

Abstract

In solving the P-gp and BCRP transporter-mediated efflux issue in a series of benzofuran-derived pan-genotypic palm site inhibitors of the hepatitis C virus NS5B replicase, it was found that close attention to physicochemical properties was essential. In these compounds, where both molecular weight (MW >579) and TPSA (>110 Å2) were high, attenuation of polar surface area together with weakening of hydrogen bond acceptor strength of the molecule provided a higher intrinsic membrane permeability and more desirable Caco-2 parameters, as demonstrated by trifluoroacetamide 11 and the benchmark N-ethylamino analog 12. In addition, the tendency of these inhibitors to form intramolecular hydrogen bonds potentially contributes favorably to the improved membrane permeability and absorption. The functional group minimization that resolved the efflux problem simultaneously maintained potent inhibitory activity toward a gt-2 HCV replicon due to a switching of the role of substituents in interacting with the Gln414 binding pocket, as observed in gt-2a NS5B/inhibitor complex cocrystal structures, thus increasing the efficiency of the optimization. Noteworthy, a novel intermolecular S=O···C=O n → π* type interaction between the ligand sulfonamide oxygen atom and the carbonyl moiety of the side chain of Gln414 was observed. The insights from these structure–property studies and crystallography information provided a direction for optimization in a campaign to identify second generation pan-genotypic NS5B inhibitors.
Keywords: Hepatitis C virus, NS5B, benzofuran, P-gp, BCRP, efflux, TPSA, hydrogen bonding
Chronic hepatitis C virus (HCV) infection, which affects around 150 million people in the world, is a liver disease that can progress into liver cirrhosis and life-threatening hepatocellular carcinoma, and is responsible for about 700,000 deaths per year globally.1 Intensive drug discovery research in the past quarter century since the characterization of the virus in 1989 has resulted in the clinical application of HCV-targeted direct-acting antiviral agents that have replaced the previous standard of care, pegylated-interferon, and ribavirin. A cure for chronic HCV infection, as measured by the sustained virological response, can be achieved with the new all oral combination therapies that offer good safety profiles, broad genotype activity, and abridged treatment duration.2,3 These small molecule drugs inhibit one of the three HCV nonstructural proteins, the NS3/4A serine protease, the NS5A replication cofactor, and the NS5B replicase, all of which play essential roles in the HCV replication cycle.4,5 The NS5B replicase is an RNA-dependent RNA polymerase that is responsible for the synthesis of HCV genomic RNA. Its tertiary structure adopts a right-hand configuration with fingers, palm, and thumb domains representative of polymerase enzymes. In addition to the active site-directed nucleoside/nucleotide inhibitors and their prodrugs, non-nucleoside inhibitors that bind to three distinct allosteric sites (thumb-I, thumb-II, and palm site) in HCV NS5B replicase have also been developed.6,7
We have reported the discovery of BMS-929075 (1), a palm site allosteric inhibitor of HCV NS5B replicase (Figure 1), that was advanced into Phase 1 clinical studies.8 In cell-based replicon assays, this benzofuran-based compound 1 exhibited potent HCV inhibitory activity toward all genotypes with the exception of gt-2 (EC50 > 100 nM).
Figure 1.

Structure of the benzofuran-derived NS5B palm site inhibitor 1.
