

Abstract
Some disulfide bonds perform important structural roles in proteins, but another group has functional roles via redox reactions. Forbidden disulfides are stressed disulfides found in recognizable protein contexts, which currently constitute more than 10% of all disulfides in the PDB. They likely have functional redox roles and constitute a major subset of all redox‐active disulfides. The torsional strain of forbidden disulfides is typically higher than for structural disulfides, but not so high as to render them immediately susceptible to reduction under physionormal conditions. Previously we characterized the most abundant forbidden disulfide in the Protein Data Bank, the aCSDn: a canonical motif in which disulfide‐bonded cysteine residues are positioned directly opposite each other on adjacent anti‐parallel β‐strands such that the backbone hydrogen‐bonded moieties are directed away from each other. Here we perform a similar analysis for the aCSDh, a less common motif in which the opposed cysteine residues are backbone hydrogen bonded. Oxidation of two Cys in this context places significant strain on the protein system, with the β‐chains tilting toward each other to allow disulfide formation. Only left‐handed aCSDh conformations are compatible with the inherent right‐handed twist of β‐sheets. aCSDhs tend to be more highly strained than aCSDns, particularly when both hydrogen bonds are formed. We discuss characterized roles of aCSDh motifs in proteins of the dataset, which include catalytic disulfides in ribonucleotide reductase and ahpC peroxidase as well as a redox‐active disulfide in P1 lysozyme, involved in a major conformation change. The dataset also includes many binding proteins.
Keywords: disulfide, redox‐active disulfide, thiol‐based redox signaling, forbidden, disulfide
Introduction
Biochemistry textbooks describe disulfides as permanent crosslinks in proteins that stabilize protein structures. Nonetheless there is a considerable body of literature that shows at least some disulfides are redox active.1 Those that are oxidized and reduced in the active site of enzymes are best known but others involved in conformational changes have also been characterized.2 The likelihood a disulfide will be reduced is a function of its physicochemical properties, which are determined by its context. Within a protein, disulfide reduction may be affected by static features—such as electrostatic environment and accessibility, as well as dynamic influences—such as conformational change and binding of ligands; external to it, the redox milieu is also important.
From a computational point of view, the influence of static features of the protein context is easier to assess. Torsional energy, which is an important determinant of the redox potential, can be calculated using quantum chemical techniques.3, 4 Studies have shown that rather than forming a continuum, disulfide torsional energies tend to form a skewed bipartite distribution, heavily populated by disulfides with low torsional energies but with a peak of higher torsional energy disulfides that are likely involved in redox processes.5 This lends some support to the concept that there are in fact two disulfide proteomes: a structurally stabilizing one and a redox‐active one.6
The disulfide‐bonded cysteine (Cys) residues of a subset of these higher torsional energy disulfides occupy recognizable motifs in secondary structure. We dubbed them “forbidden disulfides” because it was originally postulated that disulfides would not form between pairs of Cys in these motifs because they were stereochemically disfavoured.7, 8 Searches of the Protein Data Bank (PDB) at the time supported the original conjecture. However, instances of these unusual disulfides began to appear in structures and interestingly, their prevalence in the PDB is growing.1 It now seems likely that the unique stereochemical properties of forbidden disulfides are integral to their redox activity and that crystallographers are becoming increasingly adept at coaxing proteins containing these reactive disulfides into homogeneous conformations amenable to crystallization.
Previously we identified 15 major subtypes of disulfides violating all four stereochemical rules postulated by Thornton7 including disulfides that link Cys residues in helices (α, α+3; α, α+4), along strands (β, β+2), adjacent in the polypeptide chain (i, i+1) and lastly between strands.1, 9 The most prevalent of these seem to be the Between‐Strand Disulfides (BSDs), which surprisingly now constitute over 10% of the disulfides in the PDB, making them an important subclass of protein disulfide bonds.10, 11 BSDs connect two Cys residues located on adjacent strands of β‐sheet (or β‐ribbon) structures. We identified four distinct BSD types in the PDB: Cross‐Strand Disulfides (CSDs), which connect Cys residues immediately opposite each other across adjacent β‐strands (residues i, j); End‐Twist Disulfides (ETDs), which connect Cys out of register by one residue (i, j±1); β‐Diagonal Disulfides (BDDs), connecting residues i and j±2; and β‐flip Disulfides (BFDs), connecting Cys at positions i and j±3.1, 9 Representatives of each of these BSD types were found in both parallel and anti‐parallel β‐structures.
This work represents the third in a series of papers describing the stereochemical features of various BSD motifs, their impact on the surrounding β‐structure and their relevance to biological processes. The first publication10 surveyed the prevalence of various types of BSD in the PDB while the second paper characterized the most abundant subtype, described below.11
CSDs in anti‐parallel β‐sheet (aCSDs) are by far the most common BSD,10, 11 exceeding in number the better studied and well known inter‐helical CXXC motif (α, α+3). The backbone atoms of cross‐strand pairs of residues are found in two profoundly different geometries in antiparallel β‐sheet, which alternate both along the two involved strands and across the sheet, orthogonal to the strand direction in the plane of the sheet: the hydrogen‐bonded (HB) and non‐hydrogen‐bonded (NHB) sites (Fig. S1). Early investigations of cross‐strand partners in antiparallel β structures12, 13, 14, 15 showed that Cys pairs are favoured across NHB sites and often form disulfide bonds (aCSDns). As the two involved Cys residues are already held in place by the hydrogen‐bonding network between the strands, the presence of the disulfide in the secondary structure appears, at first, to be redundant. In contrast, Cys pairs in HB sites were found to be less common and disulfide formation was not seen. Hutchinson et al.15 performed molecular dynamics simulations on model peptide systems containing a cystine in the HB site in order to identify why such disulfides (aCSDhs) should be disfavoured. Minimisation gave structures with highly strained disulfide conformations with average energies ~40 kJ mol−1 higher than those of corresponding structures containing aCSDns. Furthermore, as the simulation progressed, the disulfide conformation relaxed, breaking the hydrogen bonding (H bonding) network and destroying the β‐structure. Hence, it was believed that CSDs could not form across HB sites in antiparallel β‐sheet.
This view changed in the late 2000s, when Santiveri et al.16 and Indu et al.17 studied the effects of introducing CSDs (both aCSDns and aCSDhs) into peptide and protein systems, respectively. Their experiments on mutant systems revealed aCSDh structures could form, however the introduction of the disulfide had a neutral or destabilizing effect on the system. In contrast, aCSDns were strongly stabilizing. Furthermore, Indu et al.17 identified five protein structures in the PDB containing a Cys pair oxidized to form a disulfide in an HB site, all of which were homologous. Based on the disulfide conformations adopted, they proposed that one of the factors disfavoring aCSDh formation is steric repulsion between the sulfur atom(s) and the carbonyl group(s) of the cross‐strand hydrogen bond(s).
More recently in a survey of BSD structures in a dataset of X‐ray crystal structures with resolution of 3 Å or better from PDB Archive Version 4.0 (July 2011),10 we found 4865 instances of the more common aCSDn, comprising 254 non‐homologous clusters. In contrast, there were only 114 instances of aCSDh structures, representing 20 clusters of disulfides. aCSDh disulfides were found both in the middle and at the termini of β‐ladders (pairs of adjacent strands).
Despite the relative rarity of aCSDhs compared to aCSDns, several aCSDhs are known to perform crucial redox roles in their resident proteins. One of these is found in ribonuclease reductase (PDB: 2r1r), where the redox‐active aCSDh, connecting Cys 225 and Cys 462, is essential for DNA synthesis in all forms of life.18 Another aCSDh is in the well‐studied archetype of thiol peroxidases, bacterial alkyl hydroperoxide reductase C, ahpC (PDB: 1yex A165‐B46), which protects the cell against oxidative stress by detoxifying peroxides.19
Given the proven biological importance of aCSDhs, we analyzed their properties in this third paper on BSDs. We identified disulfide conformations that allow Cys to connect across HB sites and estimated the resulting torsional strain using quantum chemical calculations. We also assessed the strain exerted by the aCSDh motif on the surrounding β‐sheet, i.e., the backbone conformations enforced on the involved Cys residues, and the twisting and shearing of the sheet due to the presence of the aCSDh. One critical property identified is how deeply the aCSDh is embedded within the β‐structure, which determines how easily the structure can be deformed to accommodate the presence of the disulfide and also how the overall strain might be accommodated in the system. In contrast, embeddedness was not so important for the more populous aCSDn; this is likely a result of the different physicochemical properties of these two motifs. The biological function of proteins bearing aCSDhs is also reviewed. The properties of aCSDhs will be regularly contrasted with the far more common aCSDns described in our previous work.11 To avoid confusion between these quite similar looking terms, whenever aCSDns are mentioned the n will be bolded.
Results
aCSDh structures: sub‐motifs and classes
A survey of a recent snapshot of the PDB (see Methods) found 240 examples of aCSDhs in protein structures (Fig. 1). These could be grouped into 37 clusters based on the involved fold. Full details of all properties we investigated for each of the aCSDh clusters found in the dataset are reported in Table 1. Details of the SCOP protein folds in which the disulfides are found, their positions within the fold and the degree of conservation of the disulfide can be found for selected structures in Table 2.
Figure 1.
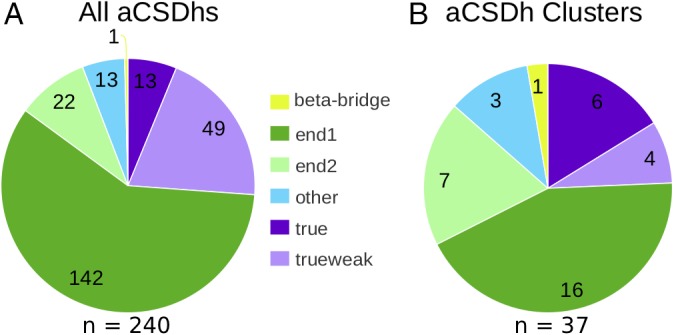
Distribution of structures containing aCSDh submotifs in the current PDB snapshot. (A) The distribution of aCSDhs in the dataset amongst the three possible sub‐motifs and their classes (see Fig. 2 for definitions). (B) The distribution of non‐homologous aCSDh clusters in the dataset amongst the sub‐motifs.
Table 1.
