

Abstract
Human MICAL1 is a member of a recently discovered family of multidomain proteins that couple a FAD‐containing monooxygenase‐like domain to typical protein interaction domains. Growing evidence implicates the NADPH oxidase reaction catalyzed by the flavoprotein domain in generation of hydrogen peroxide as a second messenger in an increasing number of cell types and as a specific modulator of actin filaments stability. Several proteins of the Rab families of small GTPases are emerging as regulators of MICAL activity by binding to its C‐terminal helical domain presumably shifting the equilibrium from the free – auto‐inhibited – conformation to the active one. We here extend the characterization of the MICAL1–Rab8 interaction and show that indeed Rab8, in the active GTP‐bound state, stabilizes the active MICAL1 conformation causing a specific four‐fold increase of k cat of the NADPH oxidase reaction. Kinetic data and small‐angle X‐ray scattering (SAXS) measurements support the formation of a 1:1 complex between full‐length MICAL1 and Rab8 with an apparent dissociation constant of approximately 8 μM. This finding supports the hypothesis that Rab8 is a physiological regulator of MICAL1 activity and shows how the protein region preceding the C‐terminal Rab‐binding domain may mask one of the Rab‐binding sites detected with the isolated C‐terminal fragment. SAXS‐based modeling allowed us to propose the first model of the free full‐length MICAL1, which is consistent with an auto‐inhibited conformation in which the C‐terminal region prevents catalysis by interfering with the conformational changes that are predicted to occur during the catalytic cycle.
Keywords: MICAL, flavoprotein, FAD‐containing monooxygenase/oxidase, enzyme kinetics, auto‐inhibition, Rab, small‐angle X‐ray scattering, regulation, protein–protein interaction
Abbreviations
- ADLTE
autosomal dominant lateral epilepsy
- bMERB
bivalent MICAL/EHBP Rab‐binding domain
- CasL
Crk (p38)‐associated substrate‐related protein
- CC
coiled‐coils
- CH
calponin homology
- CRMP
collapsin response mediator protein
- DTT
dithiothreitol
- EHBP
Eps15 homology domain‐binding proteins
- EMT
epithelial–mesenchymal transition
- ESCRT
Endosomal Sorting Complex Required for Transport
- F‐actin
filamentous actin
- G‐actin
globular actin
- GEF
GTP‐exchange factor
- GppNHp
guanosine 5′‐[β,γ‐imido]triphosphate
- ITC
isothermal titration calorimetry
- LIM
domain found in Lin11, Isl‐1, and Mec‐3 proteins
- Kd
dissociation constant
- Km
Michaelis–Menten constant
- kcat
turnover number
- MICAL
the Molecule Interacting with CasL
- MICAL‐cL
MICAL‐like protein containing only the C‐terminal domain
- MICAL‐L1 and MICAL‐L2
MICAL‐like Proteins 1 and 2
- MO
monooxygenase‐like domain
- MOCH
truncated MICAL form comprising the MO and CH domains
- MOCHLIM
truncated MICAL form comprising the MO, CH and LIM domains
- NADP(H)
(reduced) nicotinamide dinucleotide phosphate
- NDR
nuclear Dbf2‐related
- PHBH
p‐hydroxybenzoate hydroxylase
- PI3K
phosphatidylinositol‐4,5‐bisphosphate 3‐kinase
- PIP2
phosphatidylinositol bisphosphate
- pOHBz
p‐hydroxybenzoate
- RBD
Rab‐binding domain
- SAXS
small‐angle X‐ray scattering
- SDS
sodium dodecyl sulfate
- SEC
size exclusion chromatography
- SH3
Src homology domain‐3
Introduction
Human MICAL1 is a multidomain protein that couples an N‐terminal FAD‐containing domain (Residues 1–489) to a calponin homology domain (CH, Residues ~510–613), a LIM domain (from Lin‐11, Isl‐1, and Mec‐3 gene products, Residues ~688–756) and a C‐terminal region, which harbors several functional fragments and is believed to be the main site of regulation of MICAL catalytic activity (Fig. 1).
Figure 1.
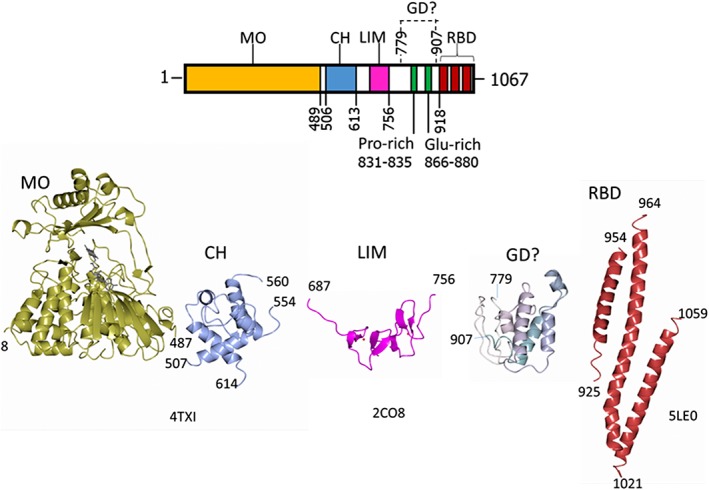
Domain organization of human MICAL1 and structures of the isolated domains. The numbering indicates the boundaries of the known domains of MICAL1: MO, monooxygenase‐like FAD‐containing domain; CH, type 2 calponin homology domain; LIM, LIM domain containing two zinc finger motifs; GD? indicates a potential globular domain identified by Robetta modelling, which contains the Pro‐rich motif typical of regions binding to SH3 domains and a Glu‐rich region; RBD, Rab‐binding domain as identified by 45, 46. The experimental (4TXI, 2CO8, 5LE0) and modeled (GD?) structures of the MICAL1 domains are also shown in ribbon. These and other structural models have been drawn with CCP4 MG.70
MICAL1 is a member of the relatively recently discovered family of MICAL proteins (from the Molecule Interacting with CasL1), which are conserved from insects to mammals and have been proposed to play multiple roles in cells, as well as to be subjected to (tight) control by interacting proteins, covalent modification and, perhaps, small molecules (see Refs. 2, 3, 4, 5, 6, 7 for recent reviews).
In mammals MICAL1, MICAL2, and MICAL3 are encoded by different genes, while there is only one gene encoding MICAL in the model organism Drosophila melanogaster.
MICALs are being implicated in a rapidly growing number of fundamental processes, including cell–cell contacts, cell division, differentiation and migration, epithelial–mesenchymal transition (EMT), axon growth and steering, formation of neuro‐muscular junctions, vasculogenesis, cardiogenesis, pathogen infection, vesicle trafficking, and gene transcription.2, 4, 6 Modulating MICAL1 and MICAL2 levels has been shown to be beneficial in reversing EMT, which is at the basis of metastatization in cancer.8, 9 MICAL1 inhibition has been proposed to promote axon regeneration following degeneration or spinal cord injury.10, 11, 12 Very recently, mutations in the gene encoding MICAL1 have been found to cause autosomal dominant lateral epilepsy (ADLTE), a rare genetic disease.13 In these processes, MICAL may serve as a scaffold to organize and modulate interacting proteins. As a result MICAL may interfere with the organization of tight junctions and intermediate filaments (through, e.g., interaction with CasL1) or with apoptosis (through, e.g., interaction with nuclear Dbf2‐related (NDR) kinases).14, 15
Several functions of MICALs require the intactness of their conserved N‐terminal FAD‐containing domain (e.g. Refs. 16, 17, 18). The latter catalyzes a NADPH oxidase reaction producing hydrogen peroxide,19 which may serve as a second messenger and lead to modulation of activity of target proteins through H2O2‐mediated oxidation, as proposed for the regulator of microtubules assembly collapsin response mediator protein‐2 (CRMP2),20 or the activation of the signaling pathway formed by phosphatidylinositol‐4,5‐bisphosphate 3‐kinase (PI3K) and the serine–threonine protein kinase Akt leading to breast cancer cell invasion.9
In the presence of filamentous actin (F‐actin), the velocity of NADPH oxidation is greatly enhanced due to a decrease of K m for NADPH and an increase of k cat for MICAL1.21, 22 Qualitatively similar effects have been observed for the isolated mouse MICAL2 flavoprotein domain,23 and the Drosophila protein.18, 24 Such NADPH‐dependent reaction catalyzed by the flavodomain causes oxidation of actin residues thereby weakening the filament and promoting its disassembly.24, 25, 26, 27 Thus, this catalytic activity makes MICAL unique among actin‐binding proteins, which typically promote actin assembly or disassembly upon binding interactions.28, 29, 30, 31 Whether oxidation is limited to conversion of Met44 and Met47 of the actin D‐loop to methionine sulfoxide residues following a switch from an oxidase to a hydroxylase activity (as proposed in Refs. 18, 26, 32) or oxidation of several residues (including the two methionine residues) takes place through in situ production of hydrogen peroxide (as supported by other data21) is still a debated issue. Both proposals are consistent with the known structure of mouse MICAL1 flavoprotein domain, which has been solved at 1.45–2.9 Å resolution (PDB ID: 2BRA, 2BRY, 2C4C, isolated domain19, 33) and 2.3–2.9 Å (Fragment 2–615 comprising the flavoprotein and the CH domains, PDB ID: 4TXI, 4TXK 34, Fig. 1). The flavoprotein domain is structurally similar to bacterial p‐hydroxybenzoate hydroxylase (PHBH), the prototype of enzymes of the class of FAD‐containing aromatic monooxygenases,35, 36, 37 which, however, shares a common fold with enzymes of the oxidase family such as, e.g., d‐amino acid oxidase.38 The similarity with PHBH, which is low at the primary structure level,3 extends to the conformational change from a “flavin out” (PDB ID: 2BRA and 2BRY 19, 33) to a “flavin in” conformation triggered by reduction by NADPH (PDB ID: 2C4C 33) and to the conservation of residues that in PHBH are implicated in NADPH binding.3, 19, 33 For this reason, we have previously referred to this domain as the MO domain from monooxygenase‐like domain21, 22 without necessarily implying that it catalyzes a monooxygenase/hydroxylase reaction. Rather, we have proposed that the MICAL1 MO domain is a bona fide NADPH oxidase and that the intrinsic flexibility of enzymes of the PHBH family is exploited to add another layer of control of MICAL's activity.2, 21
The CH domain was postulated to promote interaction with F‐actin on the basis of its presence in actin‐binding proteins. This concept is supported by work on the mouse MICAL1 MOCH form.34 However, it was found to have a modest effect on the interaction of human MICAL1 with F‐actin,21 a finding that is consistent with lack of actin binding of the isolated CH domain39 and that two tandem CH domains are found in actin‐binding proteins of the actinin family,40, 41 taken as the prototype of proteins containing CH domains. Whether the putative phosphatidylinositol bisphosphate (PIP2)‐binding site present in this domain has a modulating role has not been explored, yet. Also the specific role of the LIM domain has been poorly explored, but it may assist the C‐terminal domain for binding to some of MICAL interactors.42
The comparison of the spectroscopic and kinetic properties of different MICAL1 forms comprising the flavoprotein MO domain, the MO and CH (MOCH) and the MO, CH, and LIM (MOCHLIM) regions indicates that the CH and LIM domains cause an increase of the K m value for NADPH in the NADPH oxidase activity with little or no effect on the measured k cat.21 The K m increase was interpreted as due to an electrostatic effect, reinforcing the similarity with PHBH: at pH 7, the overall positive charge of the proteins decreases due to addition of the acidic CH and LIM domains to the basic MO weakening NADPH binding.