During the subsequent second generation campaign to identify pan-genotypic inhibitors from this series, it was discovered that incorporation of a N-(2-hydroxyethyl)methylsulfonamide group at the C6 position of the benzofuran ring led to a ∼20-fold enhancement in inhibitory activity against gt-2a (JFH-1 strain) replicon cells, as illustrated by the C6 matched molecular pair 2 and 3 (Table 1). During the lead optimization resulting in 1, it was observed in the property–activity relationship that potency toward gt-1b, gt-1a, and the pre-existing gt-1bC316N mutant generally trended with an increase in lipophilicity.8 A more desirable and balanced potency profile was achieved with the phenylcyclopropyl amide 4, which is almost one cLogP9 unit more lipophilic than the 2-pyridinyl matched pair analog 3 and has inhibitory EC50 values below 10 nM across gt-1b, gt-1a, and gt-2a replicons. However, initial in vitro profiling of these C6-N-(2-hydroxyethyl)methylsulfonamide substituted benzofuran compounds showed that they had less than desirable microsomal stability, as exemplified by the low percentage of unmetabolized 4 remaining in human and rat liver microsomes (HLM and RLM, respectively) in a high throughput assay. The inadequate metabolic stability of the compound was resolved by incorporating a 2-pyrimidine ring, a metabolic stability enhancing moiety identified during the lead optimization leading to 1, in place of the phenyl ring in 4 and removing the primary hydroxyl group in the hydroxyethyl side chain to avoid any possible metabolic modifications of this potentially vulnerable functionality. The derived analog 5 showed similar lipophilicity to 4 but a significant improvement in metabolic stability in both HLM and RLM compared to 4 while maintaining the overall antiviral potency profile (Table 1). Although the intrinsic membrane permeability of this series of potent N-ethylmethylsulfonamide analogs was respectable, as indicated by the PAMPA (parallel artificial membrane permeability assay) values, a major deficiency in the in vitro profile was the high susceptibility to efflux in Caco-2 cells, as exemplified by the high efflux ratio exhibited by 5 and its close 4′-methoxy analog 6 (>8.6 and >12 (Table 2), respectively). As a result, these compounds exhibited low oral bioavailability with poor exposure in both the plasma and liver of rats following oral dosing. In the presence of zosuquidar,10 a selective P-glycoprotein (P-gp) efflux pump inhibitor, the efflux of compound 6 in Caco-2 cells was dramatically suppressed (efflux ratio = 1.3) and the apical to basolateral (A to B) permeability was favorably increased to 168 nm/s (Table 3). The same phenomenon was also observed in the presence of KO143,11 a selective inhibitor of breast cancer resistance protein (BCRP), although to a lesser extent. These results suggested that the efflux of the sulfonamide analogs was predominantly mediated by P-gp and to some extent by the BCRP transporter as well.
Table 1. Inhibitory Potency towards HCV Replicons (EC50) and in Vitro Profiling for Compounds 2–5.

Table 2. In Vitro Profiling and Inhibitory Potency towards HCV gt-2a Replicon (EC50) for Compounds 6 and 8–12.


HBA = hydrogen bonding acceptor, relative strength on the pKBHX scale.12
TPSA = topological polar surface area.9
A-B = apical to basolateral; B-A = basolateral to apical, Data for 5: efflux ratio >8.6, A-B < 15, B-A = 130.
Replicon, JFH-1 strain, data are average values of n ≥ 2 independent experiments;
Data were obtained from a single experiment performed in duplicate.
pKBHX data for N-methylaniline. See SI for CC50 and SD.
Table 3. Caco-2 Permeability Behavior of Compound 6 in the Presence of Transporter Inhibitors.
| Caco-2 Pc (nm/s) |
||||
|---|---|---|---|---|
| Cpd | inhibitor | efflux ratio | A-B | B-A |
| 6 | Zosuquidar (P-gp) | 1.3 | 168 | 215 |
| KO143 (BCRP) | 4.8 | 67 | 324 | |
Based on the SAR of the C6-unsubstituted series,8 the N-ethylmethylsulfonamide group appeared to be responsible for the potent activity against gt-2 but also a contributor to the high efflux liability of this new series of pan-genotypic inhibitors. One approach explored to address the efflux issue was to enhance the intrinsic membrane permeability of the molecule while retaining the sulfonamide moiety to preserve potency toward gt-2 virus. This could be achieved empirically by varying the substitution pattern at the 3′- and 4′-positions on the C5-phenyl ring, with F, Cl, Me, and OMe groups examined. In this regard, a substituent at the 4′-position that could potentially engage the cyclopropyl amide NH in an intramolecular hydrogen bond to facilitate permeability appeared more favorable.13 As shown in Table 4, in general, the PAMPA membrane permeability of these compounds increased, and the apical to basolateral forward permeability in Caco-2 cells was enhanced with increasing lipophilicity, as suggested by their cLogP values. Although the 3′-methyl analog 7c appeared to be an outlier, the A to B influx of this compound was quantifiable compared to that of compound 6. The 3′-Cl-4-OMe analog 7b emerged with the highest PAMPA value in these C6-sulfonamide derivatives. Encouragingly, the Caco-2 efflux ratio of this compound also decreased substantially to a more desirable value of 2.2. However, the low metabolic stability in HLM and RLM (t1/2 = 13.9 and 26 min, respectively) and the unsatisfactory exposure in both the plasma and liver (C5h = 277 and 7180 nM, respectively) of rats following oral administration in a pharmacokinetic screen prevented this compound from being further progressed.
