

Abstract
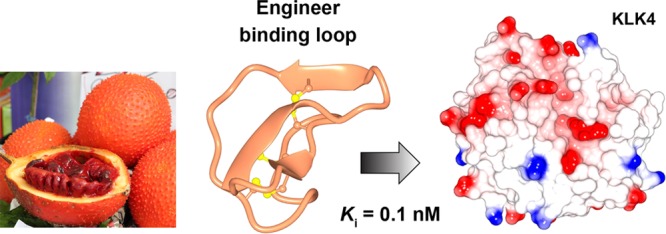
Kallikrein-related peptidase 4 (KLK4) is a serine protease that has putative intracellular and extracellular functions in prostate cancer progression. Here we show that MCoTI-II, a 34-amino acid cyclic peptide found in the seeds of red gac (Momordica cochinchinensis), is an inhibitor of KLK4. By grafting a preferred KLK4 cleavage sequence into MCoTI-II, we produced a highly potent KLK4 inhibitor (Ki = 0.1 nM) that displayed 100,000-fold selectivity over related KLKs and the ability to penetrate cells. Additionally, by substituting positively charged noncontact residues in this compound, we produced a potent and selective KLK4 inhibitor that does not penetrate cells. The inhibitors were shown to be nontoxic to human cells and stable in human serum. These KLK4 inhibitors provide useful chemical tools to further define the role(s) of both intracellular and extracellular KLK4 in prostate cancer cell lines and disease models.
Keywords: Prostate cancer, Cell penetrating peptide, Momordica cochinchinensis trypsin inhibitor-II, Kallikrein-related peptidase 4
The kallikrein-related peptidase (KLK) family comprises 15 serine proteases that have tissue-specific, hormone-regulated expression profiles and are implicated in a range of diseases, including cancers.1 The most well-known KLK is prostate-specific antigen (KLK3), which has been utilized as a serum biomarker of prostate cancer progression for decades.2 Another member of the family, KLK4, is overexpressed in prostate cancer,3−6 and ectopic expression of KLK4 in prostate cancer cell lines is associated with increased cell motility5,7 and proliferation.5,8 KLK4 can cleave extracellular proteins that modulate cytoskeletal organization, extracellular matrix adhesion, and cell migration, resulting in an epithelial to mesenchymal transition that may facilitate prostate cancer metastasis to bone.7,9 KLK4 and the protease-activated receptors (PARs) 1 and 2 colocalize in prostate cancer primary tumors and bone metastases, and KLK4 can activate PAR-1 and PAR-2 in prostate cancer cell lines.6,10,11 These findings indicate that inhibiting KLK4 might be an approach for developing new prostate cancer therapies.
In a previous study we used the 14-amino acid cyclic peptide sunflower trypsin inhibitor-1 (SFTI-1) to design a potent and selective KLK4 inhibitor.12−14 SFTI-1 is a reversible inhibitor of several serine proteases from the S1 family, including trypsin,15,16 KLKs,17 matriptase,18 and cathepsin G,19 and has been used to develop engineered inhibitors for a range of protease targets. To design the KLK4 inhibitor based on SFTI-1, we screened KLK4 against a noncombinatorial peptide substrate library to identify a preferred P4–P1 cleavage sequence (FVQR) and substituted this sequence into the contact β-strand of SFTI-1 (Figure 1).12 The resulting peptide (SFTI-FCQR) potently inhibited KLK4 and blocked KLK4-mediated PAR-2 activation in cell-based assays.12
Figure 1.
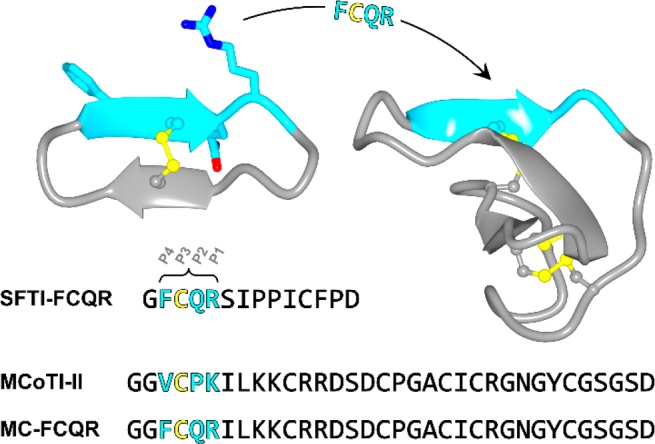
Strategy for designing KLK4 inhibitors based on MCoTI-II. Ribbon diagram of SFTI-FCQR (left, PDB ID 4K1E) and MCoTI-II (right, PDB ID 4GUX) with disulfide bonds shown in ball and stick representation. The modified residues in SFTI-FCQR are shown in stick model (P4 Phe, P2 Gln and P1 Arg) with carbon atoms in cyan. The P4–P1 segment of MCoTI-II (cyan) has the same structure as SFTI-FCQR, allowing substitution of the FCQR sequence into MCoTI-II to generate the KLK4 inhibitor MC-FCQR. The sequence of each inhibitor is shown in single letter amino acid code (colored to match the ribbon diagram).
