

Abstract
An overview is provided of the molybdenum‐ and tungsten‐containing enzymes that catalyze the interconversion of formate and CO2, focusing on common structural and mechanistic themes, as well as a consideration of the manner in which the mature Mo‐ or W‐containing cofactor is inserted into apoprotein.
Keywords: molybdenum, tungsten, formate dehydrogenase, formylmethanofuran dehydrogenase
Introduction and scope
The oxidation of formate to CO2 is a widespread metabolic process catalysed by two quite different classes of enzymes: simple metal‐independent enzymes that are NAD+‐dependent and act via a ternary E•formate•NAD+ complex with direct hydride transfer between the two substrates, and metal‐dependent enzymes containing molybdenum or tungsten that are frequently components of quite large assemblies and operate via a ping‐pong mechanism in which the metal center (in the MoVI or WVI oxidation state) reacts with formate to become reduced, with the reducing equivalents thus obtained subsequently transferred to other redox‐active centers (and eventually on to an oxidizing substrate) thereby regenerating the oxidized metal center. It is these latter enzymes that are the focus of the present account. While most metal‐containing formate dehydrogenases function physiologically in the direction of formate oxidation to CO2, at least some work in the opposite direction as CO2 reductases, taking reducing equivalents from a source such as H2 and using them to reduce CO2 to formate in a reaction of considerable interest for bioremediation of atmospheric CO2 and the energy economy.1 In addition, methanogenic archaea encode Mo‐ and/or W‐containing formylmethanofuran dehydrogenases that are now recognized to function by reducing CO2 to formate, which subsequently (and at a different site in a very large enzyme complex) condenses with methanofuran.2 As will be seen, these enzymes share profound structural and functional similarities to the formate dehydrogenases and, for this reason, the two groups of enzymes are properly considered together.
Systematics and structural/architectural overview
Molybdenum‐containing enzymes are presently understood to fall into three large families on the basis of the structures of their metal centers, as epitomized by the enzymes xanthine oxidase, sulfite oxidase and DMSO reductase.3 The tungsten‐containing enzymes fall into two families, exemplified by aldehyde:ferredoxin reductase and the formate dehydrogenases, respectively.4 It has long been recognized that members of the DMSO reductase family of Mo‐containing enzymes and the formate dehydrogenase family of W‐containing enzymes are closely related, with fundamentally the same active site metal centers, as illustrated in Figure 1. Indeed many organisms encode separate but very similar (in some cases virtually identical) formate dehydrogenases containing tungsten on the one hand or molybdenum on the other; the same is true for formylmethanofuran dehydrogenases. The metal centers of these enzyme for which X‐ray crystal structures exist are shown in Figure 2, with the metal coordinated to two equivalents of a pyranopterin‐containing cofactor that chelates the metal via an enedithiolate side chain. In all the enzymes considered here, this cofactor is elaborated as the dinucleotide of guanine, most often referred to in the literature as MGD5 (initially taken to mean molybdopterin guanine dinucleotide, but since subsequently being found in tungsten‐containing enzymes as well is presently understood to mean metallopterin guanine dinucleotide). The remainder of the metal coordination sphere is made up of a cysteine or selenocysteine residue provided by the polypeptide and, as recently shown,6 a terminal sulfido ligand (Mo=S or W=S) that is essential for catalytic activity.
Figure 1.
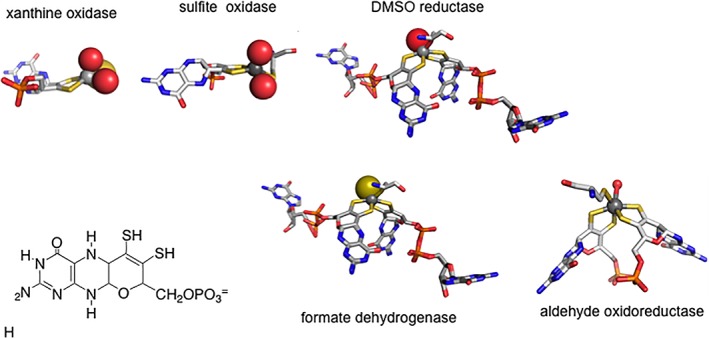
Active site structures for the three families of molybdenum‐containing enzymes and two families of tungsten‐containing enzymes. The structures in the former case are, from left to right, for xanthine oxidase, sulfite oxidase and DMSO reductase, in the latter case formate dehydrogenase and aldehyde oxidoreductase. The structure of the tetrahydro form of the pyranopterin cofactor common to all these enzymes (as well as the tungsten‐containing enzymes) is shown at bottom left, which in all the formate dehydrogenases and formylmethanofuran dehydrogenases is present as the dinucleotide of guanine.
Figure 2.
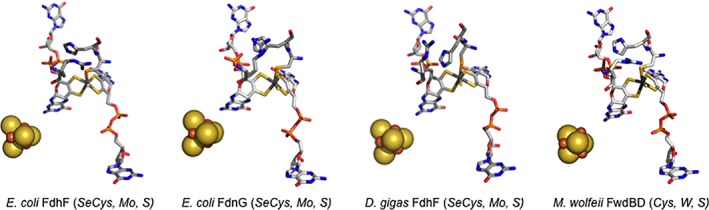
Structures of the molybdenum‐ or tungsten‐containing active sites of various formate‐reducing or CO2‐oxidizing enzymes. The structures are for E. coli FdhF formate dehydrogenase (PDB 1FDO), the FdnG subunit of the E. coli FdnGHI formate dehydrogenase (PDB 1KQF), the FdhA subunit of the D. gigas FdhAB formate dehydrogenase (PDB 1H0H) and the FwdBD subunits of the M. wolfeii formylmethanofuran dehydrogenase (PDB 5T61). The molybdenum and tungsten centers are represented in stick mode, and the [4Fe‐4S] clusters common to all four proteins in space‐filling mode.
At this point, the enzymes considered here can be subdivided on the basis of (1) whether they are formate dehydrogenases or formylmethanofuran dehydrogenases; (2) the metal present in the active site, molybdenum or tungsten; and (3) whether the ligand provided by the protein is cysteine or selenocysteine. A final level of variation in these enzymes, as discussed further below, is their overall subunit architecture, which ranges from the simple, monomeric FdhF formate dehydrogenases from E. coli (containing molybdenum)7 and Clostridium carboxidovorans (containing tungsten),8 both of which possess a single [4Fe‐4S] cluster in addition to the Mo/W center, to the astonishingly complex Mo‐(FmdABCDFG)4 and W‐(FwdABCDFG)4 formylmethanofuran dehydrogenases of Methanothermobacter wolfeii, with the holoenzymes possessing four Mo/W centers and 46 [4Fe‐4S] clusters [2, 9; see below]. The properties of a number of at least partially characterized Mo‐ and W‐containing formate dehydrogenases and formylmethanofuran dehydrogenases are summarized in Table 1, which includes references 10–227. The interested reader is also referred to recent reviews of the general properties of these enzymes.28, 29
Table 1.
