

Abstract
Peroxiredoxins are thiol‐dependent peroxidases that function in peroxide detoxification and H2O2 induced signaling. Among the six isoforms expressed in humans, PRDX1 and PRDX2 share 97% sequence similarity, 77% sequence identity including the active site, subcellular localization (cytosolic) but they hold different biological functions albeit associated with their peroxidase activity. Using recombinant human PRDX1 and PRDX2, the kinetics of oxidation and hyperoxidation with H2O2 and peroxynitrite were followed by intrinsic fluorescence. At pH 7.4, the peroxidatic cysteine of both isoforms reacts nearly tenfold faster with H2O2 than with peroxynitrite, and both reactions are orders of magnitude faster than with most protein thiols. For both isoforms, the sulfenic acids formed are in turn oxidized by H2O2 with rate constants of ca 2 × 103 M−1 s−1 and by peroxynitrous acid significantly faster. As previously observed, a crucial difference between PRDX1 and PRDX2 is on the resolution step of the catalytic cycle, the rate of disulfide formation (11 s−1 for PRDX1, 0.2 s−1 for PRDX2, independent of the oxidant) which correlates with their different sensitivity to hyperoxidation. This kinetic pause opens different pathways on redox signaling for these isoforms. The longer lifetime of PRDX2 sulfenic acid allows it to react with other protein thiols to translate the signal via an intermediate mixed disulfide (involving its peroxidatic cysteine), whereas PRDX1 continues the cycle forming disulfide involving its resolving cysteine to function as a redox relay. In addition, the presence of C83 on PRDX1 imparts a difference on peroxidase activity upon peroxynitrite exposure that needs further study.
Keywords: peroxiredoxin 1, peroxiredoxin 2, hyperoxidation, redox signaling, hydrogen peroxide, peroxynitrite, kinetics
Abbreviations
- CP
peroxidatic cysteine
- CR
resolving cysteine
- DTT
dithiotreitol
- EcTR
Escherichia coli thioredoxin reductase
- EcTrx1
Escherichia coli thioredoxin 1
- HRP
horseradish peroxidase
- MMTS
methyl‐methanethiosulfonate
- NEM
N‐ethylmaleimide
- PRDX1
human peroxiredoxin 1
- PRDX2
human peroxiredoxin 2
- Prx
peroxiredoxin(s)
Introduction
Peroxiredoxins (Prx) are thiol‐dependent peroxidases that efficiently reduce H2O2 and also organic hydroperoxides and peroxynitrous acid.1, 2 They have been mostly known as good antioxidants, scavengers of toxic levels of peroxides, and recently more attention is focused on their role in redox signaling considering their abundance and high rate constant of reaction with H2O2.3, 4
A specialized cysteine residue called peroxidatic cysteine, CP, is responsible for the reduction of the peroxide substrate (the oxidation step in the catalytic cycle, Fig. 1). The sulfenic acid formed (CP‐SOH) condenses with a cysteine residue from another subunit, the resolving cysteine, CR, to form an intermolecular disulfide (the resolution step). Thus, each dimer contains two catalytic sites. The resolution step includes a crucial conformational change (from fully folded to locally unfolded or FF→LU) that makes the CP side chain more accessible and in which both conformations are in dynamic equilibrium. The disulfide is finally reduced by an electron donor such as thioredoxin to regenerate the dithiol enzyme (the reduction step, Fig. 1). Any modification of the peroxidatic cysteine like protonation, alkylation or nitrosylation5, 6 will prevent reaction with the peroxide substrate rendering an inactive peroxidase. In addition, the reaction of CP‐SOH with another molecule of peroxide yields a sulfinic acid (Prx hyperoxidation, Fig. 1) inactivating the enzyme until sulfiredoxin (only expressed in eukaryotes) catalyzes its ATP‐dependent reduction back to CP‐SOH.7
Figure 1.
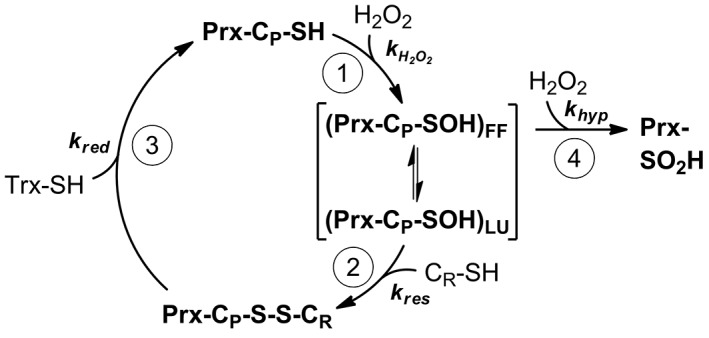
Catalytic cycle of peroxide reduction by 2‐Cys Prx. (1) Oxidation. Reaction of Prx peroxidatic cysteine CP with peroxide substrate yield the sulfenic acid. In brackets is represented the dynamic equilibrium between the LU and the FF structural conformations. (2) Resolution. Disulfide formation between CP and CR. (3) Reduction by thioredoxin to complete the cycle. (4) Hyperoxidation. Reaction of the sulfenic acid of CP with H2O2, yielding a sulfinic acid which inactivates the enzyme. For each step, the associated rate constant is represented.
Prx were originally divided into three categories depending on the number and location of cysteine residues involved in catalysis: the 1‐Cys, the typical 2‐Cys and the atypical 2‐Cys Prxs.2, 8 More recently, based on functional site sequence similarity, Prx have been classified into subgroups Prx1, Prx5, Prx6, Tpx, AhpE, and PrxQ.9 Humans express six Prx isoforms, four in the Prx1 subgroup, one Prx5 and one in the Prx6 subgroup. Human PRDX1 and PRDX2 belong to the Prx1 subgroup, both are 2‐Cys typical Prx, both are cytosolic and have more than 95% sequence similarity, even more, 77% sequence identity, but yet they hold different biological functions albeit associated with their peroxidase activity.
The redox state of these Prx has been associated with the oligomeric structure switching from the disulfide‐oxidized homodimer to the reduced decamer (doughnut‐shaped pentamer of dimers) as described before for bacterial AhpC, also a member of the Prx1 subgroup.10 In addition to the catalytic cysteine residues CP and CR (C52 and C173 for PRDX1, C51 and C172 for PRDX2), both contain another Cys residue (C71, C70, respectively) that is poorly accessible and need SDS addition to be quantified or alkylated.11 In addition, PRDX1 has a fourth Cys residue, C83, located in the dimer–dimer interface that can be alkylated and was reported glutathionylated favoring dimer formation.12
The relevance of these two cytosolic Prx isoforms in cell homeostasis, and at the same time their differential role, is underlined by the observation that knock‐out mice in PRDX1 developed malignant tumors and die prematurely,13 whereas knock‐out mice in PRDX2 were healthy and fertile but developed hemolytic anemia and splenomegalia.14.
