

Abstract
MiaB is a member of the methylthiotransferase subclass of the radical S‐adenosylmethionine (SAM) superfamily of enzymes, catalyzing the methylthiolation of C2 of adenosines bearing an N 6‐isopentenyl (i6A) group found at position 37 in several tRNAs to afford 2‐methylthio‐N 6‐(isopentenyl)adenosine (ms2i6A). MiaB uses a reduced [4Fe–4S]+ cluster to catalyze a reductive cleavage of SAM to generate a 5′‐deoxyadenosyl 5′‐radical (5′‐dA•)—a required intermediate in its reaction—as well as an additional [4Fe–4S]2+ auxiliary cluster. In Escherichia coli and many other organisms, re‐reduction of the [4Fe–4S]2+ cluster to the [4Fe–4S]+ state is accomplished by the flavodoxin reducing system. Most mechanistic studies of MiaBs have been carried out on the enzyme from Thermotoga maritima (Tm), which lacks the flavodoxin reducing system, and which is not activated by E. coli flavodoxin. However, the genome of this organism encodes five ferredoxins (TM0927, TM1175, TM1289, TM1533, and TM1815), each of which might donate the requisite electron to MiaB and perhaps to other radical SAM enzymes. The genes encoding each of these ferredoxins were cloned, and the associated proteins were isolated and shown to support turnover by Tm MiaB. In addition, TM1639, the ferredoxin‐NADP+ oxidoreductase subunit α (NfnA) from Tm was overproduced and isolated and shown to provide electrons to the Tm ferredoxins during Tm MiaB turnover. The resulting reactions demonstrate improved coupling between formation of the 5′‐dA• and ms2i6A production, indicating that only one hydrogen atom abstraction is required for the reaction.
Keywords: redox homeostasis, iron–sulfur cluster, redox potential, ferredoxin, S‐adenosylmethionine, methylthiolation
Abbreviations and symbols
- 5′‐dA•
5′‐deoxyadenosyl radical
- DTT
dithiothreitol
- FAD
flavin adenine dinucleotide
- flr
flavodoxin reductase
- flv
flavodoxin
- i6A
N 6‐(isopentenyl)adenosine
- LC–MS/MS
liquid chromatography tandem mass spectrometry
- ms2i6A
2‐methylthio‐N 6‐(isopentenyl)adenosine
- MTTase
methylthiotransferase
- NAD+
nicotinamide adenine dinucleotide
- RS
radical S‐adenosylmethionine
- SAH
S‐adenosylhomocysteine
- SAM
S‐adenosylmethionine
- Tm
Thermotoga maritima
Introduction
The radical S‐adenosylmethionine (SAM) superfamily of enzymes catalyze a wide variety of reactions, almost all of which require the intermediacy of a 5′‐deoxyadenosyl 5′‐radical (5′‐dA•), a potent oxidant. The most common role for the 5′‐dA• is to abstract hydrogen atoms from non‐covalently bound substrates, initiating substrate‐based catalysis. MiaB, a member of the methylthiotransferase (MTTase) subclass of radical SAM (RS) enzymes, uses the 5′‐dA• intermediate to catalyze the attachment of a methylthio (–SCH3) group to C2 of N 6‐(isopentenyl)adenosine, which is found in most tRNAs that decode codons with a leading uridine. The resulting 2‐methylthio‐N 6‐(isopentenyl)adenosine (ms2i6A), like many RNA modifications found within the anticodon stem loop, confers improved codon‐anticodon base‐pairing and results in decreased reading frame‐shift errors in a codon‐specific manner. Defects in the gene encoding its human homolog, CDK5RAP1, have been linked to mitochondrial myopathy and cardiac dysfunction.1, 2, 3, 4, 5, 6
Like other members of the RS MTTase subclass of enzymes, MiaB harbors two essential [4Fe–4S] clusters ligated by two strictly conserved cysteine‐containing motifs, and both clusters display 2+/1+ redox couples. The cluster ligated by the cysteine‐containing motif within the N‐terminal domain of MiaB is termed the auxiliary cluster ([4Fe–4S]aux), while the second cluster, ligated by cysteinyl residues in the hallmark CX3CX2C RS motif, binds SAM and is termed the RS cluster ([4Fe–4S]RS). In the reduced [4Fe–4S]1+ state, the RS cluster promotes the reductive cleavage of SAM to form [4Fe–4S]2+, methionine, and a 5′‐dA•, an intermediate in almost all RS reactions. In Escherichia coli (E. coli) and many other organisms, the flavodoxin reducing system, composed of flavodoxin and flavodoxin reductase, with electrons ultimately deriving from NADPH, reduces the [4Fe–4S]2+ cluster to the 1+ oxidation state. However, in organisms that lack the flavodoxin reducing system, such as the Gram‐negative hyperthermophile Thermotoga maritima, the source of electrons required to reduce the [4Fe–4S]2+ cluster is unknown.
While in vitro studies of numerous RS enzymes, including MiaB, have been conducted using artificial chemical reductants such as dithionite and reduced methyl viologen to supply electrons to the RS cluster, the use of these reductants often leads to side reactions, such as the reductive cleavage of SAM without substrate‐based catalysis.7 The use of the physiological flavodoxin/flavodoxin–reductase reducing system may decrease the extent of this abortive cleavage, as observed with other RS enzymes8, 9; however, Tm MiaB is inactive in the presence of E. coli flavodoxin, prompting our search for a homologous reducing system in the hyperthermophile's genome. Tm lacks a flavodoxin homolog; however, a number of ferredoxins are encoded in the organism's genome, some of which have already been characterized.8 Both ferredoxins and flavodoxins transport electrons in numerous cellular reactions despite their differences in size, structure, and functional cofactors. Whereas ferredoxins are small (typically <100 amino acid) proteins with Fe/S clusters, flavodoxins are larger (~170 amino acids) and contain flavin mononucleotide (FMN) or flavin adenine dinucleotide (FAD) cofactors, and neither shares similar secondary structure or tertiary folds.10 However, both proteins possess similar negatively charged surface domains near their active sites that are purported to allow for their interchangeability among binding partners.10 Indeed, work conducted with the cyanobacteria Anabaena reducing systems indicates that flavodoxins and ferredoxins are interchangeable in reducing Photosystem I and share similar active site electrostatic alignment.10
Ferredoxins are small, acidic, non‐heme Fe‐containing cytosolic proteins tasked with the role of electron transport from a donor to an acceptor partner protein. These proteins are typically expressed under conditions when iron is not limiting. They participate in cellular energy metabolism and are involved in a variety of pathways ranging from glycolysis, respiration, and nucleic acid and amino acid synthesis. Additionally, ferredoxins are involved in the assembly of iron‐sulfur (Fe/S) clusters themselves.10 Unlike the flavin‐dependent electron carriers that perform similar roles in electron transfer, ferredoxins contain one or more redox‐active Fe/S clusters in place of flavin cofactors.10 These Fe/S clusters are found in a variety of configurations. They are typically categorized into “plant” type ferredoxins, which harbor a [2Fe–2S] cluster, and “bacterial” type ferredoxins, which harbor one or more [4Fe–4S] or [3Fe–4S] clusters. They may also be further differentiated based on cluster ligation motifs. Plant type ferredoxins are widely distributed in nature and are found in eukaryotes, plants and bacteria. They typically contain a single [2Fe–2S] cluster and have a molecular weight on the order of 14 kDa, which is larger than most bacterial type ferredoxins.11 This group is typically divided into subclasses, one of which is involved in photosynthesis and present in higher plant chloroplasts and blue green algae. These ferredoxins ligate a [2Fe–2S] cluster using four cysteinyl resides found in a CX4CX2CX29C motif.12 A separate subclass of plant type ferredoxins, which are present in eukaryotic mitochondria of higher animals, transport electrons to cytochrome P450 enzymes.12 In contrast, these ferredoxins contain Fe/S cluster binding motifs in a CX5CX2CX36‐37C pattern and are typified by the E. coli ferredoxin, which has been well characterized since its identification in 1974 and is involved in the isc operon‐mediated assembly of Fe/S clusters.11, 13, 14
Bacterial‐type ferredoxins, which commonly harbor one or two [4Fe–4S] clusters, but have also been observed to contain both [4Fe–4S] and [3Fe–4S] clusters in the same protein, are widely distributed mainly among bacteria and archaea. Members of this ferredoxin class contain cysteinyl residues arranged in a CX2CX2CXNC motif, wherein the denoted cysteines ligate all four Fe ions of the cuboidal cluster.14 This ligation geometry typically results in clusters that are resistant to degradation upon oxygen exposure, which is in contrast to [4Fe–4S] clusters in which only three Fe ions are coordinated by cysteinyl residues, leaving one Fe with an open coordination site and, therefore, susceptible to dissociation. The binding site of the [4Fe–4S] cluster in bacterial ferredoxins is typically located near the surface of the protein, and the cluster is shielded from solvent access by the polypeptide backbone. The formation of [3Fe–4S] clusters in bacterial ferredoxins may arise from the interconversion of [4Fe–4S] clusters through the loss of the fourth Fe ion by degradation.9 Bacterial‐type ferredoxins possessing a [3Fe–4S] cluster typically contain a binding motif that either lacks a fourth cysteinyl residue or contains a non‐cysteinyl ligand such as an aspartate to the fourth Fe ion, which may more readily permit cluster interconversion.9 Conversely, cuboidal [4Fe–4S] clusters may also be generated by addition of an Fe ion to a [3Fe–4S] cluster as in the case of aconitase self‐activation.9, 14, 15
