CDK9 ligands, then any cross-selectivity with other protein kinases may manifest as similarity in the respective small molecule binding sites. The kinase profile of dinaciclib 3 was obtained from the publicly available HMS LINCS database (456 kinase targets),33 and we only considered kinases that exhibited a <5% difference in response compared to CDK9. This analysis left 30 distinct kinases, of which, 21 could be assessed for binding site similarity due to the availability of structural data. We retrieved structures from both the Protein Data Bank (PDB) and, where available, homology models from the Protein Model Portal (PMP),34 giving a total of 252 protein structures (SI and Table S1).
The SiteHopper pocket analysis tool was then applied to our 21 kinase data set, comparing detected pockets of each kinase with reference pockets derived from structures of CDK9. Each kinase was analyzed with respect to the maximum PatchScore observed with any of the reference CDK9 pockets. This analysis identified 11 kinases with a maximum PatchScore of >1.3 (Table 1 and SI, Table S2), a cutoff proposed to describe significant structural similarity between binding sites.27 As expected, CDK9 itself was the top-ranked hit, as the score simply represents conformation differences between different protein structures. The second and fifth ranked hits, CDK2 and CDK14 respectively, had already been confirmed as a direct protein target of 1 from our previous work.24 From the remaining hits, TAOK1 and HIPK2 (discovered from analysis of homology models) were selected for further study as they were evolutionarily more distant from CDK9 (sequence identities of 24% and 8% and sequence homology of 48% and 40%, respectively) and for which functional assays were available. The activity of aminopyrimidine 1 was assessed using radio labeled filter binding assays to give IC50 values of 490 and 30 nM for TAOK1 and HIPK2, respectively (SI, Tables S4, S5).35 Thus, despite little total sequence identity between TAOK1, HIPK2, and CDK9, each protein still shares a common kinase fold and possesses similar binding sites, resulting in intrafamily polypharmacology (Figure 1; SI, Figures S3, S4).
Table 1. SiteHopper Analysis Comparing the Kinase Binding Site Similarity with CDK9a.
| rank | kinase | max SiteHopper PatchScore |
|---|---|---|
| 1 | CDK9 | 2.66 |
| 2 | CDK2 | 1.86 |
| 3 | TAOK1 | 1.81 |
| 4 | CDC2L5 | 1.58 |
| 5 | CDK14 | 1.58 |
| 6 | HIPK2 | 1.48 |
| 7 | CDKL5 | 1.47 |
| 8 | HIPK3 | 1.42 |
| 9 | ICK | 1.38 |
| 10 | PFPK5 | 1.35 |
| 11 | CDK4-cyclinD1 | 1.31 |
Only the top 11 hits are shown using the maximum score obtained against all available conformations, see SI.
Although from these data alone we cannot conclude that either kinase will significantly contribute to the efficacy or in vivo toxicology of aminopyrimidine (1), they do suggest that binding site pocket analysis can be a useful tool for prioritizing off-target proteins for further evaluation as part of a lead optimization selectivity assessment. Particularly as neither protein kinase selected using pocket analysis displays high sequence similarity to CDK9,36 so may not have been chosen for further study based on this commonly used selectivity criterion. Our data demonstrates that in areas of drug discovery where intrafamily polypharmacology is often an issue, ligand dissimilarity cannot necessarily be used to assume different off-target profiles. Additionally, our analysis makes use of publicly available homology models retrieved from the PMP. Caution must always be exercised when considering these data, but as pocket comparison using SiteHopper does not involve precise modeling of directional interactions between proteins and ligands, homology models can be valuable. This allows proteins to be studied for polypharmacology, even when structures are not yet available.
Although we were successful in using pocket analysis to discover off-target proteins from within the kinase family with the pyrimidine series, identifying additional high affinity targets for the second bisamide series proved to be more challenging. Pirin is a member of the cupin superfamily of proteins,37 defined by the β-barrel structure at their center. Members of the cupin super family possess little sequence similarity and display a wide range of both catalytic and noncatalytic functions.38 In contrast to our CDK9 example of polypharmacology, all binding site and pocket analysis tools, as well as sequence similarity searching, yielded no protein hits for further investigation (data not shown).