The aim of this work is to evaluate the kinetic differences between human PRDX1 and PRDX2 that contribute to understand their physiological role.
Results
Reduction of H2O2 by PRDX1 and PRDX2 followed by intrinsic fluorescence
The reaction of reduced Prx with H2O2 was followed by the change in fluorescence as previously described.15, 16, 17 Two phases were observed, one rapid decrease in fluorescence and a slower increase [Fig. 2(a)]. The rate constant of the fast phase showed a linear dependence with H2O2 concentration and was assigned to the reaction of CP‐SH with H2O2, with a second‐order rate constant of k H2O2 = 1.1 × 108 M−1 s−1 for PRDX1 and 1.6 × 108 M−1 s−1 for PRDX2.17 The slow phase was previously associated with the resolution step, the formation of disulfide Prx16, 17 but at higher concentrations of H2O2 the hyperoxidation reaction is competing for the same CP‐SOH. In fact, the slow phase was fitted to a single exponential function to obtain a first‐order rate constant (k obs, s−1) that increased linearly with the concentration of H2O2 [Fig. 2(b)]. From the slope, a second‐order rate constant for the reaction of CP‐SOH with H2O2, k hyp, was obtained (1.8 × 103 M−1 s−1 for PRDX1 and 2 × 103 M−1 s−1 for PRDX2 at pH 7.4 and 25 °C). The intercepts represent the rate constant of disulfide formation, 11 s−1 for PRDX1 and 0.2 s−1 for PRDX2 [Fig. 2(b)] consistent with our previous report.17
Figure 2.
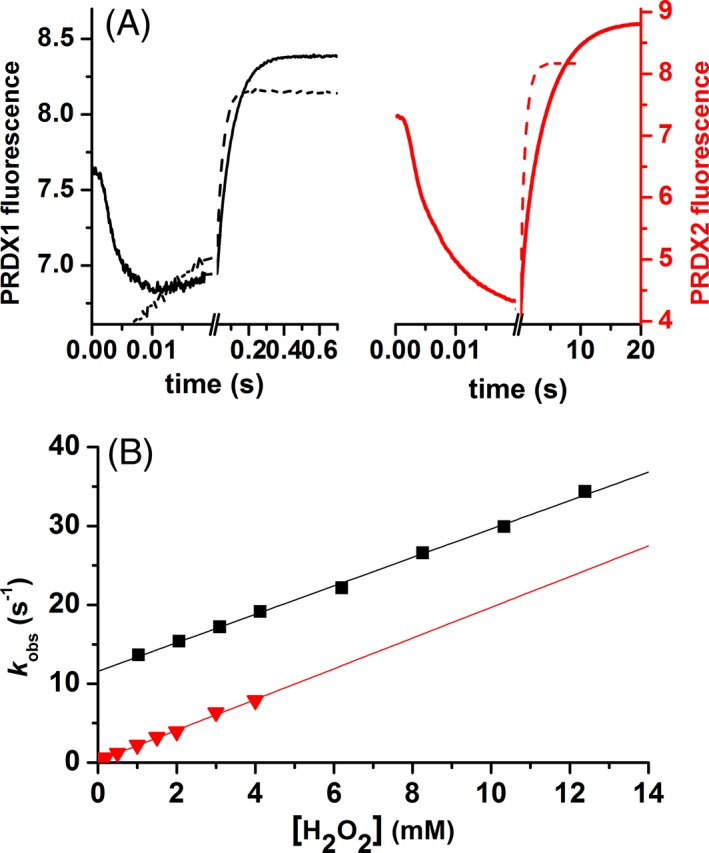
Kinetics of PRDX1 and PRDX2 oxidation by H2O2. (A) Time course of the intrinsic fluorescence of 0.25 μM PRDX1 (black line) or 0.25 μM PRDX2 (red line) upon oxidation with 1.25 μM or 1.5 μM H2O2, respectively (solid line) and 1 mM H2O2 (dashed line) in both cases, at pH 7.4 (traces are the average from 15 runs). (B) The slower phase was fitted to a single exponential function and the first‐order rate constant (k obs) plotted as a function of H2O2 concentration. PRDX1 (black squares) and PRDX2 (red triangles).
Peroxidase activity of PRDX1 and PRDX2
Kinetic parameters were determined for the NAPDH‐linked peroxidase activity using the coupled TR, Trx assay as before.18 At a fixed concentration of H2O2 (saturating but not inactivating the enzyme) and varying the concentration of thioredoxin, the oxidation of NADPH was followed at 340 nm with saturating thioredoxin reductase so that the rate‐limiting step was the reduction of Prx by Trx at low Trx concentrations. Initial rates as a function of Trx concentration [Fig. 3(a)] allowed the determination of K m for Escherichia coli thioredoxin and k cat values for both isoforms, summarized in Table I‐.
Figure 3.
