

Abstract
Mercaptoacetamide histone deacetylase inhibitors are neuroprotective agents that do not exhibit the genotoxicity associated with more commonly used hydroxamate inhibitors. Here, we present the crystal structure of a selective mercaptoacetamide complexed with the C-terminal catalytic domain of HDAC6. When compared with the structure of a mercaptoacetamide bound to the class I isozyme HDAC8, different interactions are observed with the conserved tandem histidine pair in the active site. These differences likely contribute to the selectivity for inhibition of HDAC6, an important target for cancer chemotherapy and the treatment of neurodegenerative disease.
Keywords: Protein crystallography, enzyme inhibitor, zinc-binding group, cancer chemotherapy
Metal-dependent histone deacetylases (HDACs) catalyze the hydrolysis of acetyllysine residues in histone and nonhistone protein substrates to regulate diverse cellular processes.1−4 Phylogenetic analysis divides these enzymes into the class I HDACs (1, 2, 3, and 8), the class II HDACs, further subdivided into the class IIa isozymes (4, 5, 7, and 9) and the class IIb isozymes (6 and 10), and the sole class IV isozyme HDAC11.5 These enzymes adopt the α/β fold first observed in the binuclear manganese metalloenzyme arginase,6−8 so the protein fold of an HDAC is designated the arginase-deacetylase fold.
The active site metal ion of an HDAC, typically Zn2+ but possibly Fe2+in vivo,9 is required for catalysis. In the first step of catalysis, the scissile carbonyl group of acetyllysine coordinates to Zn2+ and accepts a hydrogen bond from a nearby tyrosine;10,11 both interactions are required to polarize the scissile carbonyl for nucleophilic attack by a metal-bound water molecule, which in turn is activated by a histidine general base.12,13
As epigenetic regulators of protein structure and function, the HDACs currently serve as molecular targets in drug design programs focusing on new strategies for the treatment of cancer, neurological diseases, and immunological disorders.14,15 In particular, the class IIb isozyme HDAC6 is a prominent drug target due to the function of catalytic domain 2 as the cytosolic tubulin deacetylase.16−18 Inhibition of HDAC6 suppresses microtubule dynamics, resulting in cell cycle arrest and apoptosis.19 Additionally, HDAC6 inhibition promotes survival and regeneration of neurons, suggesting that selective HDAC6 inhibitors could be used in treating spinal cord injury and neurodegenerative diseases.20,21 However, structural and mechanistic conservation across HDAC isozymes complicates the design of isozyme-selective inhibitors.
Even so, inhibitors exhibiting better than 102–103-fold selectivity for HDAC6 have been identified,22 and the first crystal structures of HDAC6 catalytic domains enable a deeper understanding of structure–activity relationships for selective inhibitors.23,24 These structural studies indicate that the molecular basis of selectivity relies on interactions between sterically bulky capping groups and the surface of the enzyme, particularly in the region of the L1 loop flanking the active site, as well as interactions in the aromatic cleft of the substrate binding groove.23−27
To date, nearly all of the inhibitors that have been structurally characterized in complex with HDAC6 bear hydroxamate Zn2+-binding groups. The genotoxicity associated with the hydroxamate group, such as that of the classic HDAC inhibitor suberoylanilide hydroxamic acid (SAHA,28 formulated as the cancer chemotherapy drug Vorinostat29), argues against the use of a hydroxamate-containing inhibitor as a long-term therapy for diseases other than cancer.30,31 The chemical basis of genotoxicity derives from the Lossen rearrangement (Figure 1), which yields a reactive isocyanate intermediate capable of covalently modifying cellular components.30 This undesirable chemistry motivates the search for HDAC inhibitors with alternative zinc-binding groups.
Figure 1.