Using in vitro kinase recombinant protein profiling, we had previously shown that bisamide 2 possessed moderate affinity for the protein kinase, B-Raf (IC50 = 420 nM), concurrent with its high affinity for pirin (KD = 38 nM).23 The interfamily protein activity of bisamide 2 for both B-Raf and pirin is surprising, as the two proteins possess no significant binding site sequence similarity. To rationalize this result, we solved the crystal structure of bisamide 2 bound to pirin (Figure 2A; SI, Figures S8, S14, Table S6) and compared its binding mode to the docked structure of bisamide 2 in B-Raf (Figure 2B). Bisamide 2 displays a distinct binding conformation in pirin, with the two amide carbonyls forming an eclipsed conformation around the central ring. In the docked structure of bisamide 2 in B-Raf, assumed to be ATP competitive, the two amide carbonyls form a perpendicular conformation, consistent with the crystal structures of the bisamide chemotype bound to EphA3 (PDB 3DZQ) and p38 (PDB 3KQ7) kinases (SI, Figures S5, S6). This is in contrast to the BRD4/PLK1 example of interfamily polypharmacology,20 where BI-2536 adopts essentially an identical conformation against both protein targets (SI, Figure S7).
Figure 2.

Structural comparison of pirin and B-Raf. N, blue; O, red; S, yellow; Cl, green; metal, brown; H omitted for clarity. Only key residues have been shown and the solvent, except the metal-bound water, has been omitted for clarity. Hydrogen bonds are shown as yellow dashes. (A) Bisamide 2 bound to pirin demonstrating the key hydrogen bonds to Asp43 and the metal-bound water, and the amide-eclipsed conformation. The quinoline ring motif is solvent exposed. (B) Docked structure of bisamide 2 bound to B-Raf (PDB 4G9C; SI, Figures S4, S5 for additional pictures), 2 binds in a distinct staggered conformation and the quinoline ring motif is placed in close proximity to the hinge region. (C) Structure of PLX4720 (7) bound to pirin. The azaindole–ketone motif forms an equivalent hydrogen bonding array to the amide of 2. The chloro-substituent is buried deeply within the narrow binding tunnel. Because the sulfonamide moiety is disordered, it cannot be observed in the electron density but is shown here for clarity. (D) PLX4720 (7) bound to B-Raf (PDB 3C4C). The azaindole motif interacts with the hinge region and the chloro-substituent points on a solvent exposed vector. (E,F) Key interaction of PLX4720 (7) and bisamide 2, respectively, bound to pirin. Hydrogen bonds are represented by an arrow, π-stacking interactions by a dotted line. Key interactions of both ligands bound to B-Raf are shown in SI, Figure S14. (G) Chemical structure of FP-Probe 4.
Even though pocket analysis had been unable to detect any similarity between the pirin and B-Raf binding sites, we hypothesized that if the affinity of bisamide 2 to both proteins was a more general phenomenon, then it should be possible to discover a second ligand of a distinct chemotype that could bind to both proteins.
The bisamides are currently the only known high affinity chemotype for pirin but B-Raf has been extensively studied,39 resulting in multiple inhibitors of distinct chemotype being discovered. To rapidly screen B-Raf inhibitors in an efficient manner, we developed a new pirin binding assay. Fluorescence polarization (FP) assays are highly versatile and have been widely used to characterize proteins that do not possess enzymatic activity.40 The design of the FP-probe was carried out by analysis of the crystal structure of 2 bound to recombinant pirin and through strong precedent from previous pirin ligand design (Figure 2B,G; SI Figure S13),23,42 The FP-probe 4 (Figure 2G) was synthesized using a 9-atom linker in seven steps and 1.2% overall yield (see SI, Chemistry Experimental).