Ferredoxins that contain two [4Fe–4S] clusters may be further subcategorized into two broad groups based on their amino acid sequence and the electrochemical potentials of their clusters. Short ferredoxins of approximately 55–61 amino acids in length are categorized as Clostridial‐like, typified by the ferredoxin isolated from Clostridium pasteurianum, containing a CX2CX2CXNCP binding motif to coordinate each cluster.16 In contrast, the ferredoxin isolated from Allochromatium vinosum typifies the second subclass of two‐cluster ferredoxins, which are longer (approximately 82 amino acids) and are termed Alvin‐like. These ferredoxins coordinate one cluster with a with a CX2CX2CXNCP binding motif and a second cluster with a motif containing a 6 amino acid insert between the second and third cysteinyl residues, resulting in a CX2CX8CXNCP motif.16, 17 Structurally, Alvin‐like ferredoxins contain an additional C‐terminal extension forming an elongated α‐helix that is typically absent in more compact Clostridial‐like ferredoxins. However, in both subclasses of bacterial‐type ferredoxins, the distance between both clusters remains conserved at approximately 12 Å from their centers or about 6 Å from their edges, which has been proposed to facilitate electron transfer.18
The majority of proteins containing redox‐active [4Fe–4S] clusters participate in a 2+/1+ redox couple and possess reduction potentials as low as −700 mV. A smaller subset of FeS cluster‐containing proteins contains a high‐redox potential iron–sulfur protein (HiPIP) cluster that participates in a 3+/2+ transition. The HiPIP clusters have a much higher redox potential, which has been observed in the range of +50 to +450 mV.19, 20 The two [4Fe–4S] clusters in Clostridial‐like ferredoxins are isopotential and possess reduction potentials of approximately −400 mV, while those of Alvin‐like ferredoxins typically vary as much as 200 mV, with the lowest potential determined to be ~ −700 mV.16 In comparison, fewer redox potentials for the 1+/0 redox transitions in [3Fe–4S] clusters have been measured, in part due to the challenges in assigning these clusters as the native state of ferredoxins as opposed to arising from oxidative degradation of [4Fe–4S] clusters. Redox potentials for ferredoxins harboring [3Fe–4S] clusters thus far have been found to lie within the range of +100 mV to −450 mV.21
Ferredoxin TM0927 from the hyperthermophilic Gram‐negative bacterium Thermotoga maritima (Tm) contains a single cubane [4Fe–4S] cluster that is remarkably stable, even at temperatures greater than 100°C. The four iron ions of the Fe/S cluster in TM0927 are ligated by four cysteinyl residues, and the cluster has a redox potential of −388 mV.22 This ferredoxin is an electron donor in the reversible and bifurcating ferredoxin‐NADP+‐oxidoreductase pathway, in which the heterodimeric complex NfnAB couples the oxidation of two TM0927 proteins with the reduction of two molecules of NADP+ to NADPH.8, 23, 24 The NfnAB complex is found in many anaerobic bacteria and archaea, and is a complex of an α (NfnA) and a β (NfnB) subunit. In Tm, the α subunit (TM1639) contains an FAD cofactor (α‐FAD) that is bound by a six‐stranded antiparallel β‐barrel domain, an NAD(P) binding α/β domain, and a conserved [2Fe–2S] cluster‐binding motif with a consensus sequence DX4CX2CXNC in which D220, C225, C228, and C240 coordinate the Fe/S cluster (Fig. 1). The β subunit (TM1640), contains an additional FAD binding site (β‐FAD) and two [4Fe–4S] clusters.
Figure 1.
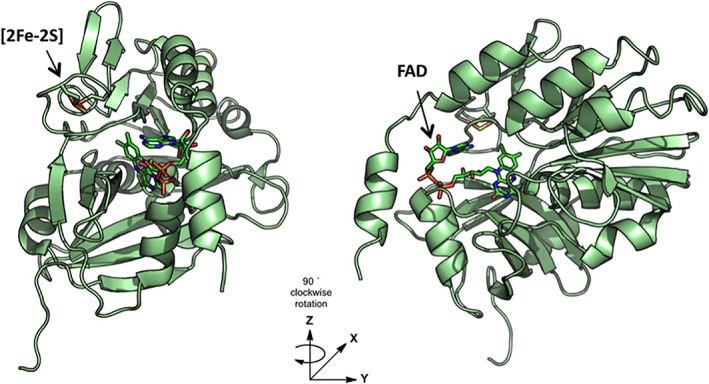
Crystal structure of the α‐subunit of ferredoxin–NAD(P)+–oxidoreductase from Tm (TM1639) (PDB 4YLF), indicating the binding site of the FAD cofactor and the [2Fe–2S] cluster ligated by D220, C225, C228, and C240.
The TM1639/TM1640 complex can catalyze the NAD+‐dependent reduction of two ferredoxins by two NADPH molecules in the forward reaction, or the NADH dependent oxidation of two ferredoxins by two NADP+ molecules in the reverse reaction25
| (1) |
The ferredoxin binding site has not been conclusively determined, but is postulated to be adjacent to the terminal [4Fe–4S] cluster of TM1640.25 In the proposed mechanism for TM1639/TM1640, NADPH binds to TM1640 near the β‐FAD, transferring two electrons via a hydride to generate β‐FADH− and a molecule of NADP+ that dissociates from the active site (Fig. 2). One electron is lost from β‐FADH−, generating β‐FAD•. The electron from β‐FADH− is transferred to the [2Fe–2S] cluster of the α‐subunit and then to the α‐FAD cofactor, generating α‐FAD•. Meanwhile, a second electron is lost from β‐FAD•, generating β‐FAD. This electron is transferred to a ferredoxin via the two [4Fe–4S] clusters in the β‐subunit. The process repeats, generating a second equivalent of NADP+ and a second reduced ferredoxin. The second electron is delivered to the α‐FAD• generated in the first round via the [2Fe–2S] cluster of the α‐subunit. The resulting α‐FADH− transfers a hydride to a molecule of NAD+ to form NADH and α‐FAD. However, TM1639 was found to be capable of reducing TM0927 in the absence of TM1640, and was therefore used in combination with NADPH and other ferredoxins found in Tm as a coupled reducing system.
Figure 2.
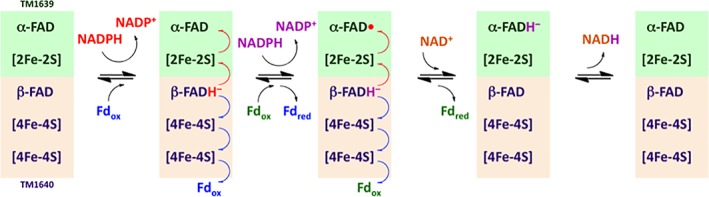
Proposed mechanism of the bifurcating ferredoxin–NAD(P)+–oxidoreductase, adapted from Demmer et al.25 The α‐subunit of the reductase (TM1639) is shown in green, while the β‐subunit (TM1640) is shown in peach. Both subunits contain an FAD binding domain and Fe/S cluster(s).
This reducing system may also function in the transport of electrons to RS enzymes such as Tm MiaB, which demands elevated temperatures in order to catalyze methylthiolation of its RNA substrates efficiently. Although previous work has demonstrated the use of E. coli flavodoxin, flavodoxin reductase and NADPH as an electron donor system in vivo and in vitro, these proteins are temperature sensitive and do not support robust catalysis at high temperature. As such, Tm MiaB activity assays were previously limited to the use of artificial chemical reductants such as dithionite and methylviologen, which support catalysis but often lead to uncoupling of 5′‐dA and ms2i6A formation. The inconsistent stoichiometry of these products can hinder the mechanistic interpretation of reactions. The use of the ferredoxin reducing system for thermophilic enzymes such as Tm MiaB in vitro may reduce the uncoupling of product formation and assist in the mechanistic investigation of Tm MiaB.
In this work, we express, purify and characterize five ferredoxins (TM0927, TM117, TM1289, TM1533, and TM1815) from Tm, the last four of which have not been studied previously. We demonstrate that each of these ferredoxins can support Tm MiaB‐catalyzed methylthiolation in in vitro reactions using the α‐subunit alone of the ferredoxin–NADH–oxidoreductase, and we show that the reductase is capable of reducing ferredoxins in instances in which dithionite cannot. Using the ferredoxin reducing system, we demonstrate improved coupling between 5′‐dA formation and ms2i6A generation relative to using an artificial chemical reductant, which provides improved insight into the stoichiometry of the reaction. In our subsequent paper, we describe the redox characteristics of the ferredoxins that may rationalize their reactivity pattern with MiaB.
Results
Bioinformatic identification of Tm ferredoxins
Genes corresponding to ferredoxins within the Tm genome were identified using the NCBI pBLAST search algorithm using the ferredoxin sequences from E. coli as a search query.26 The resulting ferredoxin sequences were confirmed based on sequence annotation using protein BLAST (pBLAST) and conservation of Fe/S cluster binding motifs determined by protein family domain (PFAM) annotation. The candidate sequences and their NCBI accession numbers were checked for redundancy by sequence alignment, resulting in the identification of five unique ferredoxins: TM0927, TM1175, TM1289, TM1533, and TM1815.
Overproduction and UV–visible absorbance of Tm ferredoxins and ferredoxin–NADP+–oxidoreductase
The relative purities of the ferredoxins and ferredoxin reductase are estimated to be at least 90% based on analysis by sodium dodecylsulfate–polyacrylamide gel electrophoresis (SDS–PAGE) (Fig. 3). The estimated sizes of the hexahistidine‐tagged ferredoxins were determined by molecular weight calculation using the ExPASy server27 as follows: TM0927 (8.2 kDa), TM1175 (9.4 kDa), TM1289 (12.3 kDa), TM1533 (12.7 kDa), TM1815 (17.7 kDa), and TM1639 (33.1 kDa). Typical yields obtained from 16 L of culture in M9 minimal media ranged from 2 to 20 mg for ferredoxins and 50 to 100 mg for ferredoxin reductase, as determined by the method of Bradford using BSA as a standard.
Figure 3.