Although KLKs are mostly associated with extracellular functions, KLK4 has also been reported to be present in the cytoplasm and nucleus in prostate cancer primary tumors, bone metastases, and cell lines.3,4,6,8,9 Thus, it has been proposed that KLK4 has both intracellular and extracellular prostate cancer-promoting functions. For example, intracellular KLK4 has been reported to be involved in activating the mammalian target of rapamycin (mTOR) pathway in prostate cancer cells.8 Activation of the mTOR pathway occurs in many cancers, including prostate cancer.20 Additionally, siRNA knockdown of intracellular KLK4 in human prostate tumor xenografts was shown to reduce tumor growth in vivo.8 However, whether the putative functions of intracellular KLK4 relate to proteolytic activity remains a subject of debate. Indeed, a KLK4 transcript variant that displays nuclear localization lacks exon 1,4 which encodes the first 19-residues of the mature enzyme (Ile16–Val34, chymotrypsin numbering). Additionally, while full-length KLK4 has been reported to show cytoplasmic localization,4 a KLK4 mutant where the catalytic Ser was substituted to Ala (S195A) was also capable of modulating intracellular signaling pathways.8 These findings suggest that inhibitors with cell penetrating properties may be useful for further exploring the intracellular functions of KLK4.
Momordica cochinchinensis trypsin inhibitor-II (MCoTI-II) is a member of the squash family of serine protease inhibitors and has been shown to internalize into cells without disrupting the cell membrane.21,22 MCoTI-II is a 34-amino acid backbone cyclic peptide with three disulfide bonds23,24 (Figure 1). It enters cells via macropinocytosis, with mutagenesis studies showing that this process is influenced by a surface patch of positively charged residues on MCoTI-II.20 MCoTI-II also has exceptional thermal, chemical, and enzymatic stability, making it an attractive scaffold for drug design.25−27 Engineered inhibitors based on MCoTI-II have been developed for a range of serine proteases, including β-tryptase,28,29 elastase,29 matriptase,18,30 and factor XIIa (FXIIa).31 Additionally, MCoTI-variants have been used to design cell penetrating peptides that target intracellular pathways implicated in cancer.32 Collectively, these findings indicate that MCoTI-II is a potential template for designing inhibitors that can target intracellular KLK4.
Screening MCoTI-II against KLK proteases revealed that it was a potent inhibitor of KLK4 (Ki = 1.6 nM), but not the closely related proteases KLK5, KLK7, and KLK14 (Table 1). Previously, we identified that FVQR is a preferred KLK4 cleavage sequence that can be used for inhibitor design by substituting the P1 Arg, P2 Gln, and P4 Phe residues into the binding loop of SFTI-1.12 The trypsin binding loop of MCoTI-II has the same structure as SFTI-1 across the P1–P4 residues, as revealed by NMR spectroscopy (in solution)23,33 and X-crystallography (in complex with trypsin).15,34 Therefore, we hypothesized that the P1, P2, and P4 residues of MCoTI-II could be modified to produce a novel inhibitor of KLK4. The trypsin binding loop of MCoTI-II has a different sequence to SFTI-1 and includes a Pro residue at P2 (Figure 1). Therefore, when designing MCoTI-II variants based on the FVQR sequence, we substituted each residue individually to generate compounds MC-K6R (P1 Arg), MC-P5Q (P2 Gln), and MC-V3F (P4 Phe). Each variant showed an increase in activity against KLK4, with the most potent variant being MC-P5Q (12-fold improved activity). This result shows that the conserved P2 Pro in the squash inhibitor family is not essential for activity and can be substituted during the design process, as we demonstrated previously for FXIIa inhibitors based on MCoTI-II.31 Combining all three substitutions in one variant produced an inhibitor (MC-FCQR) that was slightly more potent against KLK4 (Ki = 0.1 nM) and displayed more than 100,000-fold selectivity over KLK5, KLK7, KLK14, plasmin, FXIIa, thrombin, and plasma kallikrein. MC-FCQR also showed 18,000-fold weaker activity against trypsin compared to MCoTI-II and 5,000-fold selectivity over factor Xa.