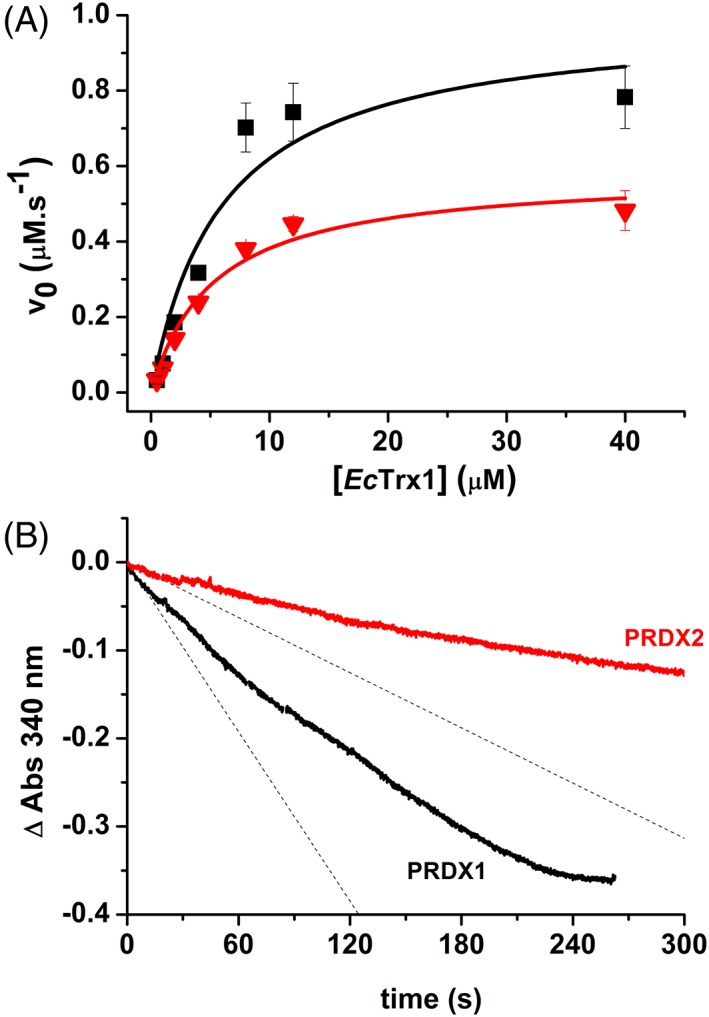
NADPH‐linked peroxidase activity. (a) Determination of kinetic parameters (k cat, K M): reduction of H2O2 catalyzed by PRDX1 (black squares) or PRDX2 (red triangles) was monitored at different initial EcTrx1 concentrations with 200 μM NADPH, 1 μM EcTR, 0.5 μM PRDX1 or PRDX2 and 10 μM H2O2 in 50 mM phosphate buffer pH 7.4, 150 mM NaCl. NADPH oxidation was monitored at 340 nm. Experimental data was fitted to Michaelis–Menten equation (solid lines). (b) Representative time trace of H2O2‐dependent NADPH oxidation by the Prx/Trx/TR system at 50 μM H2O2. Reaction mixture contained 200 μM NADPH, 1 μM EcTR, 8 μM EcTrx1, 0.5 μM PRDX1 (black trace) or PRDX2 (red trace) in 50 mM phosphate buffer pH 7.4, 150 mM NaCl and the reaction was started by addition of H2O2. Dashed lines represent the initial slope of the reaction.
Table I‐.
Summary of Kinetic Parameters for Human PRDX1 and PRDX2.
|
|
|
k res |
|
k cat |
|
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (M−1 s−1) | (M−1 s−1) | (s−1) | (μM) | (s−1) | (M−1 s−1) | (M−1 s−1) | ||||||
| PRDX1 | 3.8 × 107(16) | 1.1 × 10 7a | 9 (16) | 5.5 (48)c | 4.4 (48) | 1.77 × 10 3 | 2.8 × 10 5b | |||||
| 1.1 × 108(17) | 7.6 × 10 6b | 12.9 (17) 11–12 | 4 | 2 | ||||||||
| PRDX2 | 0.13–1 × 108(18) | 1.4 × 107(18) | 0.25 (16) | 2.7 (48)c | 2 (48) | 1.2 × 104 | 3.5 × 10 4b | |||||
| 1.6 × 108(17) | 1.1 × 10 7 b | 2 (25) | 2.4d, 4e (18) | 0.2 (18) | 1.97 × 10 3 | |||||||
| 0.64 (17) 0.2–0.3 |
3 | 1.2 |
The values in italics were determined in the present work, see “Results” section
Competition with HRP at pH 7.4.
Fluorescence time courses at pH 7.3.
Rat thioredoxin.
Human thioredoxin.
E. coli thioredoxin.
Differential susceptibility to hyperoxidation
The measurement of the activity at higher concentrations of H2O2 favors the hyperoxidation of Prx in turnover, and a slowdown of NAPDH oxidation was clearly observed, which was more pronounced for PRDX2 than for PRDX1 [Fig. 3(b) using 50 μM H2O2]. In addition, when reduced Prx was mixed with increasing concentrations of H2O2, the formation of dimers (bound by one or two disulfides) could be followed by nonreductive SDS‐PAGE [Fig. 4(a)] and the presence of hyperoxidized CP (CP‐SO2H) detected by western blot (WB) [Fig. 4(b)]. Again, higher susceptibility to hyperoxidation was observed for PRDX2 than for PRDX1 as previously observed by Cox et al.19 when comparing PRDX3 with cytosolic PRDX1 and PRDX2. Figure 4(a) shows the increase in monodisulfide dimers of PRDX2 with increasing H2O2 denoting a faster hyperoxidation (once CP is hyperoxidized it cannot form the intermolecular disulfide) that is confirmed by western blotting [Fig. 4(b)]. In addition, at high excess of H2O2, hyperoxidized monomer was detected for PRDX2 and not yet for PRDX1. It is worth noting the presence of higher oligomers (tetramers) in the case of PRDX1 and not PRDX2 exposed to H2O2 [Fig. 4(a)] probably formed by C83 disulfides as they are reduced by β‐mercaptoethanol (not shown).
Figure 4.
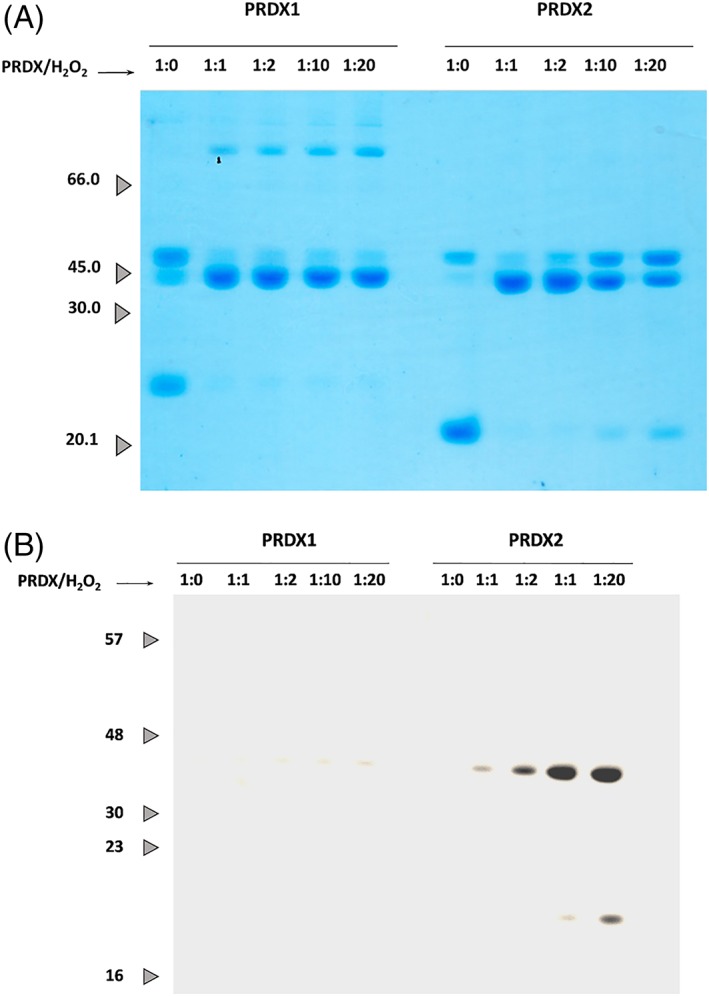
PRDX1 and PRDX2 differential hyperoxidation susceptibility. About 5 μM PRDX1 or PRDX2 were treated with increasing concentrations of H2O2. About 1 μg of protein of each sample was loaded and resolved in a 12% SDS‐PAGE and analyzed by Coomassie staining (a) or WB using anti‐Prx‐SO2/3H antibody (b).