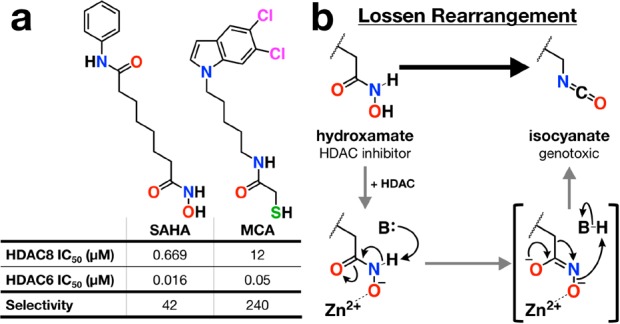
(a) Structures and selectivity data for SAHA43 and MCA alongside (b) the mechanism for the Lossen rearrangement as potentially catalyzed by the Zn2+ ion in the HDAC active site.
Notably, inhibitors bearing mercaptoacetamide zinc-binding groups are not genotoxic and exhibit superior neuroprotective properties.32 Moreover, certain mercaptoacetamide inhibitors exhibit nanomolar affinity and better than 103-fold selectivity against HDAC6.33,34 Until now, the structural basis for HDAC6 affinity and selectivity has not been defined.
Here, we present the 1.85 Å-resolution X-ray crystal structure of the complex between the mercaptoacetamide inhibitor N-(5-(5,6-dichloro-1H-indol-1-yl)pentyl)-2-mercaptoacetamide (MCA, Figure 1)34 and HDAC6 catalytic domain 2 from Danio rerio (zebrafish). The active site of the zebrafish ortholog is identical to that of human HDAC6 catalytic domain 2, and the zebrafish ortholog is superior for X-ray crystallographic studies (henceforth, HDAC6 catalytic domain 2 is simply referred to as “HDAC6”).23 MCA exhibits 240-fold selectivity for the inhibition of HDAC6 over HDAC8 (Figure 1). Full experimental details regarding the synthesis of MCA,34 IC50 determinations against HDAC6 and HDAC8, and the crystal structure determination of the HDAC6–MCA complex are outlined in the Supporting Information. Crystallographic refinement converged smoothly to Rwork/Rfree = 0.190/0.226; data collection and refinement statistics are recorded in Table S1. The overall structure of the polypeptide chain in the HDAC6–MCA complex is essentially identical to that in the unliganded enzyme (root-mean-square deviation = 0.2 Å over 292 Cα atoms), indicating that no major structural rearrangements are required for the binding of MCA.
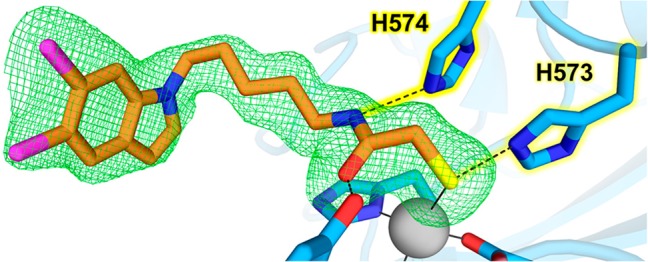
An electron density map showing the bound inhibitor is presented in Figure 2. The thiol group of MCA is presumably ionized to the negatively charged thiolate and coordinates to the active site Zn2+ ion such that the overall metal coordination geometry is a slightly distorted tetrahedral, with ligand–Zn2+–ligand angles ranging 94–127° across both monomers in the asymmetric unit of the crystal; metal coordination geometry deviates from ideal tetrahedral geometry by an average deviation of 10 ± 5° for ligand–Zn2+–ligand bond angles. For monomers A and B, the Zn2+···S separations are 2.3 Å, the C–S–Zn2+ angles are 114° and 120°, and the C–C–S–Zn2+ dihedral angles are 14° and 2°, respectively. Apart from the cisoid C–C–S–Zn2+ dihedral angle, the Zn2+ coordination geometry is ideal as outlined for thiolate–metal coordination interactions in refined protein structures.35 In comparison, the Zn2+-bound thiolate group of the cyclic depsipeptide Largazole thiol exhibits similar coordination geometry except for a more favorable C–C–S–Zn2+ dihedral angle of 92° in its complex with HDAC8.36
Figure 2.