The C‐terminal region of MICAL1 appears to be mainly responsible of the modulation of the catalytic activities of MICAL MO, and also of the scaffold functions of MICAL unrelated to its catalytic activity (see interaction with CasL or NDR kinases1, 14, 16). In the free enzyme, it gives rise to an auto‐inhibited form, which was initially proposed by Schmidt et al.42 Drosophila MICAL was identified thanks to the interaction of this region with the cytoplasmic side of plexin,16 which is at basis of the activation of MICAL1 at the axon growth cone in response to the binding of the extracellular semaphorin with its plexin receptors. This event results in local disassembly of the actin cytoskeleton in the axon path‐finding process. Its Pro‐rich region, centered on a PXXP motif, is the likely docking site of Src Homology 3 Domain (SH3)‐containing kinases, which could regulate MICAL activity by phosphorylation, as shown in the case of the non‐receptor tyrosine kinase Abl and Drosophila MICAL.7, 43 This kinase has been recently proposed to bind to the PXXP motif in the C‐terminal region of MICAL and to promote phosphorylation of Tyr 500 of the MO domain (corresponding to Tyr 458 of MICAL1) leading to a mild (approximately 30%) increase of the rate of NADPH oxidation in the presence of F‐actin. That also human MICAL1 is likely under the control of phosphorylation events is supported by data deposited in the Phosphosite Plus website (https://www.phosphosite.org/44), which reports that Tyr483 (but not Tyr458), along with Tyr906, is often found to be phosphorylated in high‐throughput post‐translational modification studies.
Kinetic studies on full‐length MICAL1 and on forms progressively lacking the C‐terminal region, the LIM domain and the CH domain indicated that the C‐terminus leads to a conformational equilibrium between a fully active (open) and a fully inactive (closed) form that lies approximately 9:1 toward the inactive species in free MICAL21 (Fig. 2). Whether binding of PlexA or other proteins to the C‐terminal region of MICAL1 actually shifts the equilibrium toward the active MICAL conformation has not been tested experimentally except for proteins of the Rab family, although not on a quantitative level.17, 45, 46
Figure 2.
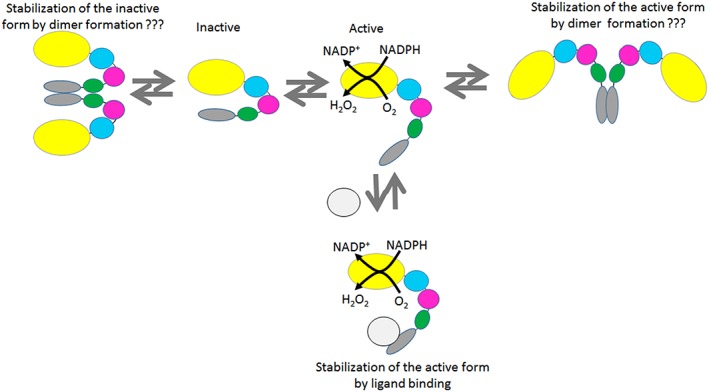
Proposed scheme of distribution between active and inactive conformations of MICAL1. The inactive conformation was calculated to prevail approximately 9:1 over the active one in free MICAL.21 (Macro)molecules binding to the C‐terminal domain of MICAL, which includes the Pro‐ and Glu‐rich regions (green) as well as the Rab‐binding domain (RBD, grey) are proposed to stabilize the active conformation. Such molecules would include, e.g., SH3‐containing proteins, Rab proteins or the cytoplasmic side of semaphorin‐activated plexin. The possibility that the active or inactive conformations of MICAL1 are stabilized by dimerization is also included.
Indeed, since the early studies on MICALs, proteins of the Rab family of Ras‐related small GTPases have been shown to interact with the C‐terminal part of the enzyme and they have been proposed to be MICAL regulators,47, 48, 49 implicating MICALs in the regulation of intracellular membrane trafficking.50, 51, 52 Rab proteins exist in the active GTP‐bound and inactive GDP‐bound conformations with the GTP/GDP exchange mediated by GTP exchange factors (GEF53) and it is the GTP‐bound active form of Rab that promotes membrane trafficking upon interaction with effector proteins.
Recently, the MICAL‐Rab interplay has been highlighted by several studies. It has been shown that MICAL1 is recruited to the cell abscission site by Rab35, leading to both actin cytoskeleton disassembly and recruitment of ESCRT (from Endosomal Sorting Complex Required for Transport) to complete cell division.46 MICAL3 has also been shown to form a complex with Rab8 and ELKS (so‐called from the high content in Glu, Leu, Lys, and Ser residues)17, 54 in the presence of Rab6, by which it is recruited to exocytic vesicles participating in their trafficking.17 Rab8 and ELKS also appear to target MICAL3 to the cell mid‐body, where MICAL3 seems to function as a protein‐binding hub organizing the membranes at the intercellular bridge in the late steps of cell division,54 rather than through its NADPH oxidase activity.
Important studies have dealt with the interaction of several proteins of the broad Rab family in their active (GTP‐bound) form with the C‐terminal fragments of human MICAL1 (Residues 918–1067) and MICAL3 (Residues 1841–1990), MICAL‐like proteins (MICAL‐L1 and MICAL‐L2, which lack the MICAL MO catalytic domain), and MICAL‐cL (a short MICAL‐like protein, which also lacks the CH and LIM domains).45 The studies extended to the analysis of Rab interaction with proteins of the family of Eps15 homology domain‐binding proteins (EHBP1 and EHBP1L1), which were found to contain a C‐terminal region similar to that of MICAL proteins. The latter, previously classified as domain of unknown function (DUF) 3585, was named bMERB from “bivalent MICAL/EHBP Rab‐Binding domain”.45 A parallel study evaluated the binding interaction between Rab35 and the C‐terminal Rab‐binding domain (RBD) of human MICAL1 following the finding of the recruitment of MICAL1 to the cell abscission site.46
Rab1 family members (Rab1a and Rab35) bound in a 1:1 stoichiometry with affinities in the 2–10 μM range to the C‐terminal Rab‐binding domain (RBD) of MICAL1, MICAL3, and MICAL‐cL,45, 46 but exhibited low affinity for EHBP1 and EHBP1L1 proteins. Proteins of the Rab8 family (Rab8a, Rab 10, Rab13, and Rab 15) bound with dissociation constants in the 100 nM range to the RBD of all the five proteins tested, as established by isothermal titration calorimetry (ITC) and co‐elution on analytical size exclusion chromatography (SEC).45 Interestingly, MICAL1 and MICAL3 exhibited a second binding site for proteins of the Rab8 family, which was characterized by a dissociation constant of approximately 0.5 μM (for MICAL1) and 4.4 μM (for MICAL3). Two Rab‐binding sites of similar affinity (160 nM) were also observed for EHBP1L1 and Rab8. The higher affinity of the proteins of the Rab8 family was ascribed to the details of their N‐terminal portion. Accordingly, the RBD of MICAL‐cL (Residues 534–683) was crystallized in complex with one copy of Rab1 (5SZH), Rab8 (5SZI), and Rab10 (5SZJ), as well as a chimeric form of Rab1 in which the first four residues were exchanged with those of Rab8. One copy of the C‐terminus of MICAL1 (here named Rab‐binding domain, RBD) was crystallized in complex with two copies of Rab10 (PDB ID: 5LPN) and that of MICAL3 (5SZG) as a dimer with the contacts between the monomers corresponding to the Rab‐binding sites of the corresponding region of MICAL145 (Fig. 3). The RBD is formed by three helices yielding a flat surface with the central helix (Helix 2) flanked by the first (Helix 1) and third one (Helix 3). Comparisons of the high resolution structures revealed that all bMERB (RBD) examined share a conserved Rab‐binding site formed mainly by the C‐terminal part of Helix 3 and some residues of Helix 2. The different affinity of this site for the different Rab proteins seems to depend on the precise position of Helices 1 and 2 as well on the ability of the N‐terminal segment of Rab to interact with a negative patch formed by Helix 1 and Helix 2. The second Rab‐binding site found for the MICAL1, MICAL3, and EHBP1L1 C‐terminal fragments in the presence of Rab8 (by ITC and SEC) and of Rab10 (by crystallography for MICAL1 RBD) was mainly formed by residues at the N‐terminus of Helix 1 and some residues of Helix 2 leading to the finding that the bMERB domain likely arose from duplication of an approximately 200 bp DNA fragment encoding a single α‐hairpin (Helix 1 and half of Helix 2) to yield the complete Helix 2 and the flanking Helices 1 and 3. Thus, one can define in MICAL1 RBD a first Rab‐binding site (Site 1 or Site N) involving the N‐terminal part of Helix 1, exhibiting a K d in the μM range, and second Rab‐binding site (Site 2 or Site C) at the C‐terminus of Helix 3, exhibiting a K d in the 100 nM range (55 nM for MICAL1 RBD). It is the second Rab‐binding site (Site 2 or Site C) that is conserved in all bMERB/RBD and its affinity for Rab proteins ranges from 160 nM (EHBP1L1) to 4.4 μM (MICAL3 RBD) depending, most likely, on the fine geometry of the structure formed by the three helices as well as the precise identity of the Rab protein.
Figure 3.
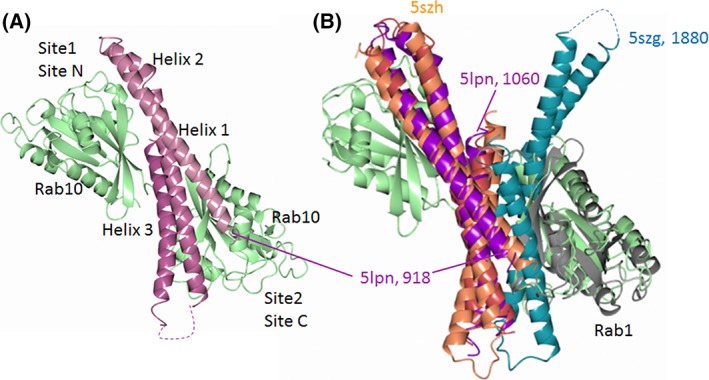
The bMERB domain of MICAL proteins and their complexes with Rab. (A) Complex between MICAL1 RBD (purple) and two copies of Rab10 in complex with GppNHp (green, PDB ID: 5LPN 45) bound to Site 1 (or Site N) and Site 2 (or Site C). (B) Superposition of the MICAL1:Rab10 complex (5LPN) with the MICAL3 RBD dimer (5SZG, red and dark cyan)45 and the complex between the RBD (bMERB) of MICAL‐cL1 (coral) and Rab1 (grey, 5SZH 45) to show the overlapping of the dimerization region of MICAL3 RBD with the Rab‐binding site located at the C‐terminus of MICAL RBD (Site 2 or Site C).