Reduction of peroxynitrous acid by PRDX1
As reported before for PRDX2, PRDX1 is able to reduce peroxynitrous acid. The second‐order rate constant of this reaction was determined by competition with horseradish peroxidase (HRP) as detailed in the section “Materials and Methods.” First, a rate constant of (2.0 ± 0.2) × 106 M−1 s−1 was determined for the oxidation of HRP by peroxynitrite under our experimental conditions (Fig. 1) consistent with previously reported data.20, 21, 22 In the presence of reduced PRDX1, the yield of peroxynitrite‐dependent oxidation of HRP to Compound I was inhibited (Fig. 5) and using Equation 1, a rate constant of peroxynitrite‐mediated PRDX1 oxidation was obtained, k ONOOH = (1.5 ± 0.3) × 107 M−1 s−1 at pH 7.4 and 25 °C.
Figure 5.
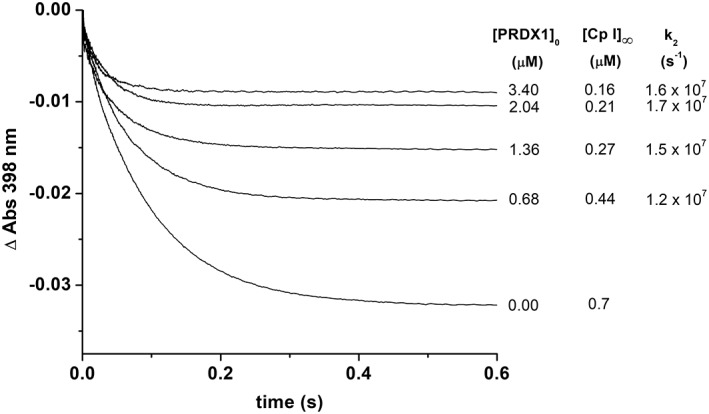
Determination of the rate constant for the reaction of peroxynitrite with PRDX1 by competition kinetics. Time courses of HRP (5 μM) oxidation by peroxynitrite (0.7 μM) in the presence of prereduced PRDX1 (from bottom to top 0, 0.68, 1.36, 2.04, and 3.4 μM) in 100 mM sodium phosphate buffer pH 7.4 plus 0.1 mM dtpa at 25 °C. From the concentration of Compound I formed at each PRDX1 concentration, a value of k2 was calculated using Equation 1.
Reduction of ONOOH by PRDX1 and PRDX2 followed by intrinsic fluorescence
Upon reaction with excess of ONOOH, the fluorescence of PRDX1 shows a time course with three phases [Fig. 6(a)]. The data fit nicely to triple exponential functions up to 13.5 μM ONOOH (Supporting Information Fig. S2). The first exponential fluorescence decay yields a rate constant that depends linearly on ONOOH concentration [Fig. 6(c)] with a slope comparable with the oxidation rate constant obtained by HRP competitive kinetics (Fig. 5). We have assigned this rate constant to (Table I‐). The linear fit has a significant non‐zero intercept (40 ± 2 s−1). The rate constant from the second ascending exponential also depended linearly on ONOOH concentration [Fig. 6(d)]. In this case, the y intercept (12 ± 0.2 s−1) coincided with k res and, analogous to the analysis of the reaction with H2O2, the slope of the linear fit was assigned to the hyperoxidation reaction by ONOOH (, Table I‐). Finally, although the third phase could be fitted to an exponential function, the rate constant obtained had a nonlinear dependence on ONOOH concentration. This phase is coincident in time with the spontaneous decay of ONOOH via isomerization to nitrate and radical formation (t1/2 = 2.2 s). Thus, it can be ascribed to a combination of several processes such as C83 and tryptophan oxidation (both first order in ONOOH), and oxidation/nitration of aromatic residues caused by radicals derived from ONOOH (zero order in ONOOH).
Figure 6.
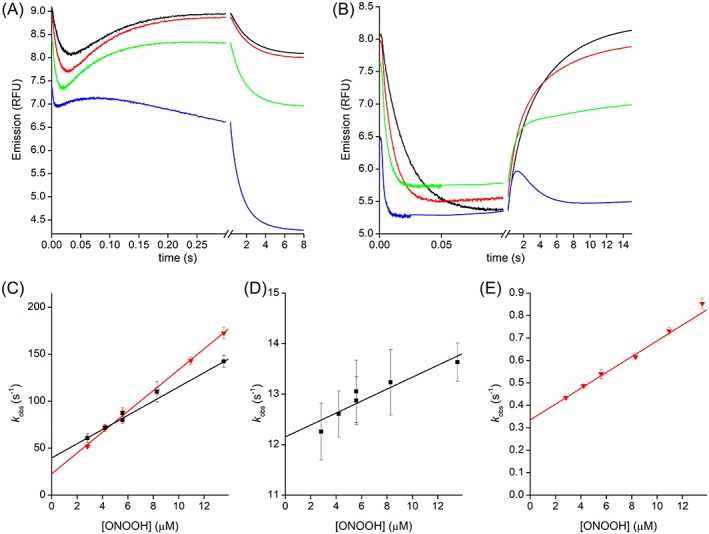
Kinetics of PRDX1 and PRDX2 oxidation by ONOOH. Time courses of the oxidation of PRDX1 (a) or PRDX2 (b) with excess ONOOH, from top to bottom 2.8, 8.3, 13.5 and 71 μM, and traces are averages of 15 runs. (c) Second‐order plot of the rate constant of the fast exponential descending part of the time courses for PRDX1 (black squares) or PRDX2 (gray triangles). Second‐order plot of the rate constant of the ascending exponential part of the time courses for PRDX1 (black squares, d) or PRDX2 (gray triangles, e). General conditions: buffer A, pH 7.3, 25 °C, λex = 280 nm, λem > 320 nm.