A Full List of Disulfide Clusters that Adopt aCSDh Motifs. In each case, a representative disulfide is given along with the number of oxidized structures and their average total torsional energy, the LonE and LatE levels observed and the disulfide conformations adopted. Also shown is an indication of whether the fold can exist without the disulfide, the Pfam family of the fold and any evidence for redox activity (either in the literature or a PDB code in which the disulfide is reduced).
| Total EkJ mol−1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cluster Name | Representative disulfide | No. Ox. | Ave | dev | LonEb | LatEa | Conf.c | Non Str?d | Pfam | Redox active?e |
| RalGDSf | 1lxd A16‐B16 | 1 | 14.6 | NA | true | 2 | lst | Yes | RA | 1lfd A16‐C16 |
| Spermidine synthase | 3bwc A 206–255 | 2 | 45.0 | 1.0 | true | 2 | rst/lst | Yes | Spermine_synth | 3bwb A206‐255 |
| DNA polymerase processivity factor, BMRF1 | 2z0l A206‐D206 | 2 | 40.7 | 3.4 | true/wktrue | 2 | rst | Yes | Herpes_DNAp_acc | |
| SIGLECsg | 1qfo A 22–79 | 32 | 14.3 | 3.5 | wktrue | 0/1h | lhk/lsd | Yes | Vset | 1qfp A 22–79i |
| CelQ lectin | 5gxy A535‐B535 | 5 | 18.0 | 4.7 | true | 2 | rhk/lst | Yes | CBM_3 | |
| Glycine receptor | 5tin B 198–209 | 10 | 11.5 | 0.2 | wktrue | 1 | lhk | Yes | Neur_chan_LBD | |
| IL17receptor a | 5n9b A 43–50 | 1 | 15.6 | NA | wktrue | 1 | lhk | No | IL17R_fnIII_D1 | |
| Lipocalin | 4a8z A 117–124 | 1 | 83.4 | NA | true | 1 | r | Yes | Lipocalin | |
| Cell entry protein | 4uf7 A 378–422 | 2 | 21.3 | 0.31 | true | 2 | lhs | Yes | HN | |
| ML domains | 1xwv A 8–119 | 67 | 14.1 | 5.5 | end1 | 1 | lhk/lGGT | Na | E1_DerP2_DerF2 | 1ktj B 8‐119i |
| Type IV pilin | 4bhr A 71–85 | 5 | 12.4 | 2.1 | end2/1 | 1 | lhk/lGGT | No | Type IV pilin | |
| IL1, Family member 5 | 1md6 A 7–153 | 3 | 17.8 | 7.0 | end1/2 | 0/1 | lGGT | Yes | IL1 | |
| Archaeal α‐amylase | 1mxg A 388–432 | 4 | 8.0 | 1.1 | end1 | 1 | lGGT | Yes | Alpha‐amylase | |
| Ribonuclease reductase, R1 subunit, C‐term domain | 2r1r A 225–462 | 11 | 30.6 | 9.5 | end1/2 | 0/1 | rst/lG′GT | Yes | Ribonuc_red_IgC | 18 |
| Alkyl hydroperoxide reductase, ahpC | 1yex A165‐B46 | 3 | 7.9 | 1.3 | end2 | 0 | lGGT | Yes | AhpC‐TSA | 28 |
| Archaeal cysteine synthase | 1wkv A 36–64 | 8 | 10.7 | 1.5 | end1 | 0 | lG′GT | Na | PALP | 5b36, 5b3a |
| Bacteriophage lysozyme | 1xjt A 13–44 | 1 | 13.3 | NA | end2 | 0 | lhk | Na | Phage_lysozyme | 30 |
| Reprolysin‐like metalloproteasesj | 1dth A 157–164i | 5 | 13.6 | 2.5 | end1/2 | 0/1 | lGGT | Na | Reprolysin | |
| ORB2K trypsin inhibitork | 2o9q C 4–14 | 1 | 16.0 | NA | end2 | 0 | lhk | Yes | peptide | |
| Ephrin receptor, FIII d | 3fl7 A 366–379 | 2 | 52.0 | 14.5 | end1/2 | 1 | rmx/lG′GT | Yes | fn3 | |
| Mast/stem cell growth factor receptor | 2o26 U 152–184 | 4 | 23.2 | 0.8 | end1 | 1h | lsd | Yes | Iset | |
| Acid‐sensing ion channel | 2qts A 173–180 | 22 | 16.6 | 5.6 | end1/2 | 1 | lhk/lsd | No | ASC | |
| CEL‐IV lectin | 3alu A41‐C151 | 10 | 10.1 | 3.1 | end1 | 0 | lGGT | Na | Lectin_C | |
| Triglyceride lipase | 3ngm A 99–105 | 2 | 28.6 | 4.9 | end1 | 0 | rst/sd | Yes | Lipase_3 | |
| DDR1 receptor Tyr kinase | 5bvw A 691–698 | 2 | 29.2 | 1.4 | end2 | 1/0 | rhk/rst | Yes | Pkinase | |
| cd45 | 5fmv A 309–321 | 4 | 12.7 | 0.9 | end1 | 2 | rG′GT | Yes | cd45 fIII | 3zos |
| 5fmv A 503–535 | 4 | 17.2 | 2.0 | end1 | 1 | lhk | Yes | |||
| Cytokine receptor factor 2 | 5j11 C 138‐168 | 1 | 22.1 | NA | end1 | 2 | rG′GT | Yes | Ck rcptr 2 fIII | |
| eff‐1 fusion protein | 4ojd H 152–164 | 2 | 18.9 | 2.0 | end2 | 1 | lGGT | No | EFF‐AFF | |
| gemin5 | 5gxi A 240–256 | 6 | 22.8 | 7.0 | end1 | 0 | lG′GT | Yes | WD40 | 5h1k |
| Peptidase M23 | 5b0h A 81–124 | 1 | 7.0 | NA | end1 | 1 | lGGT | No | Peptidase_M23 | |
| NBL1 | 4x1j B 82–123 | 2 | 7.2 | 1.4 | end1 | 0 | lGGT | No | DAN | 5yu8 |
| mpeptidase inhibitor | 5mrv C 9–23 | 1 | 12.6 | NA | end1 | 1 | lGGT | No | peptide | |
| IgK‐IgG | 1fn4 C211‐D208 | 1 | 24.7 | NA | β‐bridge | 0 | rst | No | IgL‐IgH | |
| peptidase M2 (ace) | 2iul A 352–370 | 4 | 18.6 | 1.6 | other | 0/1 | lGGT | Yes | Peptidase_M2 | |
| cutf Cu homeostasis | 2z4i 31–34 | 1 | 9.0 | NA | other | 0 | rG′GT | No | cutf Cterm | |
| TIM lipase | 5xk2 B 111–114 | 2 | 11.0 | 0.9 | other | 0 | lG′GT | Yes | Abhydrolase_8 | |
| RAET1L | 4s0u C 50–66 | 1 | 22.5 | NA | end1 | 1 | rhk | No | MHC_I | |
Levels of embeddedness shown in grey occur in less than 10% of structures.
End1 refers to Class 1 end‐aCSDhs and end2 to Class 2 end‐aCSDhs, see Fig. 2.
Prefixes “r” and “l” indicate right‐ and left‐handed conformations respectively. St = staple; hk = hook; sd = saddle one of χ2 and χ2′ between 110 and 130; mx = χ2 ~ −130°, χ2′ ~ 100° (on one of the minor maxima of the surface). Only conformations seen in at least 10% of structures are listed.
Can the protein fold exist without the disulfide? Yes = fold has been seen to exist without the disulfide; Na = the disulfide is not in the central fold of the protein; No = no crystal structures of this fold without the disulfide have yet been reported. See Table II for more details.
Numbers indicate literature references which describe the redox activity of the disulfide. PDB codes indicate structures in which the disulfide is reduced or in a mixed oxidation state.
In the crystal structures, the Cys is near the N‐terminus; in the sequences it is in the middle (i.e., the crystal structures do not contain the first domain(s)). It may not be possible for the full length protein to form the disulfide.
All SIGLECs apart from SIGLEC15.
Access to this disulfide is impeded along both edges.
The disulfide in this structure is a mixture of oxidized and reduced forms.
In snake venom metalloproteases the β‐hairpin between the Cys is of variable length, in some cases it is too short to allow the aCSDh motif to form. In the single ADAM protein crystal structure, the disulfide is conserved but the motif is not. A second disulfide prevents the two Cys from coming close enough for hydrogen bonding to form. It is likely the aCSDh motif is not found in most, if not all, ADAM proteins.
This is a β‐hairpin polypeptide.
Table 2.
Protein Fold Data for Selected Disulfide Clusters Listed in Table 1
| Cluster name | SCOP fold | Position within fold | Fold without disulfide |
|---|---|---|---|
| RalGDS | β‐Grasp (ubiquitin‐like) | Homodimer interface | c‐Raf1 (1c1y) |
| Spermidine synthase | S‐adenosyl‐l‐methionine‐dependent methyltransferases | Connecting strands 5 and 7—same side as helices 5 and 6 | Spermidine synthase in Thermus (1uir) |
| DNA polymerase processivity factor, BMRF1 | DNA clamp | Homodimer interface | Monomer |
| SIGLECs | Immunoglobulin‐like β‐sandwich | BE—inside | Immunoglobulin (1bww) |
| ML domains | Immunoglobulin‐like β‐sandwich | Connecting N‐terminal extension to G—outside | Not seen |
| Lymphocyte antigen 96 | Immunoglobulin‐like β‐sandwich | Connecting N‐terminal extension to G—outside | Not seen |
| Ganglioside GM2 activator | Ganglioside M2 (gm2) activator | Connecting N‐terminal extension to G—outside | Not seen |
| Interleukin 1 family Member 5 | β‐Trefoil | Tying termini together | Interleukin‐1β (1l2h) |
| Archaeal α‐amylase | Glycosyl hydrolase domain | CF—outside | Bacterial α‐amylase (1g94) |
| Ribonuclease reductase, R1 subunit, C‐terminal domain | PFL‐like glycyl radical enzymes | Connecting strands 1 and 6 of the αβ barrel—in core | PFL (1h16) |
| Alkyl hydroperoxide reductase, ahpC | Thioredoxin fold (glutathione peroxidase‐like) | Connecting loop between strand 1 and helix 1 to C‐terminal of another domain | Glutathione peroxidase (1gp1) |
| Archaeal Cys synthase | Tryptophan synthase β subunit‐like PLP‐dependent enzymes | In β‐ribbon in N‐terminal extension | Not seen—not in main fold |
| Bacteriophage Lysozyme | Lysozyme‐like | Connecting N‐terminal helical region to β‐sheet | Not seen—in modified N‐terminus |
| Reprolysin‐like metalloproteases | Zincin‐like | In β‐hairpin in loop between two C‐terminal helices | Not seen—modification of main fold |
| ORB2K trypsin inhibitor | β‐hairpin | Connecting ends of β‐hairpin | β‐hairpin |
| Ephrin receptors | Immunoglobulin‐like β‐sandwich | CC’—inside | Fibronectin (1fna) |
| Mast/stem cell growth factor receptor | Immunoglobulin‐like β‐sandwich | CF—outside | GCSF receptor, N‐term domain (2d9q) |
| Acid‐sensing ion channel | Core: 3 layers, β/β/α; central antiparallel β‐sheet of 5 strands, order 32145; top mixed β‐sheet of 6 strands, order 213654 | Connecting strands 1 and 2 of top sheet, inner side | Not seen—no other proteins with this fold |
| CEL‐IV lectin | C‐type lectin | Connecting β‐hairpin between helices 1 and 2 to C‐terminus of another monomer | Not seen—not in main fold |
| Lipase | α/β‐Hydrolases | In β‐hairpin elaboration within helix 2 | Triacylglycerol lipase (1tib) |
An indication is also given of the position of the disulfide within the fold and an example of a closely related structure where the BSD is not present. For structures with an immunoglobulin‐like β‐sandwich or closely related folds, the disulfide position is described in terms of the normal strand labeling scheme for immunoglobulin‐like β‐sandwiches. In all other cases, the β‐strands and α‐helices have been numbered from the N‐ to C‐terminus within the relevant domain. We note in some cases this numbering scheme may be subjective.
Some of the clusters contained members from highly divergent genes where the aCSDh is retained as a crucial part of the protein despite topological remodeling of the fold as the function of the duplicated daughter genes diverged. For example, one cluster contained structures of proteins of sialic‐acid binding lectins (SIGLECs) from six different genes while another contained 12 different genes encoding the MD‐2‐related lipid‐recognition (ML) domains. The diverse members of the ML domains, which bind lipids or glycolipids, are involved in functions ranging from chemosensing in insects to the innate immune system in higher organisms. Other clusters contained more than one aCSDh located in different parts of the protein, with examples in the angiotensin‐converting enzyme (ACE) and the extracellular portion of receptor‐type tyrosine–protein phosphatase C (PTPRC, CD45), a cell surface receptor required for T‐cell activation. Alignments for these four folds are shown in Figure S2.