Table 1. Inhibitor Sequences and Inhibition Constants (Ki).
| Inhibitor | Sequencea | Protease | Ki (nM) |
|---|---|---|---|
| MCoTI-II | GGVCPKILKKCRRDSDCPGACICRGNGYCGSGSD | KLK4 | 1.6 ± 0.1 |
| Plasmin | 28 ± 631 | ||
| Trypsin | 0.0023 ± 0.000718 | ||
| FXIIa | 750 ± 8031 | ||
| Other proteasesb | >10,000 | ||
| MC-K6R | GGVCPRILKKCRRDSDCPGACICRGNGYCGSGSD | KLK4 | 0.30 ± 0.1 |
| MC-P5Q | GGVCQKILKKCRRDSDCPGACICRGNGYCGSGSD | KLK4 | 0.13 ± 0.08 |
| MC-V3F | GGFCPKILKKCRRDSDCPGACICRGNGYCGSGSD | KLK4 | 1.3 ± 0.4 |
| MC-FCQR | GGFCQRILKKCRRDSDCPGACICRGNGYCGSGSD | KLK4 | 0.10 ± 0.03 |
| Trypsin | 31 ± 3 | ||
| FXa | 500 ± 10 | ||
| Other proteasesc | >10,000 | ||
| MC-FCQR-QK | GGFCQRILQQCQQDSDCPGACICKGNGYCGSGSD | KLK4 | 0.44 ± 0.15 |
| Trypsin | 6.9 ± 0.7 | ||
| FXa | 520 ± 10 | ||
| Other proteasesc | >10,000 | ||
| MC-K6R-QK | GGVCPRILQQCQQDSDCPGACICKGNGYCGSGSD | KLK4 | 1.8 ± 0.5 |
All peptides have a cyclic backbone with disulfide bonds between C4–C21, C11–C23, and C17–C28. Substituted residues are shown in bold, and Lys residues used for Alexa Fluor 488 labeling are underlined.
KLK5, KLK7, KLK14, FXa, thrombin, and plasma kallikrein.
KLK5, KLK7, KLK14, plasmin, FXIIa, thrombin, and plasma kallikrein.
A model of the KLK4/MC-FCQR complex was created using the KLK4/SFTI-FCQR (PDB ID: 4K1E) and trypsin/MCoTI-II (PDB ID: 4GUX) structures to explore the high potency and specificity of MC-FCQR. The KLK4/MC-FCQR model was subjected to molecular dynamics simulations, and the most prevalent conformation is shown in Figure 2. The model suggested that the modified P4–P1 sequence FCQR bound to KLK4 in an almost identical manner to the corresponding sequence in the structure of the engineered SFTI-variant in complex with KLK4 (Figure S1, RMSD of 0.6 Å across all P4–P1 atoms).14 Favorable interactions were also identified outside of the P4–P1 segment. For example, MC-FCQR residues Lys10 (P4′) and Tyr28 appeared to interact with Glu38 of KLK4 via a salt bridge and hydrogen bond, respectively. This interaction highlights one of the advantages of using MCoTI-II as a design scaffold as it contains several binding loops that are able to engage different regions of the active site cleft and can be targeted in engineering studies.
Figure 2.