The time course of PRDX2 fluorescence was somewhat simpler, as the third phase was only apparent at very high concentrations of ONOOH [Fig. 6(b)]. The first two phases were similar to what was observed using H2O2 as oxidant, namely a fast decay and a much slower recovery of the fluorescence. As with PRDX1, both the descendent and the ascendent exponential phases had rate constants that depend linearly on ONOOH concentration [Fig. 6(c,e)]. The descent phase was assigned to the oxidation step of the catalytic cycle and the ascent phase to a combination of resolution (intercept 0.34 ± 0.01 s−1) and hyperoxidation (, Table I‐).
Nitration and hyperoxidation of PRDX1 by peroxynitrous acid
Even though peroxynitrous acid is reduced by PRDX1, other enzyme residues get modified by excess of this oxidant. As observed for PRDX2,18, 23 tyrosine nitration and dityrosine crosslinks were detected [Fig. 7(a), inset]. Unlike the case of PRDX2 whose activity was observed to increase upon nitration,23 the treatment of disulfide PRDX1 with peroxynitrite resulted in a decrease of peroxidase activity [Fig. 7(a)]. The presence of an extra cysteine, C83 makes the difference. Blocking both catalytic cysteines CP‐CR (by forming the disulfide) and also C83 with methyl methanethiosulfonate before treatment with peroxynitrite, the only irreversible modification observed after peroxynitrite exposure was tyrosine nitration, but importantly, the cysteine residues could be reduced back with DTT and the activity recovered. Under these conditions, nitration of PRDX1 rendered a more active and robust peroxidase [Fig. 7(b)] in the same way as observed before with PRDX2.23
Figure 7.
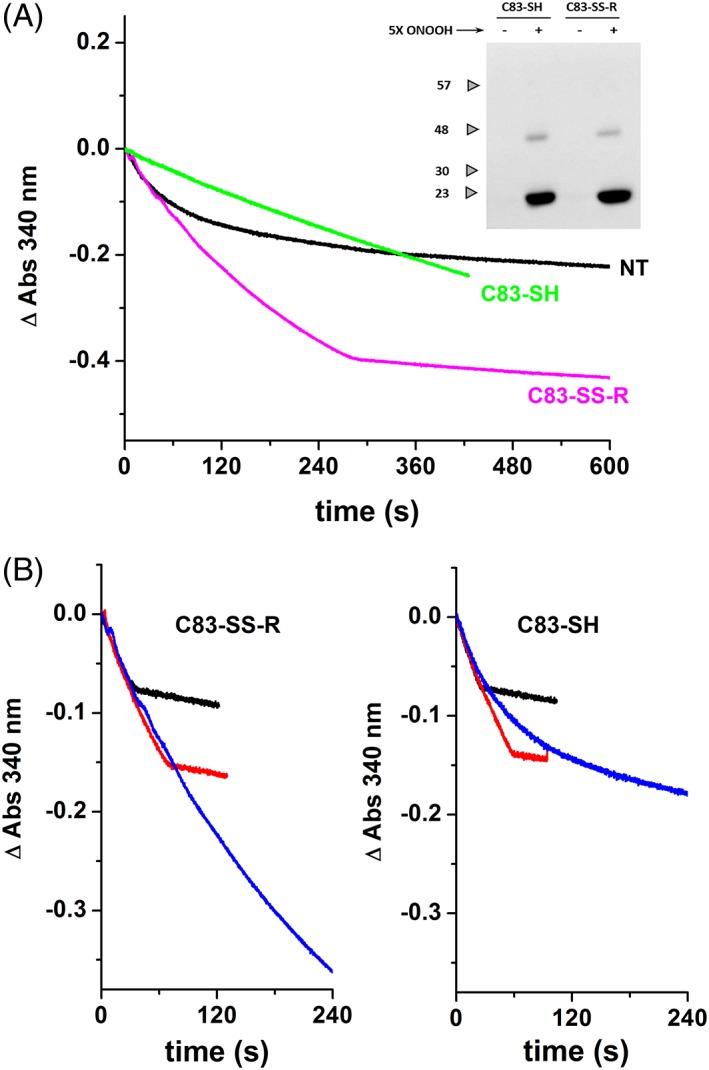
Peroxynitrite effect on PRDX1 depends on the redox state of C83. (a) Different samples of PRDX1 were assayed for peroxidase activity with 50 μM H2O2. Peroxynitrite‐treated PRDX1‐C83‐SH, green line; C83‐SS‐R, magenta line; and nontreated PRDX1 (NT, black line). Inset: WB using anti‐nitrotyrosine antibody (samples resolved on a 12% SDS‐PAGE with β‐mercaptoethanol). (b) Peroxidase activity of peroxynitrite‐treated PRDX1‐C83‐SH (right panel) and C83‐SS‐R (left panel) are compared at different H2O2 concentration (10 μM, black line; 20 μM, red line; 50 μM, blue line).
Differential near‐UV circular dichroism spectra
The circular dichroism (CD) spectra in this region are influenced by the aromatic residues environment and indicative of the protein tertiary structure. The disulfide bond absorption occurs in the range of 240–290, near 260 nm, however, given that the near‐UV CD bands of disulfides are generally broader than the corresponding to the aromatic side chains, it is very difficult to assess the contribution of the former to the protein spectrum. An important contribution of Phe (peak at 265 nm), Tyr and Tyr–Trp interactions (275–285 nm) and to a lesser extent Trp (shoulder at 295 nm) was observed for PRDX1, in the same way, as registered before for PRDX224 [Fig. 8 (a)]. Upon oxidation to disulfide, there is a dramatic conformational change around these aromatic residues for PRDX2 as previously reported,24 whereas the CD signals observed between reduced and oxidized PRDX1 were significantly smaller [Fig. 8(b)], suggesting that although similar processes take place in the active centers of PRDX1 and PRDX2, some structural differences exists between them. Far‐UV CD analysis showed no differences between the two isoforms (data not shown).
Figure 8.
