

Abstract
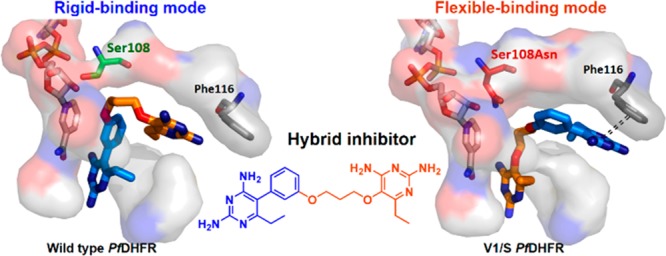
The S108N mutation of dihydrofolate reductase (DHFR) renders Plasmodium falciparum malaria parasites resistant to pyrimethamine through steric clash with the rigid side chain of the inhibitor. Inhibitors with flexible side chains can avoid this clash and retain effectiveness against the mutant. However, other mutations such as N108S reversion confer resistance to flexible inhibitors. We designed and synthesized hybrid inhibitors with two structural types in a single molecule, which are effective against both wild-type and multiple mutants of P. falciparum through their selective target binding, as demonstrated by X-ray crystallography. Furthermore, the hybrid inhibitors can forestall the emergence of new resistant mutants, as shown by selection of mutants resistant to hybrid compound BT1 from a diverse PfDHFR random mutant library expressed in a surrogate bacterial system. These results show that it is possible to develop effective antifolate antimalarials to which the range of parasite resistance mutations is greatly reduced.
Keywords: Dual binding, hybrid inhibitors, dihydrofolate reductase, resistance suppression, antimalarials
Antimalarial drugs have been important tools in the fight against malaria and will be essential in the current effort to eliminate the disease.1−4 However, a major problem is the development of parasite resistance to the drugs, which threaten their effective use.5−9Plasmodium falciparum parasites resistant to antifolate drugs such as pyrimethamine (Pyr), an inhibitor of parasite dihydrofolate reductase (PfDHFR), and combinations of antifolate and sulfa drugs are widespread throughout malaria endemic regions.10,11 Instead of abandoning PfDHFR as an antimalarial target, new approaches to develop antifolates effective against resistant parasites with mutated PfDHFR could be successful.10,12−15 The crystal structure of PfDHFR, which is joined with thymidylate synthase as a bifunctional enzyme, has been solved,16 together with its counterpart in P. vivax.17 These structures reveal features that explain the basis for mutation-induced resistance.13,14 The structures of wild-type and mutant PfDHFRs cocomplexes have been solved with inhibitors, namely, WR99210, which can still inhibit mutant enzymes effectively,18 and Pyr, to which binding has been compromised by PfDHFR mutations. Analysis of these structures has enabled target-based design of new inhibitors, some of which have very high efficacy and selectivity as antimalarial drugs.15,19 These studies reveal the key S108N mutation in PfDHFR, which introduces a larger side-chain that sterically interferes with the binding of inhibitors with a rigid p-Cl-phenyl side chain such as Pyr and cycloguanil. Antifolate resistance is amplified when the S108N mutation is accompanied by additional mutations in the inhibitor binding region of PfDHFR including N51I, C59R, and I164L. Inhibitors such as WR99210 retain tight binding affinity with mutant PfDHFRs associated with Pyr resistance and retain efficacy against parasites with the corresponding mutant PfDHFRs. WR99210 and related compounds have flexible side-chains, which can avoid steric clash with the S108N side-chain. However, these flexible inhibitors can be rendered ineffective by further PfDHFR resistance mutations, as shown by a surrogate yeast20 or bacterial system,21,22 although the frequencies and the levels of resistance against WR99210 were lower than those for Pyr. Here, we present a new design of hybrid inhibitors, with both rigid and flexible side-chains in the same molecule as a combination that can reduce the emergence of resistance through mutations. An additional rationale for making such hybrid inhibitors is that there are conflicting requirements of PfDHFR in mutations conferring resistance to rigid (Pyr-like) and flexible (WR99210-like) inhibitors.22
WR99210-resistant mutants isolated using a bacterial surrogate system are more sensitive to Pyr than Pyr-resistant mutants owing to the presence of wild-type PfDHFR residues S108, N51, C59, and I164.21,22 In general, therefore, PfDHFRs can be divided into two series, namely, the “S108 series”, which is sensitive to rigid inhibitors like Pyr but resistant to flexible inhibitors in combination with other mutations, and the “108N series” (or S108N series or N108 series22), which is sensitive to flexible inhibitors but resistant to rigid inhibitors. We hypothesize that the combination of the two types of pharmacophore (rigid and flexible) in a single molecule should be effective against parasites with both types of PfDHFR. Specifically, the rigid pharmacophore should inhibit PfDHFRs in the S108 series, whereas the flexible pharmacophore should inhibit those in the 108N series. Furthermore, we hypothesize that mutations conferring resistance to hybrid inhibitors would be extremely limited owing to constraints on residues in the substrate/inhibitor binding region of PfDHFR.