Representative molybdenum‐ and tungsten‐containing formate dehydrogenases and formylmethanofuran dehydrogenases
| Enzyme | Gene organization | Subunit composition | Localization |
|---|---|---|---|
| Formate dehydrogenases | |||
| Mo‐containing | |||
| Cys‐containing | |||
| Pectobacterium atrosepticum 10 | fdhF | α (Mo, [4Fe‐4S]) | Cytoplasmic |
| Corynebacterium glutamicum 11 | |||
| Clostridium pasteurianum 12 | fdhAB | αβ (Mo, multiple Fe/S) | Cytoplasmic |
| Methanobacterium formicicum 13 | fdhCAB | αβ (Mo, multiple [4Fe‐4S], FAD) | Cytoplasmic |
| Cupriavidus necator 14 | fdsGBACD | (αβγ)2 | Cytoplasmic |
| (Ralstonia eutropha) | α Mo, 4x[4Fe‐4S], [2Fe‐2S] | (NAD+‐dep.) | |
| Rhodobacter capsulatus 6, 15 | β FMN, [2Fe‐2S] | ||
| Methylosinus trichosporium 16 | γ [2Fe‐2S] | ||
| Pseudomonas oxalatus | |||
| Wolinella succinogenes 17, 18 | fdhEABCD | αβγ | Membrane‐ |
| α Mo, [4Fe‐4S] | Integral | ||
| β 4x[4Fe‐4S] | |||
| γ b‐type heme | |||
| SeCys‐containing | |||
| Escherichia coli (FDH‐H)7 | fdhF | α (Mo, [4Fe‐4S]) | Cytoplasmic |
| Acetobacterium woodii 1 | |||
| Desulfovibrio desulfuricans 19 | fdhABEC | αβγ | |
| Desulfovibrio vulgaris Hldnbrgh20, 21 | α Mo, [4Fe‐4S] | Periplasmic | |
| (three isozymes) | β Dd [4Fe‐4S]; Dv 3x[4Fe‐4S] | ||
| γ 4 c‐type hemes | |||
| Escherichia coli (FDH‐N, FDH‐O)22, 23 | fdnGHI, FdoGHI | (αβγ)3 | Membrane‐ |
| α Mo, [4Fe‐4S] | Integral | ||
| β 4x[4Fe‐4S] | (Mo subunit | ||
| γ 2 b‐type hemes | periplasmic) | ||
| W‐containing | |||
| Cys‐containing | |||
| Methylobacterium extorquens 24 | fdh1AB | αβ | Cytoplasmic |
| α W, [4Fe‐4S] | (NAD+‐dep.) | ||
| β 3x[4Fe‐4S] | |||
| SeCys‐containing | |||
| Clostridium carboxidovorans 8 | Unknown | α W, [4Fe‐4S] | Cytoplasmic? |
| Desulfovibrio gigas 22 | fdhAB | αβ | Periplasmic |
| Desulfovibrio alaskensis 25 | α W, [4Fe‐4S] | ||
| β 3x[4Fe‐4S] | |||
| Moorella thermoacetica 26, 27 | fdhABEC | (αβ)2 | |
| (Clostridium thermoaceticum) | α W, [4Fe‐4S] | Cytoplasmic | |
| β 3x[4Fe‐4S] | (NADP+‐dep.) | ||
| Formylmethanofuran Dehydrogenases | |||
| Mo‐containing | |||
| Cys‐containing | |||
| Methanothermobacter wolfeii 9 | fmdECB | ((αβγδεζ)2)2 | Cytoplasmic |
| α (Zn)2 | |||
| β Mo, [4Fe‐4S] | |||
| γ 2x[4Fe‐4S} | |||
| δ | |||
| ε 8x[4Fe‐4S] | |||
| ζ | |||
| W‐containing | |||
| Cys‐containing | |||
| Methanothermobacter wolfeii 2, 9 | fwdHFGDACB | ((αβγδεζ)2)2 | Cytoplasmic |
| α (Zn)2 | |||
| β Mo, [4Fe‐4S] | |||
| γ 2x[4Fe‐4S} | |||
| δ | |||
| ε 8x[4Fe‐4S] | |||
| ζ |
Most of the enzymes considered here are specific for the metal utilized, and, in many organisms, independently regulated operons encode Mo‐ and W‐utilizing enzymes that catalyze the same reaction. There are some enzymes, however, that appear to be able to accommodate either metal, depending on the growth conditions (e.g. the formylmethanofuran dehydrogenase from Methanosarcina barrkerii 30). In general, the tungsten‐containing enzymes seem to be faster (sometimes considerably so) than their molybdenum‐containing counterparts, but there is considerable overlap in the range of activities observed. Similarly, selenocysteine‐containing enzymes tend to be faster than cysteine‐containing ones, although enzymes that possess cysteine natively typically exhibit substantially faster kinetics that Sec‐to‐Cys mutants of enzyme that natively possess selenocysteine. Enzymes from hyperthermophiles invariably have tungsten, which presumably impart greater stability, but both tungsten‐ and selenocysteine‐containing enzymes tend to be much more O2‐sensitive. It has long been recognized that tungsten systems exhibit lower reduction potentials than the corresponding molybdenum systems,31 and a major factor as to which form is expressed in those organisms that encode both Mo‐ and W‐utilizing enzymes is likely the net redox poise within the cell under specific growth conditions (particularly carbon source and respiratory substrates).
The diversity described above notwithstanding, the overall protein folds of the Mo‐ and W‐containing subunits of these enzymes are remarkably similar. Figure 3 shows the structures of the Mo‐ or W‐containing subunits for the four enzymes that have to date been crystallographically characterized: the E. coli FdhF formate dehydrogenase (Mo, SeCys),7 the FdnG subunit of the E. coli FdnGHI formate dehydrogenase (Mo, SeCys),22 the FdhA subunit of the D. gigas FdhAB formate dehydrogenase (W, SeCys)32 and the FwdBD subunits of the T. wolfeii formylmethanofuran dehydrogenase (W, Cys).2 The overall folds of the proteins are clearly very similar, and the disposition of the Mo/W center (and the [4Fe‐4S] cluster relative to it) highly conserved. The most important variation in these structures is that the contiguous C‐terminal “cap” domain (in blue) found in the first three proteins exists as a separate FwdD subunit in the T. wolfeii enzyme. Also highly conserved are two additional active site residues, a His immediately to the C‐terminal side of the Cys/SeCys that co‐ordinates the metal, and an Arg that constitutes part of the substrate binding site.
Figure 3.
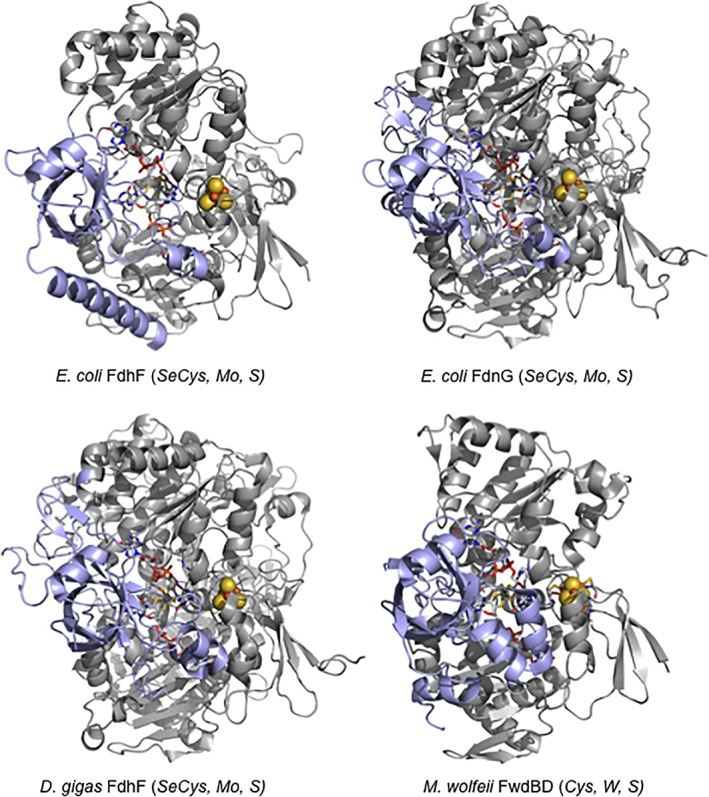
Structures of the molybdenum‐ or tungsten‐containing subunits of various formate‐reducing or CO2‐oxidizing enzymes. The structures are for E. coli FdhF formate dehydrogenase (PDB 1FDO), the FdnG subunit of the E. coli FdnGHI formate dehydrogenase (PDB 1KQF), the FdhA subunit of the D. gigas FdhAB formate dehydrogenase (PDB 1H0H) and the FwdBD subunits of the M. wolfeii formylmethanofuran dehydrogenase (PDB 5T61). The bodies of the subunits are in gray and the “cap” domain (or subunit in the case of the T. wolfeii enzyme) are in blue.