(a) Stereoview of polder omit map (4.0σ; green mesh) for MCA (orange) bound to HDAC6 (blue). Hydrogen bond and metal-coordination interactions are shown as dashed and solid lines, respectively. The active site zinc ion is represented as a gray sphere. (b) Schematic representation of active site interactions for MCA bound to HDAC6.
The zinc-bound thiolate group of MCA accepts a hydrogen bond from the side chain of H573 (Nε573···S separation = 3.1 Å), which in turn donates a hydrogen bond to D610 (Nδ573···O610 separation = 2.8 Å), thus confirming that the side chain of H573 is in the positively charged imidazolium state. H573 is the first histidine in a tandem pair found in all HDAC active sites. Similar interactions are observed in the crystal structure of Schistosoma mansoni HDAC8 (SmHDAC8; 42% identity with human HDAC8) complexed with a mercaptoacetamide analog of SAHA, in which the Zn2+-bound thiolate group accepts a hydrogen bond from H141 (average Nε141···S separation for monomers A–D = 3.3 Å) (Figure 3).37
Figure 3.
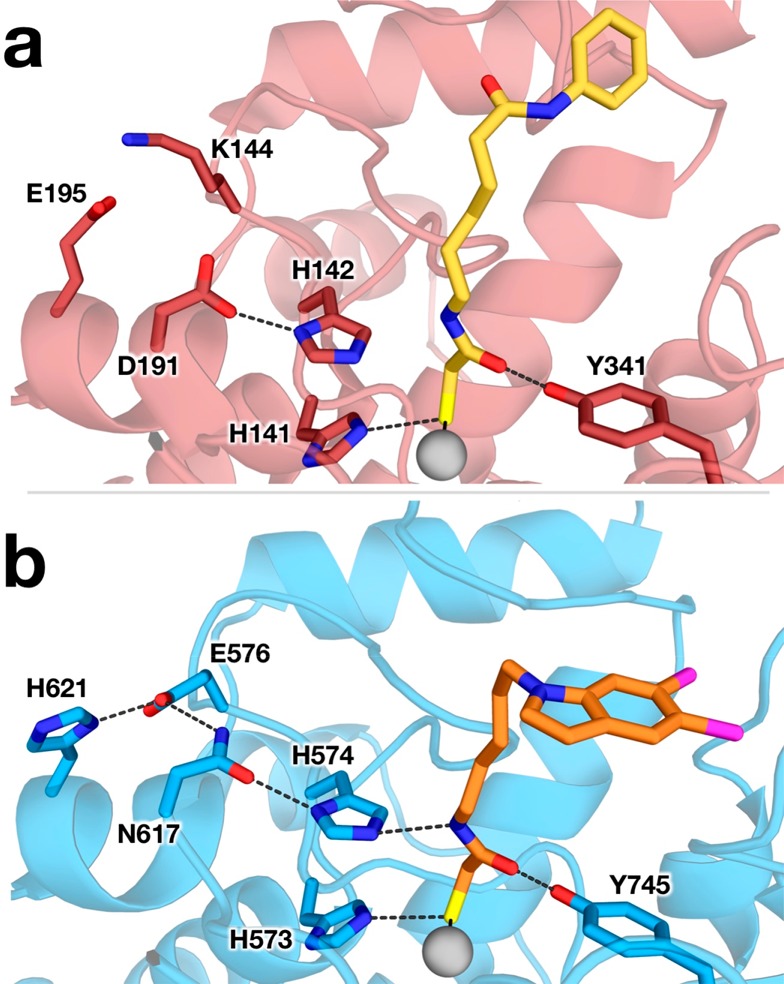
Structures of (a) S. mansoni HDAC8 (dark red; PDB ID 4CQF) complexed with a mercaptoacetamide analog of SAHA (yellow) and (b) the HDAC6–MCA complex (colors match Figure 2) showing interactions in the active site of each enzyme. Hydrogen bond and metal-coordination interactions are shown as dashed and solid lines, respectively.