Titration of recombinant pirin against a fixed concentration (2.0 nM) of FP-probe 4 revealed a very high apparent affinity for pirin (KD = 11 nM), within 5-fold of the FP-probe 4 concentration (SI, Figure S1). Using the FP-assay, bisamide 2 and our pirin chemical probe CCT251236 5(41) gave IC50 values of 44 nM (pIC50 = 7.36 ± 0.10, n = 4) and 33 nM (pIC50 = 7.48 ± 0.09, n = 46), respectively, at the tight-binding limit of the assay (for an example, see SI, Figure S2). Our negative control regioisomer 6 failed to displace the FP-probe 4 at concentrations >1 μM. These results were comparable to our previous surface plasmon resonance (SPR) data for pirin.23
A panel of seven well-validated and potent B-Raf inhibitors (entries 4, 6–11 in Table 2),43 representing several chemotypes distinct from bisamide 2 (TC = 0.38–0.67), were purchased and screened in the pirin FP-assay. No activity was observed against pirin with six of the B-Raf inhibitors. However, when we screened the ligand with the lowest TC compared to bisamide 2, the azaindole derived inhibitor, PLX4720 7, the ligand demonstrated a high affinity for pirin (IC50 = 0.67 μM, Table 2, entry 4). Thus, despite sharing little chemical similarity to bisamide 2, PLX4720 7 was also able bind both pirin and B-Raf, representing a 14% hit-rate from our very small privileged compound library.
To rationalize the high affinity of PLX4720 7 for pirin, we solved its crystal structure (Figure 2C, SI, Figure S10, S16, Table S6) and compared it to bisamide 2 (SI, Figure S12). PLX4720 7, like bisamide 2, forms no direct interactions with the metal-center of pirin, instead forming hydrogen bonds through the metal-bound water molecule and the central ketone carbonyl. The NH-group of the azaindole moiety acts as a hydrogen bond donor with Asp43, in a similar manner to the methyl-distal amide of bisamide 2. The remaining portion of the azaindole ring forms a π-stacking interaction with Phe53 and points deeply into the lipophilic narrow binding tunnel, leaving the propyl-sulfonamide moiety essentially solvent exposed and highly flexible as it could not be observed in the electron density. This is in contrast to the binding mode of PLX4720 7 bound to B-Raf (Figure 2D, PDB 3C4C), where the propyl-sulfonamide moiety is now buried deeply within a lipophilic region of the protein and it is the azaindole motif which points toward the solvent channel.
The dissimilarity in binding mode demonstrates that although our two privileged ligands (2 and 7) could bind proteins across families with high affinity, their structure–activity relationships (SAR) toward each target remain essentially distinct. Against pirin, we could exploit the solvent-exposed quinoline region of bisamide 2 to substitute the methylene group with a larger substituent. The t-butyl-piperazine bisamide analogue 8 retains its high affinity for pirin (Table 2, entry 5), but the piperazine moiety is predicted to be in close proximity to the hinge region of B-Raf, where bulky substituents should not be tolerated. This orthogonal SAR was confirmed when the B-Raf polypharmacology was lost in the case of 8 (IC50 > 10 μM). A similar contrast in SAR is observed for PLX4720 7. Against B-Raf, the chloro-substituent is on an essentially solvent-exposed vector and can be readily substituted with a phenyl ring, which gives the high affinity B-Raf ligand, vemurafinib 9 (Table 2, entry 6). However, against pirin, the chloro-substituent of 7 is buried in the lipophilic tunnel, so the phenyl substitution in 9 now results in a clear clash with the protein and the complete loss of the pirin polypharmacology, enhancing the selectivity for B-Raf.44
Understanding the role of polypharmacology in drug discovery is essential to improve the efficiency of compound development and decrease drug attrition. Using two chemotypes discovered from a high-throughput phenotypic screen, we demonstrated that binding site pocket similarity analysis can be a useful tool in selecting off-target proteins from within the same family for further investigation. This approach could offer clear benefits in lead optimization, prioritizing the study of secondary targets for their importance in efficacy or toxicity. However, when searching for proteins outside of the family of the target protein, pocket analysis was not predictive. We could demonstrate that, despite there being no detectable sequence or shape similarity in the binding sites of pirin and B-Raf, there is “inherent” or “underlying” similarity between these proteins, resulting in interfamily polypharmacology and limited SAR homology45 from two distinct chemotypes. Consistent with the BRD4/PLK1 example, there are no computational methods we are currently aware of that are able to detect, quantify, or predict this phenomenon. An analysis by Shoichet et al. of ligands that bound more than one protein from different families46 found that the majority of examples of interfamily polypharmacology were not as a result of matched residues in the ligand binding site and there was no simple code for ligand recognition. Nonetheless, using a bottom-up approach, we discovered a novel pirin ligand after screening a very small privileged compound library.