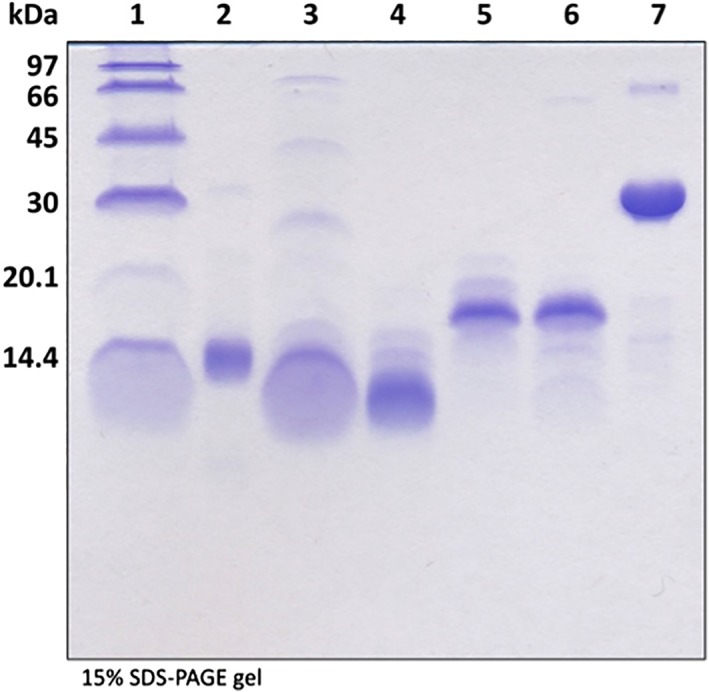
SDS‐PAGE analysis of purified Tm ferredoxins. Molecular size markers (Lane 1), TM0927 (Lane 2), TM1175 (Lane 3), TM1289 (Lane 4), TM1533 (Lane 5), and TM1815 (Lane 6). Lane 7 corresponds to TM1639, the ferredoxin reductase.
The UV–visible spectra obtained for purified ferredoxins indicate maximum absorbances at 304 nm (TM0927), 274 nm (TM1175), 283 nm (TM1289), 284 nm (TM1533), and 279 nm (TM1815), corresponding to the absorbance of aromatic amino acids within the polypeptide chain Fig. 4). The lack of Tyr or Trp residues in TM0927 results in a λ max that is contributed from the 304 nm band of the [4Fe–4S], while the presence of four Tyr residues in TM1175 results in a λ max at 274 nm. The remainder of the ferredoxins contains multiple Trp residues that contribute the most to the λ max of their absorbance spectra.27 In addition, all ferredoxins contained a broad absorbance feature centered around ~400 nm, consistent with the charge transfer bands of [4Fe–4S] clusters, as has been observed in spectra of previously reported ferredoxins.28, 29 The absorbance of this feature increased relative to the maximal absorbance of the polypeptide chain after reconstitution, which is indicative of improved incorporation of Fe/S clusters. The relative ratios of the A 280/A 400 absorbance features provide a qualitative estimate of Fe/S cluster incorporation, which decreased from 1.39 in as‐isolated TM0927 to 1.19 after chemical reconstitution. Likewise, the ratio observed for TM1175 and TM1533 decreased from 6.78 to 2.78, and 1.78 to 1.67, respectively, showing a similar effect. No significant change in the A 280/A 400 ratios was observed for TM1289 and TM1815, which decreased from 1.49 to 1.47, and 2.28 to 2.26, respectively, indicating no further increase in cluster assembly as a function of the chemical reconstitution process. This observation suggests that the recombinant E. coli expression system, along with co‐expression of genes involved in Fe/S cluster assembly on plasmid pDB1282, support ferredoxin cluster assembly in vivo.30, 31 The method of Beinert et al. was used to quantify the Fe and sulfide content per ferredoxin polypeptide chain and yielded the following values32, 33: TM0927 (4.7 ± 0.1 Fe per monomer), TM1289 (6.9 ± 0.3 Fe per monomer), TM1533 (7.4 ± 0.3 per monomer), TM1815 (7.4 ± 0.3 Fe per monomer), and TM1175 (6.3 ± 0.5 Fe per monomer). The Fe content per protein was determined based on protein concentration by the method of Bradford and was approximately equal to one cluster in TM0927 and two clusters in the remaining ferredoxins.34 However, the absence of correction factors for the Bradford protein analysis lends some uncertainty to these estimates, as was shown with Tm MiaB, which overestimates the concentration by nearly 50% (correction factor of 1.47).35 Therefore, EPR spectroscopy and electrochemistry were also used to characterize the cluster content in the isolated proteins. In addition, the UV–visible absorbance spectrum of the ferredoxin reductase was used to estimate the incorporation of FAD into the polypeptide based on its molar absorptivity (ε 450 nm = 11,300 M−1 cm−1). The fraction of the ferredoxin reductase containing the FAD cofactor was found to increase from 0.25 in the as‐isolated protein to 0.80, after anaerobic reconstitution. The concentration of FAD bound to the ferredoxin reductase after incubation for 2 h at 4°C and gel filtration to remove unbound small molecules was assumed to be the concentration of active enzyme for the ferredoxin reductase in subsequent activity assays.
Figure 4.
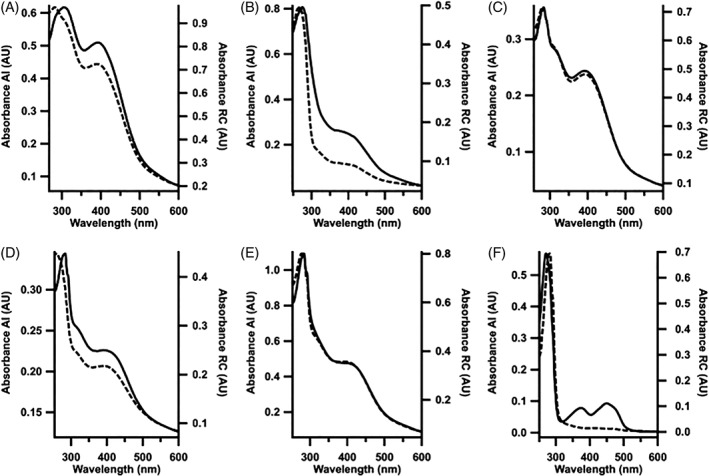
UV–visible absorption spectra of Tm ferredoxins before (dashed trace) and after (solid trace) anaerobic chemical reconstitution, respectively: (A) TM0927 (0.7 μM, 2.51 μM), (B) TM1175 (6.5 μM, 4.0 μM), (C) TM1289 (1.1 μM, 4.5 μM), (D) TM1533 (0.8 μM, 1 μM), and (E) TM1815 (4.7 μM, 3.5 μM) and (F) TM1639 as‐isolated (dashed trace, 0.9 μM) and after FAD incorporation (solid trace, 1.6 μM).
Assessing the minimum reducing system components that support MiaB activity
To determine the minimal reducing system requirements to support MiaB activity, MiaB was incubated with a ferredoxin, TM0927, the reductase, TM1639, or both, in the presence of substrate i6A tRNA and SAM. The reducing system consisting of NADPH and either ferredoxin, the reductase, or both, was incubated for 15 min at 65°C prior to the addition of MiaB. When TM1639 was omitted in the reaction, formation of 5′‐dA and ms2i6A was barely detectable. This observation is consistent with the requirement for an electron to generate the 5′‐dA• intermediate [Fig. 5(A)], which is ultimately derived from NADPH and is mediated by the reductase. Interestingly, When TM0927 was omitted from the reaction [Fig. 5(B)], small, but significant, amounts of ms2i6A and 5′‐dA were produced at equimolar concentrations and at comparable initial rates (0.7 ± 0. 1 and 0.9 ± 0.1 μM min−1, respectively), suggesting that the reductase itself can reduce MiaB directly to a considerable degree [Fig. 5(B)], but not to the same extent observed in the reactions containing the complete reducing system, which generated greater 5′‐dA and ms2i6A concentrations overall. When both TM0927 and TM1639 were supplied in the MiaB reaction [Fig. 5(C)], one equivalent each of ms2i6A and 5′‐dA was formed (61 μM and 66 μM, respectively). Additionally, the rates of formation of ms2i6A and 5′‐dA (1.7 ± 0.1 μM min−1 and 2.1 ± 0.1 μM min−1, respectively) were nearly double that in reactions containing only the reductase, indicating that the addition of ferredoxin to the reaction mixtures increased the final concentration of product. This behavior is in contrast to the results of previously reported reactions, in which dithionite was used to supply the requisite electron. In those instances, although one equivalent of ms2i6A is generated, approximately 1.3‐fold more 5′‐dA is formed, likely due to the unproductive abortive cleavage of SAM triggered by substrate binding.35
Figure 5.
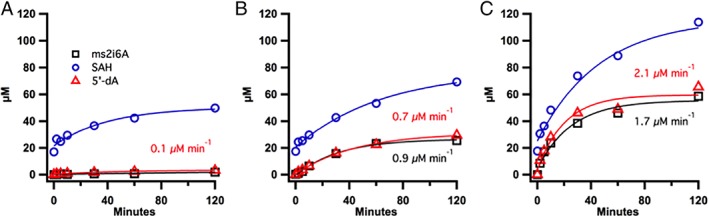
Comparison of Tm MiaB activity with components of the TM0927, TM1639, and NADPH reducing system. The activity of Tm MiaB was determined in reactions containing components of the ferredoxin reducing system: (A) ferredoxin TM0927 and NADPH, (B) TM1639 and NADPH, and (C) ferredoxin TM0927, TM1639 and NADPH, and show the formation of ms2i6A (black squares), 5′‐dA (red triangles), and SAH (blue circles) at specific time points. Reactions were conducted at 65°C and contained the following in a 100 μL volume: 50 μM Tm MiaB, 150 μM i6A tRNAPhe, 1 mM SAM, 50 μM TM0927, 55 μM TM1639, 1 mM NADPH and buffer system (50 mM HEPES (pH 7.5), 150 mM KCl, 1 mM MgCl2, and 686 μM L‐(+)‐tryptophan as an internal standard). The reducing system consisting of TM0927 and NADPH (A), TM1639 and NADPH (B), or TM0927, TM1639, and NADPH (C), was incubated for 15 min prior to addition to the reaction containing Tm MiaB.