In this Letter, we report the synthesis of hybrid inhibitors capable of inhibiting P. falciparum carrying either wild-type or multiple mutant DHFRs, and the modes of binding for these compounds. The hybrid inhibitors bind with similar affinities to both types of PfDHFR. Significantly, they bind in the active site of PfDHFRs in the expected manner, namely, binding with the rigid end to wild-type (S108) PfDHFR and with the flexible end to mutant (S108N) PfDHFR. The inhibitors show a low tendency to induce new antifolate-resistant mutants. Furthermore, in anticipation of further potential development as oral formulation, the 2,4-diaminopyrimidine scaffold was employed to take advantage of its pKa, which is suitable for oral bioavailability (pKa 6–7), as previously rationalized in P218 development.15 Hence, they should serve as good models for further development of oral antifolate antimalarials, which can forestall further development of mutation-induced resistance.
Results and Discussion
Our previous works on the synthesis of DHFR inhibitors showed that P30 (a rigid diaminopyrimidine; Pyr-like with m-Cl substituent) and P65 (a flexible diaminopyrimidine; WR99210-like) are more effective inhibitors than Pyr.15,23
In this study, we designed hybrid inhibitors through modification of the side chain of a rigid inhibitor at the m- or p-phenyl substituent, so that they carry a rigid and a flexible moiety at either end (Figure 1).
Figure 1.

Designs of inhibitors that can automatically modulate their binding modes to suit the catalytic sites which they encounter. (A) Structures of representative DHFR inhibitors. Pyr and P30 are rigid inhibitors, whereas P65 is a flexible inhibitor. (B) Our design of the dual binding mode DHFR hybrid inhibitors. The rigid part, represented by Pyr derivative, is joined with a flexible diaminopyrimidine moiety. (C) Structures of hybrid inhibitors synthesized in this study.
In our design approach, hybrid compounds were prepared as outlined in Scheme 1. Rigid diaminopyrimidines 2a–b and the flexible diaminopyrimidine 3 were prepared according to methods modified from the previous reports (Scheme 1A).15,24 Mitsunobu reaction between compounds 2a–b and 3 furnished the desired hybrid compounds BT1 and BT2 bearing two DHFR binding sites on opposite ends of the molecule. BT1 consists of the rigid end linked at the m-phenyl substituent to the flexible end, whereas BT2 has the rigid end linked at the p-phenyl substituent to the flexible end. For a hybrid bearing rigid ends on both sides of the molecule with a flexible five-atom linker, a condensation reaction of enol-ether of compound 4 with guanidine was performed to yield the desired hybrid BT3 (Scheme 1B). In this study, compounds BT2S and BT3S, which are hydrochloride salt forms of BT2 and BT3, respectively, were also prepared to give compounds with greater solubility for biological testing.
Scheme 1. Synthesis of Hybrid Inhibitors: (A) Rigid–Flexible Hybrid and (B) Rigid–Rigid Hybrid.

Reagents and conditions: (a) CH2N2, dioxane-MeOH; (b) guanidine, DMSO-MeOH, 90–100 °C; (c) cyclohexene, Pd/C, 65 °C; (d) Ph3P, DIAD, DMF, rt; (e) HCl, H2O.