Table 1 summarizes the better‐known formate dehydrogenases and formylmethanofuran dehydrogenases, and Figure 4 shows their subunit makeup and localization within the cell. This list is intended to be illustrative rather than comprehensive, in no small part because of the enormous number of putative enzymes that have been annotated in genomics studies, and the reader is referred to a recent review for additional information.29 The simplest of these enzymes are the Mo‐containing FdhF formate dehydrogenases from E. coli (containing selenocysteine coordinated to the metal)7 or Pectobacterium atrosepticum (containing cysteine),10 as well as the W‐containing FdhF from C. carboxidovorans 8 that are monomeric proteins containing a Mo or W center and a [4Fe‐4S] cluster; all three are cytosolic components of formate:hydrogen lyase systems. Next in complexity are enzymes such as the W‐containing Fdh1AB from Methylobacterium extorquens (containing cysteine)24 and the Mo‐containing FdhABC from organisms such as Desulfovibrio desulfuricans (containing selenocysteine);19 these enzyme are all localized to the periplasm rather than cytoplasm. A higher still level of complexity is exhibited by the membrane‐integral FdnGHI and FdoGHI formate dehydrogenases from, for example, E. coli,22, 23 that are (αβγ)3 trimers possessing Mo‐containing subunits resembling the above FdhF proteins that are attached to membrane‐integral, heme‐containing subunits via a subunit containing four [4Fe‐4S] clusters; FdnGHI is expressed under strictly anaerobic conditions, while FdoGHI is expressed in the transition form in aerobic to anaerobic growth; both contain selenocysteine. Next are the cytosolic, NAD+‐dependent FdsABG enzymes from Cupriavidus necator 14 and Rhodobacter capsulatus 15 that contain molybdenum coordinated by cysteine residues; both are organized as (αβγ)2 dimers of trimers. More complicated still is the cytosolic H2‐dependent CO2 reductase from, for example, Acetobacterium woodii that consists of four subunits: an iron‐only hydrogenase subunit, two different subunits each containing four [4Fe‐4S] clusters, and an FdhF‐like molybdenum‐ and [4Fe‐4S]‐containing subunit at which CO2 is reduced to formate.1 The A. woodii genome encodes tandem genes for this last subunit, one encoding a subunit that possesses selenocysteine coordinated to the molybdenum and the other cysteine. Finally, there are the Mo‐ or W‐containing formylmethanofuran dehydrogenases, FmdABCDFG and FwdABCDFG, respectively, from methanogenic archaea such as Methanothermobacter wolfeii and Methanosarcina barkerii.9 The tungsten‐containing enzyme (and presumably also the molybdenum‐containing one) from T. wolfeii has a magnificently complex ((αβγδεζ)2)2 structure with a 110‐Å long spiral chain of [4Fe‐4S] clusters connecting the two tungsten centers within each (αβγδεζ)2 protomeric dimer (with the two dimers connected via a pair of bridging [4Fe‐4S] clusters;2 both Mo‐ and W‐containing enzymes have cysteine coordinated to the metal. It is interesting that while the two enzymes are encoded by separate fmd and fwd operons, the molybdate‐inducible operon encoding the Mo‐containing enzyme is greatly truncated and consists of only three genes, including that encoding the Mo‐containing subunit and the “cap” (which is present as a C‐terminal domain of the enzyme's FmdC subunit).9 The implication is that subunits encoded by the constitutively expressed fwd operon are incorporated into the mature Mo‐containing enzyme under the conditions in which the latter is expressed.
Figure 4.
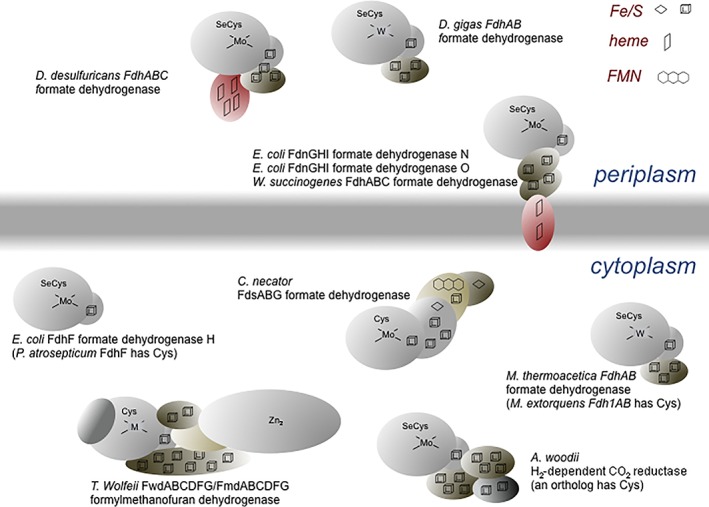
A schematic of the subunit and cofactor constitution of the formate dehydrogenases and formylmethanofuran dehydrogenases, as well as the subcellular localization. Only the makeup of the protomeric unit is shown. FdnGHI and FdoGHI are protomeric trimers, and FdsABG a protomeric dimer. FwdABCDFG and its molybdenum‐containing ortholog are protomeric tetramers.
The formate dehydrogenases and formylmethanofuran dehydrogenases are thought to be evolutionarily ancient, and both the acetogenic Wood–Ljungdahl pathway of bacteria and the version found in methanogenic archaea both begin with CO2 reduction by a Mo‐ or W‐containing enzyme (an H2‐oxidizing CO2 reductase in the former case, a formylmethanofuran dehydrogenase in the latter).33, 34 Indeed, the acetogenic and methanogenic Wood–Ljungdahl pathways share many common intermediates, passing through consecutive formyl‐, methenyl‐, methylenyl‐, and methylpterin species. The acetogenesis pathway involves tetrahydrofolate while methanogenesis utilizes methanopterin, but on the basis of the obvious similarities in pathway intermediates the two pathways were originally thought to share a common evolutionary origin. As the enzyme systems of the two pathways became better characterized, however, it became clear that the enzymes responsible for cognate steps in fact had dramatically different subunit architectures and the pathways thus appeared to represent convergent rather than divergent evolution, with the shared chemical transformations simply being coincidental. As crystal structures for the enzymes became available, however, a deep structural similarity between the participants in the acetogenic and methanogenic pathways re‐emerged, as reflected in the structures shown in Figure 3 for the Mo‐ or W‐containing sites that catalyze CO2 reduction in vivo. Recent genomics analyses indeed confirm that these pathways are indeed very ancient, likely tracing back to the Last Universal Ancestor to all extant life forms,35 an organism that utilized a “molecular toolkit” of genes encoding simple proteins that possessed a range of redox‐active cofactors that it used to mix and match in developing various metabolic capabilities.36 It is this process that is now thought to have given rise to the differences in protein architectures seen in the acetogenic and methanogenic Wood–Ljungdahl pathways.