Comparison of Tm ferredoxin activity with MiaB
To quantify the ability of the Tm ferredoxins to support Tm MiaB activity, the conversion of the synthetic Tm tRNAPhe IV (UAA) substrate containing i6A into the ms2i6A product was monitored under turnover conditions using the coupled reducing system (Fig. 6). At designated time points, aliquots of the reaction mixture were removed and treated with acid to quench the reaction, which was then titrated to an appropriate pH for further manipulation. The RNA was digested, and the nucleotides were dephosphorylated and subjected to analysis by LC/MS as previously described.35 A temperature dependence of product formation was observed, with reactions conducted at 25°C and 37°C generating a marginal amount of ms2i6A, while reactions conducted at 65°C and 80°C, which approaches the optimal growth temperature of Tm (90°C), generated more product with faster initial rates.28 However, the co‐substrate SAM was unstable in reactions conducted at 80°C within the time frame of these reactions. Therefore, reaction temperatures were limited to 65°C. Additionally, reactions that contained a 1:1 stoichiometry of ferredoxin and ferredoxin reductase generated more ms2i6A at faster initial rates than those in which either component was substoichiometric. Reactions in which the reducing system was incubated prior to addition of MiaB supported product formation at faster initial rates than when all components were added simultaneously.36 This behavior is likely due to pre‐reduction of the reductase by hydride transfer from NADPH to yield the two‐electron reduced hydroquinone state of FAD prior to successive single electron transfers to the ferredoxins, which may be rate‐limiting due to the absence of the reductase β‐subunit.
Figure 6.
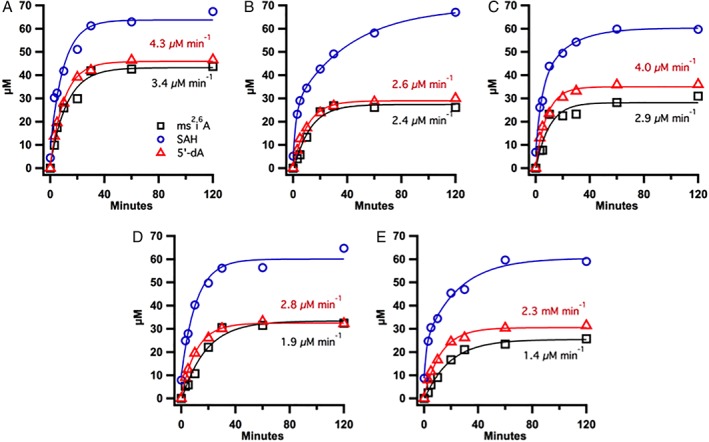
Comparison of Tm MiaB activity using the Tm ferredoxin reducing system. The activity of Tm MiaB was determined in reactions containing ferredoxin reductase, NADPH, and various ferredoxins: (A) TM0927, (B) TM1175, (C) TM1289, (D) TM1533, and (E) TM1815. ms2i6A product (black squares), 5′‐dA (red triangles) and SAH (blue circles). Reactions were conducted at 65°C and contained the following in a 100 μL volume: 50 μM Tm MiaB, 150 μM i6A tRNAPhe, 1 mM SAM, 73.4 μM Tm ferredoxin, 73.4 μM Tm ferredoxin‐NADP reductase, 1 mM NADPH, and buffer system (50 mM HEPES (pH 7.5), 150 mM KCl, 1 mM MgCl2, and 686 μM L‐(+)‐tryptophan as an internal standard). The reducing system consisting of ferredoxin, TM1639, and NADPH was incubated for 1 h prior to addition of MiaB.
In MiaB reactions containing the coupled reducing system, TM0927 was found to support the greatest turnover, generating nearly one equivalent of the ms2i6A product (~44 μM) at an estimated initial rate of 3.4 μM min−1 within 30 min of reaction initiation. The formation of 5′‐dA within this time frame was coupled to product formation, resulting in 47 μM 5′‐dA with an initial rate of 4.3 μM min−1, the fastest rate observed with the employed ferredoxins. The rates for ms2i6A and 5′‐dA formation were nearly two‐fold greater in reactions where the reductase and ferredoxins were incubated for 1 h prior to adding MiaB [Fig. 6(A)] as compared to reactions where the incubation time was limited to 15 min [Fig. 5(C)]. TM1533 supported the formation of 0.6 equivalents (32 μM) of ms2i6A with an equivalent concentration of 5′‐dA, both of which were generated at similar initial rates (2.8 μM min−1 and 1.9 μM min−1 for 5′‐dA and ms2i6A, respectively).
By contrast, TM1815 supported the formation of the least amount of ms2i6A, with 0.5 equivalents of product (26 μM) formed at a rate of 1.4 μM min−1, and 0.6 equivalents (31 μM) of 5′‐dA formed at a rate of 2.3 μM min−1. The remaining two ferredoxins supported between 0.5 and 0.6 equivalents of product formation with 0.6–0.7 equivalents of 5′‐dA. However, regardless of the ferredoxin used, a stoichiometry of approximately 1:1 between 5′‐dA and ms2i6A was observed, which is in contrast to reactions using dithionite, in which a greater degree of uncoupling was observed.
Although the coupled reducing system clearly supports the reduction of MiaB, the compatibility between the reductase and each ferredoxin may lead to differences in product formation. Therefore, in a separate series of experiments, the reductase was excluded and the ferredoxins were reduced with dithionite and then subjected to anaerobic gel‐filtration chromatography to remove excess reducing equivalents prior to adding them to MiaB reactions (Fig. 7). In these reactions, TM0927 supported the formation of 1.4 equivalents (72 μM) of ms2i6A within 30 min of reaction initiation. Here, product was formed at an initial rate of 11.7 μM min−1, which is approximately three‐fold faster than the rate obtained using the full reducing system. This behavior may indicate that either the rate of FAD reduction in TM1639 or the rate of electron transfer to TM0927 is limiting compared to electron transfer to MiaB. The formation of 5′‐dA, however, was still coupled to that of product formation (13.2 μM min−1), as observed with the full reducing system. However, product formation by MiaB varied considerably among the remaining ferredoxins. Although TM1175 supported formation of nearly one equivalent of product (48 μM) at an initial rate of 6 μM min−1, it generated 1.4 equivalents (72 μM) of 5′‐dA at a rate that was almost twice that of ms2i6A formation (10 μM min−1), indicating that decoupling occurred. Similar reactions with the remaining ferredoxins showed significantly lower support for MiaB turnover. TM1289 supported only 0.3 equivalents of ms2i6A (13 μM) formation, while TM1533 and TM1815 yielded less than 0.1 equivalents of product (approximately 5 μM each).
Figure 7.
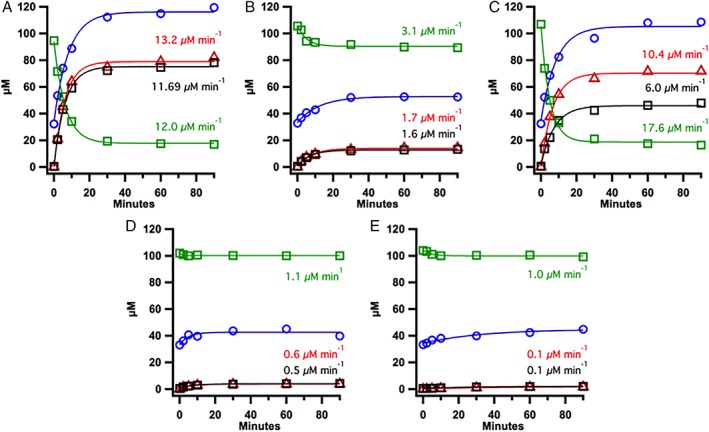
Comparison of Tm MiaB activity with dithionite‐reduced ferredoxin at 65°C. MiaB activity was determined in reactions in which dithionite‐reduced ferredoxin was supplied as reductant in the absence of ferredoxin–reductase and NADPH. Shown are MiaB reactions with (A) TM0927, (B) TM1289, (C) TM1175, (D) TM1533, and (E) TM1815, indicating the formation of ms2i6A (black squares), 5′‐dA (red triangles), and SAH (blue circles) and loss of i6A green squares) at specific time points. Reactions were conducted at 65°C and contained the following in a 100 μL volume: 50 μM Tm MiaB, 100 μM i6A tRNAPhe, 1 mM SAM, 150 μM dithionite‐reduced Tm ferredoxin, and buffer system (50 mM HEPES (pH 7.5), 150 mM KCl, 1 mM MgCl2, and 686 μM L‐(+)‐tryptophan as an internal standard).