The Cys residues in most aCSDhs are found within a single protein chain but there are a few examples of intrachain disulfides, most forming disulfide‐linked homodimers. In the majority of cases, such as RalGDS, a single Cys residue from each chain forms a one‐disulfide‐linked homodimer. However, two Cys from each chain can also form a two‐disulfide linked homodimer, with examples in ahpC (PDB: 1yex A165‐B46) and in the Cel‐IV C‐lectin (PDB: 3alu A41‐C151). There is also one example of an aCSDh‐linked heterodimer between an immunoglobulin light and heavy chain IgK‐IgG (PDB: 1fn4 C211‐D208). These will be discussed further below.
In a previous publication,10 we introduced the concepts of longitudinal and lateral embeddedness to describe the extent of secondary structure surrounding the BSD. Embeddedness influences the degree to which the β structure can be deformed to accommodate the presence of the disulfide and also how the overall strain might be accommodated in the system. It also partially determines the accessibility of the disulfide to reducing agents. Longitudinal embeddedness (LonE) is a measure of the degree of canonical hydrogen bonding on either side of each Cys residue within the disulfide‐containing β‐ladder, while lateral embeddedness (LatE) describes the presence of additional β‐chains hydrogen bonded to either side of the β‐ladder in which the disulfide is found. Three different levels of LonE were identified in existing aCSDh structures (Fig. 2): “true,” where canonical H bonding is found on both sides of the two Cys Cα atoms; “end,” where it is only found on one side; and “β‐bridge,” when the disulfide is located within an isolated β‐bridge structure such that there is H bonding only between the two involved Cys residues themselves. The definition of LonE, described in a previous publication on aCSDns,11 is adapted slightly because of the disulfide's location in an HB rather than a NHB site (see below). As before, we refer to a BSD sub‐motif with a particular level of LonE with a prefix (e.g., true aCSDh). Three levels of LatE are distinguished: disulfides located on β‐ribbon structures (β‐ribbon‐BSDs or in shorthand, using a dashed suffix, BSD‐0s), consisting of two isolated β‐strands; disulfides where one of the Cys is located on an edge strand of a β‐sheet (edge‐BSDs or BSD‐1s) and BSDs in the middle of 4+ strand β‐sheets (mid‐sheet‐BSDs or BSD‐2s). Details of the embeddedness and the level of strain in the disulfide bonds for each cluster are shown in Table 1.
Figure 2.
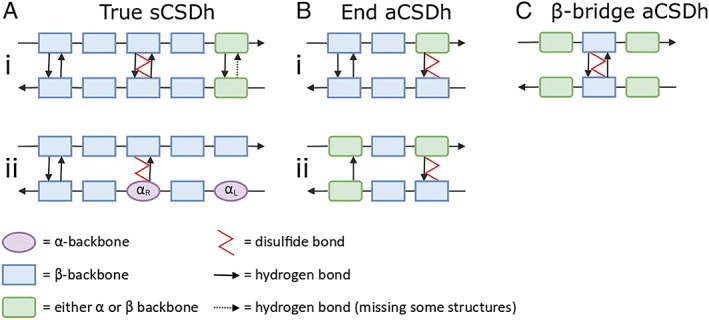
Hydrogen bonding diagrams for aCSDh sub‐motifs depicting longitudinal embeddedness. In CSDs, a Cys residue i on one strand is disulfide linked to a Cys j on the adjacent strand, where j is the residue immediately opposite i. Classes of longitudinal embeddedness are (A) true; (B) end, and (C) β‐bridge. Two classes of the true‐aCSDh sub‐motif are shown: i true‐aCSDh, with two hydrogen bonds between the Cys residues; ii weaktrue‐aCSDh, with one hydrogen bond between the Cys and one Cys relaxed to an α backbone conformation. There are also two classes of end‐aCSDhs: i at the end of a β‐ladder (end1); ii adjacent to an NHB site β‐bridge (end2). Residues which consistently adopt a β backbone conformation are shown as blue rectangles and those which are always found in α conformation are purple ovals. Any residue which can adopt either backbone conformation is shown as a green rectangle with rounded corners. Hydrogen bonds are shown as arrows. Dashed arrows mean that the hydrogen bond may be missing in some structures. The disulfide bond is described by a red zig‐zag connecting two Cys residues.
For aCSDns, the disulfide properties were not found to strongly depend on the degree of embeddedness.11 For aCSDhs the situation is very different. The two Cys in an aCSDn motif are located in an NHB site [Fig. S1(A)]. These sites are quite flexible, allowing in‐plane puckering of the sheet. Puckering reduces the Cα–Cα distance across the NHB site from the canonical value in regular β‐sheet of ~ 4.5 Å20 to ~ 4.0 Å (the median Cα–Cα distance for the staple disulfide conformation that is most commonly adopted when disulfides connect adjacent β‐strands11). In an aCSDh, however, the backbone atoms form hydrogen bonds and several different variants arise based on the extent of H bonding between them. In the true‐aCSDh, the Cys residues are connected by two hydrogen bonds, as typically found between residues in an HB site of canonical β‐sheet, plus a disulfide linkage [Fig. 2(Ai)]. The average Cα–Cα distance across canonical HB sites is 5.3 Å.20 However, compression of HB sites is impeded by the shared backbone hydrogen bonds, so the site cannot deform in the manner of an aCSDn across an NHB site. The two β‐chains are forced to tilt toward each other in order to accommodate the disulfide. A representative ribbon structure for a true‐aCSDh can be seen in Figure 3. While the sheet itself shows no obvious buckling, the side view in Figure 3(B) shows that the Cα–Cβ bonds are tilted toward each other. In canonical antiparallel β‐sheet, these bonds are perpendicular to the β‐sheet, as is also true for most aCSDns (Fig. S3).
Figure 3.
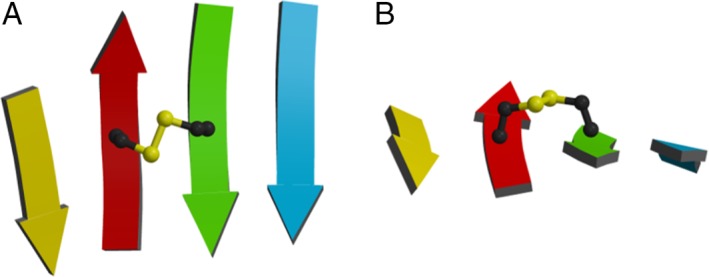
Orthogonal views of a representative disulfide in an aCSDh motif found in Trypanosoma cruzi spermidine synthase: PDB 3bwc A Cys 206 to Cys 255 (true‐aCSDh‐2). (A) From above. (B) Side view along the strands. Note: no significant buckling is seen in the sheet due to the presence of the disulfide; however, the two Cα–Cβ bonds are tilted toward each other.
Cys residue tilting puts significant strain on the backbone hydrogen bonds linking the two Cys residues in an HB site. While hydrogen bonds prefer to be linear (see Fig. S3), the average N‐O‐C angle in the true‐aCSDh structures of the dataset is ~ 40° from linearity (see Fig. 3). As a result of this strain, one of the hydrogen bonds connecting the two Cys is often broken in the crystal structures of the dataset. This can lead to end‐aCSDhs (which will be described in more detail below) or, if there is extensive H bonding between the two strands of the disulfide‐containing β‐ladder, to the class of true‐aCSDhs shown in Figure 2(Aii). In this LonE sub‐motif, dubbed a “weaktrue‐aCSDh,” one of the bonds between the two Cys is broken, with one of the Cys being twisted into an α backbone conformation.21 Despite the absence of one of the critical hydrogen bonds, this is still regarded as an example of a true‐aCSDh sub‐motif due to the presence of canonical H bonding between the two chains on both sides of the disulfide‐bonded Cys residues.
Examples of true aCSDhs
There are only 12 true‐aCSDh structures in the dataset (see Fig. 1), found in six non‐homologous protein clusters (Table 1).
In three of these clusters, the aCSDh links two monomers in a protein dimer: Epstein–Barr virus (ebv) DNA polymerase accessory protein BMRF1 (Cys 206, PDB: 2z0l), celQ endoglucanase (Cys 535, PDB: 5gxy) and the RAS‐interacting domain of RalGDS (Cys 16, PDB: 1lxd). In BMRF1 and RalGDS, a major feature of the interaction is the alignment of the two proteins along the outer strands of their β‐sheets to form an apparent continuous β‐sheet between the two monomers that may form a platform for other protein interactions, such as DNA‐binding or signaling, respectively.22, 23 In all of these true aCSDh structures, both Cys and their four neighboring residues are found in the β region of the Ramachandran plot,21 i.e., the dimer interface is well protected by H bonding surrounding the putative redox‐active disulfide.
In the remaining three clusters, the aCSDh forms an intrachain disulfide linkage. One of these is in spermidine synthase (PDB: 3bwc, Cys 206–255). Another is in a viral hemagglutinin that engages the human ephrinB2 receptor to effect cell entry, protein G from a bat paromyoxvirus (PDB: 4uf7, Cys 378–422). This will be discussed further below, along with BMRF1, in the context of forbidden disulfides and viruses. The aCSDh stabilizes the third blade of the β‐propeller and is not present in the related viral proteins NiV‐ or HeV‐G.24 The third example is in a lipocalin (PDB: 4a8z A 117–124), a lipid‐binding protein.
Interestingly, in two structures of distinct clusters, the RalGDS dimer and spermidine synthase, one of the disulfide‐forming thiols is adjacent in the polypeptide chain to another Cys in the free thiol form (see Fig. S4). If the disulfide isomerized, these two pairs of adjacent Cys could potentially form another forbidden disulfide motif—a vicinal motif (i, i+1).9, 25 An intramolecular vicinal disulfide could prevent the Cys from forming a sulfenic acid under oxidizing conditions. The vicinal disulfide could potentially then isomerize to form the aCSDh‐linked dimer observed in the crystal. In the case of RalGDS, this was suggested to be a slow process in vitro, driving the gradual formation of crystals over a period of 6 months.26
Weaktrue aCSDhs
The dataset also contains 49 weaktrue‐aCSDh structures found in five clusters. Several of these weaktrue‐aCSDhs are in the extracellular portions of cell surface receptors: the glycine receptor, IL17 receptor A, and the SIGLECs. In two of the clusters the disulfide is consistently embedded as weaktrue: the SIGLECs (e.g.. PDB: 1nko Cys 46 to Cys 106) and the glycine receptor (e.g., PDB: 5tin B 198–209), while the IL17RA structure is a singleton (PDB: 5n9b A 43–50). The SIGLECs are sialic acid‐binding Ig‐like lectins of the innate immune system, which are modified Vset Ig domains. They will be discussed later in the context of the immune system and a potential role for aCSDhs in ligand binding sites. In the remaining two clusters, the embeddedness is heterogeneous ie alternate structures have different embeddedness, suggesting conformation flexibility or different states of the proteins: the aCSDh of the viral BMRF1 is true in one dimer of the crystallographic unit and weaktrue in the other, possible reflecting forces related to crystal contacts. In the other example, the aCSDh in a single ML domain is weak‐true, whereas all the other aCSDhs in ML domains are end‐aCSDhs. Interestingly, the single weak‐true example has a molecule bound in the binding pocket, which may affect the conformation of the nearby aCSDh (PDB: 3web A 6–124). The aforementioned aCSDh in BMRF1 is intermolecular while the aCSDhs of the other clusters are intramolecular.