In the HDAC6–MCA complex, the mercaptoacetamide carbonyl oxygen accepts a hydrogen bond from the phenolic hydroxyl group of Y745 (O745···O separation = 2.4 Å). This interaction mimics the role of Y745 in polarizing the scissile carbonyl group of acetyl-l-lysine.23 However, the carbonyl oxygen of MCA is 2.0 Å away from the Zn2+ coordination site ordinarily required for substrate binding, such that the Zn2+···O separation is 3.4 Å. Since both metal coordination and hydrogen bond interactions are required to activate the scissile amide group for hydrolysis, the amide group of the mercaptoacetamide is rendered chemically inert through its binding geometry in the HDAC6 active site.
The mercaptoacetamide NH group donates a hydrogen bond to Nε of H574 (Nε574···N separation = 3.2 and 3.0 Å in monomers A and B, respectively), which requires that the side chain of H574 is in the neutral imidazole form. This is the second histidine in the tandem pair; intriguingly, the corresponding interaction with the second histidine, H142, in the mercaptoacetamide complex with SmHDAC8 is too long for hydrogen bonding (Nε···N separation range of 3.5–4.0 Å in monomers A–D). Structures of mercaptoacetamide inhibitor complexes with HDAC6 and SmHDAC8 are compared in Figure 3. The lack of a hydrogen bond with H142 in the SmHDAC8–mercaptoacetamide complex may be due to the side chain of H142 being protonated as the positively charged imidazolium cation.
The role of the second histidine in the tandem pair as a hydrogen bond acceptor, requiring a neutral imidazole side chain, is similarly required for the binding of hydroxamate inhibitors with bidentate Zn2+ coordination geometry. In bidentate hydroxamate complexes with HDAC6, the hydroxamate NH group donates a hydrogen bond to H574.23,25,26 The same is true in human HDAC8–hydroxamate inhibitor complexes, where the hydroxamate NH group donates a hydrogen bond to H143.38,39 Thus, the mercaptoacetamide moiety is, in effect, a functional mimic of a hydroxamate group in terms of its ability to make an identical constellation of metal coordination and hydrogen bond interactions in the HDAC6 active site. Even better, the mercaptoacetamide moiety is not mutagenic since it is not subject to degradation via the Lossen rearrangement.
Why, then, does the mercaptoacetamide exhibit different binding interactions in the active site of SmHDAC8, lacking a hydrogen bond with the second active site histidine in the tandem pair? The mercaptoacetamide moiety clearly does not serve as a functional mimic of a hydroxamate group in binding to HDAC8. This difference likely contributes to weaker inhibition of class I HDAC isozymes by mercaptoacetamides.33,34 Structural comparisons of HDAC6, SmHDAC8, and human HDAC8 reveal that the second histidine is in a different electrostatic environment in each isozyme. In SmHDAC8 and human HDAC8, this histidine (H142 in SmHDAC8, H143 in human HDAC8) donates a hydrogen bond to a negatively charged carboxylate side chain (D191 in SmHDAC8, D183 in human HDAC8), which elevates the histidine pKa.13 In contrast, the corresponding histidine in HDAC6, H574, donates a hydrogen bond to the neutral carboxamide side chain of N617, which would elevate the pKa of H574 but not as much as would result if the hydrogen bond were made with a negatively charged carboxylate. Accordingly, H574 of HDAC6 is less basic than H142/H143 of SmHDAC8/human HDAC8. Consequently, H142 of SmHDAC8 and H143 of human HDAC8 are more likely to be protonated than H574 of HDAC6 at a given pH.