Electron paramagnetic resonance spectroscopy
Low temperature X‐band continuous‐wave EPR was used to characterize the Fe/S cluster cofactors of ferredoxins in dithionite‐reduced and non‐reduced samples. The spectrum of the dithionite‐reduced TM0927 at pH 7.5 shows a signal with rhombic geometry and corresponding g‐tensor principle values g x = 1.88, g y = 1.93, g z = 2.07, consistent with the presence of a [4Fe–4S]1+ cluster with a non‐integer (S = ½) spin system [Fig. 8(A)]. The non‐reduced TM0927 spectrum is devoid of features within this region, where a [3Fe–4S]1+ cluster would be typically observed [Fig. 8(B)]. Similarly, the EPR spectrum obtained for dithionite‐reduced TM1175 [Fig. 8(C)] is consistent with a reduced [4Fe–4S]1+ cluster with similar g‐values as those observed for TM0927. A minor shoulder (g = 2.04) and a broader linewidth (g = 1.87) are consistent with a second half‐integer spin [4Fe–4S]1+ cluster with slightly different parameters that is overlaid in the same region. Analysis of the spectrum obtained for dithionite‐reduced TM1815 [Fig. 8(D)] also shows features corresponding to two overlapping [4Fe–4S]1+ clusters with slightly different parameters as observed in TM1175. However, one cluster (g x = 1.87, g y = 1.93, g z = 2.07) appears to be reduced to a lesser extent than the other (g x = 1.88, g y = 1.94, g z = 2.05) based on the apparent amplitudes of the signals at corresponding g‐values. The feature at g = 2.05 is more prominent in this spectrum as compared to that of the other ferredoxins. Dithionite‐reduced TM1289 [Fig. 8(E)] also displays a similar spectrum to that of TM1175 except with a broader line width at g = 1.88, possibly due to contributions from two clusters that cannot be readily resolved. The feature at g = 2.05 is more intense in this spectrum as compared to the same feature observed in the spectrum of TM1175. The non‐reduced control for TM1175 [Fig. 8(F)] lacks any features except for the resonator‐originating background signal, indicating the absence of a [3Fe–4S]1+ cluster. The EPR spectrum of TM1533 [Fig. 8(G)] also appears to be composed of two half‐integer spin clusters, with one contributing the signal at g = 2.08 and the other at g = 2.05. The two clusters present in TM1533 also appear to be more clearly resolved, with a small shoulder adjacent to the zero crossing at g = 1.94 more apparent. Additionally, the linewidth observed at g = 1.88 is sharper than that in the other dicluster ferredoxins. The spectrum of the non‐reduced TM1533 [Fig. 8(H)] indicates a sharp feature centered at g = 2.02 that corresponds to the presence of minor amounts of a [3Fe–4S]1+ cluster species in the sample.
Figure 8.
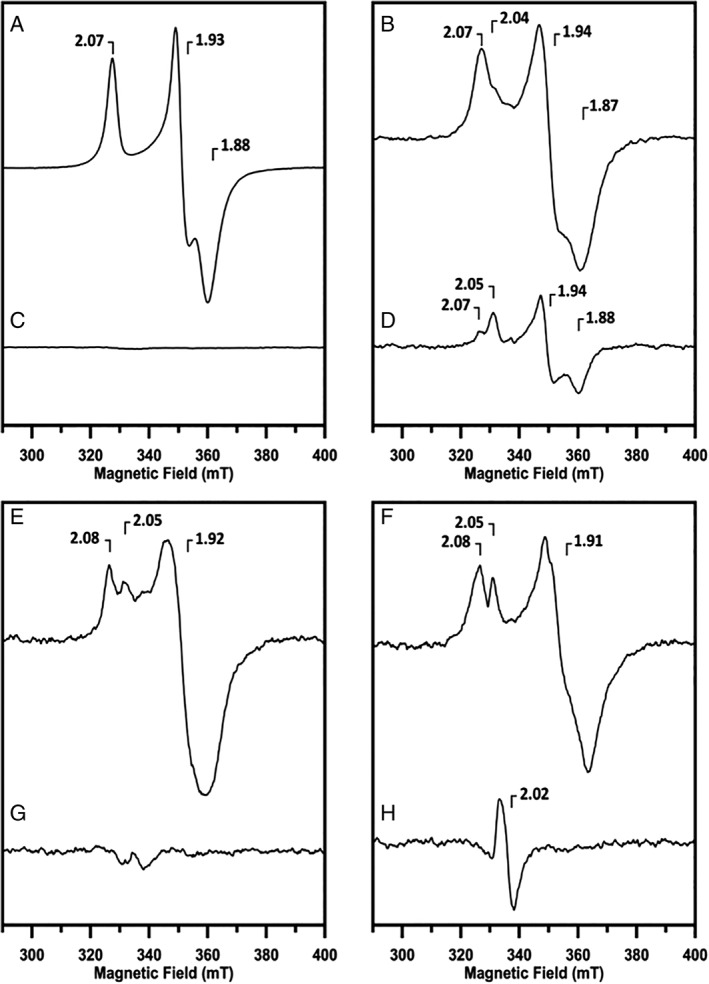
X‐band CW EPR spectra of reconstituted Tm ferredoxins at low temperature (10 K) with 0.2 mW microwave power and 9.48 GHz microwave frequency. Dithionite‐reduced samples contained 400 μM TM0927 (A); 100 μM TM1175 (C); 100 μM TM1815 (D); 100 μM TM1289 (E); and 300 μM TM1533 (G). Non‐reduced samples contained 400 μM TM0927 (B); 100 μM TM1289 (F); and 300 μM TM1533 (H).
A Cu standard solution of known concentration was analyzed under identical conditions used to analyze the ferredoxins in order to spin‐quantify the reduced [4Fe–4S]1+ clusters. The double integration of the TM0927 spectra indicated the presence of 1.3 spins per protein. The excess spin over the expected one equivalent corresponding to a single reduced cluster might suggest an underestimation of the protein concentration. Spin quantification of the reduced TM1175 indicates 0.8 spins/protein, and gave the highest spin ratio obtained of the dicluster ferredoxins analyzed. Spin quantification of the remaining ferredoxins indicated a much lower degree of cluster reduction: TM1289 resulted in 0.4 spins/protein, and both TM1533 and TM1815 resulted in 0.2 spins/protein. These results suggest that only a fraction of the [4Fe–4S] clusters in these ferredoxins are accessible to reduction by dithionite, far less than the anticipated two equivalents. Spin quantification of the [3Fe–4S]1+ cluster observed in non‐reduced TM1533 only accounted for approximately 1.9 μM, or less than 0.01 spins/protein. Such species were not observed in TM0927 and TM1533. Simulations of EPR spectra are provided in Figure 9 and Table 1.
Figure 9.

Simulation of EPR spectra of reconstituted Tm ferredoxins. Blue—experimental spectra (the same as in Fig. 8), red lines represent complete simulations, black solid lines represent simulation of Species #1 (g max = 2.07–2.08), black dashed lines represent simulations of the Species #2 (g max ~2.04–2.05).
Table 1.
Principal g‐Values of Species Identified in the Reconstituted Tm Ferredoxins (see Figure 9)
| Species #1 | Species #2 | ||||||
|---|---|---|---|---|---|---|---|
| g1 | g2 | g3 | g1 | g2 | g3 | Ratio | |
| Tm0927 | 2.070 (0.010) | 1.931 (0.010) | 1.880 (0.024) | ‐ | ‐ | ‐ | ‐ |
| Tm1175 | 2.074 (0.035) | 1.935 (0.030) | 1.868 (0.040) | 2.037 (0.001) | 1.944 (0.005) | 1.884 (0.010) | 10:1 |
| Tm1815 | 2.073 (0.001) | 1.930 (0.025) | 1.873 (0.040) | 2.049 (0.010) | 1.942 (0.005) | 1.880 (0.020) | 1:2.2 |
| Tm1289 | 2.077 (0.020) | 1.924 (0.040) | 1.860 (0.053) | 2.042 (0.041) | 1.942 (0.042) | 1.884 (0.040) | 1:1.25 |
| Tm1533 | 2.080 (0.033) | 1.905 (0.040) | 1.860 (0.03) | 2.047 (0.000) | 1.931 (0.021) | 1.908 (0.024) | 4.8:1 |
Numbers in brackets are g‐strain values used.
Discussion
MiaB, as with other members of the RS enzyme superfamily, requires an electron to generate the reduced [4Fe–4S]1+ state of the conserved RS cluster responsible for coordinating and cleaving SAM to generate the 5′‐dA• necessary for catalysis. Previous work demonstrated that RimO from Bacteriodes thetaiotaomicron, another member of the MTTase family of RS enzymes, can use the E. coli flavodoxin and flavodoxin‐reductase system to supply the requisite electrons for catalysis.6, 37, 38 However, experiments conducted with Tm MiaB or Tm RimO and the E. coli flavodoxin reducing system at 25°C indicated very limited product formation. Further, analysis of the Tm genome by BLAST sequence alignment with E. coli flavodoxin did not identify corresponding genes in this organism. In contrast, prior to our current study, the ferredoxin TM0927 had been identified and well characterized,28, 29, 39 with the implied function as a redox partner with ferredoxin–NADP+–oxidoreductase (TM1639).23, 25 However, a complete view of the ferredoxin profile of Tm had yet to be achieved, inspiring our studies of the reactivity of all Tm ferredoxins with Tm MiaB at elevated temperatures.
All five of the ferredoxins studied here (TM0927, TM1175, TM1289, TM1533, and TM1815) were initially assessed for the types of Fe–S clusters they harbor. While the structure of TM0827 was already available [Fig. 10(A)], the SWISS‐MODEL database was used to predict an overall fold and potential for [4Fe–4S] cluster binding for the other four ferredoxins [Fig. 10(B–E)]. All four additional ferredoxins were presumed to bind two [4Fe–4S] clusters due to their comparable size and sequence similarity to clostridial‐like ferredoxins that also coordinate two cubane clusters through conserved CX2CX2CXNCP binding motifs. While some of the cluster binding motifs in the Tm ferredoxins differ from clostridial orthologues, with short amino acid inserts, all contain four cysteinyl residues and no obvious alternative cluster ligands, suggesting the presence of 2 [4Fe–4S] clusters only.
Figure 10.