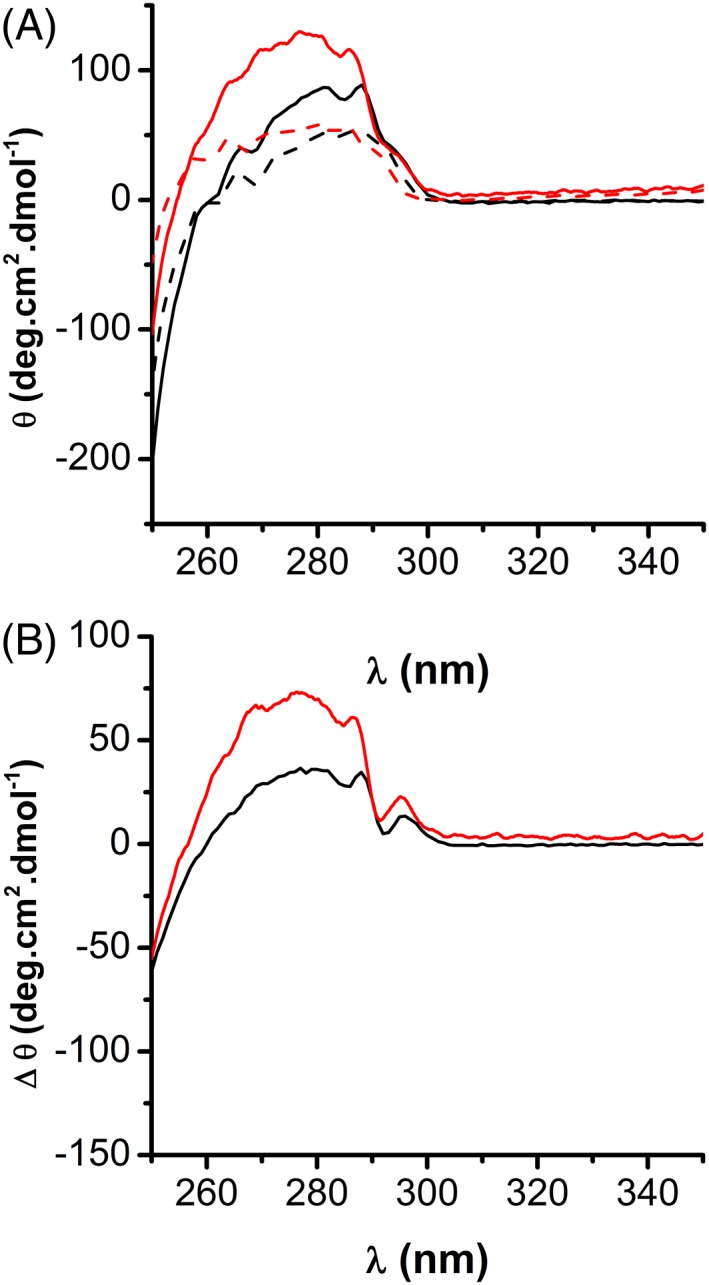
Comparison of Near‐UV CD spectra of PRDX1 and PRDX2. (a) About 50 μM PRDX1 (black) or PRDX2 (red) in their reduced or oxidized state (solid and dashed lines, respectively) was analyzed by CD in the near UV. (b) Reduced disulfide‐oxidized differential CD spectra for PRDX1 (black line) and PRDX2 (red line) showing less differences in near‐UV CD signals on PRDX1 compared to PRDX2 upon oxidation, compatible with small changes in the surroundings of aromatic residues in the former.
Discussion
Taking advantage of the change in tryptophan fluorescence with the redox state of the enzyme, the reaction of reduced Prx with H2O2 was followed (Fig. 2). The oxidation of CP‐SH to CP‐SOH by H2O2 is extremely fast for both isoforms (Table I‐), five orders of magnitude higher than the rate constant for most protein cysteine residues, denoting that they are highly sensitive to H2O2.
Once the sulfenic derivative (CP‐SOH) is formed, the conformational change and reaction with the CR to form the disulfide is slower for PRDX2 than for PRDX1, showing a critical point of difference. Herein, we determined a first‐order rate constant of disulfide formation for PRDX2 of 0.25 s−1 at pH 7.4 and 25 °C that agrees with previous reports from our group (0.64 s−1 at pH 7.4, 25 °C17) and others (0.25 s−1 at pH 7.4, 22 °C16) and is somewhat smaller than the 2 s−1 first reported.25 Importantly, the rate of disulfide formation obtained for PRDX1 is one order of magnitude higher than for PRDX2, 11 s−1, and agrees with the value recently reported.16 In addition, we recently measured similar pKa values for CP‐SOH (close to 7) for both enzymes, while a 1 unit difference was determined for CR‐SH pKas (7.4 for PRDX1 and 8.5 for PRDX2).17 Thus, at the same pH, there is more concentration of the nucleophilic thiolate in the PRDX1 isoform that could contribute to accelerate its disulfide formation compared to PRDX2.
These differences in rates of disulfide formation are in accordance with the observed differences in hyperoxidation with excess H2O2 (Fig. 4) and also in turnover (Fig. 3) where PRDX2 was more readily hyperoxidized and inactivated than PRDX1. Interestingly, this higher susceptibility to hyperoxidation of PRDX2 cannot be ascribed to a higher rate of sulfinic acid formation compared to PRDX1. The determined rate constant for the reaction of CP‐SOH with H2O2 (k hyp) was similar for both isoforms, 2 × 103 M−1 s−1 under our experimental conditions [Fig. 2(b)], almost one order of magnitude lower than the rate constant previously reported for PRDX2.25 Bolduc et al.26 recently determined novel hyperoxidation resistance motifs in 2‐Cys Prx (motif A and motif B) besides the absence of YF and GGLG (present in sensitive Prx). The presence of different structural motifs (PRDX1 presents only motif A and PRDX2 only motif B) supports the differential susceptibility to hyperoxidation that is ligated to the competing resolution process (Fig. 1). Our results indicate that the reactivity of CP‐SOH with H2O2 is the same for both isoforms, thus, the difference in hyperoxidation sensitivity is mainly laying on subtle structural differences that favor the conformational change towards the competing reaction, the oxidation to disulfide. In that sense, the near‐UV CD indicates smaller differences in tertiary structure between the reduced and disulfide‐oxidized form of PRDX1 than PRDX2 (Fig. 8).