Complementary results have been presented by Fremont et al.46 who solved the structure of the isolated MICAL1 RBD (PDB ID: 5LEO), which appeared monomeric in the crystals (Fig. 1). ITC measurements led to conclude that MICAL1 C‐terminal RBD (Residues 918–1067) forms a 1:1 complex with Rab35 (a Rab1 family member) in the active (GTP‐bound) form. The K d was in the 5–10 μM range. By site directed mutagenesis, the Rab‐binding site was identified as the second (conserved) site (Site 2 or Site C) of Rai et al.45 By directly monitoring single actin filaments in a microfluidic system, Rab35 was also shown to enhance their depolymerization caused by full‐length MICAL1 and NADPH supporting the activating role of Rab upon binding to MICAL's C‐terminal RBD. Interestingly, the same experimental approach also showed that MICAL1 is not an actin severing protein. Rather, when the filaments were incubated with MICAL1 and NADPH, release of actin monomers from the filaments’ ends was observed and it continued even after MICAL and NADPH were washed away from the capillary. This result supports the concept that MICAL flavoprotein domain causes F‐actin depolymerization through in situ production of hydrogen peroxide rather than through a switch to a methionine monooxygenase activity. Oxidation of several actin residues would destabilize the filament and eventually lead to their release, as discussed before.2, 21
In order to provide a quantitative basis to the activating effect of Rab, and to test if and to what extent the MICAL domains (and linker regions) N‐terminal to its RBD influence the interaction with Rab, we undertook the study of the effect of human Rab8 on the catalytic activity of full‐length human MICAL1. The results indicate that indeed Rab8 binds to the C‐terminal region of MICAL1 stabilizing the active (open) conformation. However, only one molecule of Rab8 binds per MICAL1 molecule with an apparent affinity in the μM range, which is consistent with a regulatory role of Rab8 in the active GTP‐bound form (Fig. 2).
The finding that MICAL3 RBD dimerizes both in solution and in crystals and that the dimerization and Rab‐binding sites largely overlap45 (Fig. 3) also suggested that MICAL1 active and/or inactive conformations may be stabilized by dimer formation through their RBD, and that Rab binding could stabilize the monomeric form (Fig. 2).
Therefore, we have carried out in‐line small angle X‐ray scattering (SAXS) measurements on MICAL forms subjected to gel filtration (size exclusion) chromatography (SEC‐SAXS) in the absence or presence of Rab8. The data indicate that MICAL1 is a compact monomer in solution that could correspond to the closed/inactive conformation of free MICAL1. SEC‐SAXS measurements also allowed to detect the formation of a MICAL1–Rab8 complex and support the 1:1 binding stoichiometry observed kinetically.
Results
Effect of Rab8.GppNHp on the absorption spectrum of MICAL1
The absorption spectrum of full‐length MICAL1 (~10 μM) in Rab buffer (20 mM Hepes/NaOH buffer, pH 7.5, 50 mM NaCl, 2 mM MgCl2, 2 mM DTT) was recorded before and up to 4.5 h after the addition of Rab8 in complex with the GTP analog GppNHp (Rab8.GppNHp, 5–15 μM) at 10°C (not shown). No changes of the absorption spectrum caused by binding of Rab8 to MICAL were observed. This result is consistent with the lack of sensitivity of the absorption spectrum of FAD bound to the MO domain to the presence of the CH and LIM domain and of the C‐terminal region,21, 22 as well as the location of the Rab‐binding site in the C‐terminal region of MICAL1.
Effect of Rab8.GppNHp and Rab8.GDP on the NADPH oxidase activity of MICAL1 forms
The presence of Rab8 forms in the assays did not alter the overall shape of the activity traces, i.e. there was no indication of time‐dependent activation/inactivation of MICAL1 in the presence of varying concentrations of Rab8.GppNHp or Rab8 in complex with GDP (Rab8.GDP, data not shown). Rab8.GppNHp caused an increase of the initial velocity of the NADPH oxidase reaction catalyzed by full‐length MICAL regardless of the presence of 50 mM NaCl in the buffer (Fig. 4). The initial velocity values, expressed as v/E, obtained in the two buffers, at different Rab8.GppNHp concentrations were best fitted with eq. (1), which assumes the formation of a 1:1 complex between MICAL and Rab8.GppNHp. The apparent dissociation constants were similar in the two buffers differing for the NaCl presence and in the 10 μM range (Fig. 4 and Table 1).
Figure 4.
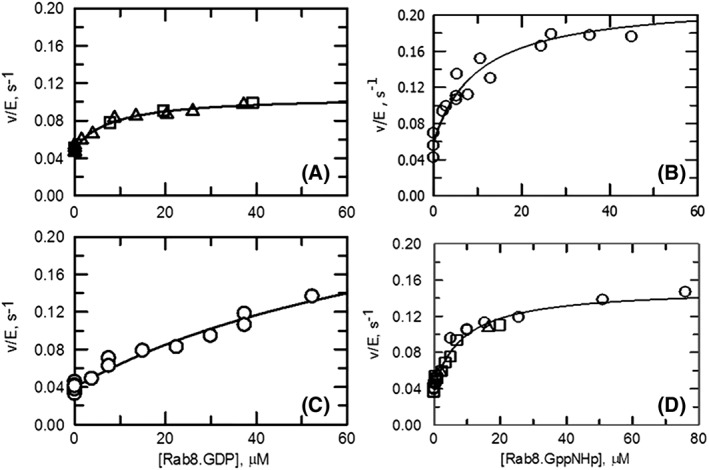
Effect of Rab8 forms on the NADPH oxidase activity of human MICAL1. Activity assays were carried out at 25°C in 20 mM Hepes/KOH, pH 7.5, 2 mM MgCl2, 2 mM DTT (panels A, B) or 20 mM Hepes/KOH, pH 7.5, 2 mM MgCl2, 2 mM DTT, 50 mM NaCl (Rab buffer, panels C, D) in the presence of 120 μM NADPH and varying concentrations of Rab8.GDP (panels A, C) or Rab8.GppNHp (panels B, D). Different symbols indicate data from different experiments. The v/E values from different experiments were fitted together to eq. (1). The values of the parameters used to draw the curves are in Table 1.
Table 1.
Effect of Rab8.GDP and Rab8.GppNHp on the NADPH Oxidase Activity of MICAL1
| MICAL, μM | Buffer | Rab8, varied | Eq. | (k +Rab–k −Rab), s−1 | k −Rab | K Rab, μM |
|---|---|---|---|---|---|---|
| 0.78 | Hepes | Rab8.GDP | 1 | 0.06 ± 0.003 | 0.05 ± 0.001 | 8.3 ± 1.6 |
| 1.5 | Rab | Rab8.GDP | 1 | 0.25 ± 0.1 | 0.04 ± 0.002 | 92 ± 49 |
| 0.9 | Hepes | Rab8.GppNHp | 1 | 0.16 ± 0.014 | 0.06 ± 0.007 | 10 ± 2.9 |
| 0.7 | Rab | Rab8.GppNHp | 1 | 0.11 ± 0.005 | 0.04 ± 0.002 | 8.8 ± 1.3 |
The NADPH oxidase activity of MICAL1 was measured in 20 mM Hepes/KOH buffer, pH 7.5, 2 mM MgCl2, 2 mM DTT (Hepes), or the same buffer containing 50 mM NaCl (Rab buffer) in the presence of varying concentrations of Rab8 forms and 120 μM NADPH. The v/E values were fitted to eq. (1), which describes a model in which the activity increases from a constant value in the absence of Rab8 (k −Rab) to a maximum value (k +Rab–k −Rab) upon formation of a 1:1 complex between MICAL1 and Rab8 whose dissociation constant is indicated as K Rab.
Rab8.GDP also increased MICAL activity (Fig. 4 and Table 1). In the absence of NaCl in the buffer, Rab8.GDP brought about only a two‐fold increase of activity with an apparent dissociation constant of the MICAL–Rab8.GDP complex similar to that observed with the active Rab8.GppNHp form (Table 1). However, the higher ionic strength Rab buffer brought about an approximately 10‐fold increase of the apparent dissociation constant of the MICAL–Rab8.GDP complex indicating that it is Rab8 in the active GTP‐bound form the likely physiological activator of MICAL1.
The effect of Rab8.GppNHp on the activity of MOCH and MOCHLIM forms was instead negligible (Fig. 5), demonstrating that MICAL RBD is necessary to bind Rab8.
Figure 5.
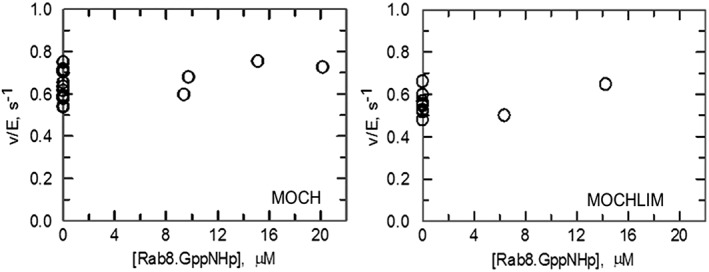
Effect of Rab8.GppNHp on the NADPH oxidase activity of MICAL1 MOCH and MOCHLIM truncated forms. Activity assays were carried out at 25°C in Rab buffer in the presence of 100 μM NADPH and varying concentrations of Rab8.GppNHp. MICAL1 MOCH or MOCHLIM forms were 0.18 μM each.
The finding that only one Rab8 molecule is sufficient to cause MICAL1 activation suggests that the MICAL1 fragment N‐terminal to the RBD (Fig. 1) masks one of the Rab‐binding sites detected with the isolated RBD (most likely Site 1/Site N), leaving only one site (most likely Site 2/Site C, which is conserved in all bMERB domains) available for binding. As an alternative, binding of Rab with high affinity to one of the sites (Site 2 or Site C) would not result in MICAL activation, which would depend on the interaction of Rab8 with Site 1 (or Site N), as suggested before.45 However, from inspection of the data of Figure 4, this case is unlikely since we did not observe any deviation from hyperbolic increase of v/E at low Rab8 concentrations where tight binding of a first Rab molecule to MICAL1 would have led to a “lag” in the curve.
The observed apparent dissociation constant of the MICAL1–Rab8.GppNHp complex in the range of 5–10 μM is one or two orders of magnitude higher than that measured for the Rab‐binding sites in the isolated MICAL1 RBD [0.5 μM for Site 1 (or Site N) and 50 nM for the C‐terminal site 2 (or site C)], but well within the range of the dissociation constant measured for MICAL1 RBD and Rab1 (2 μM)45 or Rab35 (5–10 μM).46 The increased K d can be in part rationalized assuming that Rab8 binds to the fraction of MICAL in the open/active form stabilizing it, thus taking into account a double equilibrium, and in part by taking into account possible interference of the rest of the protein with binding. In this respect, it is worth reminding that a 10‐fold increase of a K d value is associated with an increase of only 5.7 kJ/mol in the free energy change at equilibrium.