Reaction mechanism
Given the overall similarities in active site structures, it is very likely that all molybdenum‐ and tungsten‐containing formate dehydrogenases act via fundamentally the same chemical mechanism regardless of whether they possess Cys or SeCys co‐ordinated to the metal, and over the years various mechanisms have been proposed for one or another enzyme (Fig. 5).37, 38, 39, 40, 41 The initial mechanistic clue was the demonstration that oxygen from solvent was not incorporated into product CO2 in the course of the reaction, indicating that the reaction did not proceed via an oxygen atom transfer mechanism.37 Based on the original crystal structures for the E. coli FdhF enzyme,7 a mechanism was originally proposed (Fig. 5(A)) that involved formate displacing a Mo=O group from the molybdenum coordination sphere, followed by deprotonation of the Cα carbon and reduction of the molybdenum. Subsequently proposed mechanisms (Fig. 5(B) and (C)) have involved formate displacing the (seleno)cysteine instead, again followed by abstraction of the Cα proton of formate followed by transfer of a pair of electrons to the molybdenum center upon formation of the second C=O bond. A major difficulty with each of these mechanisms, however, has to do with the pK a of the Cα proton. The pK a for the Cα proton of formamide (with an ‐NH2 group rather than the ‐CO2 − of formate) is 2342 and the formate pK a will undoubtedly be much higher given the formation of the dianion. It is simply not clear how either a histidine residue or the now‐dissociated (Se)Cys could have a pK a sufficiently high to effect deprotonation of the Cα carbon of formate.
Figure 5.
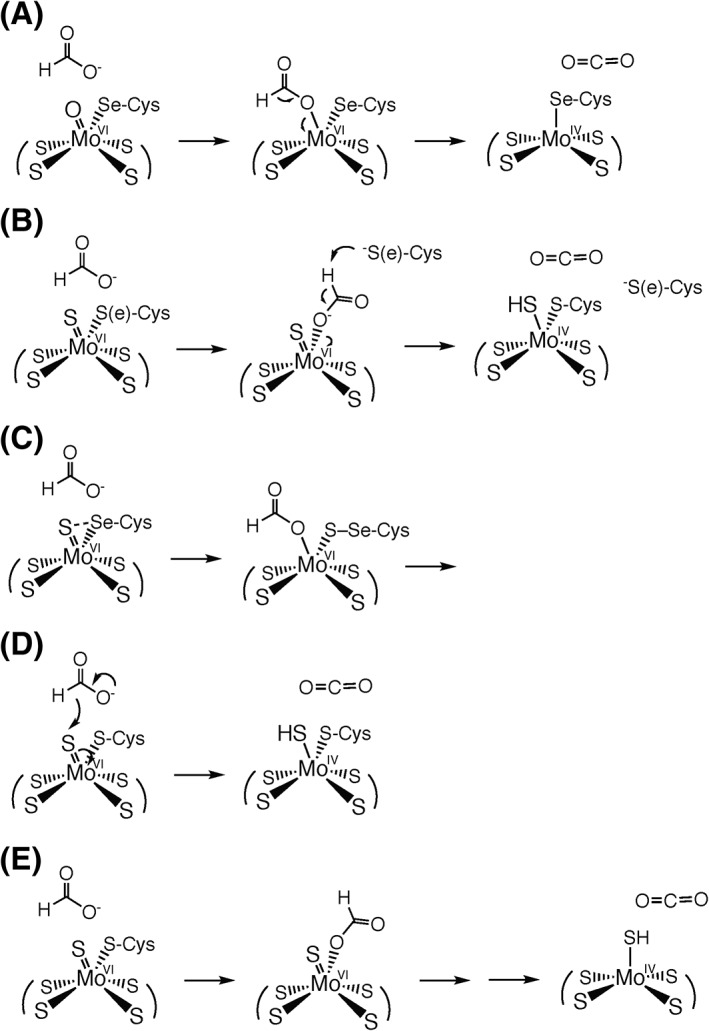
Proposed reaction mechanisms for the formate dehydrogenases. (A) The mechanism proposed originally by Boyington et al. on the basis of the initial crystallographic analysis of the enzyme, with the displaced Mo=O group lost to solvent7; (B) the mechanism suggested by Raaijmakers and Romão on the basis of the reanalysis of the original X‐ray data37; (C) the mechanism suggested by Hartmann et al. on the basis of parallels with the presumed mechanism by which the enzyme reduces nitrate38; (D) the simple hydride transfer mechanism proposed by Niks et al.39; and (E) the mechanism that proposed by Robinson et al. on the basis of the observed patterns of inhibition for nitrite and similar inhibitors seen in both the forward and reverse directions as observed electrochemically.40 In the last case, the initial intermediate is proposed to break down either by proton‐coupled electron transfer or hydride transfer processes.
A major advance in our understanding of how these enzymes work was the recent demonstration that the E. coli FdhD gene product was a sulfur transferase that, in conjunction with the IscS cysteine desulfurase, catalyzed the insertion of a Mo=S ligand into the metal coordination sphere of E. coli FdhF, and that this sulfur was required for activity.6 Although the original X‐ray crystal structure analysis of FdhF interpreted the electron density at this position as a Mo=O group,7 a subsequent reanalysis of the data has suggested that it is indeed a Mo=S37 (although a portion of the crystallographic sample likely had lost the Mo=S, presumably replaced by a Mo=O, and become inactive). Additionally, an XAS analysis of the FdsABG formate dehydrogenase from R. capsulatus has implicated a Mo=S group, and the 1.2‐Å resolution structure of the FwdABCDFG formylmethanofuran dehydrogenase2 unambiguously establishes this ligand to be a W=S. It is now generally accepted that the reaction mechanism begins with an L2MVI = S(S‐Cys) – or L2MVI = S(Se‐Cys) – active site for both Mo‐ and W‐containing enzymes, with L representing the bidentate pyranopterin cofactor.
With this structure as the starting point, the key mechanistic experiment has been the demonstration with both the SeCys‐containing E. coli FdhF37 and Cys‐containing C. necator FdsABG40 that the Cα hydrogen of formate is transferred to the molybdenum center in the course of formate oxidation and, with the metal in the Mo(V) oxidation state, is strongly coupled to the metal as determined by EPR. The hydrogen was originally proposed to be present on the His residue adjacent to the Mo‐coordinating SeCys, but the strong ~20 MHz coupling clearly indicates that the proton must be present in the immediate coordination sphere of the molybdenum. It has been proposed instead that the hydrogen is present as a Mo—SH group,40 consistent with the similarly strong hyperfine coupling of Mo—SH protons that is observed in both model complexes43 and other enzymes.44 This being the case, the Mo=S sulfur of oxidized enzyme has become protonated as the molybdenum center becomes reduced, implying that the reaction proceeds as a simple hydride transfer to give the fully reduced Mo(IV) species (with the EPR‐detectable Mo(V) valence state formed by subsequent transfer of one electron out to other redox‐active centers in the protein), as shown in Figure 5(D). Formate is known to be a good hydride donor (the reduction of NAD+ by the metal‐free formate dehydrogenases necessarily occurs via hydride transfer) and the Mo=S group of other molybdenum‐containing enzymes (notably xanthine oxidase and aldehyde oxidase45) is known to be a good hydride acceptor. An important implication of this mechanism is that the molybdenum coordination sphere is coordination stable, remaining six‐coordinate throughout the catalytic sequence with no dissociation of either the Mo=S or Mo—(Se)Cys ligands to the metal.