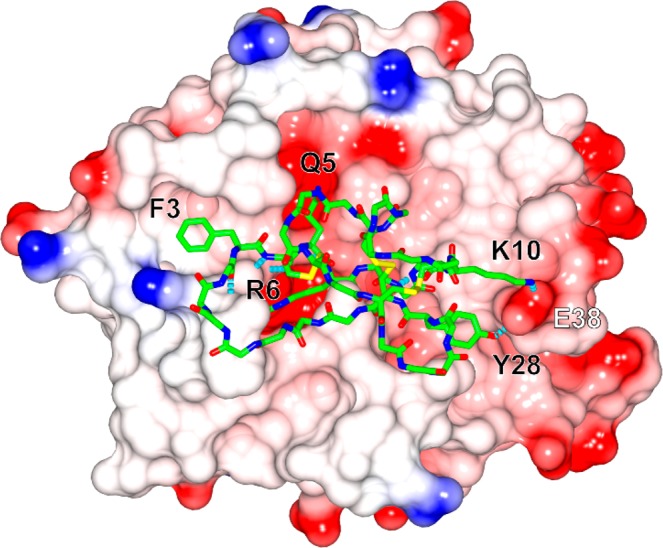
Model of the KLK4/MC-FCQR complex. MC-FCQR is displayed in stick model (backbone atoms) with selected side chains shown (carbon: green, nitrogen: blue, oxygen: red, sulfur: yellow) on the electrostatic surface of KLK4 (blue: positive, red: negative). Selected hydrogen bonds are shown as dashed lines (cyan). Residues in MC-FCQR and KLK4 are labeled in black and white, respectively.
Since KLK4 has been reported to have intracellular cancer-promoting functions,5,8 we next evaluated the cell internalization efficiencies of MCoTI-II and MC-FCQR using HeLa cells. These experiments were performed using Alexa Fluor 488 conjugated inhibitors to allow measurement of the level of internalized peptide. The fluorescence signal of HeLa cells treated with peptide (4 μM) was read before and after addition of trypan blue (a fluorescence quencher that cannot cross cell membranes). Accordingly, this experiment can distinguish between a cell penetrating peptide and a peptide that binds to the membrane without being internalized but does not reveal the peptide’s intracellular localization (whether it has escaped from endosomes). The established cell penetrating peptide HIV-Tat and its poly-Gly variant HIV-Tat-G were used as positive and negative controls, respectively. MCoTI-II internalized into cells with 14% of the efficiency of HIV-Tat compared to 23% for MC-FCQR, with this difference being statistically significant (Figure 3). The increase in cellular uptake observed for MC-FCQR may be due to the V3F and K6R substitutions promoting increased membrane binding.
Figure 3.

Peptide internalization into HeLa cells quantified by flow cytometry. The relative mean fluorescence of MCoTI-II and MCoTI-variants (4 μM) was measured before (white bars) and after (black bars) the addition of trypan blue and was normalized to the positive control HIV-TAT peptide (post-trypan blue). Statistical significance was determined using one-way ANOVA with Tukey’s post hoc test. Three independent experiments were performed, and error bars represent the standard deviation. * = p-value <0.05, ** = p-value <0.005, **** = p-value <0.00005, and ns = not significant.
Considering that KLK4 also has extracellular functions, we next focused on engineering a similarly potent KLK4 inhibitor based on MCoTI-II that is not taken up by cells. Since cellular uptake of MCoTI-II is influenced by its positive charge,21,22 we substituted Lys9, Lys10, Arg12, and Arg13 with Gln in MC-K6R and MC-FCQR to produce MC-K6R-QK and MC-FCQR-QK, respectively. Testing these inhibitors in cell internalization assays revealed that neither of these inhibitors showed detectable levels of cellular uptake (Figure 3). In enzyme inhibition assays, MC-FCQR-QK retained potent activity against KLK4, although the level of activity was 4-fold weaker compared to MC-FCQR. MC-K6R-QK also showed weaker activity compared to MC-K6R (Table 1).
For these KLK4 inhibitors to be useful as tools in experiments with cancer cells, they need to be both nontoxic to cells and stable in a serum environment. The toxicity and stability of selected peptides were evaluated using HeLa cells and human serum, respectively. As previously reported for MCoTI-II, none of the peptides tested were toxic to HeLa cells at concentrations up to 64 μM (Figure 4A). The stability of MC-FCQR and MC-FCQR-QK was investigated in human serum and, as for native MCoTI-II, these variants showed high stability in human serum for at least 24 h (Figure 4B).
Figure 4.