In terms of lateral embeddedness, our primary interest is in how rigidly the disulfide is constrained by the H bonding. Hence, our definitions require each adjacent strand to be hydrogen bonded to the disulfide‐containing β‐ladder on both sides of the relevant Cys Cα atom to be designated LatE BSD‐2 or midsheet.10 While for both the true‐ and weaktrue‐aCSDh structures in the dataset access to both sides of the disulfide is restricted by additional β‐chains, only structures with two hydrogen bonds between the Cys have sufficient H bonding with the adjacent strands to be classed as mid‐sheet‐BSDs. For the weaktrue‐aCSDhs, the H bonding to one of the neighboring strands is weak, technically giving edge‐of‐sheet LatE contexts.
LatE embeddedness could also be important for binding of protein reductants. True‐aCSDns are most commonly located on the edges of β‐sheets (BSD‐1).11 This makes them good candidates for being substrates of thioredoxin (Trx) and other related oxidoreductases, which are known to hydrogen bond to edge‐strand disulfides in order to position them for catalytic reduction. Indeed there is anecdotal evidence that aCSDns are favored substrates of Trx.27 We believe aCSDns may be cognate substrates of Trx, which would account for their proliferation in protein structures as there would be a positive selection effect for stable reducible disulfides in redox‐controlled proteins. It should be noted that a thiol oxidoreductase optimized for attacking an aCSDn in an NHB site is unlikely to be well disposed toward an aCSDh, which is out of registration for substrate binding. However, as these disulfides are highly strained, as will be described below, aCSDhs may not require the assistance of a thiol oxidoreductase to be redox‐active.
Examples of end‐aCSDhs
Given the strain on the H bonding associated with an aCSDh motif, it is not surprising that end‐aCSDhs [Fig. 2(B)], which have fewer surrounding hydrogen bonds, are far more common than “true” sub‐motifs. Twenty‐three non‐homologous examples of end‐aCSDhs (164 structures) were found in the high‐resolution dataset (see Fig. 1). End‐aCSDhs connect either the last pair of H bonding residues of a β‐ladder (where these form only one hydrogen bond; Class 1) or the pair of residues forming one side of an NHB site β‐bridge (Class 2); the former is much more common, being seen in over 85% of end‐aCSDh structures (see Fig. 1).
Unlike the case for aCSDns,11 end‐aCSDhs in each cluster in the dataset tend to be very homogeneous with respect to LonE i.e., whether they were true, weaktrue or end. Less than 3% of structures showed an alternate type of longitudinal embeddedness. As for end‐aCSDns, Cys residues involved in end‐aCSDh submotifs can adopt either α or β backbone conformations.
Examples of end‐aCSDhs, found at the terminus of a β‐sheet (Class 1), are present in almost all members of the ML domain family, which bind various lipids. Although some ML domains have been assigned to different SCOP folds, their base fold, a modified Ig Eset, is actually quite similar (see alignment in Fig. S2). ML domains belong to the same Pfam family (E1_DerP2_DerF2) and are designated as a single cluster here. The aCSDh is found between strands A and G in all structures despite slight topology differences in the vicinity, likely associated with remodeling of the binding pocket. The ML domains include house‐dust mite allergens (e.g., PDB: 1xwv Cys 8–119) and Niemann‐Pick C2 proteins from ants to mammals (e.g., PDB: 1nep Cys 8–121), as well as proteins of the innate immune system including lymphocyte antigens 86/96 (e.g., PDB: 2e56 Cys 37–148) and ganglioside M2 (GM2) activator (e.g., PDB: 1pu5 Cys 10–154).
An example of a class 2 end‐aCSDh, next to an NH β‐bridge, is the catalytic disulfide in the bacterial 2‐Cys peroxiredoxin ahpC (PDB: 1yex, 1yf0), formed between Cys 46 of one monomer and Cys 165 of another.28, 29 This enzyme provides protection from oxidative stress in bacteria by catalyzing the reduction of hydrogen peroxide and organic hydroperoxides to their corresponding alcohol and water. We note, however, that the end‐aCSDh sub‐motif is quite weak in ahpC, with the hydrogen bonds being broken in several structures. The other two well‐characterized redox‐active disulfides in the dataset were also of the end2‐aCSDh class, with the single P1 lysozyme structure and 3 of 8 ribonucleotide reductase structures adopting this motif. The other five ribonucleotide reductase structures were end1‐aCSDh.
With respect to lateral embeddedness, end‐aCSDhs are most often found on the edges of β‐sheets (> 70% of individual disulfides in the dataset), however many of these structures are redundant (i.e., in the same cluster). In stark contrast with true‐aCSDhs, only 5 end‐aCSDh structures were found in mid‐sheet LatE contexts (3%). In terms of end‐aCSDh clusters, in 11 of the 23 unique examples, the disulfides are found exclusively on the edge of a sheet and in 8, exclusively on a β‐ribbon. In addition, there are four clusters where both β‐ribbon and edge‐of‐sheet LatE contexts were seen. Interestingly, there appears to be a correlation between the end‐aCSDh class and LatE. In the majority of clusters with the end‐aCSDh at the “end of a β‐ladder” (LonE = end1‐aCSDh), it is located on the edge of the β‐sheet (latE = edge), whereas the end2‐aCSDh cluster disulfides tend to be on β‐ribbons. In six of the 11 clusters with structures exhibiting end2 embeddedness, alternate structures of the cluster have end1‐aCSDh embeddedness, suggesting these structures with relatively few hydrogen bonds may be more conformationally labile.
Several Class 2 end‐aCSDhs associated with β‐ribbons are known to be redox active. In addition to the ribonuclease reductase (RNR) and ahpC disulfides described earlier, the aCSDh in bacteriophage P1 lysozyme (PDB: 1xjt) is involved in a thiol‐disulfide isomerization reaction that activates the lysozyme.30 In contrast with ahpC, where the aCSDh must be reduced to release the catalytic Cys, in P1 lysozyme the formation of the end‐aCSDh sub‐motif between Cys 13 and Cys 44 releases the catalytic Cys 51.30 In all three characterized examples, no thiol oxidoreductase has yet been identified that mediates the reduction of the disulfide. In RNR and P1 lysozyme, the aCSDh isomerizes internally with another cysteine thiol.
It is interesting to note the contrast between the levels of lateral embeddedness seen for aCSDn and aCSDh sub‐motifs. True‐ and end‐aCSDns are spread between all three possible LatE contexts11 whereas, amongst the structures in the dataset, aCSDh sub‐motifs appear to show a strong correlation with a particular level of lateral embeddedness: true‐aCSDhs are usually found in mid‐sheet LatE contexts; Class 1 end‐aCSDhs are commonly on the edges of β‐sheets; while Class 2 end‐aCSDhs are frequently on β‐ribbons. In the case of true‐aCSDhs, it is likely that the presence of a mid‐sheet LatE context is necessary to stabilize the highly strained true‐aCSDh sub‐motif and prevent the Cys–Cys hydrogen bonds from being broken.
Only one structure was found in the dataset that could be described as a β‐bridge‐aCSDh: an inter‐chain disulfide found in a monoclonal antibody against the acetylcholine receptor (PDB: 1fn4). However, this disulfide links two C‐terminal Cys residues, with the chain‐terminating carboxylate anions being involved in both hydrogen bonds. As such, data from this structure may not be characteristic of the β‐bridge‐aCSDh sub‐motif, should it exist elsewhere in nature. There were also four structures of mutated sequences where the aCSDh disulfide had been engineered into the protein for technical purposes. These will not be discussed further here.
Disulfide conformations of aCSDhs and their interactions with the protein backbone
The disulfide conformation is used as an important descriptor of protein disulfide bonds. The conformation depends on the three critical dihedral angles: χ2 (CαCβSγSγ′), χ3 (CβSγSγ′Cβ′) and χ2′ (SγSγ′Cβ′Cα′) [see Fig. S5(A)]. The sign of the χ3 dihedral angle gives the chirality or “handedness” of the disulfide, while χ2 and χ2′ determine the conformation name, as shown in Figure S5(B). While aCSDns have a strong preference for right‐handed conformations,11 i.e., conformations with positive χ3 dihedral angles, aCSDhs have an equally strong preference to be left‐handed (Table 3). Almost all weaktrue‐ and end‐aCSDhs have negative values of χ3 (left‐handed). As for aCSDns, the source of this preference in disulfide chirality can be traced to the almost universal right‐handed twist of antiparallel β‐sheets. If a disulfide in an aCSDn motif adopts a right‐handed conformation, it can be incorporated into a β‐sheet without exerting any additional twisting strain on the structure.11 Alternately, to adopt a left‐handed conformation, an aCSDn must be associated with an unfavorable negative sheet twist. Hence, 96% of aCSDns in the dataset are right‐handed. In contrast, for aCSDhs it is a left‐handed disulfide conformation that is most compatible with a right‐handed sheet twist, while a disfavored left‐handed twist is induced by the presence of a right‐handed aCSDh. This can be seen in Figure 4, which shows optimized geometries of model compounds for the aCSDh motif. The optimized structure with a left‐handed disulfide [Fig. 4(A)] shows a twist between the two Cys residues of +26.6° [Fig. 4(Aiii)]. This is in reasonable agreement with the observed value of +20 ± 10° found by Ho and Curmi20 for HB sites in antiparallel β‐ribbons where no disulfide is present (hereafter referred to as “normal” HB sites). Conversely, calculations on a right‐handed aCSDh system [Fig. 4(B)] predict a sheet twist of −11.0° [Fig. 4(Biii)]. This negative twist is strongly disfavored. Only true aCSDhs are equally likely to be left‐ or right‐handed and these are largely associated with relatively flat β‐sheets. In practice, the few right‐handed aCSDhs in the dataset show a right‐handed twist between the two Cys, with the strain due to the non‐preferred handedness of the aCSDh being compensated for by adoption of unfavorable disulfide dihedral angles.
Table 3.
Average Disulfide Torsional Energies (DTEs), (χ1, χ1′) Torsional Energies and Total Torsional Energies (TTEs) for Left‐ and Right‐Handed aCSDh Motifs and Conformations Along with their Standard Deviations and the Number of Relevant Structures.
| Left handed | Right handed | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DTE | χ1χ1′ | TTE | DTE | χ1χ1′ | TTE | ||||||||||
| LonE | Conf. | No. | Ave | Stdev | Ave | Stdev | Ave | Stdev | No. | Ave | Stdev | Ave | Stdev | Ave | Stdev |
| true | hook | 4 | 8.6 | 0.9 | 7.3 | 0.6 | 16.0 | 0.8 | |||||||
| staple | 5 | 20.6 | 6.4 | 4.9 | 5.7 | 25.6 | 11.3 | 2 | 21.2 | 5.0 | 23.1 | 3.1 | 44.4 | 1.8 | |
| weaktrue | hook | 48 | 8.0 | 4.0 | 5.1 | 1.4 | 13.1 | 3.6 | |||||||
| staple | 1 | 15.7 | NA | 11.9 | NA | 38.3 | NA | ||||||||
| end1 | hook | 58 | 7.3 | 2.2 | 7.3 | 4.0 | 14.5 | 4.4 | 1 | 7.9 | NA | 14.6 | NA | 22.5 | NA |
| GGT | 51 | 9.7 | 4.6 | 3.8 | 3.4 | 13.4 | 6.3 | ||||||||
| G′GT | 19 | 11.2 | 5.8 | 7.6 | 5.2 | 18.8 | 9.6 | 5 | 8.1 | 1.8 | 10.1 | 4.1 | 18.2 | 2.8 | |
| saddle | 3 | 11.3 | 0.3 | 11.8 | 0.7 | 23.2 | 0.9 | ||||||||
| other | 2 | 34.4 | 4.0 | 5.1 | 2.1 | 39.6 | 6.1 | ||||||||
| minor max | 1 | 45.5 | NA | 16.6 | NA | 62.2 | NA | ||||||||
| staple | 1 | 28.0 | NA | 15.1 | NA | 43.1 | NA | ||||||||
| end2 | hook | 8 | 7.7 | 2.7 | 5.6 | 3.4 | 13.3 | 1.3 | 1 | 17.5 | NA | 10.8 | NA | 28.3 | NA |
| GGT | 8 | 14.0 | 5.7 | 1.9 | 1.9 | 15.9 | 6.7 | ||||||||
| G′GT | 3 | 18.5 | 0.0 | 10.8 | 0.0 | 29.4 | 0.0 | 5 | 8.1 | 1.8 | 10.1 | 4.1 | 18.2 | 2.8 | |
| other | 1 | 39.7 | NA | 2.1 | NA | 41.7 | NA | ||||||||
| saddle | 2 | 21.9 | 6.1 | 9.1 | 7.3 | 31.1 | 1.3 | ||||||||
Energies are given in kJ mol−1.