The hydride transfer mechanism has received considerable acceptance (see, e.g. Ref. 46), but alternate mechanisms have recently been proposed. Based on the observation that the R. capsulatus FdsABG formate dehydrogenase is able to reduce nitrate to nitrite (albeit extremely slowly, with a k cat of 0.21 min−1) and on the assumption that nitrate reduction necessarily takes place by oxygen atom transfer, a mechanism has been proposed in which nitrate displaces the Mo‐coordinating Cys 386 from the molybdenum center (in the reduced state) during turnover; by analogy, formate was proposed to displace Cys 386 (from the oxidized molybdenum center), with product CO2 being formed by abstraction of the Cα proton by the now‐dissociated Cys 386, followed by transfer of a pair of electrons to the molybdenum center,39 as shown in Figure 5(B). This mechanism is very similar to one proposed earlier,38 and suffers from the problem discussed above regarding the expected pK a for the Cα of formate. Such a general base role for the dissociated Cys becomes especially problematic when considering those enzymes containing SeCys, given its even lower pK a; the great reduction in activity on mutating the SeCys of the E. coli FdhF to Cys47 is specifically difficult to rationalize if the residue functions as a general base in the course of the reaction. This mechanism also has difficulty accounting for both the experimentally observed transfer of the Cα hydrogen to the molybdenum center37, 40 and the strong 77Se coupling in the catalytically generated Mo(V) EPR signal exhibited by enzyme isolated from cells grown on 77Se‐enriched medium that clearly demonstrates the SeCys is coordinated directly to the metal in the signal‐giving species.37 In any case, it must be recognized that nitrate can also be reduced catalytically via a variety of mechanisms in addition to oxygen atom transfer,48 and no direct proof of oxygen atom transfer in the reduction of nitrate by formate dehydrogenase (or nitrate reductase, for that matter) has yet been provided in the literature.
Further support for a mechanism involving displacement of the (Se)Cys ligand from the metal was claimed with the observation that Cys 386 of the R. capsulatus FdsABG, like the SeCys 140 of FdhF and the Cys of a Sec140Cys variant thereof,47 was found to be modified by iodoacetamide in the presence of nitrate and formate.39 The modification of Cys 386 by iodoacetamide does not necessarily prove that the Cys dissociates in the course of turnover, however. In the R. sphaeroides DMSO reductase, for example, one of the pyranopterin cofactors is known to dissociate from the metal in a way that is not catalytically relevant.49 Were the enzyme to be reacted with iodoacetamide, the dissociated pterin would undoubtedly become derivatized, yet it would be incorrect to conclude that the ligand dissociation implied by the covalent modification of the cofactor was mechanistically relevant. Still, the reanalysis of the original X‐ray diffraction data of the E. coli FdhF that suggested the terminal ligand in oxidized enzyme was a Mo=S (or Mo—SH) rather than Mo=O ligand in oxidized enzyme also identified an alternate polypeptide trace into the electron density of the reduced enzyme in which the selenocysteine had dissociated from the molybdenum and formed a loop that was in reoriented such that the both Sec 140 and His 141 were positioned well away from the molybdenum.38 The new model indeed provides an improved fit to the electron density in some ways, but it leaves unaccounted for substantial electron density in the immediate vicinity of the molybdenum, and the newly positioned His 141 in this alternate structure is in fact poorly represented in the electron density. It seems most likely that the electron density reflects two different structures, with the identified Sec 140 loop in either of two (or more) positions, and coordinated to the molybdenum in only one. There is no definitive evidence to date that the Sec‐dissociated structure, which unquestionably exists, is catalytically relevant, and it may well represent enzyme that is inactivated. It is thus difficult to say that the presently available crystallographic data unambiguously supports selenocysteine dissociation in the course of the catalytic sequence. It is also worth bearing in mind that dissociation of the Sec (or Cys) appears to be unique to the E. coli FdhF, and has not been observed in any of the other proteins that have been examined crystallographically.
X‐ray absorption spectroscopy has also been employed to ascertain the nature of the molybdenum (or tungsten) coordination sphere of the formate dehydrogenases, and has emphasized the importance of sample integrity. The initial work with the E. coli 50 and Desulfovibrio desulfuricans 40 formate dehydrogenases, both molybdenum‐ and selenocysteine‐containing enzymes, indicated that the Mo—SeCys bond was maintained in reduced as well as oxidized enzyme, but a Mo‐OH (in the E. coli enzyme) or Mo=O (in the D. desulfuricans enzyme) was found rather than the (now) expected Mo=S, suggesting that a substantial amount of the sample (as was presumably also the case in the original crystallographic work) had become inactivated. Indeed, in addition to be being quite air‐sensitive, most of these enzymes appear to be less stable in their reduced forms.51 The problem is compounded by the fact that, like many Mo‐ and W‐containing enzymes, recombinant systems typically yield protein that lacks a fully functional metal center. In the case of the cysteine‐containing R. capsulatus FdsABG formate dehydrogenase, for example, clear evidence has been found for a Mo=S in the oxidized enzyme, but only in ~50% of the molybdenum centers present in the sample.6 It is generally agreed that Mo=O and/or Mo—OH ligands are not present in the functional molybdenum coordination sphere, but are very likely to replace Mo=S and/or Mo—S(Cys) ligands should these ligands dissociate from the molybdenum in the course of inactivation. Indeed, the Mo—O ligand seen in the reduced enzyme that was interpreted as a metal‐coordinated formate may well represent an exogenous oxygen present in a population of inactivated enzyme in the XAS samples that is unrelated to functional enzyme.
Very recently, a second mechanism for the E. coli FdhF that is predicated on displacement of the protein ligand from the metal center by formate has been proposed, in which catalysis again is initiated by formate coordination to the molybdenum via its carboxylate group, displacing the selenocysteine (Fig. 5(E)).51 In this case, formate oxidation proceeds either via hydride transfer to the MoVI=S group or coupled proton electron transfer, with the Mo=S group deprotonating the Cα of formate, with electrons in the dative covalent bond between the carboxylate oxygen and molybdenum reducing the metal. The second of these alternative mechanisms involves abstraction of the formate Cα proton, and suffers the same difficulty as that discussed above given its very high pK a. This mechanism also fails to account for the strong 1H coupling of the erstwhile Cα proton that is observed by EPR. In the case of the first mechanism, it is simply not clear why tying up the electrons destined to form the second C=O double bond of product CO2 by coordinating to the molybdenum should facilitate the reaction. The basis for proposing formate displacement of the protein‐contributed selenocysteine from molybdenum to the metal came from electrochemical studies in the presence of various anionic inhibitors of the enzyme. The principal result was that ions such as N3 −, NO2 −, NO3 −, OCN−, and SCN− were all found to be strong, competitive inhibitors with respect to formate in the forward direction (with the molybdenum originally in the oxidized MoVI state), but weaker, and non‐competitive, inhibitors in the direction of CO2 reduction (where the reaction begins with the metal in the reduced MoIV state). One would indeed expect each of these anionic inhibitors to bind more effectively to more oxidized forms of the metal than reduced, but given the proximity of the substrate binding site (with its highly conserved arginine residue), the result could simply be due to electrostatics rather than dissociation of the selenocysteine. Indeed, the authors acknowledge that these same anions are competitive inhibitors with respect to formate oxidation with the metal‐independent formate dehydrogenases. Perhaps most significantly, it must be borne in mind that the structure of FdhF with one of the inhibitors used, nitrite, has been determined7 and while the nitrite is indeed found coordinated to the molybdenum, it occupies the position occupied by the Mo=S rather than the selenocysteine, which remains coordinated to the molybdenum in the structure. With the catalytically essential sulfur gone, the structure is obviously of an inactive form of the enzyme (possibly formed only in that portion of the enzyme that had already lost the Mo=S ligand), but the empirical observation is that the selenocysteine remains co‐ordinated to the molybdenum in the nitrite‐inhibited enzyme.