Table 4. In Vitro Profiling and Inhibitory Potency towards HCV gt-2a Replicon (EC50) for Compounds 7a–d Compared with 6.

Concurrently, a SAR exercise was conducted to evaluate the effect of various sulfonamide surrogates that express differential physicochemical properties related to hydrogen bonding on the permeability profile (Table 2) to gain insight for resolving the efflux issue.14,15 Interestingly, an examination of polar surface area (represented by calculated topological polar surface area (TPSA)9,16) of the molecule and hydrogen bonding acceptor (HBA) strength (relative strength on the pKBHX scale12) of the C6-functionality suggested that modulation of either physicochemical property alone was not sufficient to confer a more favorable membrane permeability profile. As shown in Table 2, TPSA progressively decreases from 6 to 8, 9, and 10, while the HBA strength increases in the reverse order from 10 to 9, 8, and 6. However, neither 6 nor 10, which are at the lower end of the pKBHX and TPSA values, respectively, provided a useful permeability profile. In these analogs where the TPSA is >110 Å2 and MW >579, values that are typically not conducive for membrane absorption, a reduced polar surface area coupled with a weaker HBA strength is required to provide a higher intrinsic permeability as well as improved Caco-2 parameters. This was manifested in the trifluoroacetamide analog 11 in which both the TPSA and HBA strength values are the lowest when compared to compounds 6–10, exhibiting the highest PAMPA value (835 nm/s) and a more favorable efflux ratio of 2.7 among these analogs. These observations led to the assessment of the weakly basic C6-N-ethyl aniline analog 12, which is also a very weak HBA, representing a compromise between minimizing both TPSA and HBA strength. Pleasingly, compound 12 maintained the high PAMPA permeability of 11. Importantly, the apical to basolateral forward flux in Caco-2 cells was dramatically improved in 12 with essentially no efflux compared to 11 and in remarkable contrast to the profile of the parent compound 6. These results are consistent with studies that demonstrated that binding to the P-gp recognition pocket is exclusively controlled by hydrogen bonding acceptors present in the substrate.17 The favorable absorption profile of 12 translated into promising exposure in both the plasma and the liver of rats with good oral bioavailability following oral administration (Table 5). The plasma AUC and the concentration in the liver at the 5 h time point obtained after oral dosing of 12 were, respectively, 10- and 5-fold higher than those from compound 6, which, in contrast to 12, was not detected in either the plasma or the liver at the 24 h time point postdosing. The undesirable high clearance after IV dosing exhibited by 6 was also substantially improved in 12.
Table 5. Pharmacokinetic Parameters in Rats for 6 and 12 (0 to 24 h)a.
| IV |
||||
|---|---|---|---|---|
| Cpd | HLM/RLMbt1/2 (min) | CL (mL/min/kg) | IV t1/2 (h) | Vss (L/kg) |
| 6 | 91/>120 | 60 | 1.6 | 14 |
| 12 | 36/39 | 11 | 4.7 | 4 |
| PO |
||||||
|---|---|---|---|---|---|---|
| Cpd | Cmax (nM) | tmax (h) | AUC (nM·h) | C5 liver (nM) | C24 liver (nM) | %F |
| 6 | 223 | 1.9 | 740 | 1177 | NDc | 20 |
| 12 | 776 | 5.3 | 7803 | 5258 | 391 | 57 |
IV, 2 mg/kg; PO, 6 mg/kg; n = 3.