Figure 4.
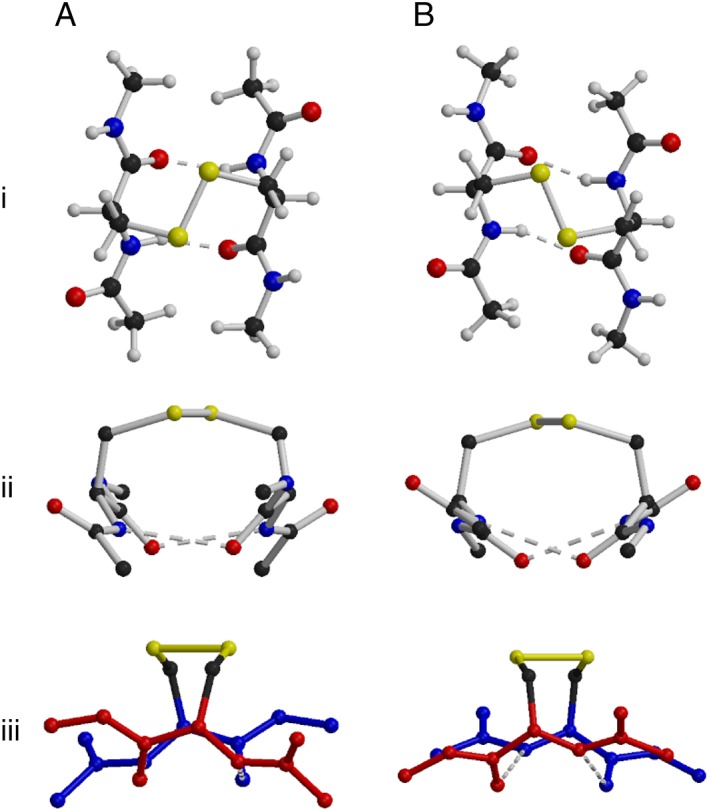
Optimized geometries for an aCSDh model compound. Atom size has been reduced and hydrogen atoms removed in the side views to improve clarity. The chains have been given different colors in iii to emphasize the sheet twist. (A) Compound with a left‐handed staple disulfide inducing a right‐handed twist in the sheet. (B) Compound with a right‐handed staple disulfide inducing a left‐handed sheet twist.
Although left‐ and right‐handed disulfides have opposite effects on sheet twist when in HB or NHB sites, the effect of the handedness of the disulfide on the sheet shear is the same for both sites. As for aCSDns, calculations on the right‐handed model compound predict a higher sheet shear (+0.80 Å) between the Cys than seen between normal HB site residues (+0.58 ± 0.25 Å) [see Fig. 4(Bi)]. In contrast, shearing in the left‐handed model acts against the natural positive sheet shear. This counteracting shear is so strong that it results in a net negative shear between the two Cys residues of −0.44 Å [Fig. 4(Ai)]. This negative shearing means the sheet loses the additional stabilization normally acquired through the formation of weak hydrogen bonding‐like interactions between the carbonyls and Hα atoms (see Fig. S1).
Left‐handed conformations do have the advantage, however, of producing a larger separation between the sulfur atoms and the carbonyl oxygens (as seen in Fig. 4i, 4ii), thereby minimizing any potential steric interactions between these atoms. These steric interactions were proposed by Indu et al.15 to be one of the factors disfavoring aCSDh motifs. While these interactions could add to the strain of an aCSDh, particularly one adopting a right‐handed conformation, the tilting of the Cys residues in these motifs (Fig. 4ii) acts to increase the S‐O distance, meaning that the contribution of these steric interactions to the instability of the motif are likely to be overshadowed by destabilization caused by sheet disruption, as described above.
The disulfide conformations adopted by aCSDhs in the dataset are shown in Figures 5(A) and 5(B). True‐aCSDhs most often adopt staple conformations, with a single cluster favoring the right‐handed hook (celQ in Table 1). Five of the true‐aCSDh structures in the dataset are left‐handed staples, i.e., they have (χ2, χ3, χ2′) ~ (+60°, −90°, +60°), and another two are right‐handed, i.e., (χ2, χ3, χ2′) ~ (−60°, +90°, −60°). In order to accommodate this disulfide conformation, which normally has r(Cα–Cα) ~ 4.0 Å, in the HB site of a β‐ladder where r(Cα–Cα) is typically ~ 5.3 Å, the χ3 dihedral angle is usually stretched beyond ±130°. Using quantum‐chemical techniques, a potential energy surface (PES) describing the changes in the strain experienced by a disulfide due to twisting around the χ2, χ3 and χ2′ dihedral angles was calculated.3 The PES can be used to predict the torsional strain of a disulfide using the dihedral angles from the crystal structure (http://www.sbinf.org/applications/pes.html).4 Reference to the PES shows that while the staple region of the PES might be expected to be associated with a local minimum, steric interactions between the Hα atoms (due to the short Cα–Cα distance) destabilize the staple conformation for most values of |χ3| [see Fig. S5(B)]. For the very high |χ3| values seen in staple aCSDhs, however, these steric interactions are reduced and a minimum is found on the PES; nevertheless, the unfavorable values of χ3 mean that these are still highly strained structures. Predictions of disulfide stability for the true‐aCSDh structures in the dataset show very high strain both in the disulfide bond (χ2, χ3, χ2′; known as the disulfide torsional energy or DTE) and also in the in χ1 and χ1′ dihedral angles (in all examples excepting one cluster). The DTE and the strain in χ1 and χ1′ can be combined to give the total torsional energy (TTE) of a disulfide. For four of the 12 true‐aCSDh structures, this is predicted to be over 40 kJ mol−1. The twist and shear experienced by true‐aCSDhs in the dataset was also assessed: those in the five clusters with positive twists have relatively low twisting strain (average twist ~ +37°), although one disulfide is associated with a large negative shear (PDB: 2z0l ~ −1.0 Å). The single cluster with apparent negative twists (CBM_3, average twist ~ −53°) also exhibit a large positive shear (~ 1.5 Å). These aCSDhs are adjacent to a β‐bulge.
Figure 5.
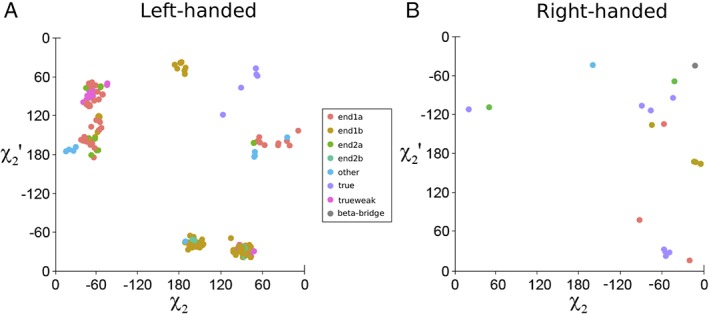
Disulfide conformations adopted by aCSDh motifs. Left‐handed conformations are shown in Map A and right‐handed conformations in Map B. Axes have been reversed for left‐handed conformations, so the same conformations are found in the same region of the PES.
Most weaktrue‐aCSDhs are found in the left‐handed hook region of the PES, ie. χ3 is negative, while one of χ2 or χ2′ is ~ +60° and the other is ~ +120°. This is a medium energy region of the surface, with DTEs between 5 and 10 kJ mol−1. Thus, although weaktrue‐aCSDhs are much less common than true‐aCSDns, the disulfide bond itself (χ2, χ3, χ2′) is actually less strained (average DTE of 8.2 kJ mol−1 vs 12.3 kJ mol−1 for aCSDns,11 see Table 3). It should be noted, however, that these structures experience higher strain in χ1 and χ1′ than true‐aCSDns, with one of these dihedral angles twisted to ~35° (semi‐eclipsed). Including an estimate of the χ1 and χ1′ contributions to the torsional strain indicates that disulfides belonging to the weaktrue‐aCSDh class experience similar levels of strain to true‐aCSDns (average TTEs 13.7 vs. 14.0 kJ mol−1 for true‐aCSDns). In addition, while these structures do not experience significant twisting strain (an average twist of +27°), a large negative shear is observed between the two Cys residues, on average−1.72 ± 0.17 Å. These large negative values are partially the result of the rotation of one of the Cys into an α backbone conformation (rotation of one of the N's away from the cross‐strand H bonding position). This alters the residue and backbone vectors and contaminates the shear calculation. Nevertheless, superposition of the true‐ and weaktrue‐aCSDh sub‐motifs [shown in Fig. 6(A)] reveals they experience very similar levels of shear, i.e., ~ − 1.0 Å. Thus, these structures experience significant disruption of the sheet, which is not seen for true‐aCSDns. If it were possible to quantify these effects, it is highly likely that weaktrue‐aCSDhs, like true‐aCSDhs, would also be found to be much less stable than true‐aCSDns.
Figure 6.
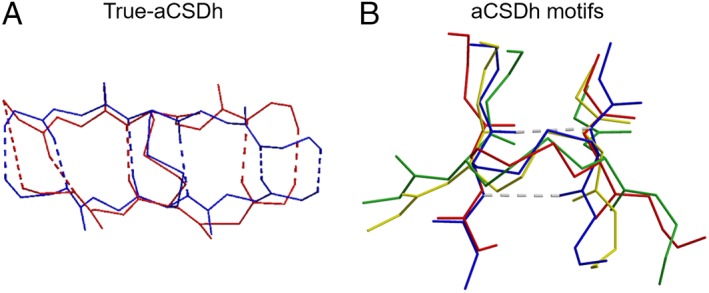
(A) Superimposition of a true‐aCSDh (PDB: 1lxd Cys A16 to Cys B16; blue) and a weaktrue‐aCSDh (PDB: 1qfo A Cys 22 to Cys 79; red), showing the similar levels of sheet shear. (B) Superimposition of disulfide conformations adopted by disulfides in aCSDh motifs. Blue: left‐handed staple (PDB: 1lxd Cys A16 to Cys B16), red: left‐handed G′GT (PDB: 1rlr A Cys 225 to Cys 462), yellow: left‐handed hook (PDB: 1xjt A Cys 13 to Cys 44), green: left‐handed GGT (PDB: 1md6 A Cys 7 to Cys 153).