Reversibility of the reaction: CO2 reduction
Apart from the documented systems that function physiologically as CO2 reductases, the literature presents a rather confusing picture with regard to whether one or another of the enzymes that function physiologically to oxidize formate is capable of catalyzing the reverse reaction, the reduction of CO2 to formate. In particular, there appears to be no clear correlation with reactivity in the reverse direction as regards metal or whether the metal is complexed with cysteine or selenocysteine. The situation is particularly unfortunate given the importance of the reaction, both from the standpoint of clearing a greenhouse gas from the atmosphere and also from the standpoint of energy storage. Particularly, if the reaction indeed proceeds via hydride transfer, it would seem that all (metal‐containing) formate dehydrogenases should be able to reduce CO2 provided sufficiently low‐potential electrons. CO2 reduction by the D. desulfuricans formate dehydrogenase has recently been comprehensively examined, with a k cat in the reverse direction being found to be 47 s−1 (as compared with the k cat for formate oxidation in the forward direction of 543 s−1).46 Similarly, in the case of the C. necator FdsABG enzyme, which had been previously reported to not be able to catalyze CO2 reduction,52 the enzyme was found to be effective in utilizing NADH to drive CO2 reduction with a k cat of 11 s−1, K m NADH of 46 μM, and K m CO2 of 2.7 mM.53 Importantly, these kinetic parameters were shown to be consistent with the expected Haldane relationship and the known reduction potentials for the NAD+/NADH and CO2/formate couples of −320 mV and − 420 mV versus SHE, respectively. Two particularly important observations came out of these studies: first, the enzyme possessed considerable diaphorase activity and it was essential to conduct experiments anaerobically; and second, it was important to use CO2‐saturated buffers in preparing substrate solutions for the reverse reaction, as bicarbonate was found to be a poor surrogate for CO2. These points likely account for the many false negative (or positive) results that have been reported in the literature, and it is likely that all the Mo‐ and W‐containing formate dehydrogenases are able catalyze the reduction of CO2.
In the case of the formylmethanofuran dehydrogenases, it was thought for many years that the reaction involved CO2 carbamoylation of methanofuran, with the adduct subsequently reduced to the product formylmethanofuran.54 The X‐ray crystal structure of the tungsten‐containing M. wolfeii FwdABCDFG enzyme, however, clearly showed that the binding site for methanofuran was some 40 Å removed from the tungsten center, with the two sites connected via a hydrophobic tunnel through which formate, once generated from CO2 at the tungsten center, diffused to the methanofuran binding site where it was incorporated into product.2 Significantly, the highly conserved His and Arg residues of the formate dehydrogenases are also conserved in the M. wolfeii formylmethanofuran dehydrogenase. This being the case, and in light of the strong structural similarities in structures of the metal centers in both Mo‐ and W‐containing formylmethanofuran dehydrogenases and formate dehydrogenases discussed above, it is likely that the two groups of enzyme operate via the same fundamental chemical mechanism.
Cofactor maturation and insertion
The FdhC and FdhD gene products are known to be required for maturation of each of the three formate dehydrogenases encoded by the E. coli genome.55, 56 The role of FdhC is presently unknown, but FdhD has been shown to be specifically involved in the transfer of sulfur from the IscS cysteine desulfurase of E. coli to the nascent molybdenum center prior to insertion into the apoproteins.57 The crystal structures of FdhD from both Desulfotalea psychrophilia (PDB 2PW9, unpublished) and E. coli (PDB 4PDE 58) have recently been determined, the latter in complex with GDP. The dimeric protein has an overall blunt conical shape, with two equivalents of GDP bound at its concave base; this location presumably represents the binding site for the bisMGD‐containing cofactor, and a model for the bisMGD form of the molybdenum cofactor indeed fits nicely at this position.58 Two cysteine residues known to be important for function,57 Cys 121 and 124 in the E. coli enzyme, are found in an unresolved loop (Residues 113–131) on the opposite side of the protein. An ~8 Å‐wide tunnel exists through the center of the protein, and in the D. psychrophilia FdhD structure, this loop, with only six residues unresolved (but unfortunately still including the conserved cysteines) extends into the tunnel. It has been proposed that this loop interacts with IscS on one side of FdhD to accept the sulfur (as a persulfide to Cys 121), then reorients itself, passing into the tunnel through to the opposite side of the protein where the distal sulfur of the cysteine persulfide is passed on to the bound molybdenum cofactor.58 Cys 124, which is not universally conserved in FdhD homologs from different organisms, is proposed to facilitate cofactor sulfuration in the case of the E. coli enzyme by forming an internal disulfide with Cys 121 in the course of the reaction.
FdhD is known to interact with FdhF in activating the enzyme,57 and likely physically inserts the now mature, sulfurated cofactor into the apo FdhF in an “open” configuration in which the cap domain has moved away from the body of the protein. The difficulty is that in the model of the molybdenum cofactor bound to FdhD, the newly incorporated Mo=S is oriented back toward the tunnel from whence it came, away from the surface of the protein, with the two pyranopterin rings extending out toward solvent. Similarly, in the body of FdhF, the Mo=S group is similarly oriented away from the face of the body that is exposed on removing the cap, which would require that the cofactor fully dissociate from FdhD in binding to apo FdhF. This would seem to be unlikely given the instability of both the pyranopterin and Mo=S moieties of the cofactor. This problem is resolved, however, if the cofactor is not passed directly to the body of FdhF but is instead transferred first to the cap domain, which then closes onto the body of FdhF with the cofactor now properly positioned. In the structure of holo FdhF, the cap domain interacts primarily with the pyranopterins of the molybdenum center, whereas the body of FdhF, like FdhD in the model of the protein in complex with the molybdenum cofactor, interacts primarily with the two guanine moieties of the cofactor. This provides a plausible means by which the sulfurated cofactor passes from FdhD once sulfurated, to the FdhF cap and then to the body of FdhF. An intriguing possibility in the context of this scheme is that sulfuration of the cofactor might not occur until formation of an FdhD:cofactor:cap ternary complex, which could ensure that only sulfurated cofactor is incorporated into FdhF.
Conclusions
Although our understanding of the structures of the microbial formate dehydrogenases and formylmethanofuran dehydrogenases has advanced considerably over the past 5 years, important structural features directly relevant to catalysis of formate oxidation and CO2 reduction remain. Most critical at present is the need to establish unambiguously by X‐ray crystallography whether the metal‐coordinating Cys or SeCys dissociates from the metal in the course of the reaction, which will indicate whether it will prove necessary to consider alternatives to a hydride transfer mechanism. Much additional mechanistic work, particularly with the tungsten‐containing enzymes and the formylmethanofuran dehydrogenases, is also needed to validate the underlying assumption of the present account: that all these enzymes function via the same basic mechanism for the reversible interconversion of formate and CO2.
Acknowledgments
Work in the authors’ laboratory is supported by a grant from the US Department of Energy (DE‐SC0010666 to R. H.).