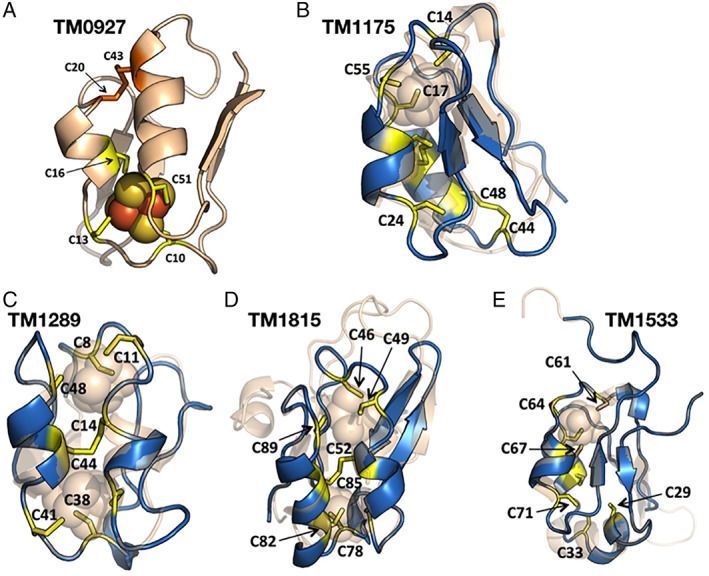
(A) Crystal structure of TM0927 (PDB 1VJW), indicating the cysteinyl ligands to the single [4Fe–4S] cluster (yellow) and the remaining two cysteinyl residues in the polypeptide that form a disulfide bond (orange). Homology models (marine) composed for (B) TM1175 based on the conserved sequence alignment with the TM0927 structure (PDB 1VJW, wheat), which share 35.1% sequence identity and 81% sequence coverage; (C) TM1289 based on the conserved sequence alignment with the dicluster ferredoxin from Pa (PDB 1DUR, wheat). The two ferredoxins share 44% sequence identity and 53% sequence coverage and contain two [4Fe–4S] clusters coordinated by conserved cysteinyl binding residue motifs for both cluster sites; (D) TM1815 based on the conserved sequence alignment with the [3Fe–4S] and [4Fe–4S] containing ferredoxin from Acidianus ambivalens (PDB 2VKR). The sequence alignment between TM1815 and the Aa ferredoxin indicates 37% sequence identity and 44% coverage; and (E) TM1533 based on the conserved sequence alignment with an electron transfer‐ubiquinone oxidoreductase (PDB 2GMH) from porcine mitochondria, which share 37.8% sequence identity and 80% sequence coverage.
The cluster content of all five ferredoxins produced in E. coli was verified by measuring the iron and sulfide content as well as by optical and EPR measurements. While the former of these experiments was consistent with full cluster loading (given the errors associated with the elemental and protein concentration determination), the optical spectra (Fig. 4) revealed the relatively featureless, broad absorption feature centered at ~400 nm associated with [4Fe–4S] clusters. Measurement of the EPR spectra required reducing such [4Fe–4S] clusters to the 1+ oxidation state. While the single cluster‐containing TM0927 could be cleanly reduced with dithionite, in all other cases, only partial reduction could be achieved. For TM1175, apparently a single [4Fe–4S] cluster could be reduced (0.8 spin/protein), while TM1289, TM1533, and TM1815 all showed only small amounts of reduction (less than 0.5 spin/protein in all cases), indicating that low redox potentials or unfavorable redox kinetics must be at work for these proteins. In the case of these last three ferredoxins (TM1289, TM1533, and TM1815), multiple species were identified in the EPR spectrum, indicating that while reduction of a single cluster is not complete, partial reduction of each cluster may be attained upon treatment with dithionite.
The apparent functionality of these ferredoxins in supporting MiaB catalysis further bore out their apparent differences in redox chemistry. The optimal reducing system required to support turnover by MiaB was a ferredoxin, a reductase (TM1639), the i6A tRNA substrate, and SAM, where the reducing equivalents that flowed through the reductase were supplied by NADPH. We note that the reductase itself has some ability to support MiaB catalysis in the absence of a ferredoxin partner. Using the crystallographically characterized TM0927 ferredoxin, it was clear that the best rates (and excellent coupling between SAM usage and product methylthiolation chemistry) could be achieved when all components of the reducing system were in place [Fig. 5(C)]. Side‐by‐side comparisons were then conducted by examining the kinetics and yield of product formation for both 5′‐dA and ms2i6A by keeping all of the components the same except for the identity of the ferredoxin component. In these experiments (Fig. 6), the single [4Fe–4S] cluster protein TM0927 was found to support the largest yield of products (effectively one equivalent) at the fastest rate; however, all ferredoxins were able to support catalysis to some extent. The next most capable was TM1533, which supported catalysis with rates that were diminished by less than 50%, and still supported nearly the equivalent amount of final ms2i6A product. The other three ferredoxins, which all contained 2 [4Fe–4S] clusters, supported turnover at rates approximately 50% slower than TM0927 and produced only 50% as much product.
These initial experiments suggested that the coupling system was nearly invariant in terms of the ferredoxin employed. To probe this issue further, we then turned toward pre‐reducing the ferredoxins initially with dithionite (Fig. 7). In this context, the extent of turnover (and the associated rate) appears to scale with the extent of reduction of each ferredoxin, as determined by EPR. TM0927, the most reduced Fd (TM0927), supported the formation of 1.4 equivalents of the tRNA product, which was fully coupled to the cleaved product of SAM, and at rates three‐fold faster than the full reductase‐based system. The four 2 [4Fe–4S] ferredoxins (all poorly reduced by dithionite) also supported turnover poorly. Ferredoxin TM1175 was the most successful. It was the most reduced of the 2 [4Fe–4S] ferredoxins as determined by EPR (0.8 spin/protein), and it yielded 1.0 equivalent of product, but 1.4 equivalents of 5′‐dA (at a rate unsurprisingly higher than that of ms2i6A formation), indicating a loss of coupling. The other three ferredoxins fared worse. TM1289 could only be reduced to the level of 0.4 spins/protein, and both TM1533 and TM1815 were barely reduced (0.2 spins/protein) after incubation with dithionite. Likewise, the ms2i6A product formed in reactions with these ferredoxins was unsurprisingly low, hovering between 0.1 and 0.3 equivalents of the tRNA‐based product.
We note that in contrast to the low methylthiolation activity observed with dithionite‐reduced ferredoxins, particularly those containing 2 [4Fe–4S] clusters, reactions with the same proteins yielded more product formation (about 0.5–0.6 equivalents) when the reductase was present. This conclusion is supported by the inability of dithionite–reduced TM1289, TM1533 and TM1815 to catalyze MiaB turnover, as demonstrated in Figure 7, as well as the low fraction of these reduced ferredoxins upon incubation with dithionite as determined by EPR spin quantification in Figure 8. While the direct reduction of MiaB by the reductase itself is likely accounting for some of the ms2i6A formed in these reactions, the rates of methylathiolation are 2–4‐fold greater when the ferredoxins are supplied to the reaction as compared to reactions with the reductase alone. This observation leads us to hypothesize that the reductase is indeed capable of reducing even the 2 [4Fe–4S] cluster ferredoxins.
Conclusion
Together, these data hint at a preferential role of some of the ferredoxins (TM0927 and possibly TM1175) for supporting MiaB turnover. We presume that the other ferredoxins play other specific roles in the physiology of Tm. Here, their poor reactivity may be due to specificity or to the diminished ability to be reduced. The redox potential of dithionite is known to vary based on concentration and pH. For example, it ranges from −540 mV at 1 mM to −590 mV at 10 mM, and from −240 mV at pH 5, to −675 mV at pH 9 for 1 mM dithionite.40 As such, the predicted redox potential of dithionite under the conditions employed was approximately −525 mV. Although studies of the redox potentials of the 2 [4Fe–4S] cluster bearing Tm ferredoxins are absent in the literature, the potentials of similar ferredoxins from Pseudomonas nautica are as low as −715 mV, prompting our interest in the further characterization of the Fe/S clusters of each ferredoxin.41 In our following paper, we describe their redox characteristics using protein electrochemistry as a means of corroborating the reactivity and spectroscopy reported here.
Materials and Methods
Cloning and overproduction of Tm ferredoxins and ferredoxin reductase
Five genes (TM0927, TM1175, TM1289, TM1533, and TM1815) from the hyperthermophillic bacterium Tm were identified as ferredoxins based on gene function annotation, amino acid sequence alignment, and conserved PFAM domains.42 The ferredoxin‐NADP(+) reductase subunit α (TM1639) was similarly identified from the national center for biotechnology information (NCBI) basic local alignment search tool (BLAST) database.43 All genes were amplified using the polymerase chain reaction (PCR) from Tm genomic DNA using forward primers containing a 9‐nucleotide GC clamp, an NdeI restriction enzyme (RE) site, and the first 24 nucleotides of the gene. Reverse primers contained the last 26 nucleotides of the gene, a translation stop codon, and a NotI RE site followed by a 9 base pair GC clamp (Table 2). The genes were amplified by PCR, and the resulting products were purified by aragose gel electrophoresis according to standard procedures.44 The PCR products and vector were digested with NdeI and NotI, ligated together, and transformed into Ec DH5 α. The resulting constructs were sequence verified at the Penn State Genomics Core Facility (University Park, PA), then co‐transformed into the E. coli BL21 (DE3) expression strain with the pDB1282 plasmid containing genes from the isc Fe/S cluster assembly machinery from Azotobacter vinelandii as described elsewhere.31, 45 Bacterial cultures were grown at 37°C with 180 rpm shaking in M9 minimal media supplemented with 25 μM FeCl3 and 100 μg/mL ampicillin and 50 μg/mL kanamycin for selection.
Table 2.