The lifting of the structural constraints imposed by the H bonding of true‐BSD sub‐motifs might be expected to allow relaxation of end‐BSDs into more favorable disulfide conformations. While such relaxation is not seen for end‐aCSDns,11 almost none of the end‐aCSDh structures in the dataset adopt the staple conformation seen in their true‐aCSDh analogues. Instead, for end‐aCSDhs relaxation occurs by several different mechanisms, resulting in disulfide structures that are scattered between different regions of conformational space (see Fig. 5). These different modes of relaxation are shown in Figure 6(B) in comparison with a true‐aCSDh (in blue).
In the first class of structures (shown in yellow in the figure), the N of one of the Cys is rotated away from its normal position in the β‐chain, allowing twisting of χ1 and χ2 (or χ1′ and χ2′) to give a hook conformation. This is the same mode of relaxation seen in the weaktrue‐aCSDh structures. Some of the disulfides that use this relaxation mode include those found in ML domains (mentioned earlier) and the disulfide connecting Cys 173 to Cys 180 in acid‐sensing ion Channel 1 (PDB: 2qts). In these end‐aCSDhs the Cys residues have a slightly higher relative sheet twist in comparison with their weaktrue‐aCSDh analogues (+37 ± 7° vs. +27 ± 5°), while the (contaminated) measured relative shear is marginally reduced (−1.46 ± 0.43 Å vs. −1.73 ± 0.14 Å).
In a second class of structures, it is the carbonyl of one of the Cys that twists to give a G′GT disulfide conformation [red in Fig. 6(B); see also Fig. S5]. This mode of relaxation is seen in the catalytic disulfide of ribonuclease reductase (RNR) and also in the end‐aCSDh between Cys 36 and Cys 64 of Archaeon Aeropyrum pernix Cys synthase (PDB: 1wkv). This movement actually works against the natural twisting of the sheet, resulting in an inter‐residue twist in the disulfide ~ below +10° (+9.5 ± 6.1° for the 7 RNR structures). (For some of these disulfides, twisting of both peptide bonds on the unbound side of the disulfide away from a H bonding conformation contaminates the calculated sheet twist. However, superposition of these structures reveals that the actual twist is similar to that of other G′GT end‐aCSDhs. This group of structures has therefore been excluded from the average twist calculation.) While this very low twist indicates that the conformation exerts twisting strain on the system, it does allow relaxation of the sheet shear. Although measurement of the shear will again be affected by the carbonyl movement (as described above for the movement of the N in true‐aCSDhs), the calculations do reveal a more natural positive shear for the G′GT end‐aCSDhs (measured average +0.90 ± 0.21 Å).
For the third class of structures [green in Fig. 6(B)] both the N and the carbonyl twist, resulting in a GGT conformation (see Fig. S5). The redox active end‐aCSDh in ahpC belongs in this category, along with (in most cases) the disulfide between Cys 10 and 154 in Ganglioside M2 activator (PDB: 1pu5). In these structures the two Cys residues are no longer even approximately parallel, so calculations of relative twist and shear are often not meaningful. In general, GGT structures have similar calculated twists to the hook end‐aCSDhs (+43 ± 15°) but less negative measured shears (−0.77 ± 0.58 Å).
Generally, structures in the GGT minima have χ3 greater than −88°, structures in the hook regions have χ3 between −88° and −100°, while in those that adopt G′GT conformations χ3 is less than −100°. This is consistent with the way the PES changes with χ3.4 When |χ3| is less than 90° the GGT conformation is more stable than the hook. Between 90° and 110° the hook is more stable, but for |χ3| ≥ 110° the surface begins to level out, so there is no significant difference in energy between the three regions. There does not appear to be any correlation between the end‐aCSDh class and the relaxation mode adopted (and hence also no correlation with the torsional strain of the disulfide).
The different relaxation modes available to end‐aCSDhs result in a very wide distribution of DTEs (see Table 3). For left‐handed examples, the overall average DTE is 9.8 kJ mol−1, with the more abundant hook (mean: 7.3 kJ mol−1, n = 66) and GGT end‐aCSDhs (mean: 10.2 kJ mol−1, n = 59) being more stable, and the G′GT structures being less so (mean: 12.2 kJ mol−1, n = 22). For aCSDns, both Cys tend to adopt a gauche+ χ1 conformer (χ1 ~ −60°).11 This means there is minimal strain on χ1 and χ1′ and 80–90% of the dihedral strain is instead accommodated in the three central dihedral angles (χ2, χ3, χ2′). In order to connect across an HB site, however, the Cys residues must instead have gauche‐ (positive) χ1 conformations. Ideally both Cys would have χ1 ~ +60°, however this would place the Cβ–Sγ bond roughly perpendicular to the chain direction and result in a displacement between the two Cα atoms (a sheet shear) of ~2 Å. A compromise must therefore be made between the shearing strain in the sheet and non‐optimal (somewhat eclipsed) χ1 dihedral angles. As a result, the Cys must adopt higher energy χ1 values in addition to the strained conformations in χ2, χ3 and χ2′. Thus, in aCSDhs, the strain in the disulfide is more equally accommodated between χ1 and χ1′ and the three central dihedral angles compared to aCSDns. Including χ1 and χ1′ contributions in the calculations of the TTE raises the mean energy to 15.7 kJ mol−1 (hook 14.3 kJ mol−1, GGT 13.8 kJ mol−1, G′GT 20.3 kJ mol−1). Thus, the hook and GGT end‐aCSDhs are actually more stable than the corresponding end‐aCSDns (average TTE 16.0 kJ mol−1), however end‐aCSDhs with a G′GT conformation are quite unstable and likely to be more susceptible to reduction.
Discussion
Redox activity of aCSDhs
Reviewing the literature relating to proteins containing aCSDh motifs revealed that in three of the 37 clusters, the disulfide has been identified as performing an important biochemical redox function (see Table 1), while redox activity has been ascribed to a fourth. Two of the disulfides are catalytic while redox activity of a third is associated with a conformational change. As noted above, all three of these disulfides are associated with end‐aCSDh Class 2 sub‐motifs. The catalytic disulfide of ribonucleotide reductase (Cys 225–462) is responsible for the reduction of ribonucleotides to deoxyribonucleotides.18 This disulfide mostly adopts the left‐handed G′GT conformation and, as might be expected for such a reactive disulfide, experiences very high levels of strain (average TTE 31 kJ mol−1, DTE 19 kJ mol−1, χ1 and χ1′ strain 11 kJ mol−1). The disulfide isomerizes spontaneously to a surface aCSDn in ribonucleotide reductase that is reset by Trx, a mechanism reminiscent of the arsenate reductase reaction cycle.31 Another catalytic disulfide that forms an aCSDh motif which is known to be functionally important is formed between the catalytic and resolving Cys of the ahpC peroxiredoxin. Curiously, the use of both relaxation modes for the end‐aCSDh sub‐motif (to give a left‐handed GGT conformation) has resulted in a disulfide with a very low TTE (7.0 and 8.8 kJ mol−1 in the two structures in the database). We note, however, that X‐ray structures of this protein are complicated by flexibility of the C‐terminus that contains the resolving Cys (Cys 165). In many instances no residues beyond this Cys can be modeled. Although an aCSDh motif cannot be identified in these cases where the relevant flanking residues are missing, the mean total strain for the Cys 165–Cys 46 interchain disulfides in these structures is ~25 kJ mol−1. In light of this, too great a significance should not be placed on the very low strain energies predicted for the redox active ahpC end‐aCSDhs in the dataset. In contrast, the aCSDh that is formed in order to activate P1 lysozyme is intermediate in energy. In the single structure of the active form of this protein in the dataset, the disulfide adopts the left‐handed hook conformation with a TTE of 13.4 kJ mol−1, with the energy evenly split between (χ2, χ3, χ2′) and (χ1, χ1′). With the additional backbone strain due to the presence of the aCSDh motif, it is not unreasonable to expect that this disulfide may be involved in further redox reactions. In a fourth case, Cutf—an enzyme of copper homeostasis (PDB: 2z4i) —the disulfide is disrupted by Cu.32 We note that although redox activity has not yet been reported for other aCSDh clusters in the dataset, in many cases such functionality has not yet been looked for. It is extremely likely that other aCSDhs, particularly the highly strained ‘true’ sub‐motifs, will be susceptible to reduction under physiological conditions.
Aside from literature evidence, there were also a number of structures where the aCSDh was reduced in other chains or alternate structures, which might be due to the presence of reductants such as DTT in the crystal solution or radiation damage acquired during the structure solution process, particularly in high‐intensity X‐ray sources. These are noted in the last column of Table 1.
There were also some interesting patterns and trends in how the aCSDh is disposed in the overall fold. Several aCSDhs span almost their entire domain in sequence, often joining the first and last β‐strands. Examples include interleukin 36 receptor antagonist (PDB: 1md6) and the previously mentioned ML domains. Other aCSDhs span only short segments of sequence straddling a β‐hairpin (PDBs: 5bvw, 3fl7, 1htd, 3ngm, 4odj). Several aCSDhs were also located close to other forbidden disulfide motifs. An example in two non‐homologous clusters, where one of the aCSDh Cys could alternately form a vicinal disulfide that may protect and promote dimerization under oxidizing conditions, was discussed above (see Fig. S4). In another example from non‐homologous clusters, the two Cys of an aCSDh disulfide are located on the same strands as the Cys of an aCSDn motif, with the aCSDn Cys nested within the aCSDh Cys in sequence (PDBs: 3fl7, 4x1j) (see Fig. S7). We can only speculate on the mechanism of what is likely to be a compound redox switch. Reduction of some aCSDns modulates the conformation of the hairpin sequence they straddle.1 If a ligand is engaged is this process by these proteins, reduction of the distal aCSDh may be required to release it.
aCSDhs were also found in folds that were previously identified as bearing other forbidden disulfides including β‐propellers and transthyretin.1 There are two examples of aCSDhs in fusion proteins in the dataset, in the henipivirus HN (PDB: 4uf7) and in the Caenorhabditis elegans cell–cell fusion protein eff‐1 (PDB: 4ojd), which is homologous to Class ii viral fusion proteins. We have previously observed that viral fusion proteins often contain multiple forbidden disulfides within a single fold9 and eff‐1 is similar. In addition to the aCSDh, eff‐1 contains an aCSDn between Cys 131–187 as well as other isomerized forbidden disulfide motifs.