As more examples of interfamily polypharmacology become available, we will be able to better analyze and understand what factors control the underlying similarity in protein binding sites and the conservative evolution that nature has used to generate them. We will then be better able to predict efficacy and idiopathic toxicities before compounds reach the clinic and design more efficient high-throughput screening libraries of privileged structures possessing the appropriate complexity so that they are likely to hit targets across the proteome.
Acknowledgments
J.M. is supported by Wellcome Trust grant 102361/Z/13/Z. This work was also supported by Cancer Research UK grants C309/A11566 and C2739/A22897. We acknowledge CRUK-ICR Centre funding and support from Cancer Research Technology Pioneer Fund and Battle Against Cancer Investment Trust. P.W. is a Cancer Research UK Life Fellow.
Glossary
Abbreviations
- PROTAC
proteolysis targeting chimera
- HDAC
histone deacetylase
- IDO1
indoleamine 2,3-dioxygenase
- hERG
human ether-à-go-go-related gene
- PLK1
polo-like kinase 1
- BRD4
bromodomain-containing protein 4
- HSF1
heat shock transcription factor 1
- CDK9
cyclin-dependent kinase 9
- Tc
Tanimoto coefficient
- SPR
surface plasmon resonance spectroscopy
- MOA
mechanism of action
- PDB
Protein Data Bank
- PMP
protein model portal
- HIPK2
homeodomain-interacting protein kinase 2.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00364.
Pocket analysis, B-Raf docking study, chemistry experimental, pirin fluorescence polarization assay, pirin and B-Raf crystallography analysis, kinase assay data, crystallography experimental (PDF)
Accession Codes
Atomic coordinates and structure factors for the crystal structures of pirin with 2 and 7 can be accessed using PDBs 6H1I and 6H1H, respectively. The authors will release the atomic coordinates and experimental data upon publication.
Author Contributions
All authors have given approval to the final manuscript. J.M., N.Y.M., M.C., J.B., and N.B. carried out and designed computational studies. N.E.A.C., B.W., A.E.P., M.J.T., L.E.E., C.S.R, P.W., K.J., and M.D.C. designed and synthesized small molecules and chemical probes. S.A., Y.B., and R.L.M.V.M. designed and carried out crystallography experiments. M.R., L.E.E., L.O.F., and R.B. designed and carried out FP-assay experiments. J.M. and M.D.C. wrote the manuscript.
The authors declare the following competing financial interest(s): The authors are employees/students of ICR and ICR has a commercial interest in inhibitors of the HSF1 pathway, CDKs and BRAF. The ICR operates a Rewards to Discoverers scheme.