Differences in the basicity of the second histidine in the tandem pair also have implications for catalysis. In human HDAC8, enzymological and structural studies indicate that the second histidine, H143, functions as a single general base–general acid.13 Although similar enzymological studies have not yet been performed with HDAC6, deletion of the second histidine by mutagenesis in the H574A variant enables the crystallization and structure determination of an intact enzyme–substrate complex as the tetrahedral intermediate in the catalytic mechanism.23 This observation implies that the general base functionality in the active site of H574A HDAC6 is preserved; formation of the tetrahedral intermediate requires a sufficiently nucleophilic water molecule activated by Zn2+ coordination and a general base. Thus, the remaining active site histidine, H573 in H574A HDAC6, might function as the general base in this isozyme. H574 must serve as the general acid since its deletion leads to the trapped intermediate. The tetrahedral intermediate of amide hydrolysis cannot collapse without a proton donor to the leaving amino group, so the deletion of the general acid in H574A HDAC6 results in the formation of the tetrahedral intermediate as a dead-end complex.
Other aspects of MCA binding to HDAC6 contribute to its selectivity as well. The aliphatic linker packs into the aromatic groove in the substrate binding cleft with distances of 3.7 and 3.5 Å to the phenyl rings of F583 and F643, respectively. The capping group of the inhibitor is situated within the previously characterized L1 loop pocket, an interaction that confers HDAC6 selectivity.27 The chlorine atoms of the dichloroindole capping group pack against the side chains of H463 and P464. Additionally, the capping group of the inhibitor bound to monomer A packs against R636, D638, and F642 of monomer B. Meanwhile, the molecule bound to monomer B forms lattice contacts against D460 and H462 of monomer A.
It is worthwhile to note that thiol-containing drug candidates such as MCA can be subject to oxidation chemistry or reaction with an unintended electrophile in vivo, which could compromise their inhibitory function. The thiol-containing HDAC inhibitors Romidepsin and Largazole evolved to exist as thioester and disulfide-linked prodrugs, respectively.8 Romidepsin is activated by reduction of its internal disulfide linkage to yield the active inhibitor Romidepsin thiol, and Largazole is activated by hydrolysis of its thioester linkage to yield Largazole thiol. That being said, HDAC6 is localized in the cell cytosol,40 which is highly reducing, so this would favor the free thiol form of such inhibitors, including MCA. Moreover, there is precedent for the efficacy of a thiol-functionalized drug as exemplified by Captopril, which contains a thiol group targeting Zn2+ coordination in the active site of angiotensin converting enzyme.41,42
In summary, the present study highlights a chemical difference in the binding of mercaptoacetamides and hydroxamates to HDAC6 and HDAC8, specifically with regard to interactions with the tandem histidine pair in the active site. While each class of inhibitor contains a functional group that directly coordinates to Zn2+ (C–S– and N–O–, respectively), interactions with nearby active site residues differ. When bound to either enzyme, the hydroxamate N–O– group interacts with both histidine side chains. However, mercaptoacetamides exhibit different interactions in HDAC6 and HDAC8, specifically with regard to the second histidine. This highlights the importance of the tandem histidine pair in each of these enzymes; differences in the basicity of the second histidine residue can influence inhibitor binding and catalysis, which in turn can be exploited to enhance inhibitor selectivity.
Acknowledgments
We thank B. Havlinova for excellent technical assistance. Additionally, we thank Dr. R. Rajashankar and synchrotron beamline staff at the Northeastern Collaborative Access Team beamlines (supported by Grants P1 GM103403 and S10 RR02905) at the Advanced Photon Source (APS), a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory (ANL) under contract DE-AC02-06CH11357.
Glossary
ABBREVIATIONS
- HDAC
histone deacetylase
- MCA
N-(5-(5,6-dichloro-1H-indol-1-yl)pentyl)-2-mercaptoacetamide
- SAHA
suberoylanilide hydroxamic acid
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00487.
Materials and Methods; Table S1, data collection and refinement statistics (PDF)
Accession Codes
The atomic coordinates and crystallographic structure factors of the HDAC6–MCA complex have been deposited in the Protein Data Bank (www.rcsb.org) with accession code 6MR5.