Effect of Rab8.GppNHp on the apparent steady‐state kinetic parameters of the NADPH oxidase reaction of MICAL1
The steady‐state kinetic parameters of the NADPH oxidase reaction of MICAL1 were measured in Rab buffer in the absence and in the presence of Rab8.GppNHp (10 μM). As shown in Figure 6, Rab8 does not alter the hyperbolic dependence of v/E as a function of NADPH, and it mainly has an effect on the apparent k cat of the reaction. From the k cat value (1.1 s−1), by taking into account that Rab8.GppNHp was used at a concentration (10 μM), i.e. close to the apparent K d value of the MICAL–Rab complex (5–10 μM), we may conclude that at saturating Rab8.GppNHp concentrations, k cat could reach a value of ~1.7 (for a K d value of 5 μM) or ~2.2 s−1 (for a K d value of 10 μM) so that most (if not all) MICAL would be converted to the catalytically active form. Indeed, k cat values in the range of 2–3 s−1 have been consistently measured for the MO, MOCH, and MOCHLIM forms.21, 22 The K m value for NADPH is high due to both the high K m for NADPH measured for full‐length MICAL1 at low ionic strength (375 μM)21 and its increase to approximately 1.1 mM is brought about by 50 mM NaCl in Rab buffer. The hypothesized shift from the inactive/closed to the active/open conformation brought about by Rab8 binding does not alter the apparent affinity for NADPH (Fig. 6). This result is in agreement with the concept that in the active/open conformation of MICAL1, the affinity for NADPH should be that of the truncated MOCHLIM form.21 With this protein species, the K m for NADPH was 230 μM in low ionic strength buffer, but increased to 0.97 ± 0.07 mM in the presence of 50 mM KCl.21
Figure 6.
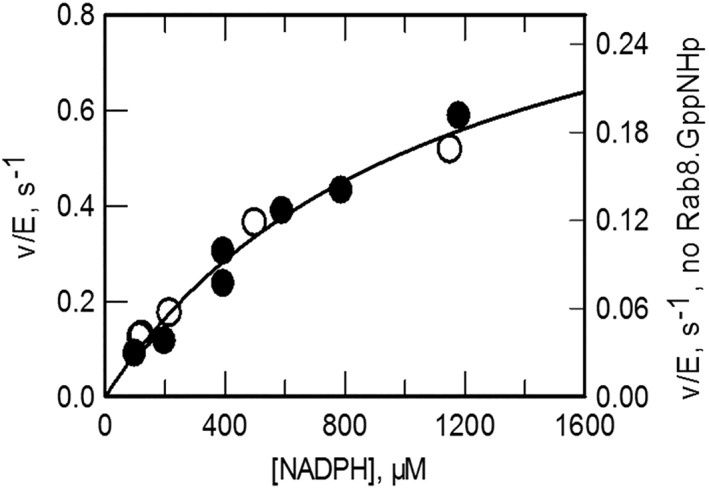
Effect of Rab8.GppNHp on the steady‐state kinetic parameters of the NADPH oxidase activity catalyzed by MICAL1. Assays were carried out at 25°C in Rab buffer in the absence (open circles, right axis) and in the presence of Rab8.GppNHp (10 μM, closed circles, left axis). The curve shown has been obtained by fitting the data collected in the absence and presence of Rab after dividing those obtained with Rab by 3.07. The K m value for NADPH was 1.1 ± 0.25 mM. The apparent k cat was 0.35 ± 0.05 s−1, which corresponds to a value of 1.1 ± 0.05 s−1 when the normalization factor (3.07) is taken into account.
Searching for a high affinity Rab8‐binding site
By working with the isolated MICAL1 Rab‐binding domain, Rai et al.45 observed the formation of a Rab8/MICAL–RBD 2:1 complex. By ITC, one binding site (Site 2 or Site C, Fig. 3) was characterized by a dissociation constant in the nM range (55 nM), while the second (Site 1 or Site N) by an apparent K d of approximately 0.5 μM. Proteins of other Rab families (i.e., Rab1 and Rab35) formed a 1:1 complex with the isolated MICAL RBD at Site 2 (or Site C) and bound more weakly with dissociation constants in the 2–10 μM range.45, 46
The finding, by kinetic analyses, of an apparent dissociation constant of 5–10 μM for a 1:1 complex between MICAL and Rab8.GppNHp would be consistent with either the hypothesis that the N‐terminal fragment comprising the MO, CH and LIM domains, as well as part of the C‐terminal region of MICAL1, abolishes the low affinity binding site at the N‐terminus of the RBD (Site 1), while the second Rab‐binding site, at the very C‐terminus of MICAL1, is the one we detect although with weakened binding.
However, binding of Rab8 to a site with nM affinity may be slow and may not be detected in our activity assays. To test this hypothesis, we measured the NADPH oxidase activity of MICAL1 that had been incubated with Rab8.GppNHp for up to 18 h (Table 2). No activity increase other than that observed immediately after MICAL addition to an assay mixture containing Rab8 and NADPH took place.
Table 2.
Effect of Long‐Term Incubation of MICAL1 with Rab8.GppNHp
| Time, h | v/E, s−1 | |
|---|---|---|
| −Rab | + Rab | |
| 0 | 0.037 | 0.102 |
| 1 | 0.042 | 0.094 |
| 17.3 | 0.042 | 0.088 |
| 18.5 | 0.041 | 0.099 |
1.5 μM MICAL1 was incubated in the absence or in the presence of 9.8 μM Rab8.GppNHp in Rab buffer, on ice, for the indicated times. 120 μl aliquots were transferred in a microcuvette, equilibrated at 25°C before starting the reaction with the addition of NADPH (140 μM).
To further test the possibility that MICAL1 and Rab8 could form a tight complex, which cannot be detected by activity measurements, we turned to SEC in house and, later, coupled to SAXS measurements in the SEC‐SAXS format described below. Co‐chromatography of Rab8 and the isolated MICAL1 RBD had been indeed observed.45
Samples (200 μl) of MICAL1 (13 μM) or Rab8.GppNHp (25 μM) alone and of MICAL1 (13 μM) and Rab8.GppNHp (25 μM), after preincubation on ice for 4 h, were loaded on a Superose 12 column in Rab buffer [Fig. 7(A)]. The MICAL‐containing fractions (0.5 ml) were analyzed by absorption spectroscopy, activity and SDS‐PAGE. No indication of co‐elution of Rab8 with MICAL1 was obtained from SDS‐PAGE or the specific activity values measured on fractions collected during the chromatography (not shown), supporting the kinetic data that indicate that only a low affinity Rab‐binding site is present in full‐length MICAL1. However, that MICAL1 and Rab8 had interacted prior to gel filtration was evident from the comparison of the chromatograms shown in Figure 7(A). When MICAL1 alone is chromatographed (Fig. 7, thin line), the main peak is preceded by a shoulder, which also contains MICAL. Rab8 alone elutes as a well‐defined peak [Fig. 7(A), dashed line]. When the MICAL1‐Rab8 mixture was chromatographed [Fig. 7(A,) thick line] the leading shoulder of the MICAL peak disappeared and MICAL peak was sharper, although at the same elution position as in the previous chromatogram. Rab8 eluted at a lower volume in a broader peak than when chromatographed alone indicating an equilibrium between free and MICAL‐bound Rab. The results have been confirmed and extended by carrying out SEC‐SAXS measurements using higher protein concentrations (see below).
Figure 7.
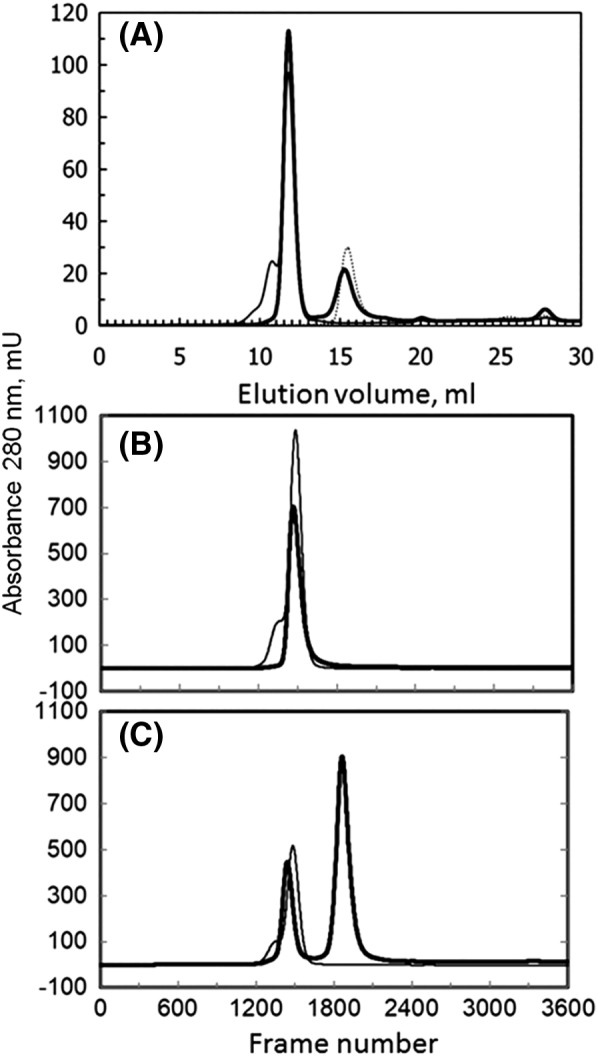
Size exclusion chromatography of MICAL1and Rab8.GppNHp. Panel A: Samples (200 μl) of MICAL1 (~13 μM, thin line) or Rab8.GppNHp (~25 μM, dashed line) alone and of MICAL1 (~13 μM) and Rab8.GppNHp (~25 μM), after preincubation on ice for 4 h (thick line) were chromatographed on a Superose 12 column in Rab buffer at a flow rate of 0.5 ml/min. Panel B: Samples (100 μl) of MOCHLIM (~80 μM, thick line) and MICAL1 (~90 μM, thin line) were injected on the Superose 12 column in Rab buffer with in line UV and SAXS detection. Panel C. Comparison of the elution profiles of MICAL1 (thin line, ~90 μM) and MICAL1 (~39 μM) and Rab8.GppNHp (~450 μM) after 7 h preincubation (thick line) from the Superose 12 column equilibrated in Rab buffer during the SEC‐SAXS experiment. For better comparison of the two chromatograms, the absorbance values of the chromatogram of the MICAL1 sample were divided by two to take into account the different concentration of MICAL1 in the two samples.
Assuming that MICAL aggregates are stable when the protein is diluted in the assays and that they are inactive, their dissociation caused by Rab8 is not sufficient to justify the 3–4‐fold increase of activity we observed. By calculating the ratio of the areas underlying the shoulder and the main peak with the Unicorn software controlling the AKTA apparatus, we calculated that the shoulder contains only 23% of the eluted protein, which may lead to only a 23% increase of activity.
During gel filtration chromatography on Superose 12 or Superose 6 column that had been calibrated with standard proteins, full‐length MICAL1 (main peak) eluted with an apparent mass of ~160 kDa regardless of the buffer (50 mM sodium phosphate, pH 7.5, 100 mM NaCl, 5–10% glycerol, 1 mM DTT, 1 mM EDTA or Rab buffer) and the gel filtration column (Superose 12 or Superose 6) indicating that the protein may exist in solution as a mixture of monomers (112 kDa) and dimers (224 kDa) in rapid equilibrium, as a non‐globular monomer or as a compact dimer.
The possibility that MICAL1 dimerizes and whether the dimerization may stabilize the inactive or the active conformations of MICAL and/or be influenced by the presence of Rab8 was studied by SAXS.