With regard to the biological functions of the resident proteins, a number of weaktrue aCSDhs are located close to functional binding sites. These are an aCSDh in the glycine receptor, bridging the glycine binding site (PDB: 5tin), another in IL‐17 receptor A near a sugar‐binding site (PDB: 5n9b), a third in the lipocalin myelin‐binding protein near the palmitic acid‐binding site (PDB: 4a8z), a fourth near the lid of the lipases (PDB: 3ngm) and a fifth in the previously mentioned SIGLEC domains, which are Vset Ig domains that have evolved to specifically bind sialic acid. Literature on these disulfides often cites their role is fashioning the shape of the binding site. The aCSDh, which is specific to the SIGLEC Vset domains, widens the interior of the β sandwich, making it accessible for interaction with sialic acid.33 The aCSDh in the glycine receptor straddles a β‐hairpin known as the orthosteric binding site where strychnine and other ligands are bound.34 Given the known redox activity of aCSDhs and their co‐location with binding sites, it seems likely that redox‐activity has a role in the binding or release of ligands in these proteins. This notion is supported by the aCSDh in the ML domains, where the domain has evolved from a simple cholesterol‐binding carrier molecule to a molecule of the immune system that, in its lipopolysaccharide‐bound state, engages a Toll receptor. In both ants and dust mites, it is a carrier molecule of a chemo‐sensing system. Accommodation of the different ligands has been achieved by evolutionary remodeling of several elements of secondary structure. Despite the changes to the fold, the aCSDh near the ligand‐binding site has been conserved, suggesting it is crucial to this function. The aCSDh in dust mite allergen (Table 1, Cys 8–119), as well as a second disulfide between Cys 73–78, are critical to the IgE‐binding capacity of Der f 2 in sera of several mite‐allergic patients.35
Some proteins with functions previously associated with forbidden disulfides were also represented in the aCSDh dataset,1, 9 specifically molecules of the immune system as well as proteins from pathogens. One aCSDh is in a viral HN that gains cell entry through recognition of the human ephrinB2 receptor while another is in a DNA processivity factor, BMRF1. With regard to the immune system, multiple molecules containing aCSDhs were found in proteins of both the innate and adaptive immune systems. The ubiquitous Ig/FIII fold also featured repeatedly among the folds in the dataset: Ig‐like folds represented included the modified Vset of the SIGLECs, the modified Eset of the ML domains as well as the Fibronectin type III domains of CD45 and IL17RA. These folds are often employed in tandem as spacers in their resident proteins. The innate immune system is represented by the ML domains, which respond to foreign lipopolysaccharides in vertebrates, and IL Family 5. Perhaps unsurprisingly, the aCSDhs are frequently found in modified versions of the Ig fold that are associated with the “business end” of the molecule, suggesting the aCSDh is crucial to an evolved function. Canonical Ig domains contain an inter‐sheet disulfide between Cys residues on strands B and F. In the SIGLECs there is instead an intra‐sheet aCSDh between Cys 22 and 79, located on the B and E strands of the ABED sheet. The standard Ig Cys residue on the F strand is replaced by Phe‐96, which packs against the aCSDh. The conversion of the standard intersheet disulfide in the Ig domain of the SIGLECs into a forbidden disulfide is somewhat reminiscent of CD4 where the canonical Ig intersheet disulfide has been modified to form an aCSDn between strands C and F.36 This disulfide has been demonstrated to be reduced during the process of T‐cell signaling, and its reduction is also co‐opted during entry of the HIV viral envelope glycoprotein (gp120) into the cell.36
Finally, some proteins in the dataset contained more than one aCSDh located in different parts of the protein. The dual aCSDhs in the angiotensin‐converting enzyme (ACE) have likely arisen by gene duplication. Nonetheless, it is important to note that despite sequence divergence of the N‐ and C‐domains, the aCSDh has been retained, supporting its functional importance. The dual aCSDhs in tandem FIII domains of the extracellular portion of receptor‐type tyrosine‐protein phosphatase C (PTPRC, CD45), a cell surface receptor required for T‐cell activation, are not so easily explained. They are not located in homologous positions of the fold. The more N‐terminal aCSDh (PDB: 5fmv Cys 309–320) straddles strands A and B of the FIII domain whereas the aCSDh in the more C‐terminal FIII domain (Cys 503–535) is between strands B and E. Nor are the specific locations of the two B strand Cys homologous: they are out of register in the two domains and at opposite ends of the B strand. Thus, the aCSDhs in these two tandem FIII domains appear to have evolved completely independently, which is intriguing. Previously, in noting the reuse of certain folds for redox‐active functions, we speculated it may be related to the redox‐responsive promoters being retained during gene duplication. While this might be the case for the duplicated promoter in ACE, it cannot explain the dual aCSDhs in PTPRC. The reuse of the very same type of forbidden disulfide in a non‐homologous position in the fold suggests some positive evolutionary pressure on the protein to include aCSDhs specifically. This would be the case if reduction of both aCSDh disulfides is affected by the same protein reductant.
Previously we postulated that aCSDns may be cognate substrates of the redox homeostasis protein Trx, which would account for their prevalence in protein structures.1, 11 In general, the low abundance of aCSDhs in the PDB argues against a common reductant like Trx. Indeed, some aCSDhs have such high torsional energies that they are instead reduced spontaneously in their resident proteins by internal isomerization and other mechanisms. However, another interesting feature of many of the proteins containing aCSDhs in the dataset is that they cycle through endosomes. Thus many of these disulfides may rely on a change in solvent milieu, rather than a cognate ligand, to reduce the disulfide. For example, the carrier proteins that cycle through endosomes may release their cargo upon reduction of the disulfide.
Conclusion
Like aCSDns, the two Cys of an aCSDh motif are found as cross‐strand partners in antiparallel β‐sheet but differ, in that the two Cys residues of the disulfide occupy an HB site and are additionally linked by one or two backbone hydrogen bonds. This places significant strain on the β‐sheet, with the two β‐chains tilting toward each other in order to allow the Cys Cα atoms into close enough proximity for the disulfide bond to form. These high levels of strain mean that either the disulfide or the hydrogen bonds that form the aCSDh motif (or both) are highly susceptible to cleavage. Here, we investigated the populations, embeddedness, disulfide conformations and torsional strain of this class of BSDs.
aCSDhs in HB sites of antiparallel β‐sheet are far less common than their aCSDn analogues in NHB sites of protein β‐sheets. They are represented by 240 protein structures in 37 unique clusters in comparison to 20564 aCSDn structures in over 252 different clusters. Unlike aCSDns, end‐aCSDhs are far more common than those with “true” levels of LonE. A mid‐sheet LatE context appears necessary to stabilise a true‐aCSDh sub‐motif, with the additional β‐chains flanking the β‐ladder containing the true‐aCSDh helping to constrain the two Cys residues to the β region of the Ramachandran plot. When these extra strands are not present, either or both of the Cys tend to twist away from β‐conformation, breaking one of the hydrogen bonds between the Cys and often leading to end‐aCSDh sub‐motifs. This allows relaxation of the highly strained conformations seen for true‐aCSDhs to lower energy hook, GGT or G′GT disulfides.
Ab initio quantum chemical calculations give important insights into the interactions between aCSDh motifs and the protein backbone. Left‐handed aCSDh conformations produce a positive (right‐handed) twist between the two Cys residues while right‐handed aCSDhs have a strongly disfavored left‐handed twist. As β‐sheets have an inherent right‐handed twist, only left‐handed aCSDhs can be incorporated into proteins without additional strain. There is a complication, however, as left‐handed aCSDh conformations also produce large negative shears between the two Cys residues. This means that aCSDhs experience high levels of shearing strain, which further destabilizes the motifs.
As for other forbidden disulfides, aCSDhs have been shown to be involved in several reactive processes. The most important example involves the end‐aCSDh connecting Cys 225 to Cys 462 of ribonucleotide reductase. This highly strained disulfide (average TTE of 29 kJ mol−1) is responsible for the reduction of ribonucleotides to deoxyribonucleotides and is therefore essential for the biosynthesis of DNA. The generally higher torsional strains associated with aCSDhs likely make them less stable then aCSDns and more easily reduced. Unlike aCSDns, which are known to undergo assisted reduction by Trx‐like thiol oxidoreductases, the higher energies of aCSDhs may allow them to be reduced without the assistance of a thiol oxidoreductase. In the case of ribonucleotide reductase, ahpC and P1 lysozyme, internal isomerization with another thiol may affect this process. For other instances, changes in the redox milieu may be sufficient. As a large number of the proteins containing aCSDhs in the dataset are known to cycle through endosomes, this trafficking may be sufficient to reduce the disulfide. In several instances the aCSDh is near a ligand‐binding site, suggesting that reduction of the disulfide may allow release of the ligand.
Methods
Construction of reference database and non‐redundant dataset
The construction of the reference dataset of disulfide bonds from the PDB and the clustering of homologous disulfides in BSD motifs to form a non‐redundant dataset has been described in detail elsewhere.10 In brief, the dataset contains details of all disulfide bonds in X‐ray crystal structures with resolutions of 3 Å or better from a PDB snapshot of June 8 2018, comprising 140, 547 structures. Custom programs were developed to identify BSD motifs within this dataset and to incorporate information on secondary structure motifs from the SCOP database.37 The torsional strain on the disulfide bond, as well as the twist and shear between the Cys residues were also predicted, as described below. Properties of the disulfides, such as their SCOP family, Cys residue numbers and inter‐residue distance were used to cluster homologous disulfides together to give a non‐redundant set. Structures not assigned to a SCOP family were grouped with the appropriate cluster using the protein structure comparison service “Fold at European Bioinformatics Institute” (http://www.ebi.ac.uk/msd-srv/ssm)38 and the 3D Similarity function from the RCSB PDB website (based on the jFATCAT‐rigid algorithm),39 as well as fold data from the InterPro40 and Pfam41 databases. Swiss‐PDBViewer42 was used to superimpose and view structures to quality check the clustering process.
Ab initio calculations
Estimation of torsional strain in disulfide bonds
The relative stabilities of disulfide bonds were interpolated from a potential energy surface (PES) for torsion around χ2, χ3 and χ2′ (10° resolution). The PES was calculated using the model compound diethyl disulfide at the M05‐2X43/6‐31G(d)44 level of computational theory with complete geometry optimizations for each data point. Zero‐point energies were not included as these are too computationally expensive to calculate. All energies are quoted relative to the lowest energy minimum on the PES, which corresponds to disulfides adopting the spiral conformation.
Linear interpolation of the points on the PES was used to predict the torsional strain in χ2, χ3, and χ2′ experienced by each disulfide structure in the PDB. Throughout the analysis this is referred to as the disulfide torsional energy (DTE). The effects of strain in χ1 and χ1′ were estimated with empirical terms from the Amber force field.45 The sum of the disulfide strain/energy and the χ1, χ1′ strain is referred to as the total torsional energy (TTE).
Disulfide conformations
The χ2 and χ2′ dihedral angles (the disulfide conformations) adopted by left‐handed and right‐handed aCSDhs have been plotted separately in Figure 5(A) and (B). The axes are reversed for left‐handed disulfides so that the same conformations appear in the same regions of the plots.
Investigations of aCSDh handedness preferences
aCSDh motifs demonstrate a strong preference for left‐handed disulfide conformations. In order to investigate the reasons for this preference, M05‐2X/6‐31G(d) calculations were performed on model compounds for the protein disulfide bond. This model was designed to include all atoms of the Cys residues plus the carbonyl and Cα of the previous residue, and the N and Cα of the subsequent residue in each protein chain, i.e., the peptide linkages before and after each Cys. The Cα atoms of these additional residues were substituted by methyl groups to terminate the chains. This model allowed one hydrogen bond to be formed on each side of the disulfide, giving a reasonable small‐scale model of a CSD (Fig. 3). This model compound was fully optimized for both left‐ and right‐handed disulfide conformations. All computational chemistry calculations were performed using the Gaussian03 suite of programs46 on the AC and XE clusters at the NCI National Facility.
Sheet twist and shear
Insertion of a BSD into a β‐sheet or chain often results in distortion of the surrounding β structure. In order to obtain a measure of this distortion, the relative twist and shear between β‐partners in the region of the disulfide was calculated. Following the method of Ho and Curmi,20 each residue was described by a vector (the residue backbone vector) connecting the midpoints of its two peptide bonds (see Fig. S4). A chain backbone vector could thus be defined as the difference (in the case of antiparallel sheet) or sum (for parallel sheet) of the two residue vectors. The relative shear of the two β‐partners was calculated by projecting the positions of the Cα atoms of each residue onto the chain backbone vector and finding the distance between these projected points. For the purposes of this work, it was necessary to modify the method of Ho and Curmi for determining the relative twist between two β‐partners: instead of using the angle between the two residue backbone vectors, we used the dihedral angle between them, with the vector connecting the midpoints of the residue backbone vectors being the axis of the dihedral angle. Ho and Curmi were interested in residue pairs within a β‐sheet for which the two methods should give comparable results; we were additionally interested in obtaining a measure of the twist between residues at the end of β ladders, where the two chains may begin to diverge. The use of the dihedral angle reduces the influence of this divergence on the calculated twist.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Supporting information
Appendix S1: Supporting Information
Acknowledgments
The authors would like to thank the NCI national facility for a grant of computing time that allowed the quantum chemical calculations mentioned in this paper to be performed.