We determined the inhibition constants, anti-Plasmodial activities, and toxicities against mammalian (Vero) cells of the hybrid inhibitors. Table 1 shows the inhibition constants (Ki) against PfDHFR enzymes of the hybrid inhibitors, together with Pyr, P30, and P65 as comparators. BT1 shows very high affinities for both wild-type and mutant PfDHFRs, with much higher Ki values for human DHFR (hDHFR), giving high Ki ratio (selectivity) value, as compared with P30 and P65. However, BT2 and BT2S show lower affinities for PfDHFRs and hDHFR, giving lower selectivity. In contrast, Pyr shows weak binding with the mutant enzymes, whereas P30 shows stronger binding with both wild-type and mutant enzymes. However, these compounds gave very low selectivity values. Unexpectedly, the bi-rigid BT3S also has high affinities for all PfDHFRs with high selectivity.
Table 1. Inhibition Constants (Ki) of the Compounds against DHFR Enzymes.
|
Ki PfDHFRe (nM, ±
SD) |
|||||||
|---|---|---|---|---|---|---|---|
| compd | wild-type (NCSI) | double mutation (NRNI) | triple mutation 1 (IRNI) | triple mutation 2 (NRNL) | quadruple mutation (QM; IRNL) | Ki hDHFR (nM) | selectivity ratio (Ki h/Ki QM) |
| Pyr | 0.60 ± 0.20a | 53.9 ± 6.5a | 67.1 ± 4.2b | 112 ± 17b | 390 ± 160b | 28.2 ± 2.5c | 0.07 |
| P30 | 0.80 ± 0.10a | 1.40 ± 0.00a | 2.10 ± 0.22b | 2.71 ± 0.43b | 3.30 ± 0.40b | 1.60 ± 0.20c | 0.5 |
| P65 | 0.49 ± 0.10 | 3.15 ± 0.13 | 2.61 ± 0.22 | 4.21 ± 0.23 | 5.59 ± 0.10d | 47.6 ± 4.0 | 8.5 |
| BT1 | 0.44 ± 0.04 | 0.53 ± 0.07 | 0.71 ± 0.14 | 1.25 ± 0.10 | 2.32 ± 0.24 | 22.5 ± 2.9 | 9.7 |
| BT2f | 0.35 ± 0.02 | 1.03 ± 0.12 | 1.00 ± 0.15 | 6.32 ± 0.87 | 6.96 ± 0.91 | 18.1 ± 1.3 | 2.6 |
| BT2Sg | 0.31 ± 0.05 | 1.10 ± 0.19 | 0.74 ± 0.09 | 5.72 ± 0.90 | 5.8 ± 1.5 | 20.2 ± 3.3 | 3.5 |
| BT3Sg | 0.40 ± 0.03 | 1.34 ± 0.23 | 1.35 ± 0.27 | 2.66 ± 0.35 | 4.1 ± 1.2 | 33.6 ± 10.6 | 8.1 |
Table 2 shows the anti-Plasmodial activities (IC50) and toxicities against mammalian (Vero) cells of the inhibitors. As anticipated from the Ki values, BT1 shows good IC50 (<5 μM) against both wild-type and Pyr-resistant mutant P. falciparum. The corresponding IC50 value is higher against Vero cells, which translates to moderate to high selectivity. BT2S shows lower IC50 than BT1 for wild-type; however, it has comparable IC50 against mutant parasite and selectivity to P65, but with lower IC50 than BT1. Interestingly, the bi-rigid inhibitor BT3S shows good IC50 against both wild-type and mutant parasite with better selectivity than BT1 and BT2S. In contrast, P30 and P65 show markedly lower selectivity owing to either poor inhibition of mutant parasite strains or similar inhibition of parasites and Vero mammalian cells.
Table 2. Anti-Plasmodial Activity (IC50) against P. falciparum Carrying Wild-Type or Mutant DHFR and Cytotoxicity against Mammalian (Vero) Cells of the Compounds.