The quaternary structure of MICAL1 and the MICAL1–Rab8 complex
Batch SAXS measurements on MOCH (0.8–12 mg/ml) confirmed that the protein is monomeric and the scattering data were consistent with the scattering computed from the published high resolution structure of the mouse MICAL1 MOCH form (PDB ID: 4TXI 34; Fig. 8 and Table 3). In this respect, it should be noted that batch SAXS measurements were essential to establish the relative positioning of the MO and CH domains in crystals in the absence of electron density connecting them.34
Figure 8.
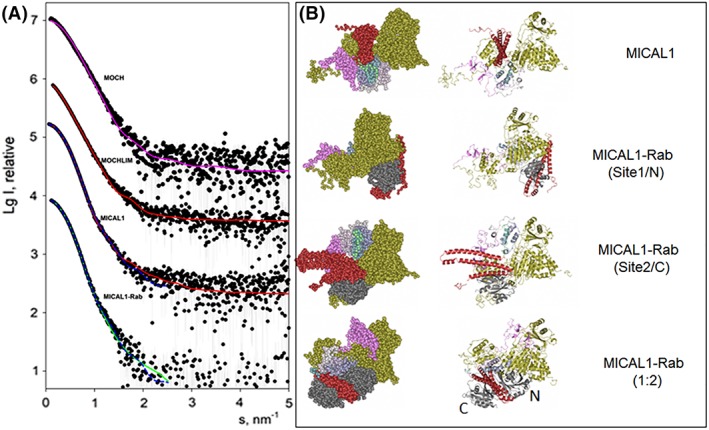
SAXS‐based modelling of MICAL1 and its complexes with Rab8.GppNHp. Panel A presents the experimental SAXS data depicted as dots, and the computed curves from the structural models in panel B shown as lines. The fit of the experimental scattering from MOCH by that computed from the monomeric structure of MOCH (PDB ID: 4TXI 34, Fig. 1) is displayed as magenta line. The simultaneous fits of the scattering from MOCHLIM and MICAL1 by a model of MICAL1 (panel B, top) are shown as red solid lines. The results of the simultaneous fitting of the scattering from MICAL1 and MICAL1‐Rab8.GppNHp complex assuming 1:1 stoichiometry are displayed as blue dashed lines; the two complexes with Rab8 binding to Sites 1 and 2 are shown in (B, middle). The best fit from a MICAL1‐Rab8.GppNHp with 1:2 stoichiometry (B, bottom) is displayed as a green solid line. The successive curves in panel A are displaced down by one logarithmic unit for better visualization. In the right panels the color scheme is as follows: gold, MOCH (4TXI); pink, LIM (2CO8); shaded from light to dark sea green: globular domain predicted by Robetta modeling (GD in Fig. 1); red, MICAL1 RBD (extracted from 5LPN); grey: Rab (extracted from 5LPN). In the lower panel, N and C indicate the position of Rab with respect to the two binding sites detected with the isolated RBD (5LPN).
Table 3.
SAXS Data Collection Statistics and Summary of Structural Results
| Construct | MOCH | MOCHLIM | MICAL | MICAL+Rab |
|---|---|---|---|---|
| Data collection parameters | ||||
| Beam line | P12 BioSAXS | P12 BioSAXS | P12 BioSAXS | P12 BioSAXS |
| Wavelength (nm) | 0.124 | 0.124 | 0.124 | 0.124 |
| s range (nm−1) | 0.09–6.7 | 0.14–5.0 | 0.06–5.0 | 0.11–5.0 |
| Exposure time (s) | 1 | 1 | 1 | 1 |
| Temperature (K) | 293 | 293 | 293 | 293 |
| Mode | Batch | Sec‐SAXS | Sec‐SAXS | Sec‐SAXS |
| Software employed for SAXS data reduction, analysis, and interpretation | ||||
| SAXS data reduction | Sasflow 63 | Sasflow 63 | Sasflow 63 | Sasflow 63 |
| SAXS data reduction | Primusqt 64
Gnom 67 |
Chromix55, Primusqt 64
Gnom 67 |
Chromix55, Primusqt 64
Gnom 67 |
Chromix55, Primusqt 64
Gnom 67 |
| Atomic structure based/hybrid modelling | Crysol 68 | Coral 56 | Coral 56 | Coral 56 |
| Structural parameters | ||||
| R g, nm | 3.4 ± 0.1 | 4.0 ± 0.2 | 3.7 ± 0.1 | 3.7 ± 1 |
| D max, nm | 12 ± 1 | 18 ± 2 | 12 ± 1 | 12 ± 1 |
| Porod volume estimate (nm3) | 100 ± 15 | 145 ± 20 | 212 ± 20 | 234 ± 25 |
| Displaced volume calculated from monomer (nm3) | 81 | 116 | 157 | 180 |
MOCHLIM and full‐length MICAL1, as well as MICAL1‐Rab8.GppNHp mixtures, were also analyzed in batch mode, but the samples demonstrated a tendency to partial multimerization (data not shown). In order to remove the influence of the small fraction of aggregated protein that could be present in solution (see Fig. 7), SAXS measurements were performed directly on the eluate of the gel filtration (size exclusion) chromatography column (Fig. 7, panels B and C).55 In such SEC‐SAXS mode, measurements are conducted every second, i.e. at a flow rate of 0.5 ml/min scattering signals are obtained every 8.3 μl. With this resolution, protein multimers in the early part of the MICAL peak are reliably separated from the later monomers and only the frames corresponding to the monomeric state can be used for the further analysis.
Data obtained with MOCHLIM and MICAL1 samples chromatographed in phosphate buffer and Rab buffer were indistinguishable from each other. Analyses of SAXS measurements across the protein peak indicated that the proteins were homogeneous. Thus, the SAXS measurements across each peak could be averaged, after correction for protein concentration as revealed by in‐line measurement of the refractive index of the eluent. The oligomeric states of the proteins were assessed from the excluded particle (so‐called Porod) volumes Vp in nm3, which are numerically about 1.6 times the molecular mass in kDa. The Vp value increased from 145 nm3 (MOCHLIM, 86.5 kDa) to 212 nm3 (MICAL1, 119 kDa, Table 3) suggesting that both MOCHLIM and MICAL in the main chromatographic peaks are present as monomers.
A somewhat less correlated behavior of the R g values (Table 3) could be explained by a higher mobility of the domains in the MOCHLIM construct, which is also reflected in the lower elution volume of MOCHLIM, as compared to MICAL1 [Fig. 7(B)].
The study of a mixture of full‐length MICAL (4.6 mg/ml, 39 μM) and Rab8.GppNhp (11 mg/ml, 450 μM) that had been pre‐incubated in the cold for 7 h yielded the chromatographic profile shown in Figure 7(C). As observed in the previous “in house” experiment with lower MICAL and Rab8 concentrations [Fig. 7(A)], pre‐incubation with Rab8 led to the disappearance of the aggregated species preceding MICAL main peak and to a broad Rab8.GppNHp peak. Interestingly, the first peak appeared to elute at a smaller volume than when MICAL was chromatographed alone [compare thin and thick traces in Fig. 7(C)].
The SAXS measurements on the eluate corresponding to the first main protein peak again showed that only one species was present, while the characteristics of the protein species in the second peak fully confirmed that the unbound Rab8 is monomeric under these conditions.
The excluded volume of the species in the main peak was 234 nm3, thus higher than that measured for free MICAL1 (212 nm3) pointing to the formation of a MICAL1–Rab8 complex.
To further test this hypothesis and to gain structural information on MOCHLIM, MICAL1, and the hypothetical MICAL–Rab complex, an attempt was made to construct a consensus model fitting simultaneously all three scattering curves from MOCHLIM, MICAL1, and MICAL1–Rab8.GppNHp (Fig. 8). For this purpose, the program Coral, which allows flexible fitting of the SAXS profiles by hybrid models of multidomain proteins, was employed.56
For the MOCHLIM analysis, the high resolution structures of mouse MICAL1 MOCH (PDB ID: 4TXI 34), and a model of the human MICAL1 LIM domain solved by NMR (PDB ID: 2CO8) were used (Fig. 1). For the full‐length MICAL1 and MICAL1–Rab8.GppNHp complex, in addition to the domains used to model MOCHLIM, the crystallographic model of the human MICAL1 RBD (Residues 918–1060; PDB ID: 5LPN, Chain B45), as well as a structural template made from RBD domain bound to one or two Rab10.GppNHp (from 5LPN), were utilized. The 779–907 region between the LIM and the RBD was modeled as a globular all‐α domain according to Robetta,57 which predicted it to be related to part of protein TM0771 from Thermotoga maritima msb8 (PDB ID: 2GNO, Fig. 1). The other missing parts were represented as potentially flexible chains of dummy residues.
It was not possible to fit the three curves simultaneously (data not shown), but both combinations of the scattering data from two constructs (i.e., MOCHLIM and MICAL1, or MICAL1 and MICAL1‐Rab8.GppNHp) could be neatly fitted (Fig. 8). This result suggested that a conformational change occurs in the MOCHLIM domain upon Rab binding, which preserves the overall shape of MICAL1.
A common model of free MOCHLIM and MICAL1 fits the two datasets with discrepancy χ 2 = 1.63 and 1.86, respectively [for the shorter construct, the reduced number of domains is taken into consideration during the fitting; Fig. 8(A), red line in MOCHLIM and MICAL1 curves] yielding the MICAL model in Figure 8(B). MICAL1 and MICAL1–Rab8.GppNHp were fitted together, while assuming 1:1 MICAL1–Rab8.GppNHp complexes in the two alternative positions of Rab (Site 1/Site N or Site 2/Site C) on the surface of RBD, and also in a 1:2 stoichiometry as in the crystallographic structure of MICAL1 RBD in complex with Rab10 (PDB ID: 5LPN, Fig. 3). No good fit could be obtained for the latter stoichiometry with two curves fitting (data not shown). In contrast, both 1:1 complexes built by Coral yielded good fits to the observed data (Fig. 8, blue lines MICAL1 and MICAL1–Rab curves). When Rab was modeled bound to Site 1 (or Site N) we obtained χ 2 values of 1.52 and 1.28 for full‐length MICAL1 (Fig. 8) and the MICAL–Rab complex, respectively; for Rab bound to the second position (Site 2 or Site C), χ 2 values of 1.37 and 1.39 were obtained.
These findings support a 1:1 stoichiometry of the MICAL1–Rab8.GppNHp. Still, the modelling of the 1:2 complex against its scattering curve alone was possible, but with larger discrepancy (χ 2 = 1.5) as compared to either 1:1 case (Fig. 8, MICAL1–Rab curve, green line). This result suggests that the 1:2 configuration is less likely than either one of the 1:1 complexes.
The MICAL model derived by fitting the MOCHLIM and free MICAL1 SAXS measurements [Fig. 8(B)] reveals a monomeric compact structure in which the RBD may inhibit the catalytic activity of MO by preventing the conformational changes that are part of the catalytic cycle.21, 22 In this conformation, the conserved Rab‐binding site (Site 2 or Site C) is accessible so that Rab8 can bind and destabilize the auto‐inhibited conformation of MICAL1. Both Coral‐derived models of possible MICAL1–Rab8 1:1 complexes show compact structures and appear to imply important conformational rearrangements in MICAL1.