HLM/RLM denotes human and rat liver microsomes, respectively.
ND = not detected.
Compound 12 maintained potent activity toward gt-1b, 1a, and gt-1bC316N mutant replicons (EC50 = 1.2, 2.3, and 3.6 nM, respectively); moreover, it exhibited the same level of inhibitory potency against gt-2a replicon as the parent C6-sulfonamide compound 6 (Table 2), a result that was not anticipated based on the apparent requirement of a hydrogen bond accepting moiety at C6 for activity toward gt-2 virus. The optimization toward a pan-genotypic NS5B inhibitor in the benzofuran and related 7-azabenzofuran series18,19 followed the recognition of this key SAR point that a HBA was dispensable. In the cocrystal structure of compound 5, a close analog of 6, in complex with gt-2a NS5B L30S protein, the N-ethylmethylsulfonamide moiety assumes a conformation that appears to be partly driven by the electronic repulsion between the sulfonamide oxygen atoms and the central C5-phenyl ring. The sulfonamide methyl group projects into a hydrophobic pocket formed by the side chains from the Pro197, Leu384, and Tyr415 residues of the protein (Figure 2a). The ethyl group is not in direct interaction with the protein, but rather in intramolecular van der Waals contact with the C5-phenyl ring within compound 5. One of the sulfonamide oxygen atoms forms a hydrogen bond with the guanidine moiety in the side chain of Arg200. In addition, the pyrimidine moiety π-stacks with the phenyl ring of Tyr452 and the pyrimidinylcyclopropyl amide carbonyl forms a network of water-bridged hydrogen bonds to nearby residues including Tyr452 and Trp550. In the absence of the SO2 group, the N-ethylamino side chain can adopt a conformation in which the ethyl group flips to fill the volume of the hydrophobic pocket, as revealed by a cocrystal structure of the 5-fluoropyrimidine derivative 13, a close analog of compound 12, bound to gt-2a NS5B L30S protein (Figure 2b). In this cocrystal structure, the carbonyl oxygen atom of the cyclopropylamide in compound 13 forms water-bridged hydrogen bonds to the side chain of Arg200 and the backbone carbonyl of Tyr195 through a single water molecule. The N–H of the ethylamino moiety in compound 13 forms a hydrogen bond with the carbonyl oxygen atom of the primary amide in the side chain of Gln414 with a N–H···O=C distance of 2.1 Å. Compound 5 also forms direct interactions with the side chain of Gln414. In this case, one of the pyrimidinyl nitrogen atoms hydrogen bonds to the Gln414 side chain amide NH2 moiety. Interestingly, one of the sulfonamide oxygen atoms is positioned such that an n → π* interaction20,21 with the Gln414 side chain amide carbonyl is possible. The S=O···C=O distance is 3.5 Å, which is close to the sum of the van der Waals radii of oxygen and carbon (3.22 Å), and the O···C=O angle is 108.0°, a value consistent with Bürgi–Dunitz trajectory (Figure 3). These compensatory interactions in the vicinity of Gln414, a key residue that differentiates gt-2 NS5B protein from that of gt-1, as observed in the structures of the complexes of 5 and 13 with the gt-2a NS5B L30S protein, may account for the improved coverage of gt-2a observed for the C6-sulfonamide and C6-N-ethylamino des-sulfonamide analogs.
Figure 2.

(a) Structure and close-up view of the Gln414 binding pocket region in gt-2a NS5B L30S/5 complex cocrystal structure with key interacting residues highlighted. (b) Structure and close-up view of the Gln414 binding pocket region in gt-2a NS5B L30S/13 complex cocrystal structure with key interacting residues highlighted. Hydrogen bonds are denoted with dashed yellow lines, π-stacking, and possible n → π* interactions are shown as dashed blue and magenta lines, respectively, and water molecules are depicted as red spheres. Image generated with The PyMOL Molecular Graphics System (v. 2.0, Schrödinger, LLC).