| IC50P. falciparume (μM,
± SD) |
|||||||
|---|---|---|---|---|---|---|---|
| compd | TM4/8.2 (NCSI) | K1CB1 (NRNI) | W2 (IRNI) | CSL-2 (NRNL) | V1/S (IRNL) | cytotoxicity Vero cells (μM) | selectivity ratio (Vero/V1/S) |
| Pyr | 0.08 ± 0.01a | 30.9 ± 8.4a | 73.5 ± 8.5b | 42 ± 15b | >100b | >100 | NAc |
| P30 | 0.42 ± 0.10a | 1.31 ± 0.45a | 3.94 ± 0.80b | 2.53 ± 0.38b | 9.1 ± 2.8b | 7a | 0.8 |
| P65 | 0.23 ± 0.07d | 1.92 ± 0.53 | 2.27 ± 0.65 | 4.4 ± 2.1 | 3.5 ± 1.6d | 4.03 ± 0.41 | 1.2 |
| BT1 | 0.29 ± 0.05 | 0.22 ± 0.06 | 0.25 ± 0.05 | 0.51 ± 0.08 | 1.72 ± 0.69 | 5.94 ± 0.65 | 3.4 |
| BT2Sf | 0.063 ± 0.014 | 1.84 ± 0.31 | 2.35 ± 0.10 | 3.95 ± 0.36 | 4.40 ± 0.71 | 6.3 ± 1.5 | 1.4 |
| BT3Sf | 0.035 ± 0.0033 | 0.33 ± 0.038 | 0.30 ± 0.03 | 0.31 ± 0.02 | 0.33 ± 0.023 | 2.53 ± 0.44 | 7.7 |
We assessed the potential of PfDHFR variants conferring resistance to BT1 using a bacterial surrogate system. From a library of 1.5 × 105 PfDHFR variants, BT1-resistant cells were obtained. Fourteen independent BT1-resistant colonies were characterized by DNA sequencing, which showed novel resistance mutations K97N, S108T, and E199V in addition to the Pyr-resistant mutations N51I, C59R, and I164L. However, all BT1-resistant mutants shared the same haplotype (combination of mutations) of IRNTLV, in contrast with the wild-type NCKSIE and quadruple mutant IRKNLE haplotypes. From these data, we infer that the diversity of BT1-resistant PfDHFR variants is much lower than that of variants resistant to rigid or flexible antifolates, each with 12 different haplotypes.22 However, the diversity of PfDHFR resistance mutations in parasites could be affected by other factors, such as GTP cyclohydrolase gene copy number,27 which are not modeled in the bacterial surrogate.
Cocrystal structures of the hybrid inhibitors with the enzymes were investigated. According to the binding modes of wild-type and mutant PfDHFRs with rigid and flexible pyrimidine inhibitors,16 the rigid end of BT1 with a m-phenyl substituent on the pyrimidine nucleus would be expected to bind preferentially to the active site pocket containing S108 of the wild-type PfDHFR with no steric hindrance. However, it was possible that the bulky p-phenyl substituent of BT2 might encounter steric clash even with S108, and BT2 might preferentially bind to the wild-type active site with the flexible end. However, the flexible ends of both BT1 and BT2 should bind preferentially to the active site of mutant PfDHFR containing the bulky S108N. Notably, BT3 can only bind to the active sites of wild-type and mutant PfDHFR in the same way. To investigate the effects and the interaction mode of the three inhibitors to PfDHFR enzyme, we determined BT1, BT2, and BT3 cocrystal structures of PfDHFR-TS, both wild-type and quadruple mutant DHFR domains at 2.2–2.38 and 2.38–2.6 Å resolutions, respectively (Figure 2 and Table S1 in the Supporting Information). Moreover, BT1 and BT2 cocrystal structures with hDHFR were determined in order to visualize the difference in the binding modes of the hybrid inhibitors (see details in the Supporting Information).
Figure 2.

Inhibition modes of hybrid inhibitors in PfDHFR. Detailed interactions of (A) BT1, (B) BT2, and (C) BT3 with wild-type PfDHFR, quadruple mutant PfDHFR, and superposition of wild-type and quadruple mutant PfDHFRs. Displacement of NADPH was observed in quadruple mutant due to steric clash with S108N upon binding of BT3. Inhibitors and key amino acids are shown as a stick model. Important interactions are highlighted as dashed lines in black, and π–π interaction is drawn as a double dashed line.
As expected, BT1 showed distinctive binding modes between wild-type and quadruple mutant PfDHFRs (Figure 2A). It binds to the wild-type enzyme with the rigid end in the active site, through the pyrimidine moiety interacting with D54 and the rigid phenyl in the vicinity of S108. In contrast, the flexible end of BT1 binds to the quadruple mutant enzyme at D54, with the flexible alkoxy group avoiding steric clash with S108N in close proximity (3.4 Å). The nicotinamide moiety of NADPH was not perturbed by binding of BT1 and the pyrimidine core of the rigid end formed π–π interaction with F116. BT1 with m-phenyl substitution is therefore a potentially good template for inhibitors, which would forestall further resistance mutations in both the S108 and 108N series.