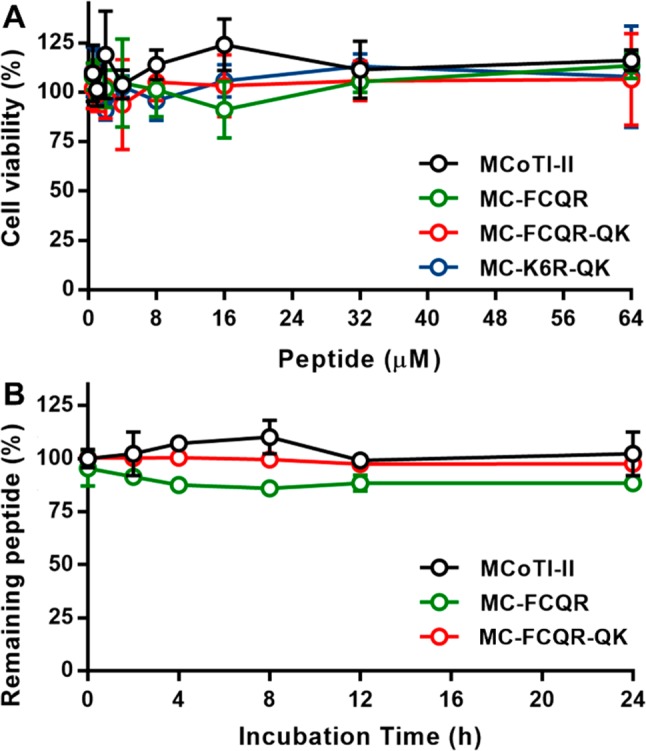
Cellular toxicity and serum stability of MCoTI-II variants. (A) HeLa cells were incubated with MCoTI-II analogues for 2 h prior to incubation with resazurin for 18 h at 37 °C. The absorbance of the metabolite product resorufin was read on a microplate spectrophotometer at 540 and 620 nm. The cell viability percentage was quantified as the absorbance ratio (A620 nm/A540 nm). (B) Serum stability of selected MCoTI-II variants. The stability was calculated as a percentage of the area under the curve for the peptide peak in serum (detected by analytical HPLC) at each time point compared to the peptide peak in PBS at 0 h. The error bars represent standard deviation values from three independent experiments.
In conclusion, this study presents two promising lead compounds that have been designed to target KLK4 in either the intracellular (MC-FCQR) or extracellular (MC-FCQR-QK) space. Both compounds are highly potent, and selective inhibitors of KLK4, show no toxicity to HeLa cells and are highly stable in human serum. One potential drawback is that both MCoTI-variants show slight off-target activity against FXa. However, this study has focused predominantly on the P1–P4 residues of MCoTI-II and the scaffold has additional contact residues that can be engineered to improve the inhibitor’s activity and selectivity in future studies. Nonetheless, these inhibitors are suitable for further experiments using human prostate cancer cell lines, or in vivo prostate cancer models, to further identify the potential roles of KLK4 in cancer progression.
Acknowledgments
We thank Peta Harvey for assistance with NMR spectroscopy experiments as well as Olivier Cheneval and Joachim Weidmann for peptide synthesis support.
Glossary
Abbreviations
- FXa
activated factor X
- FXIIa
activated factor XII
- KLK
kallikrein-related peptidase
- MCoTI-II
Momordica cochinchinensis trypsin inhibitor-II
- mTOR
mammalian target of rapamycin
- SFTI-1
sunflower trypsin inhibitor-1
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00422.
Experimental procedures, compound characterization, molecular modeling, and assay conditions (PDF)
This study was funded in part by a grant from the National Health and Medical Research Council (NHMRC) [GNT1084965]. J.E.S. and S.J.D. were supported by NHMRC Early Career Fellowships [GNT1069819 and GNT1120066], and D.J.C. is an Australian Research Council Laureate Fellow [FL150100146].
The authors declare no competing financial interest.