Author Present Address
§ Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States
Author Contributions
N.J.P., A.P.K., and D.W.C. designed the project. S.S. synthesized the inhibitor MCA. C.B. assayed the inhibitory potency of MCA against HDAC6. N.J.P. determined the crystal structure of the HDAC6–MCA complex. All authors interpreted results and prepared the manuscript.
This research was supported by NIH grants GM49758 to D.W.C. and NS079183 to A.P.K. N.J.P. received financial support from the NIH through Chemistry–Biology Interface Training Grant T32 GM071339. This work was in part supported by the Czech Science Foundation (15–19640S), the Czech Academy of Sciences (RVO: 86652036), and the project “BIOCEV” (CZ.1.05/1.1.00/02.0109) from the European Regional Development Fund.
The authors declare no competing financial interest.
Supplementary Material
References
- López J. E.; Sullivan E. D.; Fierke C. A. Metal-dependent deacetylases: cancer and epigenetic regulators. ACS Chem. Biol. 2016, 11, 706–716. 10.1021/acschembio.5b01067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S.; Xu W.; Jiang W.; Yu W.; Lin Y.; Zhang T.; Yao J.; Zhou L.; Zeng Y.; Li H.; Li Y.; Shi J.; An W.; Hancock S. M.; He F.; Qin L.; Chin J.; Yang P.; Chen X.; Lei Q.; Xiong Y.; Guan K. L. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Zhang Y.; Yang C.; Xiong H.; Lin Y.; Yao J.; Li H.; Xie L.; Zhao W.; Yao Y.; Ning Z. B.; Zeng R.; Xiong Y.; Guan K. L.; Zhao S.; Zhao G. P. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 2010, 327, 1004–1007. 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C.; Weinert B. T.; Nishida Y.; Verdin E.; Mann M. The growing landscape of lysine acetylation links metabolism and cell signaling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- Gregoretti I. V.; Lee Y. M.; Goodson H. V. Molecular evolution of the histone deacetylase family: functional implication of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Kanyo Z. F.; Scolnick L. R.; Ash D. E.; Christianson D. W. Structure of a unique binuclear manganese cluster in arginase. Nature 1996, 383, 554–557. 10.1038/383554a0. [DOI] [PubMed] [Google Scholar]
- Ash D. E.; Cox J. D.; Christianson D. W. Arginase: a binuclear manganese metalloenzyme. Met. Ions Biol. Syst. 2000, 37, 407–428. [PubMed] [Google Scholar]
- Lombardi P. M.; Cole K. E.; Dowling D. P.; Christianson D. W. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 2011, 21, 735–743. 10.1016/j.sbi.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantt S. L.; Gattis S. G.; Fierke C. A. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry 2006, 45, 6170–6178. 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- Vannini A.; Volpari C.; Gallinari P.; Jones P.; Mattu M.; Carfi A.; De Francesco R.; Steinkühler C.; Di Marco S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007, 8, 879–884. 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling D. P.; Gantt S. L.; Gattis S. G.; Fierke C. A.; Christianson D. W. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrates and inhibitors. Biochemistry 2008, 47, 13554–13563. 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantt S. L.; Joseph C. G.; Fierke C. A. Activation and inhibition of histone deacetylase 8 by monovalent cations. J. Biol. Chem. 2010, 285, 6036–6043. 10.1074/jbc.M109.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantt S. M.; Decroos C.; Lee M. S.; Gullett L. E.; Bowman C. M.; Christianson D. W.; Fierke C. A. General base-general acid catalysis in human histone deacetylase 8. Biochemistry 2016, 55, 820–832. 10.1021/acs.biochem.5b01327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellmeier W.; Seiser C. Histone deacetylase function in CD4+ T cells. Nat. Rev. Immunol. 2018, 18, 617–634. 10.1038/s41577-018-0037-z. [DOI] [PubMed] [Google Scholar]