Interestingly, in contrast to the BT1 structures, BT2 binds to both wild-type and quadruple mutant PfDHFR with its flexible ends (Figure 2B). An explanation for this finding is that in the wild-type enzyme, although there is no steric clash of the rigid end at S108, the p-substituted phenyl moiety would collide with the hydrophobic pocket of residues 111–116 if the rigid end of BT2 was docked at 54. For the quadruple mutant enzyme, the flexible end can avoid the steric clash with S108N and is preferred to the rigid end at D54 site, while the rigid end of the molecule caused some conformational adjustment of the enzyme (Figure 2B).
As for compound BT3, comprising rigid moieties on both ends, its good binding to quadruple mutant as well as wild-type PfDHFR was at first surprising. However, the high affinity binding can be explained by the X-ray structures of BT3 complexed with both wild-type and quadruple mutant PfDHFR, which showed well adapted accommodation of the active sites to BT3 (Figure 2C). The side chain of S108N greatly swung away from the active pocket to make room for accommodation of BT3 as compared to BT1 and BT2. Interestingly, the binding geometry of BT3 is very similar for both wild-type and quadruple mutant PfDHFR; however, in the PfDHFR quadruple mutant, BT3 binding causes a significant displacement of the nicotinamide moiety, resulting from a severe steric clash (Figure 3). From these data, we infer that the enhanced inhibition of BT3 in PfDHFR quadruple mutant occurs through displacement of NADPH cofactor, which is insignificant in the wild-type enzyme. A high temperature factor of NADPH was observed in monomer A of quadruple mutant PfDHFR, whereas in monomer B NADPH could not be fully presented owing to very weak electron density. It is noteworthy that no or very little NADPH perturbation was observed where the flexible part of the compounds was docked at D54. Therefore, the displacement of the nicotinamide ring of NADPH is important for inhibition of the PfDHFR quadruple mutant by BT3.
Figure 3.
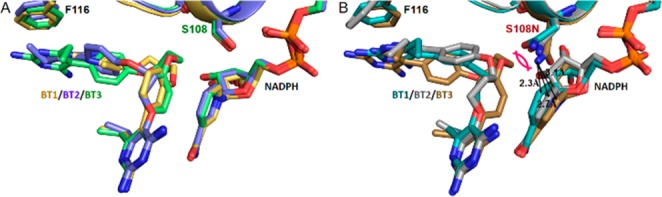
Superposition of cocomplex BT1, BT2, and BT3 structures of PfDHFRs. (A) Wild-type PfDHFR. (B) Quadruple mutant PfDHFR. The nicotinamide ring of NADPH was perturbed by the binding of the hybrid inhibitors to different levels in quadruple mutant PfDHFR structures, while insignificant displacement of NADPH occurred in wild-type. The most severe perturbation of the NADPH was observed with the binding of BT3, which coincided with the displacement of S108N side chain.
In conclusion, hybrid compounds such as BT1 with rigid and flexible pharmacophores in the same molecule can bind with high affinity to either wild-type/S108 series or quadruple mutant/108N series PfDHFR in the expected manner, consequently with favorable IC50 and selectivity. Significantly, the diversity of mutants resistant to BT1 is extremely limited, in accordance with our prediction from our earlier finding22 that there are conflicting requirements for resistance mutations against rigid and flexible inhibitors that tightly constrain resistance mutations against hybrid compounds. It is noted that another hybrid compound, BT2, binds both wild-type and mutant PfDHFR with the flexible end. Yet another compound, BT3, which is rigid at both ends, binds both wild-type and mutant PfDHFR with good affinities. These apparent anomalies are, however, fully explained by the X-ray structures of the complexes, which show conformational changes of the enzyme to accommodate the inhibitor. The high antimalarial activity of BT1 and, importantly, its ability to forestall resistance mutations, makes it a good template for further design and synthesis of antifolates, which are less vulnerable to resistance. This family of rigid-flexible hybrid compounds can be added to the list of new antifolates such as P218 with good activity against pyrimethamine-resistant P. falciparum.15
Acknowledgments
We thank the experimental facility and the technical services at beamline 13B1 provided by the “Synchrotron Radiation Protein Crystallography Facility of the National Core Facility Program for Biotechnology, Ministry of Science and Technology” and the “National Synchrotron Radiation Research Center”, a national user facility supported by the Ministry of Science and Technology, Taiwan, ROC. We gratefully acknowledge the Synchrotron Light Research Institute, Thailand for the experimental facility and technical services at beamline 7.2W.