Figure 3.
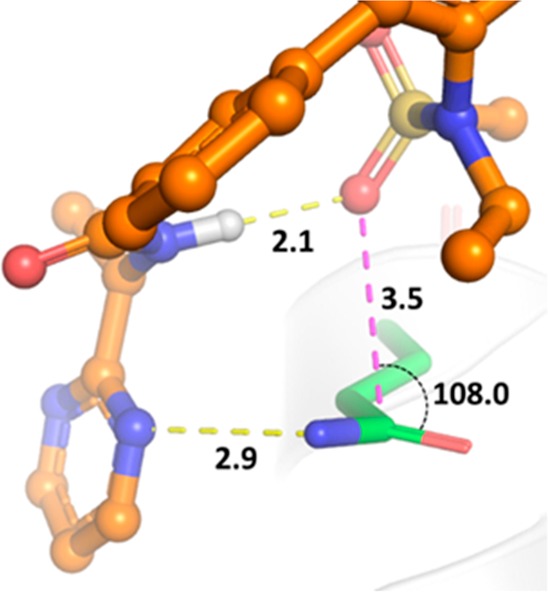
Detailed view of intra- and intermolecular hydrogen bonds involving the ligand pyrimidinylcyclopropyl amide and N-ethylmethylsulfonamide moieties of 5 and the putative n → π* interaction formed between Gln414 and the ligand in the gt-2a NS5B L30S/5 complex. Hydrogen bonds and the n → π* interaction are depicted as dashed yellow and magenta lines, respectively. Distances are in angstroms and the interaction angle is reported in degrees. Image created with The PyMOL Molecular Graphics System (v. 2.0, Schrödinger, LLC).
Interestingly, as depicted in the structure of the gt-2a NS5B L30S/5 complex (Figure 2a), the inhibitor adopts a conformation in which the pyrimidinylcyclopropyl amide N–H engages in an intramolecular hydrogen bond with one of the oxygen atoms of the C6-sulfonamide group (N–H···O=S distance = 2.1 Å) forming an unusual 11-membered ring pseudoheterocycle. Without the strong hydrogen bonding acceptor SO2 group, the pyrimidinylcyclopropyl amide N–H points toward the 4′-methoxy oxygen atom, forming an alternative intramolecular hydrogen bond, as represented in compound 13 in the gt-2a NS5B L30S/13 complex structure (Figure 2b). The propensity of both the sulfonamide and the N-ethylamino compounds to form intramolecular hydrogen bonds likely contributes to the good to high intrinsic membrane permeability, as suggested by their PAMPA values described above, that potentially facilitate the absorption process.13,15,22,23 However, it is interesting to note that such potential to form intramolecular hydrogen bonds in compounds 6 to 10 did not alleviate the transporter-mediated efflux. In this example, it is the combination of the TPSA and HBA strength properties of the molecule that determines the efflux susceptibility.
In conclusion, the poor absorption problem presented by P-gp and BCRP transporter-mediated efflux of the benzofuran class of pan-genotypic HCV NS5B replicase palm site inhibitors, in which both the MW and TPSA are high, was resolved by reduction of polar surface area in conjunction with decreasing hydrogen bond acceptor strength. The permeability of these inhibitors was also enhanced by their ability to form intramolecular hydrogen bonds. The benchmark compound 12 emerging from this study exhibited promising pharmacokinetic parameters and oral bioavailability in rats. The interactions of the N-ethylmethylsulfonamide group in the gt-2a Gln414 binding pocket were fulfilled by a simpler N-ethylamino moiety, enabling the preservation of potent activity toward gt-2a in 12 and functional group efficiency in optimization. These SAR insights and the structural information derived from NS5B/inhibitor cocrystals provided a clear path for the optimization of second generation pan-genotypic inhibitors.18,19
Acknowledgments
We would like to thank colleagues in Lead Profiling for collecting in vitro profiling data, and the Discovery Chemistry Synthesis group for scale-up synthesis of intermediates. Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. X-ray data for gt-2a NS5B/5 were collected by Rick Walter and Gina Ranieri of Shamrock Structures LLC using beamline 08ID-1 at the Canadian Light Source, which is supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00379.