Peroxynitrous acid is also a peroxide substrate for PRDX1. A second‐order rate constant of 1.5 ± 0.3 × 107 M−1 s−1 (by competition with HRP) and 1.1 × 107 M−1 s−1 by intrinsic fluorescence at pH 7.4 and 25 °C were determined (Figs. 5 and 6), similar to the previously reported value for PRDX2.18 Thus, both PRDX1 and PRDX2 isoforms reduce H2O2 more efficiently than ONOOH, like all members of Prx1 subgroup, which is contrary to the expected trend based on SN2 reactivity between thiolates and peroxides. The reaction is expected to be faster if it produces a less basic leaving group27, 28 and as NO2 − is significantly less basic than OH−, reactions of thiolates with ONOOH are usually much faster than with H2O2. Therefore, both PRDX1 and PRDX2 are not only specialized in peroxide sensing and reduction but more specifically in H2O2, in contrast with human Prx5 that reduces peroxynitrite faster than H2O2. Urate hydroperoxide was also found as a substrate for both isoforms with a lower rate constant (5–23 × 105 M−1 s−1 16).
We recently discussed the possibility of using the ratio of the rates of reactions 2 over 1 (k res/k ROOH, from Table I‐) as a measure of the sensing ability of each Prx toward a peroxide substrate17 as it represents the threshold concentration of substrate at which the protein switches from mostly dithiol to mostly sulfenic. The same quotient can be calculated to compare the sensing ability toward different ROOH substrates and yields that PRDX2 is more sensitive in each case, with threshold concentrations of 1.6, 23, and 110 nM for H2O2, ONOOH, and urate hydroperoxide, respectively. The concentrations in the case of PRDX1 are significantly higher (0.1, 1.45, and 22 μM, respectively). This differential reinforces the idea of the two Prx working as stepwise sensors, each switching in specific concentration ranges of each specific peroxide substrate.
Changes in intrinsic fluorescence also allowed us to follow the hyperoxidation of PRDX1 and PRDX2 by peroxynitrite (Fig. 6). Interestingly, the hyperoxidation with ONOOH is faster than with H2O2 (3.5 × 104 M−1 s−1 vs 2 × 103 M−1 s−1) in accordance with the SN2 expected trend and as observed before by western blotting for PRDX2. 18 The rate constant determined for PRDX1 hyperoxidation with peroxynitrite is nearly 10‐fold higher than for PRDX2 (Table I‐). It is unlikely that C83 oxidation contributes to the process herein interpreted as hyperoxidation, as a rate constant in the order of 103 M−1 s−1 is expected for a nonperoxidatic cysteine reacting with ONOOH.29 Although is higher for PRDX1 than for PRDX2, the hyperoxidation efficiency in a single turnover is related to the ratio [ONOOH]/k res, that is, the competition between the rates of hyperoxidation and resolution. Such ratio is higher for PRDX2 at all concentrations of ONOOH.
Disulfide‐oxidized PRXD1 treated with an excess of peroxynitrite resulted in nitration of the enzyme and aggregation to oligomers [Fig. 7(a), inset] as reported before for PRDX2 isolated from erythrocytes.18 However, the effect of peroxynitrite‐dependent nitration of PRDX1 on peroxidase activity was different from what was previously observed for PRDX2 depending on the redox state of its C83 (Fig.7). This extra cysteine, even though not involved in the catalytic cycle, needs to be reduced for PRDX1 to display its full peroxidase activity. As pointed out earlier, this C83 represents a critical differential element between these isoforms.16, 30, 31
For both PRDX1 and PRDX2, the preferential peroxide substrate is H2O2 (over ONOOH or alkyl hydroperoxides).32 Together they are estimated to consume most of the H2O2 in the cytosol from endogenous and extracellular sources.33 They are very sensitive sensors of cytosolic H2O2 but once the sulfenic derivative is formed, they can take different pathways. PRDX2‐CP‐SOH lasts longer and even though this increases the chance to get inactivated by hyperoxidation, it also offers the chance to condense with another cysteine residue of a redox protein (forming a mixed disulfide via its CP). Conversely, PRDX1‐CP‐SOH is more prone to react with its own CR‐SH to form the disulfide dimer and the redox signal relay depends on this disulfide oxidizing a redox protein (in this case, the mixed disulfide should involve the PRDX1‐CR). In fact, at the cellular level, different transient disulfide interactions were detected for each isoform, for example, PRDX1 with kinase ASK134 while PRDX2 with transcription factor STAT3.35 The kinetics of Prx disulfide reduction, the last step in the catalytic cycle is the least studied and only thioredoxin has been assayed as a reductant so far, but other partners need to be explored. Further studies on this reduction step will shed more light on the mechanisms underlying Prx‐dependent redox signaling.
Materials and methods
Chemicals
Dithiothreitol (DTT), N‐ethyl maleimide (NEM), and reduced nicotinamide adenine dinucleotide phosphate (NADPH) were purchased from AppliChem GmbH (Darmstadt Germany). Hydrogen peroxide (H2O2), HRP, and diethylenetriaminepentaacetic acid (dtpa) were purchased from Sigma‐Aldrich (St Louis, MO). 4,4′‐Dithiodipyridine (DTDPy) was from Acros Organics (Fisher Scientific, Hampton, NH). Peroxynitrite was synthesized as in Reference 36. All other reagents were of analytical grade and used as received.
Recombinant proteins expression and purification
Human recombinant PRDX1 and PRDX2 were produced in E. coli without any affinity tag. The use of the mature protein is crucial since there is evidence that extra residues can influence 2‐Cys Prx structure/activity relationship.37, 38 Recombinant PRDX1 was expressed with a pET17b plasmid and purified as previously described6 with minor modifications. After anionic exchange chromatography, a size‐exclusion chromatography was performed in a HiLoad 16/60 Superdex 200 column (GE Healthcare, Chicago, IL) equilibrated with 50 mM sodium phosphate pH 7.4, 150 mM sodium chloride, and 0.1 mM dtpa (Buffer A). Recombinant PRDX2 was expressed with a pET17b plasmid and purified as reported in Reference 38. Both, PRDX1 and PRDX2 were expressed without any extra residue in their primary structure. Expression vector pET9a (Novagen, Darmstadt, Germany) with the E. coli Trx1 gene (EcTrx1) was expressed and purified according to Reference 39. E. coli thioredoxin reductase (EcTR) was expressed in E. coli BL21(DE3) transformed with the plasmid pTrR301 and purified as reported in Reference 40. Protein purity was evaluated by SDS‐PAGE.