Glossary
ABBREVIATIONS
- PfDHFR
Plasmodium falciparum dihydrofolate reductase
- hDHFR
human dihydrofolate reductase
- PfDHFR-TS
Plasmodium falciparum dihydrofolate reductase-thymidylate synthase
- Pyr
pyrimethamine
- DIAD
diisopropyl azodicarboxylate
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00389.
Result of cocrystal structures of hybrid inhibitors with hDHFR; materials and methods; Figure S1 and Tables S1 and S2 (PDF)
Accession Codes
The atomic coordinates and structure factor amplitudes have been deposited in the Protein Data Bank: PfDHFR-TS with accession codes 6A2K (TM4-BT1), 6A2L (V1/S-BT1), 6A2M (TM4-BT2), 6A2N (V1/S-BT2), 6A2O (TM4-BT3), and 6A2P (V1/S-BT3), and hDHFR with accession codes 6A7C (BT1) and 6A7E (BT2).
Author Present Address
† Faculty of Medical Technology, Rangsit University, Pathumthani 12000, Thailand.
Author Contributions
‡ These authors contributed equally. The manuscript was written by Y.Y., B.T., P.C., P.J.S., J.V., and S.K. with contributions of all authors.
We gratefully acknowledge the financial support from Grand Challenges Explorations—The Bill & Melinda Gates Foundation (Grant ID # 52992). Protein crystallography was financially supported by grants from Cluster Program and Management Office, National Science and Technology Development Agency, Thailand (CPMO-P-13-00835 and P-14-50883), and Synchrotron Light Research Institute, Thailand (P-10-10168).
The authors declare no competing financial interest.
Supplementary Material
References
- Diagana T. T. Supporting malaria elimination with 21st century antimalarial agent drug discovery. Drug Discovery Today 2015, 20, 1265–1270. 10.1016/j.drudis.2015.06.009. [DOI] [PubMed] [Google Scholar]
- Rabinovich R. N.; Drakeley C.; Djimde A. A.; Hall B. F.; Hay S. I.; Hemingway J.; Kaslow D. C.; Noor A.; Okumu F.; Steketee R.; Tanner M.; Wells T. N. C.; Whittaker M. A.; Winzeler E. A.; Wirth D. F.; Whitfield K.; Alonso P. L. malERA: An updated research agenda for malaria elimination and eradication. PLoS Med. 2017, 14, e1002456. 10.1371/journal.pmed.1002456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okombo J.; Chibale K. Recent updates in the discovery and development of novel antimalarial drug candidates. MedChemComm 2018, 9, 437–453. 10.1039/C7MD00637C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooft van Huijsduijnen R.; Wells T. N. The antimalarial pipeline. Curr. Opin. Pharmacol. 2018, 42, 1–6. 10.1016/j.coph.2018.05.006. [DOI] [PubMed] [Google Scholar]
- Imwong M.; Suwannasin K.; Kunasol C.; Sutawong K.; Mayxay M.; Rekol H.; Smithuis F. M.; Hlaing T. M.; Tun K. M.; van der Pluijm R. W.; Tripura R.; Miotto O.; Menard D.; Dhorda M.; Day N. P. J.; White N. J.; Dondorp A. M. The spread of artemisinin-resistant Plasmodium falciparum in the Greater Mekong subregion: a molecular epidemiology observational study. Lancet Infect. Dis. 2017, 17, 491–497. 10.1016/S1473-3099(17)30048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straimer J.; Gnadig N. F.; Witkowski B.; Amaratunga C.; Duru V.; Ramadani A. P.; Dacheux M.; Khim N.; Zhang L.; Lam S.; Gregory P. D.; Urnov F. D.; Mercereau-Puijalon O.; Benoit-Vical F.; Fairhurst R. M.; Menard D.; Fidock D. A. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 2015, 347, 428–431. 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- malERA: An updated research agenda for insecticide and drug resistance in malaria elimination and eradication. PLoS Med. 2017, 14, e1002450. 10.1371/journal.pmed.1002450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodrow C. J.; Dahlstrom S.; Cooksey R.; Flegg J. A.; Le Nagard H.; Mentre F.; Murillo C.; Menard D.; Nosten F.; Sriprawat K.; Musset L.; Quashie N. B.; Lim P.; Fairhurst R. M.; Nsobya S. L.; Sinou V.; Noedl H.; Pradines B.; Johnson J. D.; Guerin P. J.; Sibley C. H.; Le Bras J. High-throughput analysis of antimalarial susceptibility data by the WorldWide Antimalarial Resistance Network (WWARN) in vitro analysis and reporting tool. Antimicrob. Agents Chemother. 