Supplementary Material
References
- Prassas I.; Eissa A.; Poda G.; Diamandis E. P. Unleashing the Therapeutic Potential of Human Kallikrein-related Serine Proteases. Nat. Rev. Drug Discovery 2015, 14, 183–202. 10.1038/nrd4534. [DOI] [PubMed] [Google Scholar]
- Lawrence M. G.; Lai J.; Clements J. A. Kallikreins on Steroids: Structure, Function, and Hormonal Regulation of Prostate-specific Antigen and the Extended Kallikrein Locus. Endocr. Rev. 2010, 31, 407–446. 10.1210/er.2009-0034. [DOI] [PubMed] [Google Scholar]
- Xi Z.; Klokk T. I.; Korkmaz K.; Kurys P.; Elbi C.; Risberg B.; Danielsen H.; Loda M.; Saatcioglu F. Kallikrein 4 is a Predominantly Nuclear Protein and is Overexpressed in Prostate Cancer. Cancer Res. 2004, 64, 2365–2370. 10.1158/0008-5472.CAN-03-2025. [DOI] [PubMed] [Google Scholar]
- Dong Y.; Bui L. T.; Odorico D. M.; Tan O. L.; Myers S. A.; Samaratunga H.; Gardiner R. A.; Clements J. A. Compartmentalized Expression of Kallikrein 4 (KLK4/hK4) Isoforms in Prostate Cancer: Nuclear, Cytoplasmic and Secreted Forms. Endocr.-Relat. Cancer 2005, 12, 875–889. 10.1677/erc.1.01062. [DOI] [PubMed] [Google Scholar]
- Klokk T. I.; Kilander A.; Xi Z.; Waehre H.; Risberg B.; Danielsen H. E.; Saatcioglu F. Kallikrein 4 is a Proliferative Factor that is Overexpressed in Prostate Cancer. Cancer Res. 2007, 67, 5221–5230. 10.1158/0008-5472.CAN-06-4728. [DOI] [PubMed] [Google Scholar]
- Kryza T.; Silva L. M.; Bock N.; Fuhrman-Luck R. A.; Stephens C. R.; Gao J.; Samaratunga H.; Lawrence M. G.; Hooper J. D.; Dong Y.; Risbridger G. P.; Clements J. A. Kallikrein-related Peptidase 4 Induces Cancer-associated Fibroblast Features in Prostate-derived Stromal Cells. Mol. Oncol. 2017, 11, 1307–1329. 10.1002/1878-0261.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veveris-Lowe T. L.; Lawrence M. G.; Collard R. L.; Bui L.; Herington A. C.; Nicol D. L.; Clements J. A. Kallikrein 4 (hK4) and Prostate-specific Antigen (PSA) are Associated with the Loss of E-cadherin and an Epithelial-Mesenchymal Transition (EMT)-like Effect in Prostate Cancer Cells. Endocr.-Relat. Cancer 2005, 12, 631–643. 10.1677/erc.1.00958. [DOI] [PubMed] [Google Scholar]
- Jin Y.; Qu S.; Tesikova M.; Wang L.; Kristian A.; Maelandsmo G. M.; Kong H.; Zhang T.; Jeronimo C.; Teixeira M. R.; Yuca E.; Tekedereli I.; Gorgulu K.; Alpay N.; Sood A. K.; Lopez-Berestein G.; Danielsen H. E.; Ozpolat B.; Saatcioglu F. Molecular Circuit Involving KLK4 Integrates Androgen and mTOR Signaling in Prostate Cancer. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E2572–2581. 10.1073/pnas.1304318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Collard R. L.; Bui L.; Herington A. C.; Nicol D. L.; Clements J. A. Kallikrein 4 is a Potential Mediator of Cellular Interactions Between Cancer Cells and Osteoblasts in Metastatic Prostate Cancer. Prostate 2007, 67, 348–360. 10.1002/pros.20465. [DOI] [PubMed] [Google Scholar]
- Ramsay A. J.; Dong Y.; Hunt M. L.; Linn M.; Samaratunga H.; Clements J. A.; Hooper J. D. Kallikrein-related Peptidase 4 (KLK4) Initiates Intracellular Signaling via Protease-activated Receptors (PARs). KLK4 and PAR-2 are Co-expressed During Prostate Cancer Progression. J. Biol. Chem. 2008, 283, 12293–12304. 10.1074/jbc.M709493200. [DOI] [PubMed] [Google Scholar]
- Mize G. J.; Wang W.; Takayama T. K. Prostate-specific Kallikreins-2 and −4 Enhance the Proliferation of DU-145 Prostate Cancer Cells Through Protease-activated Receptors-1 and −2. Mol. Cancer Res. 2008, 6, 1043–1051. 10.1158/1541-7786.MCR-08-0096. [DOI] [PubMed] [Google Scholar]