As a caveat, we would like to note that the calculations made here represent tentative overall models and by no means should be considered as high resolution atomic structures.
Discussion
The interaction between MICAL and Rab8 was studied in order to contribute to understand, on a quantitative basis, the effect of Rab8 on MICAL1 activity.
As predicted by the binding of Rab proteins at the C‐terminus of MICAL1,45, 46 and by the initial model of the auto‐inhibition of MICAL activity by its C‐terminal region (Fig. 2),42 the active form of Rab8 caused an increase of the NADPH oxidase activity of full‐length MICAL1, but not of the truncated MOCH and MOCHLIM forms, indicating that also in the full‐length protein the Rab‐binding site is confined to the recently identified RBD.45, 46 Under our experimental conditions, Rab8.GDP also forms a complex with MICAL1, but with a 10‐fold lower affinity with respect to the Rab8.GppNHp form when nonspecific interactions are removed with the inclusion of 50 mM NaCl in the buffer.
The activating effect of Rab8.GppNHp on MICAL1 activity was consistent with the formation of a MICAL1–Rab complex in a 1:1 stoichiometry with an apparent dissociation constant of 5–10 μM. In contrast with results obtained with the isolated MICAL1 RBD,45 no evidence for a high affinity Rab‐binding site was obtained kinetically and by size‐exclusion chromatography of a mixture containing MICAL1 and Rab8.GppNHp in a 1:2 molar ratio. Thus, it appears that the MOCHLIM fragment and part of the C‐terminal region upstream of the RBD may mask one of the Rab‐binding sites found in the isolated MICAL1 RBD.
The K d measured kinetically is approximately two orders of magnitude higher than that measured for the conserved Site 2 (or Site C) in the isolated RBD (~ 50 nM),45 but it is in the range of the dissociation constants measured for Rab1 and Rab35.45, 46 The decrease of apparent affinity may be in part justified by the fact that Rab may bind to the fraction of MICAL1 in the open conformation and in part by subtle differences in the precise geometry of the complex. The precise arrangement of Helices 1 and 2 and the interaction of Rab8 N‐terminal residues with Helix 2 appear to be responsible for the higher affinity of Rab8 than that of Rab1 to bMERB Site 2 (Site C).45
Previous work on MICAL‐RBD/Rab interaction 45 prompted us to analyze in greater detail the oligomeric state of MICAL1. The isolated MICAL1 RBD chromatographed as a monomer45 and appeared monomeric also in crystals (PDB ID: 5LEO).46 However, the MICAL3 RBD was dimeric.45 In order to establish if MICAL1 could dimerize and if dimers could stabilize its active or inactive conformations we turned to SEC‐SAXS. The experiments indicated that MICAL1 is monomeric, thus making it unlikely that dimerization stabilizes one of the MICAL conformations (Fig. 2). Using the known high resolution structures of the MO, CH, LIM, and RBD of MICAL1, and a model of part of the region linking the LIM and the RBD, flexible fitting of the SAXS data led to a first tentative model of full length MICAL1 [Fig. 8(B)]. Within the limits of the analysis, the model is consistent with an inactive state of the catalytic flavoprotein (MO) domain of MICAL1 in which the conformational changes that are believed to be part of the catalytic cycle21, 22, 33 are prevented. In this model, the Rab‐binding site located at the C‐terminus of the protein (Site 2 or Site C), which is conserved in all bMERB domains, is on the protein surface and, therefore, accessible to Rab8. At the high MICAL1 and Rab8 concentrations used for the SEC‐SAXS experiment, MICAL1 appears to elute in complex with Rab8. Data analysis supports the formation of a MICAL1–Rab8 1:1 complex, which would be consistent with the results of the kinetic analyses. Unexpectedly, modeling also suggests that Rab8 binding may imply important conformational rearrangements of MICAL1. Thus, the simple scheme of Figure 2, which involves just repositioning of the C‐terminal region in the transition between the auto‐inhibited and the active conformation may need to be revised, should these models be confirmed by high resolution structural work.
In conclusion, this series of experiments confirms and extends the identification of the Rab‐binding domain at the C‐terminus of MICAL1,45, 46 but indicates that in the full‐length protein, only one Rab molecule, regardless of its subclass, binds with an affinity in the μM range. This finding supports a modulatory role of this class of proteins on MICAL1 activity in the cell by making MICAL1 sensitive to small changes of the concentration of their active forms. By binding to the MICAL's C‐terminal region, Rab relieves MICAL's auto‐inhibition shifting the conformational equilibrium toward the active MICAL form (Fig. 2) in which the flavoprotein domain can express its full catalytic power in the H2O2‐producing NADPH oxidase reaction and the NADPH‐dependent actin depolymerizing activity, regardless of the mechanism.
Finally, besides extending the first in vitro characterization of the interaction of MICAL1 with one of its protein effectors, these experiments allowed us to present a first tentative model of full‐length MICAL1, in the isolated – mainly inactive – (closed) conformation.
Materials and Methods
Chemicals
Chemicals were purchased from Sigma‐Aldrich s.r.l (Milano, Italy).
MICAL and Rab8 production and handling
Human full‐length MICAL1 and the MOCHLIM, MOCH, and MO truncated forms were produced and purified as described in Refs. 21, 22. Prior to each experiment aliquots of purified enzyme solutions (100–500 μl enzyme solution in 50 mM sodium phosphate buffer, pH 7.5, 100 mM NaCl, 10% glycerol, 1 mM DTT, 1 mM EDTA) were thawed, centrifuged at 12,000g for 10 min at 4°C. The supernatant was gel filtered through Sephadex G25 (medium) (PD10 ‐prepacked disposable columns, GE Healthcare, Little Chalfont, United Kingdom), Superose 12 (GE Healthcare) or BioSpin (Bio‐Rad Laboratories, Segrate, Italy) columns equilibrated and eluted with 20 mM Hepes/NaOH buffer, pH 7.5, 50 mM NaCl, 2 mM MgCl2, 2 mM DTT (Rab buffer). The latter buffer was chosen to reproduce the conditions of Rai et al.45 MICAL‐containing fractions were pooled and stored on ice. If needed, MICAL was concentrated by ultrafiltration with an Amicon ultrafiltration apparatus equipped with a YM30 membrane or a Microcon‐30 centrifugal concentrator (Merck Millipore, Milano, Italy). MICAL was stored on ice in the dark up for to 2 days. Under these conditions, the enzyme was stable as judged by no loss of NADPH oxidase activity, no change in the absorption spectrum and no protein degradation upon SDS‐PAGE analysis.58
MICAL concentration was determined from its extinction coefficient at 458 nm of 8.1 mM −1 cm−1. For diluted solutions, the calculated extinction coefficient at 280 nm of 145.79 mM −1 cm−1, which takes into account the presence of the bound FAD coenzyme, was used. A mass of 119,000 was used to calculate the protein concentration in mg/ml. For MICAL MOCH and MOCHLIM, the extinction coefficient in the visible (458 nm) was 8.1 mM −1 cm−1 and those at 280 nm were 110.84 mM −1 cm−1 and 120.81 mM −1 cm−1, respectively. The mass was 68,469 and 86,479 for MOCH and MOCHLIM, respectively (see summary table in Ref. 2).
Rab8 in complex with GDP or with guanosine 5′‐[β,γ‐imido]triphosphate (GppNHp), a non‐hydrolyzable GTP analog used to stabilize the active Rab conformation, was produced and purified as previously described.45, 59, 60 Prior to each experiment, aliquots (30 μl) of human Rab8 in complex with GppNHp (~1 mM) or with GDP (~1 mM) in Rab buffer were thawed on ice and centrifuged at 12,000g for 10 min at 4°C in a microfuge. The protein concentration was determined from the absorbance at 280 nm using the calculated extinction coefficient of 27.705 mM −1 cm−1, which takes into account the presence of the bound nucleotide.60 A mass of 23,862 was used to calculate the protein concentration in mg/ml.
NADPH oxidase activity assays
The NADPH oxidase activity of MICAL forms was measured spectrophotometrically in a HP8453 diode array spectrophotometer (Agilent Technologies, Santa Clara, CA, USA) at 25°C in quartz cuvettes. The initial velocity of NADPH oxidation was calculated by using the NADPH extinction coefficient at 340 nm of 6.23 mM −1 cm−1. Standard activity assays (in 125–1000 μl) contained 20 mM Hepes/NaOH buffer, pH 7.0, and 0.1 mM NADPH.22 MICAL forms (< 20 μl in 1 ml assays) were added to start the reaction at a final concentration of 0.03–1.5 μM, depending on the enzyme form.
To determine the effect of Rab forms on MICAL's NADPH oxidase activity, assays (125–250 μl) were carried out in 20 mM Hepes/NaOH buffer, pH 7.0 (standard buffer), 20 mM Hepes/NaOH buffer, pH 7.5, 2 mM MgCl2, 2 mM DTT or Rab buffer (20 mM Hepes/NaOH buffer, pH 7.5, 50 mM NaCl, 2 mM MgCl2, 2 mM DTT). Rab8 forms (0–50 μl) were added to the cuvette containing buffer and equilibrated at 25°C for 2–3 min. Recording of the absorption spectrum allowed us to verify the actual concentration of Rab8 in each assay. NADPH was then added from a 10 mM stock solution in 20 mM unbuffered Tris to a final concentration of 0.1–0.15 mM. After recording the absorption spectrum, the reaction was initiated by adding MICAL. At the end of several assays, aliquots of the reaction mixture (30 μl) were withdrawn, mixed with 10 μl of 4× SDS‐sample buffer, denatured at 100°C for 5 min or overnight at room temperature and analyzed by SDS‐PAGE. No proteolytic degradation of either MICAL or Rab was observed.
To determine the effect of Rab8.GppNHp on the apparent k cat and K m for NADPH of the NADPH oxidase reaction, NADPH was varied between 80 and 1300 μM. Activity assays were carried out at 25°C with the HP8453 diode array spectrophotometer, which allowed us to measure the decrease of absorbance associated with NADPH oxidation at different wavelengths. Thus, we could use the absorbance changes at 340 nm (ε, 6.23 mM −1 cm1), 374 nm (ε, 2.1 mM −1 cm−1), 380 nm (ε, 1.3 mM −1 cm−1), or 390 nm (ε, 0.4 mM −1 cm−1) for the calculation of initial velocities over the broad range of NADPH concentrations used.
Data analysis
Initial velocity values were expressed as apparent turnover numbers (v/E) in s−1 by dividing the calculated initial velocity of NADPH oxidation (v) by MICAL concentration (E) in the assay.
The effect of Rab8 forms on the NADPH oxidase activity of MICAL was determined by fitting the initial reaction velocity values (expressed as v/E) to eq. (1), which describes a single site saturation curve with a non‐zero constant value in the absence of the ligand:
| (1) |
When the concentration of Rab8 forms was varied, (k +Rab–k −Rab) is the maximum effect of Rab on v/E, i.e. the extrapolated maximum rate observed in the presence of Rab minus the v/E value measured in the absence of Rab (k −Rab), L is the Rab concentration in μM, K is the apparent dissociation constant of the MICAL–Rab complex (K Rab).