Supplementary Material
References
- Reddy A. S.; Zhang S. Polypharmacology: drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41. 10.1586/ecp.12.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anighoro A.; Bajorath J.; Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874. 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- Antolin A. A.; Workman P.; Mestres J.; Al-Lazikani B. Polypharmacology in precision oncology: Current applications and future prospects. Curr. Pharm. Des. 2016, 22, 6935. 10.2174/1381612822666160923115828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudmann D. G. On-target and off-target-based toxicologic effects. Toxicol. Pathol. 2013, 41, 310. 10.1177/0192623312464311. [DOI] [PubMed] [Google Scholar]
- Muller P. Y.; Milton M. N. The determination and interpretation of the therapeutic index in drug development. Nat. Rev. Drug Discovery 2012, 11, 751. 10.1038/nrd3801. [DOI] [PubMed] [Google Scholar]
- Evans B. E.; Rittle K. E.; Bock M. G.; DiPardo R. M.; Freidinger R. M.; Whitter W. L.; Lundell G. F.; Veber D. F.; Anderson P. S.; Chang R. S. L.; Lotti V. J.; Cerino D. J.; Chen T. B.; Kling P. J.; Kunkel K. A.; Springer J. P.; Hirshfield J. Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988, 31, 2235. 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- Schneider P.; Schneider G. Privileged structures revisited. Angew. Chem., Int. Ed. 2017, 56, 7971. 10.1002/anie.201702816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A. C.; Crews C. M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16, 101. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L.; Mustafa N.; Tan E. C.; Poulsen A.; Singh P.; Duong-Thi M.; Lee J. X. T.; Ramanujulu P. M.; Chng W. J.; Yen J. J. Y.; Ohlson S.; Dymock B. W. Design and synthesis of ligand efficient dual inhibitors of janus kinase (JAK) and histone deacetylase (HDAC) based on ruxolitinib and vorinostat. J. Med. Chem. 2017, 60, 8336. 10.1021/acs.jmedchem.7b00678. [DOI] [PubMed] [Google Scholar]
- Fang K.; Dong G.; Li Y.; He S.; Wu Y.; Wu S.; Wang W.; Sheng C. Discovery of novel indoleamine 2,3-dioxygenase 1 (IDO1) and histone deacetylase (hdac) dual inhibitors. ACS Med. Chem. Lett. 2018, 9, 312. 10.1021/acsmedchemlett.7b00487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Sun T.; Liang L.; Wu T.; Wang Q. Drug promiscuity of P-glycoprotein and its mechanism of interaction with paclitaxel and doxorubicin. Soft Matter 2014, 10, 438. 10.1039/C3SM52499J. [DOI] [PubMed] [Google Scholar]
- Bowes J.; Brown A. J.; Hamon J.; Jarolimek W.; Sridhar A.; Waldron G.; Whitebread S. Reducing safety-related drug attrition: The use of in vitro pharmacological profiling. Nat. Rev. Drug Discovery 2012, 11, 909. 10.1038/nrd3845. [DOI] [PubMed] [Google Scholar]
- Haupt V. J.; Daminelli S.; Schroeder M. Drug promiscuity in PDB: Protein binding site similarity is key. PLoS One 2013, 8, e65894. 10.1371/journal.pone.0065894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnash K. D.; James L. I.; Frye S. V. Target class drug discovery. Nat. Chem. Biol. 2017, 13, 1053. 10.1038/nchembio.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Z. A.; Lin H.; Shokat K. M. Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer 2010, 10, 130. 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpfe D.; Tinivella A.; Rastelli G.; Bajorath J. Promiscuity of inhibitors of human protein kinases at varying data confidence levels and test frequencies. RSC Adv. 2017, 7, 41265. 10.1039/C7RA07167A. [DOI] [Google Scholar]
- Tanramluk D.; Schreyer A.; Pitt W. R.; Blundell T. L. On the origins of enzyme inhibitor selectivity and promiscuity: A case study of protein kinase binding to staurosporine. Chem. Biol. Drug Des. 2009, 74, 16. 10.1111/j.1747-0285.2009.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an example see:Bavetsias V.; Crumpler S.; Sun C.; Avery S.; Atrash B.; Faisal A.; Moore A. S.; Kosmopoulou M.; Brown N.; Sheldrake P. W.; Bush K.; Henley A.; Box G.; Valenti M.; de Haven Brandon A.; Raynaud F. I.; Workman P.; Eccles S. A.; Bayliss R.; Linardopoulos S.; Blagg J. Optimization of imidazo[4,5-b]pyridine-based kinase inhibitors: identification of a dual FLT3/aurora kinase inhibitor as an orally bioavailable preclinical development candidate for the treatment of acute myeloid leukemia. J. Med. Chem. 2012, 55, 8721. 