- Swedberg J. E.; Nigon L. V.; Reid J. C.; de Veer S. J.; Walpole C. M.; Stephens C. R.; Walsh T. P.; Takayama T. K.; Hooper J. D.; Clements J. A.; Buckle A. M.; Harris J. M. Substrate-guided Design of a Potent and Selective Kallikrein-related Peptidase Inhibitor for Kallikrein 4. Chem. Biol. 2009, 16, 633–643. 10.1016/j.chembiol.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Swedberg J. E.; de Veer S. J.; Sit K. C.; Reboul C. F.; Buckle A. M.; Harris J. M. Mastering the Canonical Loop of Serine Protease Inhibitors: Enhancing Potency by Optimising the Internal Hydrogen Bond Network. PLoS One 2011, 6, 1–11. 10.1371/journal.pone.0019302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley B. T.; Ilyichova O.; Costa M. G.; Porebski B. T.; de Veer S. J.; Swedberg J. E.; Kass I.; Harris J. M.; Hoke D. E.; Buckle A. M. Direct and Indirect Mechanisms of KLK4 Inhibition Revealed by Structure and Dynamics. Sci. Rep. 2016, 6, 35385. 10.1038/srep35385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckett S.; Garcia R. S.; Barker J. J.; Konarev A. V.; Shewry P. R.; Clarke A. R.; Brady R. L. High-resolution Structure of a Potent, Cyclic Proteinase Inhibitor From Sunflower Seeds. J. Mol. Biol. 1999, 290, 525–533. 10.1006/jmbi.1999.2891. [DOI] [PubMed] [Google Scholar]
- de Veer S. J.; Li C. Y.; Swedberg J. E.; Schroeder C. I.; Craik D. J. Engineering Potent Mesotrypsin Inhibitors Based on the Plant-derived Cyclic Peptide, Sunflower Trypsin Inhibitor-1. Eur. J. Med. Chem. 2018, 155, 695–704. 10.1016/j.ejmech.2018.06.029. [DOI] [PubMed] [Google Scholar]
- de Veer S. J.; Swedberg J. E.; Akcan M.; Rosengren K. J.; Brattsand M.; Craik D. J.; Harris J. M. Engineered Protease Inhibitors Based on Sunflower Trypsin Inhibitor-1 (SFTI-1) Provide Insights Into the Role of Sequence and Conformation in Laskowski Mechanism Inhibition. Biochem. J. 2015, 469, 243–253. 10.1042/BJ20150412. [DOI] [PubMed] [Google Scholar]
- Quimbar P.; Malik U.; Sommerhoff C. P.; Kaas Q.; Chan L. Y.; Huang Y. H.; Grundhuber M.; Dunse K.; Craik D. J.; Anderson M. A.; Daly N. L. High-affinity Cyclic Peptide Matriptase Inhibitors. J. Biol. Chem. 2013, 288, 13885–13896. 10.1074/jbc.M113.460030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swedberg J. E.; Li C. Y.; de Veer S. J.; Wang C. K.; Craik D. J. Design of Potent and Selective Cathepsin G Inhibitors Based on the Sunflower Trypsin Inhibitor-1 Scaffold. J. Med. Chem. 2017, 60, 658–667. 10.1021/acs.jmedchem.6b01509. [DOI] [PubMed] [Google Scholar]
- Hsieh A. C.; Liu Y.; Edlind M. P.; Ingolia N. T.; Janes M. R.; Sher A.; Shi E. Y.; Stumpf C. R.; Christensen C.; Bonham M. J.; Wang S.; Ren P.; Martin M.; Jessen K.; Feldman M. E.; Weissman J. S.; Shokat K. M.; Rommel C.; Ruggero D. The Translational Landscape of mTOR Signalling Steers Cancer Initiation and Metastasis. Nature 2012, 485, 55–61. 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascales L.; Henriques S. T.; Kerr M. C.; Huang Y. H.; Sweet M. J.; Daly N. L.; Craik D. J. Identification and Characterization of a New Family of Cell-penetrating Peptides: Cyclic Cell-penetrating Peptides. J. Biol. Chem. 2011, 286, 36932–36943. 10.1074/jbc.M111.264424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza C.; Henriques S. T.; Wang C. K.; Craik D. J. Structural Parameters Modulating the Cellular Uptake of Disulfide-rich Cyclic Cell-penetrating Peptides: MCoTI-II and SFTI-1. Eur. J. Med. Chem. 2014, 88, 10–18. 10.1016/j.ejmech.2014.06.047. [DOI] [PubMed] [Google Scholar]