2013, 57, 3121–3130. 10.1128/AAC.02350-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey V. C.; Lukens A. K.; Istvan E. S.; Lee M. C.; Franco V.; Magistrado P.; Coburn-Flynn O.; Sakata-Kato T.; Fuchs O.; Gnadig N. F.; Goldgof G.; Linares M.; Gomez-Lorenzo M. G.; De Cozar C.; Lafuente-Monasterio M. J.; Prats S.; Meister S.; Tanaseichuk O.; Wree M.; Zhou Y.; Willis P. A.; Gamo F. J.; Goldberg D. E.; Fidock D. A.; Wirth D. F.; Winzeler E. A. A broad analysis of resistance development in the malaria parasite. Nat. Commun. 2016, 7, 11901. 10.1038/ncomms11901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregson A.; Plowe C. V. Mechanisms of resistance of malaria parasites to antifolates. Pharmacol. Rev. 2005, 57, 117–145. 10.1124/pr.57.1.4. [DOI] [PubMed] [Google Scholar]
- Venkatesan M.; Alifrangis M.; Roper C.; Plowe C. V. Monitoring antifolate resistance in intermittent preventive therapy for malaria. Trends Parasitol. 2013, 29, 497–504. 10.1016/j.pt.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nzila A. The past, present and future of antifolates in the treatment of Plasmodium falciparum infection. J. Antimicrob. Chemother. 2006, 57, 1043–1054. 10.1093/jac/dkl104. [DOI] [PubMed] [Google Scholar]
- Yuthavong Y.; Kamchonwongpaisan S.; Leartsakulpanich U.; Chitnumsub P. Folate metabolism as a source of molecular targets for antimalarials. Future Microbiol. 2006, 1, 113–125. 10.2217/17460913.1.1.113. [DOI] [PubMed] [Google Scholar]
- Yuthavong Y.; Yuvaniyama J.; Chitnumsub P.; Vanichtanankul J.; Chusacultanachai S.; Tarnchompoo B.; Vilaivan T.; Kamchonwongpaisan S. Malarial (Plasmodium falciparum) dihydrofolate reductase-thymidylate synthase: structural basis for antifolate resistance and development of effective inhibitors. Parasitology 2015, 130, 249–259. 10.1017/S003118200400664X. [DOI] [PubMed] [Google Scholar]
- Yuthavong Y.; Tarnchompoo B.; Vilaivan T.; Chitnumsub P.; Kamchonwongpaisan S.; Charman S. A.; McLennan D. N.; White K. L.; Vivas L.; Bongard E.; Thongphanchang C.; Taweechai S.; Vanichtanankul J.; Rattanajak R.; Arwon U.; Fantauzzi P.; Yuvaniyama J.; Charman W. N.; Matthews D. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 16823–16828. 10.1073/pnas.1204556109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuvaniyama J.; Chitnumsub P.; Kamchonwongpaisan S.; Vanichtanankul J.; Sirawaraporn W.; Taylor P.; Walkinshaw M. D.; Yuthavong Y. Insights into antifolate resistance from malarial DHFR-TS structures. Nat. Struct. Mol. Biol. 2003, 10, 357–365. 10.1038/nsb921. [DOI] [PubMed] [Google Scholar]
- Kongsaeree P.; Khongsuk P.; Leartsakulpanich U.; Chitnumsub P.; Tarnchompoo B.; Walkinshaw M. D.; Yuthavong Y. Crystal structure of dihydrofolate reductase from Plasmodium vivax: pyrimethamine displacement linked with mutation-induced resistance. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 13046–13051. 10.1073/pnas.0501747102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canfield C. J.; Milhous W. K.; Ager A. L.; Rossan R. N.; Sweeney T. R.; Lewis N. J.; Jacobus D. P. PS-15: a potent, orally active antimalarial from a new class of folic acid antagonists. Am. J. Trop. Med. Hyg. 1993, 49, 121–126. 10.4269/ajtmh.1993.49.121. [DOI] [PubMed] [Google Scholar]
- Yuthavong Y.; Vilaivan T.; Kamchonwongpaisan S.; Tarnchompoo B.; Thongpanchang C.; Chitnumsub P.; Yuvaniyama J.; Matthews D.; Charman W.; Charman S.; Vivas L.; Katiyar S. B.. Antimalarial Compounds with Flexible Side-Chains. U.S. Patent 8,503,491. September 10, 2013.