10.1021/jm300952s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciceri P.; Muller S.; O'Mahony A.; Fedorov O.; Filippakopoulos P.; Hunt J. P.; Lasater E. A.; Pallares G.; Picaud S.; Wells C.; Martin S.; Wodicka L. M.; Shah N. P.; Treiber D. K.; Knapp S. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat. Chem. Biol. 2014, 10, 305. 10.1038/nchembio.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ember S. W. J.; Zhu J.; Olesen S. H.; Martin M. P.; Becker A.; Berndt N.; Georg G. I.; Schönbrunn E. Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem. Biol. 2014, 9, 1160. 10.1021/cb500072z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antolín A. A.; Mestres J. Distant polypharmacology among MLP chemical probes. ACS Chem. Biol. 2015, 10, 395. 10.1021/cb500393m. [DOI] [PubMed] [Google Scholar]
- Steegmaier M.; Hoffmann M.; Baum A.; Lénárt P.; Petronczki M.; Krssák M.; Gürtler U.; Garin-Chesa P.; Lieb S.; Quant J.; Grauert M.; Adolf G. R.; Kraut N.; Peters J. M.; Rettig W. J. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007, 17, 316. 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- Cheeseman M. D.; Chessum N. E.; Rye C. S.; Pasqua A. E.; Tucker M. J.; Wilding B.; Evans L. E.; Lepri S.; Richards M.; Sharp S. Y.; Ali S.; Rowlands M.; O’Fee L.; Miah A.; Hayes A.; Henley A. T.; Powers M.; Te Poele R.; De Billy E.; Pellegrino L.; Raynaud F.; Burke R.; van Montfort R. L.; Eccles S. A.; Workman P.; Jones K. Discovery of a chemical probe bisamide (CCT251236): an orally bioavailable efficacious pirin ligand from a heat shock transcription factor 1 (HSF1) phenotypic screen. J. Med. Chem. 2017, 60, 180. 10.1021/acs.jmedchem.6b01055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rye C. S.; Chessum N. E. A.; Lamont S.; Pike K. G.; Faulder P.; Demeritt J.; Kemmitt P.; Tucker J.; Zani L.; Cheeseman M. D.; Isaac R.; Goodwin L.; Boros J.; Raynaud F.; Hayes A.; Henley A.; de Billy E.; Lynch C. J.; Sharp S. Y.; te Poele R.; O’ Fee L.; Foote K. M.; Green S.; Workman P.; Jones K. Discovery of 4,6-disubstituted pyrimidines as potent inhibitors of the heat shock factor (HSF1) stress pathway and CDK9. MedChemComm 2016, 7, 1580. 10.1039/C6MD00159A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J. G.; Vincent F.; Lee J. A.; Eder J.; Prunotto M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discovery 2017, 16, 531. 10.1038/nrd.2017.111. [DOI] [PubMed] [Google Scholar]
- Moffat J. G.; Rudolph J.; Bailey D. Phenotypic screening in cancer drug discovery - past, present and future. Nat. Rev. Drug Discovery 2014, 13, 588. 10.1038/nrd4366. [DOI] [PubMed] [Google Scholar]
- Batista J.; Hawkins P. C.; Tolbert R.; Geballe M. T. SiteHopper: A unique tool for binding site comparison. J. Cheminf. 2014, 6, P57. 10.1186/1758-2946-6-S1-P57. [DOI] [Google Scholar]
- Chen Y.; Tolbert R.; Aronov A. M.; McGaughey G.; Walters W. P.; Meireles L. Prediction of protein pairs sharing common active ligands using protein sequence, structure, and ligand similarity. J. Chem. Inf. Model. 2016, 56, 1734. 10.1021/acs.jcim.6b00118. [DOI] [PubMed] [Google Scholar]
- Le Guilloux V.; Schmidtke P.; Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinf. 2009, 10, 168. 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers J.; Brown N.; Blagg J. Mapping the 3D structures of small molecule binding sites. J. Cheminf. 2016, 8, 70. 10.1186/s13321-016-0180-0. [DOI] [Google Scholar]
- Paruch K.; Dwyer M. P.; Alvarez C.; Brown C.; Chan T.; Doll R. J.; Keertikar K.; Knutson C.; McKittrick B.; Rivera J.; Rossman R.; Tucker G.; Fischmann T.; Hruza A.; Madison V.; Nomeir A. A.; Wang Y.; Kirschmeier P.; Lees E.; Parry D.; Sgambellone N.; Seghezzi W.; Schultz L.; Shanahan F.; Wiswell D.; Xu X.; Zhou Q.; James R. A.; Paradkar V. M.; Park H.; Rokosz L. R.; Stauffer T. M.; Guzi T. J. Discovery of dinaciclib (SCH 727965): A potent and selective inhibitor of cyclin-dependent kinases. ACS Med. Chem. Lett. 2010, 1, 204. 10.1021/ml100051d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers D.; Hahn M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- KINOMEscan data; Library of Integrated Network-based Cellular Signatures, 2018; http://lincs.hms.harvard.edu/kinomescan/ (accessed April 2018).