Forward and Reverse Primer Sequences used for PCR Amplification of Ferredoxins from Tm Genomic DNA
| Primer name | Primer sequence |
|---|---|
| TM0927_fwd | 5′‐CGCGGCGTCCATATGAAGGTAAGAGTTGACGCAGATGCC‐3′ |
| TM0927_rev | 5′‐cgcggcgtc GCGGCCGCTCACTCTTCTACGCTGATAGCTCCGG‐3′ |
| TM1175_fwd | 5′‐CGCGGCGTCCATATGGCAAAGAACTGGTACCCCGTGATCG‐3′ |
| TM1175_rev | 5′‐caatgattc GCGGCCGCTTACCCATCGGCGCTCACCTCC‐3′ |
| TM1289_fwd | 5′‐CGCGGCGTCCATATGCCCTGGGTGAATTCGAAGTGTGTTGG‐3′ |
| TM1289_rev | 5′‐cgcggcgtc GCGGCCGCTTAGAACTCATCCCATCTTCCTCG‐3′ |
| TM1533_fwd | 5′‐CGCGGCGTCCATATGAGGATCGAAGATAAACTCTATTTGAAC‐3′ |
| TM1533_rev | 5′‐catagatttc GCGGCCGCTCACCCGAATTTGTAGAGC‐3′ |
| TM1815_fwd | 5′‐CGCGGCGTCCATATGGCAGAGGCAAAGAACGCTCCGTTGATTGG‐3′ |
| TM1815_rev | 5′‐cgaagaatc GCGGCCGCTCAGGGTTCGGGTTTGGTTTCCGTTTCC‐3′ |
| TM1639_fwd | 5′‐CGCGGCGTCCATATGATGGGGGGGACGGCTTTGAACG‐3′ |
| TM1639_rev | 5′‐cgcggcgtc GCGGCCGCTCATTCTGATTCACCTGCCG‐3′ |
The primer sequences contain an underlined region corresponding to NdeI restriction cleavage site for forward primers, or NotI restriction cleavage sites for reverse primers, and are preceded by a 9‐base pair GC clamp sequence.
At an optical density at 600 nm (OD600) of 0.3, 300 μM of L‐cysteine and an additional 25 μM FeCl3 were added. Additionally, 0.2% (w/v) of L‐(+)‐arabinose was added to the media to induce expression of the pDB1282 plasmid. At an OD600 of 0.6, the flasks were cooled in an ice bath for 20 min. Recombinant protein over‐production was induced by the addition of 400 μM IPTG to the media, and the cultures were incubated for 16 h at 18°C and 180 rpm prior to harvesting the cells by centrifugation at 6,000 × g for 15 min at 4°C.
Purification and reconstitution of Tm ferredoxins and ferredoxin–NADP reductase
Cell paste was stored in liquid N2 and brought into a Coy (Grass Lake, MI) anaerobic chamber and thawed in lysis buffer (50 mM HEPES pH 7.5, 300 mM KCl, 10 mM β‐mercaptoethanol, 20 mM imidazole) then treated with 0.5 mg/mL PMSF and 1 mg/mL egg white lysozyme for 20 min. The cells were disrupted on ice by sonication in 45 sec bursts for a total of 4.5 min, with 5 min pausing between bursts to maintain the cell suspension temperature. The crude lysate was transferred into centrifuge tubes, sealed with vinyl tape, removed from the anaerobic chamber and subjected to heat denaturation at 75°C for 15 min. Afterwards, the lysate was clarified by centrifugation at 40,000 × g at 4°C for 1 h then returned to the anaerobic chamber. The supernatant was retained and loaded onto Ni‐NTA resin equilibrated in lysis buffer, then washed with 100 mL of wash buffer (lysis buffer containing 40 mM imidazole and 10% (v/v) glycerol) and eluted with 50 mL of elution buffer (wash buffer containing 250 mM imidazole). The dark brown and light‐yellow fractions were collected for ferredoxins and the reductase containing Fe/S clusters and FAD, respectively. The recovered proteins were concentrated to approximately 2.5 mL using an amicon ultra‐15 centrifugation device with a 3,000 NMWL cutoff. The Fe/S clusters of the ferredoxins were chemically reconstituted under anaerobic conditions according to previously published procedures.31, 46 Similarly, the FAD cofactor of the reductase, TM1639, was loaded by incubating 1 mM of TM1639 with 2.5 mM FADox at 37°C for 4 h under anaerobic conditions. The resulting proteins were applied to a pre‐poured PD‐10 column containing sephadex G‐25 resin and exchanged into storage buffer (50 mM HEPES pH 7.5, 300 mM KCl, 1 mM DTT, 20% glycerol) to remove either excess Fe and sulfide or unbound FAD. The [2Fe–2S] cluster of TM1639 was then chemically reconstituted anaerobically in a similar manner as previously described.31, 46 The ferredoxins and ferredoxin reductase were further purified by size exclusion chromatography using an Akta FPLC purification system equipped with a sephadex S200 16/60 column housed in an anaerobic chamber. The fractions corresponding to monomeric protein were collected, pooled, and concentrated to approximately 3 mL, flash frozen, and stored in liquid N2. were applied to determine the Iron and sulfide content was determined using the methods of Beinert et al. and the concentration of FAD incorporated per polypeptide was determined based on molar absorptivity (FADox, ε450 = 11,300 M−1 cm−1) and compared against the protein concentration determined by the method of Bradford.32, 33, 34, 36
MiaB activity assays utilizing the ferredoxin reducing system
The purification of Tm MiaB and generation of i6A tRNA substrate were conducted as previously described.35 Activity assays monitoring turnover of Tm MiaB using the ferredoxin reducing system as the electron donor were conducted under anaerobic conditions at 65°C. Assay mixtures contained the following in a 100 μL volume: 50 μM Tm MiaB, 150 μM i6A tRNA, 1 mM SAM, 73.4 μM Fd, 73.4 μm FdR, 1 mM NADPH, and assay buffer (50 mM HEPES pH 7.5, 300 mM KCl, 5 mM MgCl2, and 686 μM tryptophan as an internal standard). Assay mixtures were overlaid with degassed mineral oil in order to prevent evaporation at elevated incubation temperatures. The reducing system consisting of 200 μM ferredoxin, 200 μM TM1639, and 5 mM NADPH, was incubated separately for 1 h at 65°C to pre‐reduce the ferredoxin to the one‐electron reduced semi‐quinone state prior to addition to the assay mixture containing MiaB, tRNA substrate, and SAM that had been equilibrated at 65°C for 5 min. At designated time points, 10 μL of the reaction mixture was withdrawn and quenched in an equal volume of 200 mM H2SO4, then prepared for LC/MS analysis as previously described.35
Electron paramagnetic resonance spectroscopy of ferredoxin clusters
Reconstituted ferredoxin samples for EPR analysis were prepared in an anaerobic chamber at ambient temperature (approximately 25°C). Samples contained 400 μM (TM0927), 300 μM (TM1533), or 100 μM (TM1175, TM1289, and TM1815) ferredoxin in 50 mM HEPES pH 7.5, 200 mM KCl, and 10 mM MgCl2 in a total volume of 300 μL. Reduced samples were incubated with 1 mM sodium dithionite for approximately 15 s prior to transferring the solution into a 3.8 mm quartz EPR tube and freezing by immersion into cryogenic liquid isopentane. Control samples were prepared without reductant. EPR measurements were taken using a Bruker Elexsys E580 X‐band spectrometer equipped with an ER 4122 SHQE SuperX microwave cavity, an Oxford Instruments ER 4112‐HV helium cryostat and SuperX‐FT microwave bridge. Continuous wave (CW) EPR analysis of ferredoxins were taken at 10 K with 0.2 mW microwave power, 10 G modulation amplitude and a 9.48 GHz microwave frequency. Spectra for TM0927 and TM1533 were collected using a receiver gain setting of 10,000, while those for TM1175, TM1289, and TM1815 were collected at 40,000.
Acknowledgments
This work has been supported by the NIH (GM‐101957 (S.J.B. and S.J.E.), GM‐122595 (S.J.B.) and GM‐120283 (S.J.E.)) and the Howard Hughes Medical Institute (S.J.B.).