- Falkenberg K. J.; Johnstone R. W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discovery 2014, 13, 673–691. 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- Hubbert C.; Guardiola A.; Shao R.; Kawaguchi Y.; Ito A.; Nixon A.; Yoshida M.; Wang X. F.; Yao T. P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Haggarty S. J.; Koeller K. M.; Wong J. C.; Grozinger C. M.; Schreiber S. L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 4389–4394. 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Li N.; Caron C.; Matthias G.; Hess D.; Khochbin S.; Matthias P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P. H.; Zhang L.; Zhang Y. J.; Zhang J.; Xu W. F. HDAC6: physiological function and its selective inhibitors for cancer treatment. Drug Discoveries Ther. 2013, 7, 233–242. 10.5582/ddt.2013.v7.6.233. [DOI] [PubMed] [Google Scholar]
- Rivieccio M. A.; Brochier C.; Willis D. E.; Walker B. A.; D’Annibale M. A.; McLaughlin K.; Siddiq A.; Kozikowski A. P.; Jaffrey S. R.; Twiss J. L.; Ratan R. R.; Langley B. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 19599–19604. 10.1073/pnas.0907935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões-Pires C.; Zwick V.; Nurisso A.; Schenker E.; Carrupt P. A.; Cuendet M. HDAC6 as a target for neurodegenerative diseases: what makes it different from the other HDACs?. Mol. Neurodegener. 2013, 8, 7. 10.1186/1750-1326-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler K. V.; Kalin J.; Brochier C.; Vistoli G.; Langley B.; Kozikowski A. P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, Tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai Y.; Christianson D. W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake Y.; Keusch J. J.; Wang L.; Saito M.; Hess D.; Wang X.; Melancon B. J.; Helquist P.; Gut H.; Matthias P. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 2016, 12, 748–754. 10.1038/nchembio.2140. [DOI] [PubMed] [Google Scholar]
- Porter N. J.; Mahendran A.; Breslow R.; Christianson D. W. Unusual zinc binding mode of HDAC6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 13459–13464. 10.1073/pnas.1718823114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter N. J.; Wagner F. F.; Christianson D. W. Entropy as a driver of selectivity for inhibitor binding to histone deacetylase 6. Biochemistry 2018, 57, 3916–3924. 10.1021/acs.biochem.8b00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter N. J.; Osko J. D.; Diedrich D.; Kurz T.; Hooker J. M.; Hansen F. K.; Christianson D. W. Histone deacetylase 6-selective inhibitors and the influence of capping groups on hydroxamate-zinc denticity. J. Med. Chem. 2018, 61, 8054–8060. 10.1021/acs.jmedchem.8b01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon V. M.; Webb Y.; Merger R.; Sheppard T.; Jursic B.; Ngo L.; Civoli F.; Breslow R.; Rifkind R. A.; Marks P. A. Second generation hybrid polar compounds are potent inducers of transformed cell differentiation. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 5705–5708. 10.1073/pnas.93.12.5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks P. A. Discovery and development of SAHA as an anticancer agent. Oncogene 2007, 26, 1351–1356. 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- Shen S.; Kozikowski A. P. Why hydroxamates may not be the best histone deacetylase inhibitors – what some may have forgotten or would rather forget?. ChemMedChem 2016, 11, 15–21. 10.1002/cmdc.201500486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr J. S.; Galloway S.; Lagrutta A.; Armstrong M.; Miller T.; Richon V. M.; Andrews P. A. Nonclinical safety assessment of the histone deacetylase inhibitor vorinostat. Int. J. Toxicol. 2010, 29, 3–19. 10.1177/1091581809352111. [DOI] [PubMed] [Google Scholar]