The Michaelis–Menten equation (eq. (2)) was used to fit the data obtained by varying NADPH in the presence or absence of Rab. In this equation, k cat is the v/E value extrapolated at infinite substrate concentration; K m is the Michaelis–Menten constant for the given substrate, which will be indicated as a subscript
| (2) |
In all cases, the data were first analyzed in the double reciprocal form and also fitted to alternative equations to test deviations from simple hyperbolic curves, but the best fit remained that to eq. (1) or (2).
The Grafit program (Erythacus Software Ltd. Horley, UK) was used for data fitting. Propagation of error was done according to Ref. 61.
Gel filtration (size exclusion) chromatography
For in house experiments, aliquots (250 μl) of MICAL (~10 μM), Rab forms (~25–50 μM) or MICAL (~10 μM) in Rab buffer were preincubated with Rab8 forms (25–50 μM) for 2–4 h on ice. 200 μl aliquots were injected onto a Superose 12 column (GE Healthcare) connected to a AKTA FPLC system (GE Healthcare). The column was equilibrated and eluted with Rab buffer at 0.5 ml/min. 0.5 ml fractions were collected and analyzed spectrophotometrically (to quantify MICAL present), by measuring the NADPH oxidase activity of MICAL‐containing fractions and by SDS‐PAGE. For SEC‐SAXS experiments, 100 μl aliquots of MOCHLIM (~90 μM) or MICAL1 (~90 μM) were injected onto the Superose 12 column equilibrated and eluted at 0.5 ml/min in phosphate (50 mM sodium phosphate buffer, pH 7.5, 100 mM NaCl, 5% glycerol, 1 mM DTT, and 1 mM EDTA) or Rab buffer. A mixture of MICAL1 (~39 μM) and Rab8.GppNHP (~450 μM) was chromatographed on the Superose 12 column in Rab buffer after 7 h incubation in the cold.
Small‐angle X‐ray scattering
Synchrotron SAXS measurements were performed at the P12 beamline of the EMBL (Petra‐3 storage ring, DESY Hamburg).62 Full‐length MICAL1 and the MOCHLIM and MOCH truncated forms were transferred to 50 mM sodium phosphate buffer, pH 7.5, 100 mM NaCl, 5–10% glycerol, 1 mM DTT, and 1 mM EDTA or Rab buffer by gel filtration. Protein samples were concentrated to ~10 mg/ml and frozen for transfer to EMBL‐Hamburg. After thawing and centrifugation the samples were diluted to 1–10 mg/ml and centrifuged again at 13,000 rpm for 15 min in a microfuge at 4°C prior to measurements. Protein concentration of each sample was determined spectrophotometrically prior to measurements. Parameters of SAXS experiments are summarized in Table 3.
The scattering data from MOCH were collected in batch mode using an automated sample changer at 4°C on a flowing sample (20 frames with the exposure time of 50 ms/frame). The SAXS data from MOCHLIM, MICAL1, and a MICAL1‐Rab mixture were obtained using an in‐line size exclusion chromatography (SEC‐SAXS) set‐up at room temperature. SAXS curves were measured every second on the eluate of the Superose 12 column (see previous paragraph for conditions).
For both the batch‐ and the SEC‐SAXS, the data reduction, radial averaging, and statistical analysis (e.g., to detect radiation damage, or scaling issues between frames) were performed using the SASFLOW pipeline.63 Statistically similar SAXS profiles were averaged and the buffer scattering subtracted to produce I(s) versus s scattering profiles (here, s = 4πsinθ/λ, 2θ is the scattering angle and λ is the wavelength). The SEC‐SAXS data were processed with CHROMIXS 55 to select the elution peak and the buffer scattering regions in the chromatographic profile.
SAXS data were analyzed using the ATSAS software package,64 and the primary data processing was performed using PRIMUS.65 The forward scattering I(0) and the radii of gyration (Rg) were evaluated using the Guinier approximation,66 assuming that at very small angles (s < 1.3/Rg), the intensity is represented as I(s) = I(0) exp(− (sRg)2/3. The maximum dimensions (D max) were computed using the indirect transform program GNOM,67 which also provides the distance distribution function p(r). Hybrid modelling was performed with Coral,56 which represents domains with known structure as rigid bodies and the linkers between them as chains of dummy residues. Multiple curves fitting using the data collected from the full‐length protein and from its deletion variant (MOCHLIM) was performed. Scattering from intact models was made by Crysol,68 which searches for the optimal values of hydration shell contrast, average atomic group radius and the total excluded volume to fit the experimental data I exp (s) by minimizing the discrepancy χ 2:
| (3) |
where N is the number of experimental points, c is a scaling factor, and I calc (s) and σ(s j ) are the calculated intensity and the experimental error at the momentum transfer s j , respectively.
Data deposition
The experimental SAXS data and models were deposited into SASBDB database (www.sasbdb.org 69) with accession codes SASDDR9 (full length human MICAL1), SASDDS9 (MOCHLIM), SASDDT9 (MICAL1–Rab8.GppNHp complex), and SASDDU9 (MOCH).
Conflict of interest
Authors declare no conflict of interest.
Short statement to use for broader audience
Steady‐state kinetics and small‐angle X‐ray scattering on human MICAL1, the multidomain flavoenzyme participating in actin cytoskeleton dynamics, showed that active Rab8 binds to the C‐terminus of the full‐length protein with a 1:1 stoichiometry leading to an increase of k cat of its H2O2‐producing NADPH oxidase reaction with an apparent K d of ~8 μM. The results are consistent with a physiological role of Rab8 in stabilizing the active MICAL conformation. The latter is in equilibrium with the auto‐inhibited form in which the C‐terminal region may prevent the conformational changes of the flavodomain taking place during catalysis.
Acknowledgments
Work in the Milano laboratory was supported in part by funds from the Department of Biosciences (University of Milano) and a generous donation from Mrs L. Farina (to MAV). This work was also supported in part by the Russian Federal Agency for Scientific Organizations (Agreement no. 007GZ/Ch3363/26, by the Russian Foundation for Basic Research (projects 18‐54‐74001 EMBL_T and KOMFI 17‐00‐00487) and the Max‐Planck‐Society. D. S. acknowledges the support by the Horizon 2020 program of the European Union, iNEXT Grant # 653706. We are most grateful to Prof. Roger Goody (MPI Dortmund) and Matthias Müller for helpful discussions. The excellent assistance of Dr. Alexey Kikhney (EMBL‐Hamburg Unit) during SAXS data acquisition is gratefully acknowledged.
Alessandro Esposito, Valeria Ventura, and Maxim V. Petoukhov contributed equally to the work.
References
- 1. Suzuki T, Nakamoto T, Ogawa S, Seo S, Matsumura T, Tachibana K, Morimoto C, Hirai H (2002) MICAL, a novel CasL interacting molecule, associates with vimentin. J Biol Chem 277:14933–14941. [DOI] [PubMed] [Google Scholar]
- 2. Vanoni MA (2017) Structure‐function studies of MICAL, the unusual multidomain flavoenzyme involved in actin cytoskeleton dynamics. Arch Biochem Biophys 632:118–141. [DOI] [PubMed] [Google Scholar]
- 3. Vanoni MA, Vitali T, Zucchini D (2013) MICAL, the flavoenzyme participating in cytoskeleton dynamics. Int J Mol Sci 14:6920–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fremont S, Romet‐Lemonne G, Houdusse A, Echard A (2017) Emerging roles of MICAL family proteins – from actin oxidation to membrane trafficking during cytokinesis. J Cell Sci 130:1509–1517. [DOI] [PubMed] [Google Scholar]
- 5. Giridharan SS, Caplan S (2014) MICAL‐family proteins: complex regulators of the actin cytoskeleton. Antioxid Redox Signal 20:2059–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yoon J, Terman JR (2018) MICAL redox enzymes and actin remodeling: new links to classical tumorigenic and cancer pathways. Mol Cell Oncol 5:e1384881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoon J, Terman JR (2018) Common effects of attractive and repulsive signaling: further analysis of Mical‐mediated F‐actin disassembly and regulation by Abl. Commun Integr Biol 11:e1405197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mariotti S, Barravecchia I, Vindigni C, Pucci A, Balsamo M, Libro R, Senchenko V, Dmitriev A, Jacchetti E, Cecchini M, Roviello F, Lai M, Broccoli V, Andreazzoli M, Mazzanti CM, Angeloni D (2016) MICAL2 is a novel human cancer gene controlling mesenchymal to epithelial transition involved in cancer growth and invasion. Oncotarget 7:1808–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deng W, Wang Y, Gu L, Duan B, Cui J, Zhang Y, Chen Y, Sun S, Dong J, Du J (2016) MICAL1 controls cell invasive phenotype via regulating oxidative stress in breast cancer cells. BMC Cancer 16:489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schmidt ER, Pasterkamp RJ, van den Berg LH (2009) Axon guidance proteins: novel therapeutic targets for ALS? Prog Neurobiol 88:286–301. [DOI] [PubMed] [Google Scholar]
- 11. Kolk SM, Pasterkamp RJ (2007) MICAL flavoprotein monooxygenases: structure, function and role in semaphorin signaling. Adv Exp Med Biol 600:38–51. [DOI] [PubMed] [Google Scholar]
- 12. Pasterkamp RJ, Dai HN, Terman JR, Wahlin KJ, Kim B, Bregman BS, Popovich PG, Kolodkin AL (2006) MICAL flavoprotein monooxygenases: expression during neural development and following spinal cord injuries in the rat. Mol Cell Neurosci 31:52–69. [DOI] [PubMed] [Google Scholar]
- 13. Dazzo E, Rehberg K, Michelucci R, Passarelli D, Boniver C, Vianello Dri V, Striano P, Striano S, Pasterkamp RJ, Nobile C (2018) Mutations in MICAL‐1cause autosomal‐dominant lateral temporal epilepsy. Ann Neurol 83:483–493. [DOI] [PubMed] [Google Scholar]
- 14. Zhou Y, Adolfs Y, Pijnappel WW, Fuller SJ, Van der Schors RC, Li KW, Sugden PH, Smit AB, Hergovich A, Pasterkamp RJ (2011) MICAL‐1 is a negative regulator of MST‐NDR kinase signaling and apoptosis. Mol Cell Biol 31:3603–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loria R, Bon G, Perotti V, Gallo E, Bersani I, Baldassari P, Porru M, Leonetti C, Di Carlo S, Visca P, Brizzi MF, Anichini A, Mortarini R, Falcioni R (2015) Sema6A and Mical1 control cell growth and survival of BRAFV600E human melanoma cells. Oncotarget 6:2779–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Terman JR, Mao T, Pasterkamp RJ, Yu HH, Kolodkin AL (2002) MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin‐mediated axonal repulsion. Cell 109:887–900. [DOI] [PubMed] [Google Scholar]
- 17. Grigoriev I, Yu KL, Martinez‐Sanchez E, Serra‐Marques A, Smal I, Meijering E, Demmers J, Peranen J, Pasterkamp RJ, van der Sluijs P, Hoogenraad CC, Akhmanova A (2011) Rab6, Rab8, and MICAL3 cooperate in controlling docking and fusion of exocytotic carriers. Curr Biol 21:967–974. [DOI] [PubMed] [Google Scholar]