References
- 1. Hagervall TG, Ericson JU, Esberg KB, Li JN, Bjork GR (1990) Role of tRNA modification in translational fidelity. Biochim Biophys Acta 1050:263–266. [DOI] [PubMed] [Google Scholar]
- 2. Urbonavicius J, Stahl G, Durand JM, Ben Salem SN, Qian Q, Farabaugh PJ, Bjork GR (2003) Transfer RNA modifications that alter +1 frameshifting in general fail to affect −1 frameshifting. RNA 9:760–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, Walters GB, Styrkarsdottir U, Gretarsdottir S, Emilsson V, Ghosh S, Baker A, Snorradottir S, Bjarnason H, Ng MC, Hansen T, Bagger Y, Wilensky RL, Reilly MP, Adeyemo A, Chen Y, Zhou J, Gudnason V, Chen G, Huang H, Lashley K, Doumatey A, So WY, Ma RC, Andersen G, Borch‐Johnsen K, Jorgensen T, van Vliet‐Ostaptchouk JV, Hofker MH, Wijmenga C, Christiansen C, Rader DJ, Rotimi C, Gurney M, Chan JC, Pedersen O, Sigurdsson G, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K (2007) A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet 39:770–775. [DOI] [PubMed] [Google Scholar]
- 4. Jenner LB, Demeshkina N, Yusupova G, Yusupov M (2010) Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nat Struct Mol Biol 17:555–560. [DOI] [PubMed] [Google Scholar]
- 5. Reiter V, Matschkal DM, Wagner M, Globisch D, Kneuttinger AC, Muller M, Carell T (2012) The CDK5 repressor CDK5RAP1 is a methylthiotransferase acting on nuclear and mitochondrial RNA. Nucleic Acids Res 40:6235–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Landgraf BJ, Booker SJ (2016) Stereochemical course of the reaction catalyzed by RimO, a radical SAM methylthiotransferase. J Am Chem Soc 138:2889–2892. [DOI] [PubMed] [Google Scholar]
- 7. Lotierzo M, Raux E, Tse Sum Bui B, Goasdoue N, Libot F, Florentin D, Warren MJ, Marquet A (2006) Biotin synthase mechanism: mutagenesis of the YNHNLD conserved motif. Biochemistry 45:12274–12281. [DOI] [PubMed] [Google Scholar]
- 8. Schut GJ, Adams MW (2009) The iron‐hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. J Bacteriol 191:4451–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shirakawa T, Takahashi Y, Wada K, Hirota J, Takao T, Ohmori D, Fukuyama K (2005) Identification of variant molecules of Bacillus thermoproteolyticus ferredoxin: crystal structure reveals bound coenzyme A and an unexpected [3Fe‐4S] cluster associated with a canonical [4Fe–4S] ligand motif. Biochemistry 44:12402–12410. [DOI] [PubMed] [Google Scholar]
- 10. Ullmann GM, Hauswald M, Jensen A, Knapp EW (2000) Structural alignment of ferredoxin and flavodoxin based on electrostatic potentials: implications for their interactions with photosystem I and ferredoxin‐NADP reductase. Proteins 38:301–309. [PubMed] [Google Scholar]
- 11. Cambillau C, Frey M, Mosse J, Guerlesquin F, Bruschi M (1988) Model of a complex between the tetrahemic cytochrome c3 and the ferredoxin I from Desulfovibrio desulfuricans (Norway strain). Proteins 4:63–70. [DOI] [PubMed] [Google Scholar]
- 12. Ta DT, Vickery LE (1992) Cloning, sequencing, and overexpression of a [2Fe–2S] ferredoxin gene from Escherichia coli . J Biol Chem 267:11120–11125. [PubMed] [Google Scholar]
- 13. Meyer J (1988) The evolution of ferredoxins. Trends Ecology Evol 3:222–226. [DOI] [PubMed] [Google Scholar]
- 14. Kim JH, Frederick RO, Reinen NM, Troupis AT, Markley JL (2013) [2Fe–2S]‐ferredoxin binds directly to cysteine desulfurase and supplies an electron for iron‐sulfur cluster assembly but is displaced by the scaffold protein or bacterial frataxin. J Am Chem Soc 135:8117–8120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Robbins AH, Stout CD (1989) Structure of activated aconitase: formation of the [4Fe–4S] cluster in the crystal. Proc Natl Acad Sci USA 86:3639–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giastas P, Pinotsis N, Efthymiou G, Wilmanns M, Kyritsis P, Moulis JM, Mavridis IM (2006) The structure of the 2[4Fe–4S] ferredoxin from Pseudomonas aeruginosa at 1.32‐A resolution: comparison with other high‐resolution structures of ferredoxins and contributing structural features to reduction potential values. J Biol Inorgan Chem 11:445–458. [DOI] [PubMed] [Google Scholar]
- 17. Moulis JM (1996) Molecular cloning and expression of the gene encoding Chromatium vinosum 2[4Fe–4S] ferredoxin. Biochim Biophys Acta 1308:12–14. [DOI] [PubMed] [Google Scholar]
- 18. Page CC, Moser CC, Chen X, Dutton PL (1999) Natural engineering principles of electron tunnelling in biological oxidation–reduction. Nature 402:47–52. [DOI] [PubMed] [Google Scholar]
- 19. Cowan JA, Lui SM (1998) Structure–function correlations in high‐potential iron proteins. Adv Inorg Chem 45:313–350. [Google Scholar]
- 20. Pandelia ME, Nitschke W, Infossi P, Giudici‐Orticoni MT, Bill E, Lubitz W (2011) Characterization of a unique [FeS] cluster in the electron transfer chain of the oxygen tolerant [NiFe] hydrogenase from Aquifex aeolicus . Proc Natl Acad Sci USA 108:6097–6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bak DW, Elliott SJ (2014) Alternative FeS cluster ligands: tuning redox potentials and chemistry. Curr Opin Chem Biol 19:50–58. [DOI] [PubMed] [Google Scholar]
- 22. Smith ET, Blamey JM, Zhou ZH, Adams MW (1995) A variable‐temperature direct electrochemical study of metalloproteins from hyperthermophilic microorganisms involved in hydrogen production from pyruvate. Biochemistry 34:7161–7169. [DOI] [PubMed] [Google Scholar]
- 23. Buckel W, Thauer RK (2013) Energy conservation via electron bifurcating ferredoxin reduction and proton/Na(+) translocating ferredoxin oxidation. Biochim Biophys Acta 1827:94–113. [DOI] [PubMed] [Google Scholar]
- 24. Eram MS, Wong A, Oduaran E, Ma K (2015) Molecular and biochemical characterization of bifunctional pyruvate decarboxylases and pyruvate ferredoxin oxidoreductases from Thermotoga maritima and Thermotoga hypogea . J Biochem 158:459–466. [DOI] [PubMed] [Google Scholar]
- 25. Demmer JK, Huang H, Wang S, Demmer U, Thauer RK, Ermler U (2015) Insights into flavin‐based electron bifurcation via the NADH‐dependent reduced ferredoxin:NADP oxidoreductase rtructure. J Biol Chem 290:21985–21995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. States DJ, Gish W (1994) Combined use of sequence similarity and codon bias for coding region identification. J Comput Biol 1:39–50. [DOI] [PubMed] [Google Scholar]
- 27. Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 112:531–552. [DOI] [PubMed] [Google Scholar]
- 28. Pfeil W, Gesierich U, Kleemann GR, Sterner R (1997) Ferredoxin from the hyperthermophile Thermotoga maritima is stable beyond the boiling point of water. J Mol Biol 272:591–596. [DOI] [PubMed] [Google Scholar]
- 29. Sterner R (2001) Ferredoxin from Thermotoga maritima . Methods Enzymol 334:23–30. [DOI] [PubMed] [Google Scholar]
- 30. Johnson DC, Unciuleac MC, Dean DR (2006) Controlled expression and functional analysis of iron–sulfur cluster biosynthetic components within Azotobacter vinelandii . J Bacteriol 188:7551–7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lanz ND, Grove TL, Gogonea CB, Lee KH, Krebs C, Booker SJ (2012) RlmN and AtsB as models for the overproduction and characterization of radical SAM proteins. Methods Enzymol 516:125–152. [DOI] [PubMed] [Google Scholar]
- 32. Beinert H (1978) Micro methods for the quantitative determination of iron and copper in biological material. Methods Enzymol 54:435–445. [DOI] [PubMed] [Google Scholar]
- 33. Beinert H (1982) Semi‐micro methods for analysis of labile sulfide and of labile sulfide pluse sulfane sulfur in unusually stable iron–sulfur proteins. Analyt Biochem 131:373–378. [DOI] [PubMed] [Google Scholar]
- 34. Bradford MM (1976) Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein–dye binding. Analyt Biochem 72:248–254. [DOI] [PubMed] [Google Scholar]
- 35. Landgraf BJ, Arcinas AJ, Lee KH, Booker SJ (2013) Identification of an intermediate methyl carrier in the radical S‐adenosylmethionine methylthiotransferases RimO and MiaB. J Am Chem Soc 135:15404–15416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Macheroux P (1999) UV–visible spectroscopy as a tool to study flavoproteins. Methods Mol Biol 131:1–7. [DOI] [PubMed] [Google Scholar]
- 37. Grove TL, Benner JS, Radle MI, Ahlum JH, Landgraf BJ, Krebs C, Booker SJ (2011) A radically different mechanism for S‐adenosylmethionine–dependent methyltransferases. Science 332:604–607. [DOI] [PubMed] [Google Scholar]
- 38. Lanz ND, Lee KH, Horstmann AK, Pandelia ME, Cicchillo RM, Krebs C, Booker SJ (2016) Characterization of lipoyl synthase from Mycobacterium tuberculosis . Biochemistry 55:1372–1383. [DOI] [PubMed] [Google Scholar]
- 39. Macedo‐Ribeiro S, Darimont B, Sterner R, Huber R (1996) Small structural changes account for the high thermostability of 1[4Fe–4S] ferredoxin from the hyperthermophilic bacterium Thermotoga maritima . Structure 4:1291–1301. [DOI] [PubMed] [Google Scholar]
- 40. Mayhew SG (1978) The redox potential of dithionite and SO‐2 from equilibrium reactions with flavodoxins, methyl viologen and hydrogen plus hydrogenase. Eur J Biochem 85:535–547. [DOI] [PubMed] [Google Scholar]
- 41. Macedo AL, Besson S, Moreno C, Fauque G, Moura JJ, Moura I (1996) Characterization of a 7Fe ferredoxin isolated from the marine denitrifier Pseudomonas nautica strain 617: spectroscopic and electrochemical studies. Biochem Biophys Res Commun 229:524–530. [DOI] [PubMed] [Google Scholar]
- 42. Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador‐Vegas A, Salazar GA, Tate J, Bateman A (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. [DOI] [PubMed] [Google Scholar]
- 44. Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Laboratory Press, 1989. [Google Scholar]
- 45. Cicchillo RM, Iwig DF, Jones AD, Nesbitt NM, Baleanu‐Gogonea C, Souder MG, Tu L, Booker SJ (2004) Lipoyl synthase requires two equivalents of S‐adenosyl‐L‐methionine to synthesize one equivalent of lipoic acid. Biochemistry 43:6378–6386. [DOI] [PubMed] [Google Scholar]
- 46. Lee KH, Saleh L, Anton BP, Madinger CL, Benner JS, Iwig DF, Roberts RJ, Krebs C, Booker SJ (2009) Characterization of RimO, a new member of the methylthiotransferase subclass of the radical SAM superfamily. Biochemistry 48:10162–10174. [DOI] [PMC free article] [PubMed] [Google Scholar]