- Felizmenio-Quimio M. E.; Daly N. L.; Craik D. J. Circular Proteins in Plants: Solution Structure of a Novel Macrocyclic Trypsin Inhibitor From Momordica cochinchinensis. J. Biol. Chem. 2001, 276, 22875–22882. 10.1074/jbc.M101666200. [DOI] [PubMed] [Google Scholar]
- Heitz A.; Hernandez J. F.; Gagnon J.; Hong T. T.; Pham T. T.; Nguyen T. M.; Le-Nguyen D.; Chiche L. Solution Structure of the Squash Trypsin Inhibitor MCoTI-II. A New Family for Cyclic Knottins. Biochemistry 2001, 40, 7973–7983. 10.1021/bi0106639. [DOI] [PubMed] [Google Scholar]
- de Veer S. J.; Weidmann J.; Craik D. J. Cyclotides as Tools in Chemical Biology. Acc. Chem. Res. 2017, 50, 1557–1565. 10.1021/acs.accounts.7b00157. [DOI] [PubMed] [Google Scholar]
- Gould A.; Camarero J. A. Cyclotides: Overview and Biotechnological Applications. ChemBioChem 2017, 18, 1350–1363. 10.1002/cbic.201700153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. K.; Craik D. J. Designing Macrocyclic Disulfide-rich Peptides for Biotechnological Applications. Nat. Chem. Biol. 2018, 14, 417–427. 10.1038/s41589-018-0039-y. [DOI] [PubMed] [Google Scholar]
- Sommerhoff C. P.; Avrutina O.; Schmoldt H. U.; Gabrijelcic-Geiger D.; Diederichsen U.; Kolmar H. Engineered Cystine Knot Miniproteins as Potent Inhibitors of Human Mast Cell Tryptase Beta. J. Mol. Biol. 2010, 395, 167–175. 10.1016/j.jmb.2009.10.028. [DOI] [PubMed] [Google Scholar]
- Thongyoo P.; Bonomelli C.; Leatherbarrow R. J.; Tate E. W. Potent Inhibitors of Beta-tryptase and Human Leukocyte Elastase Based on the MCoTI-II Scaffold. J. Med. Chem. 2009, 52, 6197–6200. 10.1021/jm901233u. [DOI] [PubMed] [Google Scholar]
- Glotzbach B.; Reinwarth M.; Weber N.; Fabritz S.; Tomaszowski M.; Fittler H.; Christmann A.; Avrutina O.; Kolmar H. Combinatorial Optimization of Cystine-knot Peptides Towards High-affinity Inhibitors of Human Matriptase-1. PLoS One 2013, 8, e76956 10.1371/journal.pone.0076956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swedberg J. E.; Mahatmanto T.; Abdul Ghani H.; de Veer S. J.; Schroeder C. I.; Harris J. M.; Craik D. J. Substrate-Guided Design of Selective FXIIa Inhibitors Based on the Plant-Derived Momordica cochinchinensis Trypsin Inhibitor-II (MCoTI-II) Scaffold. J. Med. Chem. 2016, 59, 7287–7292. 10.1021/acs.jmedchem.6b00557. [DOI] [PubMed] [Google Scholar]
- Ji Y.; Majumder S.; Millard M.; Borra R.; Bi T.; Elnagar A. Y.; Neamati N.; Shekhtman A.; Camarero J. A. In Vivo Activation of the p53 Tumor Suppressor Pathway by an Engineered Cyclotide. J. Am. Chem. Soc. 2013, 135, 11623–11633. 10.1021/ja405108p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsinczky M. L.; Schirra H. J.; Rosengren K. J.; West J.; Condie B. A.; Otvos L.; Anderson M. A.; Craik D. J. Solution Structures by 1H NMR of the Novel Cyclic Trypsin Inhibitor SFTI-1 From Sunflower Seeds and an Acyclic Permutant. J. Mol. Biol. 2001, 311, 579–591. 10.1006/jmbi.2001.4887. [DOI] [PubMed] [Google Scholar]
- Daly N. L.; Thorstholm L.; Greenwood K. P.; King G. J.; Rosengren K. J.; Heras B.; Martin J. L.; Craik D. J. Structural Insights Into the Role of the Cyclic Backbone in a Squash Trypsin Inhibitor. J. Biol. Chem. 2013, 288, 36141–36148. 10.1074/jbc.M113.528240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.