- Ferlan J. T.; Mookherjee S.; Okezie I. N.; Fulgence L.; Sibley C. H. Mutagenesis of dihydrofolate reductase from Plasmodium falciparum: analysis in Saccharomyces cerevisiae of triple mutant alleles resistant to pyrimethamine or WR99210. Mol. Biochem. Parasitol. 2001, 113, 139–150. 10.1016/S0166-6851(01)00207-9. [DOI] [PubMed] [Google Scholar]
- Chusacultanachai S.; Thiensathit P.; Tarnchompoo B.; Sirawaraporn W.; Yuthavong Y. Novel antifolate resistant mutations of Plasmodium falciparum dihydrofolate reductase selected in Escherichia coli. Mol. Biochem. Parasitol. 2002, 120, 61–72. 10.1016/S0166-6851(01)00440-6. [DOI] [PubMed] [Google Scholar]
- Japrung D.; Leartsakulpanich U.; Chusacultanachai S.; Yuthavong Y. Conflicting requirements of Plasmodium falciparum dihydrofolate reductase mutations conferring resistance to pyrimethamine-WR99210 combination. Antimicrob. Agents Chemother. 2007, 51, 4356–4360. 10.1128/AAC.00577-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnchompoo B.; Sirichaiwat C.; Phupong W.; Intaraudom C.; Sirawaraporn W.; Kamchonwongpaisan S.; Vanichtanankul J.; Thebtaranonth Y.; Yuthavong Y. Development of 2,4-diaminopyrimidines as antimalarials based on inhibition of the S108N and C59R+S108N mutants of dihydrofolate reductase from pyrimethamine-resistant Plasmodium falciparum. J. Med. Chem. 2002, 45, 1244–1252. 10.1021/jm010131q. [DOI] [PubMed] [Google Scholar]
- Holsworth D. D.; Jeremy M. S.; Edmunds J.; He W.; Place S.; Maiti S. An Expeditious Synthesis of 6-Alkyl-5-(4′-amino-phenyl)-pyrimidine-2,4-diamines. Synth. Commun. 2003, 33, 3467–3475. 10.1081/SCC-120024725. [DOI] [Google Scholar]
- Kamchonwongpaisan S.; Quarrell R.; Charoensetakul N.; Ponsinet R.; Vilaivan T.; Vanichtanankul J.; Tarnchompoo B.; Sirawaraporn W.; Lowe G.; Yuthavong Y. Inhibitors of multiple mutants of Plasmodium falciparum dihydrofolate reductase and their antimalarial activities. J. Med. Chem. 2004, 47, 673–680. 10.1021/jm030165t. [DOI] [PubMed] [Google Scholar]
- Kamchonwongpaisan S.; Vanichtanankul J.; Tarnchompoo B.; Yuvaniyama J.; Taweechai S.; Yuthavong Y. Stoichiometric selection of tight-binding inhibitors by wild-type and mutant forms of malarial (Plasmodium falciparum) dihydrofolate reductase. Anal. Chem. 2005, 77, 1222–1227. 10.1021/ac0487597. [DOI] [PubMed] [Google Scholar]
- Heinberg A.; Kirkman L. The molecular basis of antifolate resistance in Plasmodium falciparum: looking beyond point mutations. Ann. N. Y. Acad. Sci. 2015, 1342, 10–18. 10.1111/nyas.12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.