- 18. Hung RJ, Pak CW, Terman JR (2011) Direct redox regulation of F‐actin assembly and disassembly by Mical. Science 334:1710–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nadella M, Bianchet MA, Gabelli SB, Barrila J, Amzel LM (2005) Structure and activity of the axon guidance protein MICAL. Proc Natl Acad Sci USA 102:16830–16835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morinaka A, Yamada M, Itofusa R, Funato Y, Yoshimura Y, Nakamura F, Yoshimura T, Kaibuchi K, Goshima Y, Hoshino M, Kamiguchi H, Miki H (2011) Thioredoxin mediates oxidation‐dependent phosphorylation of CRMP2 and growth cone collapse. Sci Signal 4:ra26. [DOI] [PubMed] [Google Scholar]
- 21. Vitali T, Maffioli E, Tedeschi G, Vanoni MA (2016) Properties and catalytic activities of MICAL1, the flavoenzyme involved in cytoskeleton dynamics, and modulation by its CH, LIM and C‐terminal domains. Arch Biochem Biophys 593:24–37. [DOI] [PubMed] [Google Scholar]
- 22. Zucchini D, Caprini G, Pasterkamp RJ, Tedeschi G, Vanoni MA (2011) Kinetic and spectroscopic characterization of the putative monooxygenase domain of human MICAL‐1. Arch Biochem Biophys 515:1–13. [DOI] [PubMed] [Google Scholar]
- 23. McDonald CA, Liu YY, Palfey BA (2013) Actin stimulates reduction of the MICAL‐2 monooxygenase domain. Biochemistry 52:6076–6084. [DOI] [PubMed] [Google Scholar]
- 24. Hung RJ, Yazdani U, Yoon J, Wu H, Yang T, Gupta N, Huang Z, van Berkel WJ, Terman JR (2010) Mical links semaphorins to F‐actin disassembly. Nature 463:823–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grintsevich EE, Yesilyurt HG, Rich SK, Hung RJ, Terman JR, Reisler E (2016) F‐actin dismantling through a redox‐driven synergy between Mical and cofilin. Nat Cell Biol 18:876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hung RJ, Spaeth CS, Yesilyurt HG, Terman JR (2013) SelR reverses Mical‐mediated oxidation of actin to regulate F‐actin dynamics. Nat Cell Biol 15:1445–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giridharan SS, Rohn JL, Naslavsky N, Caplan S (2012) Differential regulation of actin microfilaments by human MICAL proteins. J Cell Sci 125:614–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dominguez R, Holmes KC (2011) Actin structure and function. Annu Rev Biophys 40:169–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aguda AH, Burtnick LD, Robinson RC (2005) The state of the filament. EMBO Rep 6:220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Puius YA, Mahoney NM, Almo SC (1998) The modular structure of actin‐regulatory proteins. Curr Opin Cell Biol 10:23–34. [DOI] [PubMed] [Google Scholar]
- 31. Dent EW, Gupton SL, Gertler FB (2011) The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harb Perspect Biol 3:a001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee BC, Peterfi Z, Hoffmann FW, Moore RE, Kaya A, Avanesov A, Tarrago L, Zhou Y, Weerapana E, Fomenko DE, Hoffmann PR, Gladyshev VN (2013) MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol Cell 51:397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Siebold C, Berrow N, Walter TS, Harlos K, Owens RJ, Stuart DI, Terman JR, Kolodkin AL, Pasterkamp RJ, Jones EY (2005) High‐resolution structure of the catalytic region of MICAL (molecule interacting with CasL), a multidomain flavoenzyme‐signaling molecule. Proc Natl Acad Sci USA 102:16836–16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alqassim SS, Urquiza M, Borgnia E, Nagib M, Amzel LM, Bianchet MA (2016) Modulation of MICAL monooxygenase activity by its calponin homology domain: structural and mechanistic insights. Sci Rep 6:22176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Palfey BA, McDonald CA (2010) Control of catalysis in flavin‐dependent monooxygenases. Arch Biochem Biophys 493:26–36. [DOI] [PubMed] [Google Scholar]
- 36. Huijbers MM, Montersino S, Westphal AH, Tischler D, van Berkel WJ (2014) Flavin dependent monooxygenases. Arch Biochem Biophys 544:2–17. [DOI] [PubMed] [Google Scholar]
- 37. Entsch B, Cole LJ, Ballou DP (2005) Protein dynamics and electrostatics in the function of p‐hydroxybenzoate hydroxylase. Arch Biochem Biophys 433:297–311. [DOI] [PubMed] [Google Scholar]
- 38. Mattevi A, Vanoni MA, Todone F, Rizzi M, Teplyakov A, Coda A, Bolognesi M, Curti B (1996) Crystal structure of d‐amino acid oxidase: a case of active site mirror‐image convergent evolution with flavocytochrome b2. Proc Natl Acad Sci USA 93:7496–7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun H, Dai H, Zhang J, Jin X, Xiong S, Xu J, Wu J, Shi Y (2006) Solution structure of calponin homology domain of human MICAL‐1. J Biomol NMR 36:295–300. [DOI] [PubMed] [Google Scholar]
- 40. Gimona M, Winder SJ (1998) Single calponin homology domains are not actin‐binding domains. Curr Biol 8:R674–R675. [DOI] [PubMed] [Google Scholar]
- 41. Gimona M, Djinovic‐Carugo K, Kranewitter WJ, Winder SJ (2002) Functional plasticity of CH domains. FEBS Lett 513:98–106. [DOI] [PubMed] [Google Scholar]
- 42. Schmidt EF, Shim SO, Strittmatter SM (2008) Release of MICAL autoinhibition by semaphorin‐plexin signaling promotes interaction with collapsin response mediator protein. J Neurosci 28:2287–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yoon J, Kim SB, Ahmed G, Shay JW, Terman JR (2017) Amplification of F‐actin disassembly and cellular repulsion by growth factor signaling. Dev Cell 42(117–129):e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43:D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rai A, Oprisko A, Campos J, Fu Y, Friese T, Itzen A, Goody RS, Gazdag EM, Muller MP (2016) bMERB domains are bivalent Rab8 family effectors evolved by gene duplication. Elife 5:e18675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fremont S, Hammich H, Bai J, Wioland H, Klinkert K, Rocancourt M, Kikuti C, Stroebel D, Romet‐Lemonne G, Pylypenko O, Houdusse A, Echard A (2017) Oxidation of F‐actin controls the terminal steps of cytokinesis. Nat Commun 8:14528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fischer J, Weide T, Barnekow A (2005) The MICAL proteins and rab1: a possible link to the cytoskeleton? Biochem Biophys Res Commun 328:415–423. [DOI] [PubMed] [Google Scholar]
- 48. Weide T, Teuber J, Bayer M, Barnekow A (2003) MICAL‐1 isoforms, novel rab1 interacting proteins. Biochem Biophys Res Commun 306:79–86. [DOI] [PubMed] [Google Scholar]
- 49. Fukuda M, Kanno E, Ishibashi K, Itoh T (2008) Large scale screening for novel rab effectors reveals unexpected broad Rab binding specificity. Mol Cell Proteomics 7:1031–1042. [DOI] [PubMed] [Google Scholar]
- 50. Grosshans BL, Ortiz D, Novick P (2006) Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci USA 103:11821–11827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goody RS, Muller MP, Wu YW (2017) Mechanisms of action of Rab proteins, key regulators of intracellular vesicular transport. Biol Chem 398:565–575. [DOI] [PubMed] [Google Scholar]
- 52. Mott HR, Owen D (2015) Structures of Ras superfamily effector complexes: What have we learnt in two decades? Crit Rev Biochem Mol Biol 50:85–133. [DOI] [PubMed] [Google Scholar]
- 53. Muller MP, Goody RS (2018) Molecular control of Rab activity by GEFs, GAPs and GDI. Small GTPases 9:5–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu Q, Liu F, Yu KL, Tas R, Grigoriev I, Remmelzwaal S, Serra‐Marques A, Kapitein LC, Heck AJ, Akhmanova A (2016) MICAL3 flavoprotein monooxygenase forms a complex with centralspindlin and regulates cytokinesis. J Biol Chem 291:20617–20629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Panjkovich A, Svergun DI (2018) CHROMIXS: automatic and interactive analysis of chromatography‐coupled small‐angle X‐ray scattering data. Bioinformatics 34:1944–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV, Svergun DI (2012) New developments in the ATSAS program package for small‐angle scattering data analysis. J Appl Cryst 45:342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim DE, Chivian D, Baker D (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 32:W526–W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. [DOI] [PubMed] [Google Scholar]
- 59. Bleimling N, Alexandrov K, Goody R, Itzen A (2009) Chaperone‐assisted production of active human Rab8A GTPase in Escherichia coli . Protein Expr Purif 65:190–195. [DOI] [PubMed] [Google Scholar]
- 60. Smith SJ, Rittinger K (2002) Preparation of GTPases for structural and biophysical analysis. Methods Mol Biol 189:13–24. [DOI] [PubMed] [Google Scholar]
- 61. Bevington PR. Data Reduction and Error Analysis for the Physical Sciences. New York: McGraw‐Hill, 1969. [Google Scholar]
- 62. Blanchet CE, Spilotros A, Schwemmer F, Graewert MA, Kikhney A, Jeffries CM, Franke D, Mark D, Zengerle R, Cipriani F, Fiedler S, Roessle M, Svergun DI (2015) Versatile sample environments and automation for biological solution X‐ray scattering experiments at the P12 beamline (PETRA III, DESY). J Appl Cryst 48:431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Franke DKA, Svergun DI (2012) Automated acquisition and analysis of small angle X‐ray scattering data. Nucl Instrum Meth Phys Res A 689:52–59. [Google Scholar]
- 64. Franke D, Petoukhov MV, Konarev PV, Panjkovich A, Tuukkanen A, Mertens HDT, Kikhney AG, Hajizadeh NR, Franklin JM, Jeffries CM, Svergun DI (2017) ATSAS 2.8: a comprehensive data analysis suite for small‐angle scattering from macromolecular solutions. J Appl Cryst 50:1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI (2003) PRIMUS: a Windows PC‐based system for small‐angle scattering data analysis. J Appl Cryst 36:1277–1282. [Google Scholar]
- 66. Guinier A (1939) La diffraction des rayons X aux très petits angles : application à l'étude de phénomènes ultramicroscopiques. Ann Phys 11:161–237. [Google Scholar]
- 67. Svergun D (1992) Determination of the regularization parameter in indirect‐transform methods using perceptual criteria. J Appl Cryst 25:495–503. [Google Scholar]
- 68. Svergun D, Barberato C, Koch MHJ (1995) CRYSOL – a program to evaluate X‐ray solution scattering of biological macromolecules from atomic coordinates. J Appl Cryst 28:768–773. [Google Scholar]
- 69. Valentini E, Kikhney AG, Previtali G, Jeffries CM, Svergun DI (2015) SASBDB, a repository for biological small‐angle scattering data. Nucleic Acids Res 43:D357–D363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McNicholas S, Potterton E, Wilson KS, Noble ME (2011) Presenting your structures: the CCP4mg molecular‐graphics software. Acta Cryst D67:386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]