Anti-HCV activity profile of compound 12, compound synthesis and characterization, biological methods, CC50 and assay standard deviation (SD) data, in vivo rat pharmacokinetic studies, Caco-2 permeability assay, production of the recombinant protein, crystallization conditions, crystallographic methods, crystallographic statistics for complexes of HCV gt-2a NS5B protein, and a figure depicting the complexes with ligand electron density contours (PDF)
Accession Codes
The PDB codes for the X-ray crystal structures of HCV gt-2a NS5B L30S in complex with 5 and 13 are 5QJ0 and 5QJ1, respectively.
The authors declare no competing financial interest.
Supplementary Material
References
- Manns M. P.; Buti M.; Gane E.; Pawlotsky J.-M.; Razavi H.; Terrault N.; Younossi Z. Hepatitis C virus infection. Nature. Rev. Dis. Primers 2017, 3, 17006 10.1038/nrdp.2017.6. [DOI] [PubMed] [Google Scholar]
- Götte M.; Feld J. J. Direct-acting antiviral agents for hepatitis C: structural and mechanistic insights. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 338–351. 10.1038/nrgastro.2016.60. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. 2015 Philip S. Portoghese Medicinal Chemistry Lectureship. Curing hepatitis C virus infection with direct-acting antiviral agents: The arc of a medicinal chemistry triumph. J. Med. Chem. 2016, 59, 7311–7351. 10.1021/acs.jmedchem.6b00915. [DOI] [PubMed] [Google Scholar]
- Scheel T. K. H.; Rice C. M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 2013, 19, 837–849. 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartenschlager R.; Lohmann V.; Penin F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 2013, 11, 482–496. 10.1038/nrmicro3046. [DOI] [PubMed] [Google Scholar]
- Sofia M. J.; Chang W.; Furman P. A.; Mosley R. T.; Ross B. S. Nucleoside, nucleotide, and non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA-polymerase. J. Med. Chem. 2012, 55, 2481–2531. 10.1021/jm201384j. [DOI] [PubMed] [Google Scholar]
- Watkins W. J.; Ray A. S.; Chong L. S. HCV Polymerase inhibitors. Curr. Opin. Drug Discovery Dev. 2010, 13, 441–465. [PubMed] [Google Scholar]
- Yeung K.-S.; Beno B. R.; Parcella K.; Bender J. A.; Grant-Young K. A.; Nickel A.; Gunaga P.; Anjanappa P.; Bora R. O.; Selvakumar K.; Rigat K.; Wang Y.-K.; Liu M.; Lemm J.; Mosure K.; Sheriff S.; Wan C.; Witmer M.; Kish K.; Hanumegowda U.; Zhuo X.; Shu Y.-Z.; Parker D.; Haskell R.; Ng A.; Gao Q.; Colston E.; Raybon J.; Grasela D. M.; Santone K.; Gao M.; Meanwell N. A.; Sinz M.; Soars M. G.; Knipe J. O.; Roberts S. B.; Kadow J. F. Discovery of a hepatitis C virus NS5B replicase palm site allosteric inhibitor (BMS-929075) advanced to phase 1 clinical studies. J. Med. Chem. 2017, 60, 4369–4385. 10.1021/acs.jmedchem.7b00328. [DOI] [PubMed] [Google Scholar]
- cLogP and TPSA were calculated by using the molecular property calculator JChem-15.9.7.0 from ChemAxon.