- Kozikowski A. P.; Chen Y.; Gaysin A.; Chen B.; D’Annibale M. A.; Suto C. M.; Langley B. C. Functional differences in epigenetic modulators – superiority of mercaptoacetamide-based histone deacetylase inhibitors relative to hydroxamates in cortical neuron neuroprotection studies. J. Med. Chem. 2007, 50, 3054–3061. 10.1021/jm070178x. [DOI] [PubMed] [Google Scholar]
- Segretti M. C. F.; Vallerini G. P.; Brochier C.; Langley B.; Wang L.; Hancock W. W.; Kozikowski A. P. Thiol-based potent and selective HDAC6 inhibitors promote tubulin acetylation and T-regulatory cell suppressive function. ACS Med. Chem. Lett. 2015, 6, 1156–1161. 10.1021/acsmedchemlett.5b00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv W.; Zhang G.; Barinka C.; Eubanks J. H.; Kozikowski A. P. Design and synthesis of mercaptoacetamides as potent, selective, and brain permeable histone deacetylase 6 inhibitors. ACS Med. Chem. Lett. 2017, 8, 510–515. 10.1021/acsmedchemlett.7b00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti P. Geometry of interaction of metal ions with sulfur-containing ligands in protein structures. Biochemistry 1989, 28, 6081–6085. 10.1021/bi00440a052. [DOI] [PubMed] [Google Scholar]
- Cole K. E.; Dowling D. P.; Boone M. A.; Phillips A. J.; Christianson D. W. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J. Am. Chem. Soc. 2011, 133, 12474–12477. 10.1021/ja205972n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolfa D. A.; Marek M.; Lancelot J.; Hauser A.-T.; Walter A.; Leproult E.; Melesina J.; Rumpf T.; Wurtz J.-M.; Cavarelli J.; Sippl W.; Pierce R. J.; Romier C.; Jung M. Molecular basis for the antiparasitic activity of a mercaptoacetamide derivative that inhibits histone deacetylase 8 (HDAC8) from the human pathogen Schistosoma mansoni. J. Mol. Biol. 2014, 426, 3442–3453. 10.1016/j.jmb.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Somoza J. R.; Skene R. J.; Katz B. A.; Mol C.; Ho J. D.; Jennings A. J.; Luong C.; Arvai A.; Buggy J. J.; Chi E.; Tang J.; Sang B.-C.; Verner E.; Wynands R.; Leahy E. M.; Dougan D. R.; Snell G.; Navre M.; Knuth M. A.; Swanson R. V.; McRee D. E.; Tari L. W. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004, 12, 1325–1334. 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Vannini A.; Volpari C.; Filocamo G.; Casavola E. C.; Brunetti M.; Renzoni D.; Chakravarty P.; Paolini C.; De Francesco R.; Gallinari P.; Steinkühler C.; Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 15064–15069. 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertos N. R.; Gilquin B.; Chen G. K. T.; Yen T. J.; Khochbin S.; Yang X.-J. Role of the tetradecapeptide repeat domain of human histone deacetylase 6 in cytoplasmic retention. J. Biol. Chem. 2004, 12, 48246–48254. 10.1074/jbc.M408583200. [DOI] [PubMed] [Google Scholar]
- Ondetti M. A.; Rubin B.; Cushman D. W. Design of specific inhibitors of angiotensin-converting enzyme: new class of orally active antihypertensive agents. Science 1977, 196, 441–444. 10.1126/science.191908. [DOI] [PubMed] [Google Scholar]
- Cushman D. W.; Ondetti M. A. History of the design of Captopril and related inhibitors of angiotensin converting enzyme. Hypertension 2001, 17, 589–592. 10.1161/01.HYP.17.4.589. [DOI] [PubMed] [Google Scholar]
- Lee J.-H.; Yao Y.; Mahendran A.; Ngo L.; Venta-Perez G.; Choy M. L.; Breslow R.; Marks P. A. Creation of a histone deacetylase 6 inhibitor and its biological effects. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 12005–12010. 10.1073/pnas.1515882112. [DOI] [PMC free article] [PubMed] [Google Scholar]; 2015, 112, E5899. 10.1073/pnas.1519546112
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.