Contributor Information
Naomi L. Haworth, Email: naomi.haworth@sydney.edu.au.
Lixia Ma, Email: lixiama77@gmail.com.
Merridee A. Wouters, Email: mwouters@cmri.org.au.
References
- 1. Wouters MA, Fan SW, Haworth NL (2009) Disulfides as redox switches: From molecular mechanisms to functional significance. Antioxid Redox Signal 12:53–91. [DOI] [PubMed] [Google Scholar]
- 2. Fan SW, George RA, Haworth NL, Feng LL, Liu JY, Wouters MA (2009) Conformational changes in redox pairs of protein structures. Protein Sci 18:1745–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haworth NL, Gready JE, George RA, Wouters MA (2007) Evaluating the stability of disulfide bridges in proteins: a torsional potential energy surface for diethyl disulfide. Mol Simul 33:475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haworth NL, Liu JY, Fan SW, Gready JE, Wouters MA (2010) Estimating relative disulfide energies: an accurate ab initio potential energy surface. Aust J Chem 63:379–387. [Google Scholar]
- 5. Wouters MA, Lau KK, Hogg PJ (2004) Cross‐strand disulphides in cell entry proteins: poised to act. BioEssays 26:73–79. [DOI] [PubMed] [Google Scholar]
- 6. Yang Y, Song Y, Loscalzo J (2007) Regulation of the protein disulfide proteome by mitochondria in mammalian cells. Proc Natl Acad Sci USA 104:10813–10817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thornton JM (1981) Disulfide bridges in globular proteins. J Mol Biol 151:261–287. [DOI] [PubMed] [Google Scholar]
- 8. Richardson JS (1981) The anatomy and taxonomy of protein structure. Adv Protein Chem 34:167–339. [DOI] [PubMed] [Google Scholar]
- 9. Wouters MA, George RA, Haworth NL (2007) Forbidden disulfides: their role as redox switches. Curr Protein Pept Sci 8:484–495. [DOI] [PubMed] [Google Scholar]
- 10. Haworth NL, Wouters MA (2013) Between‐strand disulfides: forbidden disulfides linking adjacent β‐strands. RSC Adv 3:24680–24705. [Google Scholar]
- 11. Haworth NL, Wouters MA (2015) Cross‐strand disulfides in the non‐hydrogen bonding site of antiparallel β‐sheet (aCSDns): poised for biological switching. RSC Adv 5:86303–86321. [Google Scholar]
- 12. Wouters MA, Curmi PMG (1995) An analysis of side chain interactions and pair correlations within antiparallel β‐sheets: the differences between backbone hydrogen‐bonded and non‐hydrogen‐bonded residue pairs. Proteins Struct Funct Genet 22:119–131. [DOI] [PubMed] [Google Scholar]
- 13. Gunasekaran K, Ramakrishnan C, Balaram P (1997) β‐Hairpins in proteins revisited: lessons for de novo design. Protein Engin 10:1131–1141. [DOI] [PubMed] [Google Scholar]
- 14. Mandel‐Gutfreund Y, Zaremba SM, Gregoret LM (2001) Contributions of residue pairing to β‐sheet formation: conservation and covariation of amino acid residue pairs on antiparallel β‐strands. J Mol Biol 305:1145–1159. [DOI] [PubMed] [Google Scholar]
- 15. Hutchinson EG, Sessions RB, Thornton JM, Woolfson DN (1998) Determinants of strand register in antiparallel β‐sheets of proteins. Protein Sci 7:2287–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Santiveri CM, León E, Rico M, Jiménez MA (2008) Context‐dependence of the contribution of disulfide bonds to β‐hairpin stability. Chemistry 14:488–499. [DOI] [PubMed] [Google Scholar]
- 17. Indu S, Kochat V, Thakurela S, Ramakrishnan C, Varadarajan R (2011) Conformational analysis and design of cross‐strand disulfides in antiparallel β‐sheets. Proteins Struct Funct Bioinformat 79:244–260. [DOI] [PubMed] [Google Scholar]
- 18. Eriksson M, Uhlin U, Ramaswamy S, Ekberg M, Regnström K, Sjöberg B‐M, Eklund H (1997) Binding of allosteric effectors to ribonucleotide reductase protein r1: reduction of active‐site cysteines promotes substrate binding. Structure 5:1077–1092. [DOI] [PubMed] [Google Scholar]
- 19. Ellis HR, Poole LB (1997) Roles for the two cysteine residues of AhpC in catalysis of peroxide reduction by alkyl hydroperoxide reductase from Salmonella typhimurium . Biochemistry 36:13349–13356. [DOI] [PubMed] [Google Scholar]
- 20. Ho BK, Curmi PMG (2002) Twist and shear in β‐sheets and β‐ribbons. J Mol Biol 317:291–308. [DOI] [PubMed] [Google Scholar]
- 21. Ramachandran GN, Ramakrishnan C, Sasisekharan V (1963) Stereochemistry of polypeptide chain configurations. J Mol Biol 7:95–99. [DOI] [PubMed] [Google Scholar]
- 22. Vetter IR, Linnemann T, Wohlgemuth S, Geyer M, Kalbitzer HR, Herrmann C, Wittinghofer A (1999) Structural and biochemical analysis of Ras‐effector signaling via RalGDS. FEBS Lett 451:175–180. [DOI] [PubMed] [Google Scholar]
- 23. Murayama K, Nakayama S, Kato‐Murayama M, Akasaka R, Ohbayashi N, Kamewari‐Hayami Y, Terada T, Shirouzu M, Tsurumi T, Yokoyama S (2009) Crystal structure of Epstein‐Barr virus DNA polymerase processivity factor BMRF1. J Biol Chem 284:35896–35905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee B, Pernet O, Ahmed AA, Zeltina A, Beaty SM, Bowden TA (2015) Molecular recognition of human ephrinB2 cell surface receptor by an emergent African henipavirus. Proc Natl Acad Sci USA 112:E2156–E2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carugo O, Čemažar M, Zahariev S, Hudáky I, Gáspári Z, Perczel A, Pongor S (2003) Vicinal disulfide turns. Protein Eng Des Sel 16:637–639. [DOI] [PubMed] [Google Scholar]
- 26. Huang L, Weng X, Hofer F, Martin GS, Kirn S‐H (1997) Three‐dimensional structure of the Ras‐interacting domain of RalGDS. Nat Struct Biol 4:609–615. [DOI] [PubMed] [Google Scholar]
- 27. Maeda K, Hagglund P, Finnie C, Svensson B, Henriksen A (2006) Structural basis for target protein recognition by the protein disulfide reductase thioredoxin. Structure 14:1701–1710. [DOI] [PubMed] [Google Scholar]
- 28. Hillas PJ, del Alba FS, Oyarzabal J, Wilks A, de Montellano PRO (2000) The AhpC and AhpD antioxidant defense system of Mycobacterium tuberculosis . J Biol Chem 275:18801–18809. [DOI] [PubMed] [Google Scholar]
- 29. Parsonage D, Youngblood DS, Sarma GN, Wood ZA, Karplus PA, Poole LB (2005) Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry 44:10583–10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu M, Arulandu A, Struck DK, Swanson S, Sacchettini JC, Young R (2005) Disulfide isomerization after membrane release of its SAR domain activates P1 lysozyme. Science 307:113–117. [DOI] [PubMed] [Google Scholar]
- 31. Messens J, Martins JC, Van Belle K, Brosens E, Desmyter A, De Gieter M, Wieruszeski J‐M, Willem R, Wyns L, Zegers I (2002) All intermediates of the arsenate reductase mechanism, including an intramolecular dynamic disulfide cascade. Proc Natl Acad Sci USA 99:8506–8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hirano Y, Hossain MM, Takeda K, Tokuda H, Miki K (2007) Structural studies of the Cpx pathway activator NlpE on the outer membrane of Escherichia coli . Structure 15:963–976. [DOI] [PubMed] [Google Scholar]
- 33. May AP, Robinson RC, Vinson M, Crocker PR, Jones EY (1998) Crystal structure of the N‐terminal domain of sialoadhesin in complex with 3′ sialyllactose at 1.85 Å resolution. Mol Cell 1:719–728. [DOI] [PubMed] [Google Scholar]
- 34. Huang X, Chen H, Michelsen K, Schneider S, Shaffer PL (2015) Crystal structure of human glycine receptor‐α3 bound to antagonist strychnine. Nature 526:277–280. [DOI] [PubMed] [Google Scholar]
- 35. Nishiyama C, Fukada M, Usui Y, Iwamoto N, Yuuki T, Okumura Y, Okudaira H (1995) Analysis of the IgE‐epitope of Der f 2, a major mite allergen, by in vitro mutagenesis. Mol Immunol 32:1021–1029. [DOI] [PubMed] [Google Scholar]
- 36. Mohanasundaram KA, Haworth NL, Grover MP, Crowley TM, Goscinski A, Wouters MA (2015) Potential role of glutathione in evolution of thiol‐based redox signaling sites in proteins. Front Pharmacol 6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Murzin AG, Brenner SE, Hubbard T, Chothia C (1995) SCOP – a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247:536–540. [DOI] [PubMed] [Google Scholar]
- 38. Krissinel E, Henrick K (2004) Secondary‐structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst 60:2256–2268. [DOI] [PubMed] [Google Scholar]
- 39. Ye Y, Godzik A (2003) Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics 19:ii246–ii255. [DOI] [PubMed] [Google Scholar]
- 40. Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, de Castro E, Coggill P, Corbett M, Das U, Daugherty L, Duquenne L, Finn RD, Fraser M, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, McMenamin C, Mi H, Mutowo‐Muellenet P, Mulder N, Natale D, Orengo C, Pesseat S, Punta M, Quinn AF, Rivoire C, Sangrador‐Vegas A, Selengut JD, Sigrist CJA, Scheremetjew M, Tate J, Thimmajanarthanan M, Thomas PD, Wu CH, Yeats C, Yong S‐Y (2012) InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res 40:D306–D312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer ELL, Eddy SR, Bateman A, Finn RD (2012) The Pfam protein families database. Nucleic Acids Res 40:D290–D301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guex N, Peitsch MC (1997) SWISS‐MODEL and the Swiss‐PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723. [DOI] [PubMed] [Google Scholar]
- 43. Zhao Y, Schultz NE, Truhlar DG (2006) Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J Chem Theory Comput 2:364–382. [DOI] [PubMed] [Google Scholar]
- 44. Hehre WJ, Ditchfield R, Pople JA (1972) Self‐consistent molecular orbital methods. XII. Further extensions of gaussian‐type basis sets for use in molecular orbital studies of organic molecules. J Chem Phys 56:2257–2261. [Google Scholar]
- 45. Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, DeBolt S, Ferguson D, Seibel G, Kollman P (1995) AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun 91:1–41. [Google Scholar]
- 46. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, JAM J, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al‐Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, revision e.01. Wallingford CT: Gaussian Inc, 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information