References
- 1. Schuchmann K, Müller V (2013) Direct and reversible hydrogenation of CO2 to formate by a bacterial carbon dioxide reductase. Science 342:1382–1385. [DOI] [PubMed] [Google Scholar]
- 2. Wagner T, Ermler U, Shima S (2016) The methanogenic CO2 reducing‐and‐fixing enzyme is bifunctional and contains 46 [4Fe‐4S] clusters. Science 354:114–117. [DOI] [PubMed] [Google Scholar]
- 3. Hille R (1996) The mononuclear molybdenum enzymes. Chem Rev 96:27575–22816. [DOI] [PubMed] [Google Scholar]
- 4. Johnson MK, Rees DC, Adams MWW (1996) Tungstoenzymes. Chem Rev 96:2817–2839. [DOI] [PubMed] [Google Scholar]
- 5. Mendel R, Leimkühler S (2015) The biosynghesis of the molybdenum cofactors. J Biol Inorg Chem 20:337–347. [DOI] [PubMed] [Google Scholar]
- 6. Schrapers P, Hartmann T, Kositzki R, Dau H, Reshke S, Schulzke C, Leimkühler S, Haumann M (2015) Sulfido and cysteine ligation changes at the molybdenum cofactor during substrate conversion by formate dehydrogenase (FDH) from Rhodobacter capsulatus . Inorg Chem 54:3260–3271. [DOI] [PubMed] [Google Scholar]
- 7. Boyington JC, Gladyshev VN, Khangulov SV, Stadtman T, Sun PD (1997) Crystal structure of formate dehydrogenase H: catalysis involving Mo, molybdopterin, selenocysteine and an Fe4S4 cluster. Science 275:1305–1308. [DOI] [PubMed] [Google Scholar]
- 8. Alissandratos A, Kim HK, Matthews HK, Hennessey JE, Philbrook A, Easton CJ (2013) Clostridium carboxidovorans strain P7T recombinant formate dehydrogenase catalyzes reduction of CO2 to formate. Appl Environ Micorobiol 79:741–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hochheimer A, Hedderich R, Thauer RK (1998) The formylmethanofuran dehydrogenase isozymes in Methanobacterium wolfeii and Methanobacterium terhmoautotrophicum: induction of the molybdenum isozyme by molybdate and constitutive synthesis of the tungsten isozyme. Arch Microbiol 170:389–393. [DOI] [PubMed] [Google Scholar]
- 10. Babujee L, Apodaca J, Balakrishnan V, Liss P, Kiley PJ, Charkowski AO, Glasner JD, Perna NT (2012) Evolution of the metabolic and regulatory networks associated with oxygen availability in two phytopathogenic enterobacteria. BMC Genomics 13:110 10.1186/1471-2164-13-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Witthoff S, Eggeling L, Bott M, Polen T (2012) Corynebacterium glutamicum harbours a molybdenum cofactor‐dependent formate dehydrogenase which alleviates growth inhibition in the presence of formate. Microbiol 158:2428–2439. [DOI] [PubMed] [Google Scholar]
- 12. Liu CL, Mortenson LE (1984) Formate dehydrogenase of Clostridium pasteurianum . J Bacteriol 159:375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schauer NL, Ferry JG (1982) Properties of formate dehydrogenase in Methanobacterium formicicum . J Bacteriol 150:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oh JI, Bowien B (1998) Structural analysis of the fds operon encoding the NAD+‐linked formate dehydrogenase from Ralstonia eutropha . J Biol Chem 273:26349–26360. [DOI] [PubMed] [Google Scholar]
- 15. Hartmann T, Leimkühler S (2013) The oxygen‐tolerant and NAD+‐dependent formate dehydrogenase from Rhodobacter capsulatus is able to catalyze the reduction of CO2 to formate. FEBS J 280:6083–6096. [DOI] [PubMed] [Google Scholar]
- 16. Jollie DR, Lipscomb JN (1991) Formate dehydrogenase from Methylosinus trichosporium OB3b. Purification and spectroscopic characterization of the cofactors. J Biol Chem 266:21853–21863. [PubMed] [Google Scholar]
- 17. Kröger A, Winkler E, Innerhof A, Hackenberg H, Schagger H (1979) The formate dehydrogenase involved in electron transport from formate to fumarate in Vibrio succinogenes . FEBS J 94:465–475. [DOI] [PubMed] [Google Scholar]
- 18. Bokranz M, Gutmann M, Kortner C, Kojro E, Fahrenholz F, Lauterbach F, Kröger A (1991) Cloning and nucleotide sequence of the structural genes encoding the formate dehydrogenase from Wolinella succinogenes . Arch Microbiol 156:30–34. [DOI] [PubMed] [Google Scholar]
- 19. Costa C, Teixeira M, LeGall J, Moura JJG, Moura I (1997) Formate dehydrogenase from Desulfovibrio desulfuricans ATCC 27774: isolation and spectroscopic characterization of the active sites (heme, iron‐sulfur centers and molybdenum). J Biol Inorg Chem 2:198–208. [Google Scholar]
- 20. Bursakov S, Liu MY, Payne WJ, LeGall J, Moura JJG (1995) Isolation and preliminary characterization of a soluble nitrate reductase from the sulfate‐reducing organism Desulfovibrio desulfuricans ATCC‐27774. Anaerobe 1:55–60. [DOI] [PubMed] [Google Scholar]
- 21. da Silva SM, Pimentel C, Valente FMA, Rodrigues‐Pousada C, Pereira IAC (2011) Tungsten and molybdenum regulation of formate dehydrogenase expression in Desulfovibrio vulgaris Hildenborough. J Bacteriol 193:2909–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jormakka M, Törnroth S, Byrne B, Iwata S (2002) Molecular basis of proton motive force generation: structure of formate dehydrogenase‐N. Science 2295:1863–1868. [DOI] [PubMed] [Google Scholar]
- 23. Wang H, Gunsalus RP (2003) Coordinate regulation of the Escherichia coli formate dehydrogenase fdnGHI and fdhF genes in response to nitrate, nitrite and fomate: roles fro NarL and NarP. J Bacteriol 185:5076–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Laukel M, Christoserdova L, Lidstrom ME, Vorholt JA (2003) The tungsten‐containing formate dehydrogenase from Methylopacterium extorquens AM1: purification and properties. Eur J Biochem 270:325–333. [DOI] [PubMed] [Google Scholar]
- 25. Brondino CD, Passeggi MC, Caldeira J, Almendra MJ, Feio MJ, Moura JJG, Moura I (2004) Incorporation of either molybdenum or tungsten into formate dehydrogenase from Desulfovibrio alaskensis NCIMB 13491: EPR assignment of the proximal iron‐sulfur cluster to the pterin cofactor in formate dehydrogenases from sulfate‐reducing bacteria. J Biol Inorg Chem 9:145–151. [DOI] [PubMed] [Google Scholar]
- 26. Andreese JR, Ljungdahl LG (1974) Nicotinamide adenine‐dinucleotide phosphate‐dependent formate dehydrogenase from Clostridium thermoaceticum – purification and properties. J Baceriol 120:6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ljungdahl LG, Andreesen JR (1978) Formate dehydrogenase, a selenium‐tungsten enzyme from Clostridium thermoaceticum . Methods Enzymol 53:360–372. [DOI] [PubMed] [Google Scholar]
- 28. Hartmann T, Schwanhold N, Leimkühler S (2015) Assembly and catalysis of molybdenum or tungsten‐containing formate dehydrogenases from bacteria. Biochim Biophys Acta 1854:1090–1100. [DOI] [PubMed] [Google Scholar]
- 29. Maia LB, Moura I, Moura JJG (2017) Molybdenum and tungsten‐containing formate dehydrogenases: aiming to inspire a catalyst for carbon utilization. Inorg Chem Acta 455:350–363. [Google Scholar]