- Haas J.; Roth S.; Arnold K.; Kiefer F.; Schmidt T.; Bordoli L.; Schwede T. The Protein Model Portal - a comprehensive resource for protein structure and model information. Database 2013, 2013, bat031. 10.1093/database/bat031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Centre for Kinase Profiling; MRC Protein Phosphorylation and Ubiquitylation Unit, 2017; http://www.kinase-screen.mrc.ac.uk/ (accessed April 2018).
- a RCSB PDB Protein Comparison Tool; RCSB Protein Data Bank, 2018; http://www.rcsb.org/pdb/workbench/workbench.do (accessed April 2018).; b BLAST; UniProt, 2018; https://www.uniprot.org/blast/ (accessed April 2018).
- Dunwell J. M.; Culham A.; Carter C. E.; Sosa-Aguirre C. R.; Goodenough P. W. Evolution of functional diversity in the cupin superfamily. Trends Biochem. Sci. 2001, 26, 740. 10.1016/S0968-0004(01)01981-8. [DOI] [PubMed] [Google Scholar]
- Dunwell J. M.; Purvis A.; Khuri S. Cupins: the most functionally diverse protein superfamily?. Phytochemistry 2004, 65, 7. 10.1016/j.phytochem.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Rahman M. A.; Salajegheh A.; Smith R. A.; Lam A. K. BRAF inhibitors: From the laboratory to clinical trials. Crit. Rev. Oncol. Hematol. 2014, 90, 220. 10.1016/j.critrevonc.2013.12.008. [DOI] [PubMed] [Google Scholar]
- Burke T. J.; Loniello K. R.; Beebe J. A.; Ervin K. M. Development and application of fluorescence polarization assays in drug discovery. Comb. Chem. High Throughput Screening 2003, 6, 183. 10.2174/138620703106298365. [DOI] [PubMed] [Google Scholar]
- CCT251236; ChemicalProbes.org, 2018; http://www.chemicalprobes.org/cct251236 (accessed April 2018).
- Chessum N. E. A.; Sharp S. Y.; Caldwell J. J.; Pasqua A. E.; Wilding B.; Colombano G.; Collins I.; Ozer B.; Richards M.; Rowlands M.; Stubbs M.; Burke R.; McAndrew P. C.; Clarke P. A.; Workman P.; Cheeseman M. D.; Jones K. Demonstrating in-cell target engagement using a pirin protein degradation probe (CCT367766). J. Med. Chem. 2018, 61, 918. 10.1021/acs.jmedchem.7b01406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selleckchem.com, Inhibitor Expert; Selleck Chemicals: Houston, TX, 2013; http://www.selleckchem.com/ (accessed April 2018).
- Schuffenhauer A.; Brown N.; Selzer P.; Ertl P.; Jacoby E. Relationships between molecular complexity, biological activity, and structural diversity. J. Chem. Inf. Model. 2006, 46, 525. 10.1021/ci0503558. [DOI] [PubMed] [Google Scholar]
- Frye S. V. Structure-activity relationship homology (SARAH): a conceptual framework for drug discovery in the genomic era. Chem. Biol. 1999, 6, R3. 10.1016/S1074-5521(99)80013-1. [DOI] [PubMed] [Google Scholar]
- Barelier S.; Sterling T.; O’Meara M. J.; Shoichet B. K. The Recognition of identical ligands by unrelated proteins. ACS Chem. Biol. 2015, 10, 2772. 10.1021/acschembio.5b00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- KINOMEscan data; Library of Integrated Network-based Cellular Signatures, 2018; http://lincs.hms.harvard.edu/kinomescan/ (accessed April 2018).
- b BLAST; UniProt, 2018; https://www.uniprot.org/blast/ (accessed April 2018).