- Dantzig A. H.; Shepard R. L.; Cao J.; Law K. L.; Ehlhardt W. J.; Baughman T. M.; Bumol T. F.; Starling J. J. Reversal of P-glycoprotein-mediated multidrug resistance by a potent cyclopropyldibenzosuberane modulator, LY335979. Cancer Res. 1996, 56, 4171–4179. [PubMed] [Google Scholar]
- Allen J. D.; van Loevezijn A.; Lakhai J. M.; van der Valk M.; van Tellingen O.; Reid G.; Schellens J. H. M.; Koomen G.-J.; Schinkel A. H. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 2002, 1, 417–425. [PubMed] [Google Scholar]
- Laurence C.; Brameld K. A.; Graton J.; Le Questel J.-Y.; Renault E. The pKBHX Database: Toward a better understanding of hydrogen-bond basicity for medicinal chemists. J. Med. Chem. 2009, 52, 4073–4086. 10.1021/jm801331y. [DOI] [PubMed] [Google Scholar]
- Kuhn B.; Mohr P.; Stahl M. Intramolecular hydrogen bonding in medicinal chemistry. J. Med. Chem. 2010, 53, 2601–2611. 10.1021/jm100087s. [DOI] [PubMed] [Google Scholar]
- Hitchcock S. A. Structural modifications that alter the P-glycoprotein efflux properties of compounds. J. Med. Chem. 2012, 55, 4877–4895. 10.1021/jm201136z. [DOI] [PubMed] [Google Scholar]
- Desai P. V.; Raub T. J.; Blanco M.-J. How hydrogen bonds impact P-glycoprotein transport and permeability. Bioorg. Med. Chem. Lett. 2012, 22, 6540–6548. 10.1016/j.bmcl.2012.08.059. [DOI] [PubMed] [Google Scholar]
- Ertl P.; Rohde B.; Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- Li-Blatter X.; Seelig A. Exploring the P-glycoprotein binding cavity with polyoxyethylene alkyl ethers. Biophys. J. 2010, 99, 3589–3598. 10.1016/j.bpj.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman K. J.; Parcella K.; Yeung K.-S.; Grant-Young K. A.; Zhu J.; Wang T.; Zhang Z.; Yin Z.; Beno B. R.; Sheriff S.; Kish K.; Tredup J.; Jardel A. G.; Halan V.; Ghosh K.; Parker D.; Mosure K.; Fang H.; Wang Y.-K.; Lemm J.; Zhuo X.; Hanumegowda U.; Rigat K.; Donoso M.; Tuttle M.; Zvyaga T.; Haarhoff Z.; Meanwell N. A.; Soars M. G.; Roberts S. B.; Kadow J. F. The discovery of a pan-genotypic, primer grip inhibitor of HCV NS5B polymerase. MedChemComm 2017, 8, 796–806. 10.1039/C6MD00636A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parcella K.; Eastman K.; Yeung K.-S.; Grant-Young K. A.; Zhu J.; Wang T.; Zhang Z.; Yin Z.; Parker D.; Mosure K.; Fang H.; Wang Y.-K.; Lemm J.; Zhuo X.; Hanumegowda U.; Liu M.; Rigat K.; Donoso M.; Tuttle M.; Zvyaga T.; Haarhoff Z.; Meanwell N. A.; Soars M. G.; Roberts S. B.; Kadow J. F. Improving metabolic stability with deuterium: The discovery of BMT-052, a pan-genotypic HCV NS5B polymerase inhibitor. ACS Med. Chem. Lett. 2017, 8, 771–774. 10.1021/acsmedchemlett.7b00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry R. W.; Raines R. T. The n→π* Interaction. Acc. Chem. Res. 2017, 50, 1838–1846. 10.1021/acs.accounts.7b00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla R.; Chopra D. Characterization of the short O = C···O = C π-hole tetrel bond in the solid state. CrystEngComm 2018, 20, 3308–3312. 10.1039/C8CE00697K. [DOI] [Google Scholar]
- Whitty A.; Zhong M.; Viarengo L.; Beglov D.; Hall D. R.; Vajda S. Quantifying the chameleonic properties of macrocycles and other high-molecular-weight drugs. Drug Discovery Today 2016, 21, 712–717. 10.1016/j.drudis.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alex A.; Millan D. S.; Perez M.; Wakenhut F.; Whitlock G. A. Intramolecular hydrogen bonding to improve membrane permeability and absorption in beyond rule of five chemical space. MedChemComm 2011, 2, 669–674. 10.1039/c1md00093d. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.