- 30. Bertram PA, Karrasch M, Schmitz RA, Böcher R, Albracht SPJ, Thauer RK (1994) formylmethanofuran dehydrogenases from methanogenic Archaea. Substrate specificity, EPR properties and reversible inactivation by cyanide of the molybdenum or tungsten iron‐sulfur proteins. Eur J Biochem 220:477–484. [DOI] [PubMed] [Google Scholar]
- 31. Stiefel EI (1977) The coordination and bioinorganic chemistry of molybdenum. Prog Inorg Chem 22:1–223. [Google Scholar]
- 32. Raaijmakers H, Macieira S, Dias JM, Texierira S, Bursakov S, Huber R, Moura JJG, Moura I, Romão MJ (2002) Gene sequence and the 1.8Å crystal structure of the tungsten‐containing formate dehydrogenase from Desulfovibrio gigas . Structure 10:1261–1272. [DOI] [PubMed] [Google Scholar]
- 33. Ragsdale SW, Pierce W (2008) Acetogenesis and the Wood–Ljungdahl pathway of CO2 fixation. Biochim Biophys Acta 1784:1873–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R (2008) Methanogenic arhcaea: ecologically relevant differences in energy conservation. Nature Rev Microbiol 6:579–591. [DOI] [PubMed] [Google Scholar]
- 35. Weiss MC, Sousa FL, Mrnjavic N, Neukirchen S, Roettger M, Nelson‐Sathi S, Martin WF (2016) The physiology and habitat of the last universal common ancestor. Nature Microbiol 1:16116 10.1038/nmicorbiol.2016.116. [DOI] [PubMed] [Google Scholar]
- 36. Baymann F, Lebrun E, Brugna M, Schoepp‐Cothenet B, Giudici‐Orticoni MT, Nitschke W (2003) The redox construction kit:pre‐last universal ancestor evolution of energy‐conserving enzymes. Func Genomics Evol 358:267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Khangulov SVV, GLadyshev VN, Dismukes GC, Stadtman TC (1998) Selenium‐containing formate dehydrogenase H from Escherichia coli: a molybdenum enzyme that catalyzes formate oxidation without oxygen transfer. Biochemistry 37:3518–3528. [DOI] [PubMed] [Google Scholar]
- 38. Raaijmakers HCA, Romão MJ (2006) Formate‐reduced E. coli formate dehydrogenase H: the reinterpretation of the crystal structure suggests a new reaction mechanism. J Biol Inorg Chem 11:849–854. [DOI] [PubMed] [Google Scholar]
- 39. Hartmann T, Schrapers P, Utesch T, Nimtz M, Rippers Y, Dau H, Mroginski MA, Haumann M, Leimkühler S (2016) The molybdenum active site of formate dehydrogenase is capable of catalyzing C‐H bond cleavage and oxygen atom transfer reactions. Biochemistry 55:2381–2389. [DOI] [PubMed] [Google Scholar]
- 40. Niks D, Duvvuru J, Escalona M, Hille R (2016) Spectroscopic and kinetic properties of the molybdenum‐containing, NAD+‐dependent formate dehydrogenase from Ralstonia eutropha . J Biol Chem 291:1162–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cerqueira NMFSA, Fernandes PA, Gonsalez PJ, Moura JJG, Ramos MJ (2013) The sulfur shift: an activation mechanism for periplasmic nitrate reductase and formate dehydrogenase. Inorg Chem 52(19):10766–10772. [DOI] [PubMed] [Google Scholar]
- 42. Bordwell FG, Partness JE, Hautala JA (1978) Alkyl effects on equilibrium acidities of carbon acids in protic and apolar media and the gas phase. J Org Chem 43:3113–3116. [Google Scholar]
- 43. Wilson GL, Greenwood RJ, Pilbrow JR, Spence JT, Wedd AG (1991) Molybdenum(V) sites in xanthine oxidase and relevant analogue complexes: comparison of molybdenum‐95 and sulfur‐33 coupling. J Am Chem Soc 113:6803–6812. [Google Scholar]
- 44. Bray RC, Gutteridge S, Stotter DA, Tanner SJ (1979) The mechanism of xanthine oxidase. The relationship between the rapid and very rapid electron‐paramagnetic‐resonance signals. Biochem J 177:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xia M, Dempski R, Hille R (1999) The reductive half‐reaction of xanthine oxidase. Reaction with aldehydes and identification of the catalytically labile oxygen. J Biol. Chem 274:3323–3330. [DOI] [PubMed] [Google Scholar]
- 46. Maia LB, Fonseca L, Moura I, Moura JJG (2016) Reduction of carbon dioxide by a molybdenum‐containing formate dehydrogenase: a kinetic and mechanistic study. J Am Chem Soc 138:8834–8846. [DOI] [PubMed] [Google Scholar]
- 47. Axley MJ, Böck A, Stadtman TC (1991) Catalytic properties of an Escherichia coli formate dehydrogenase in which sulfur replaces selenium. Proc Natl Acad Sci USA 88:8450–8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fanning JC (1999) The chemical reduction of nitrate in aqueous solution. Coord Chem Rev 199:159–179. [Google Scholar]
- 49. HK LI, Temple C, Rajagopalan KV, Schindelin H (2000) The 1.3 Å crystal structure of Rhodobacter sphaeroides dimethyl sulfoxide reductase reveals two distinct molybdenum coordination environments. J Am Chem Soc 122:7673–7680. [Google Scholar]
- 50. George GN, Colangelo CM, Dong J, Scott RA, Khangulov SV, Gladyshev VN, Stadtman TC (1998) X‐ray absorption spectroscopy of the molybdenum site of Escherichia coli formate dehydrogenase. J Am Chem Soc 120:1267–1273. [Google Scholar]
- 51. Robinson WE, Bassegoda A, Reisner E, Hirst J (2017) Oxidation‐state‐dependent binding properties of the active site in a Mo‐containing formate dehydrogenase. J Am Chem Soc 139:9927–9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Friedebold J, Bowien B (1993) Physiological and biochemical characterization of the soluble formate dehydrogenase, a molybdoenzyme from Alcaligenes eutrophus . J Bacteriol 175:4719–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu X, Niks D, Mulchandani A, Hille R (2017) Efficient reduction of CO2 by the molybdenum‐containing formate dehydrogenase from Cupriavidus necator (Ralstonia eutropha). J Biol Chem 292:16872–16879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vorholt JA, Thauer RK (1997) The active species of ‘CO2’ utilized by formylmethanofuran dehydrogenase from methanogenic archaea. Eur J Biochem 248:919–924. [DOI] [PubMed] [Google Scholar]
- 55. Mandrand‐Berthelot MA, Couchoux‐Luthand G, Santini CLGiordano G (1988) Mutants of Escherichia coli specifically deficient in respiratory formate dehydrogenase activity. J Gen Microbiol 134:3129–3139. [DOI] [PubMed] [Google Scholar]
- 56. Böhmer N, Hartmann T, Leimkühler S (2014) The chaperone FdsC for Thodobacter capsulatus formate dehydrogenase binds the bis‐molybdeopterin guanine dinucleotide cofactor. FEBS Lett 588:531–537. [DOI] [PubMed] [Google Scholar]
- 57. Thomé R, Gust A, Toci R, Mendel R, Bittner F, Magalon A, Walburger A (2012) A sulfurtransferase is essential for activity of formate dehydrogenases in Escherichia coli . J Biol Chem 287:4671–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Arnoux P, Ruppelt C, Oudouhou F, Lavergne J, Siponen MI, TOci R, Mendel RR, Bittner F, Pignol D, Magalon A, Walburger A (2015) Sulphur shuttling across a chaperone during molybdenum cofactor maturation. Nature Commun 6 10.1038/ncomms7148. [DOI] [PubMed] [Google Scholar]