Peroxide and protein quantification
The concentration of H2O2 was measured at 240 nm (ε240 = 39.4 M−1 cm−1),41 peroxynitrite concentration was determined at alkaline pH at 302 nm (ε302 = 1670 M−1 cm−1),42 HRP concentration was measured at 403 nm (ε403 = 1.03 × 105 M−1 cm−1)20 and EcTR at 280 nm (ε280 = 51700 M−1 cm−1). 43 For the rest of the proteins, concentration was measured by the absorption at 280 nm, using the corresponding ε determined with https://web.expasy.org/protparam/: ε280 (PRDX1)SS = 18,700 M−1 cm−1, ε280 (PRDX1)SH = 18,450 M−1 cm−1, ε280 (PRDX2)SS = 21,555 M−1 cm−1, ε280 (PRDX2)SH = 21,430 M−1 cm−1, ε280 (EcTrx1) = 14,060 M−1 cm−1.
Kinetics followed by intrinsic fluorescence
Oxidation of prereduced PRDX1 and PRDX2 by H2O2 was registered following the change in their intrinsic fluorescence as described in Reference 17, in an Applied Photophysics SX20 stopped‐flow fluorimeter with a mixing time of <2 ms, with λex = 280 nm, measuring the total emission above 320 nm. 0.25 μM Prx in buffer A was mixed with different concentrations of H2O2 (1 μM–12 mM) diluted in buffer A. At low oxidant concentration (<10 μM), the time courses were biphasic (a fast drop, followed by a slow increase in the signal) and adjustable to a double exponential function. At low oxidant concentration, the constant associated with the slower phase does not change noticeably with the concentration of H2O2. However, working at high H2O2 concentrations, the downward phase became faster than the dead time of the stopped flow, and the slow phase showed a linear dependence on H2O2 concentration. From the curve k obs versus [H2O2], the bimolecular rate constant of hyperoxidation, k hyp (slope) and the first‐order rate constant of resolution, k res (y‐axis intercept) for PRDX1 and PRDX2 were determined.
The kinetics of ONOOH reduction by PRDX1 and PRDX2 were performed in an SX20 stopped flow fluorimeter following the fluorescence of the proteins with λex = 280 nm, λem > 320 nm. Reduced PRDX1 or PRDX2 (0.5 μM) in buffer A (2X), pH 7.25, were mixed with an equal volume of ONOO− (5.6–142 μM) in 1.5 mM NaOH. The pH after mixing, measured at the outlet was 7.3. The fluorescence was followed for up to 15 s and the time courses were fitted to triple (PRDX1) or double (PRDX2) exponential functions according to the number of phases observed.
NADPH‐linked peroxidase activity
Peroxidase activity was measured spectrophotometrically following the NADPH consumption at 340 nm (ε340 = 6.22 mM−1 cm−1) in a cuvette containing 200 μM NADPH, 1 μM EcTR, 8 μM EcTrx1 (except in Fig. 4 where 0.5–40 μM Trx is used) and 0.5 μM PRDX1 or PRDX2 in buffer A. The reaction was started adding the specified amount of H2O2. All spectrophotometric measurements were made with a Cary 50 spectrophotometer (Varian, Australia).
Treatment of reduced PRDX1 with excess H2O2 (SDS‐PAGE and WB)
About 50 μL of 5 μM prereduced PRDX1 or PRDX2 were treated with 2 μL of a H2O2 solution so that the final concentration of peroxide corresponds to the desired excess (0–20). After 15 min, 2 mM NEM was added to the mixture to block reduced cysteines. Electrophoresis loading buffer (0.125 M Tris‐HCl, 4% SDS, 20% glycerol, 0.053% bromophenol blue, pH 6.8) was added and samples were resolved in a 12% polyacrylamide gel electrophoresis with sodium dodecyl sulfate (12% SDS‐PAGE). In WB experiments, the proteins resolved on the gel were transferred to PVDF membrane, blotted with rabbit polyclonal antibody against hyperoxidized Prx (Anti‐Peroxiredoxin‐SO3, AbFrontier, Seoul, Korea) and detection performed with fluorescent goat anti‐mouse IRDye 680RD (LI‐COR Biosciences, Lincoln, NE) secondary antibodies. Blots were scanned using a G:BOX Chemi XRQ (Syngene, Cambridge, UK).
Reaction rate constant with peroxynitrite determined by competence with HRP
PRDX1 capacity to reduce peroxynitrite was corroborated and characterized by competition assay with HRP as previously reported.18, 22, 44 Time courses of formation of Compound I upon oxidation of 5 μM HRP by 0.7 μM peroxynitrite were recorded in the absence or in the presence of reduced PRDX1 (0.68–3.4 μM). The reaction was followed at 398 nm, the isosbestic point for HRP‐compounds I and II (Δε398 = 4.2 × 104 M−1 cm−1 45 in an Applied Photophysics SX20 stopped‐flow spectrophotometer. HRP and PRDX1 solutions were prepared in 100 mM sodium phosphate pH 7.4 with 0.1 mM dtpa and stock peroxynitrite diluted in 10 mM NaOH. For each registered temporal course, k 2 (corresponding to the bimolecular rate constant of PRDX1 oxidation by peroxynitrite = k ONOOH) was calculated using Equation 1:
| (1) |
where kHRP is the rate constant of peroxynitrite‐mediated HRP oxidation obtained herein (2.0 × 106 M−1 s−1at pH 7.4 and 25 °C, consistent with previous report.20, 21, 46 [HRP]0 is the initial concentration of HRP (5 μM), [PRDX1‐SH]0, the initial concentration of reduced PRDX1 (0.7–3.4 μM), [CpI]∞ the concentration of Compound I at completion of reaction, [PRDX1‐S2]∞, the concentration of PRDX1 oxidized at completion of reaction, calculated as [CpI]∞ in the absence minus in the presence of the indicated concentration of PRDX1.44
PRDX1‐controlled oxidation to disulfide
To assure the oxidation of the catalytic site but keeping C83 reduced, PRDX1 was treated with 10 mM DTT for 30 min at room temperature. Residual DTT was removed passing the mixture through a Bio‐Spin column (BioRad, Hercules, CA) and protein as well as thiol concentration were immediately determined (thiol concentration was determined with DTDPy as reported in Reference 47). After reduction, three of the four thiols present in PRDX1 could be detected by this method. As two of these thiols belong to CP and CR, 1 mol of H2O2 was added for every three moles of thiol quantified so that to yield the disulfide in the catalytic site and avoiding C83 oxidation.
Treatment of disulfide‐oxidized PRDX1 with excess of peroxynitrite
Treatment of PRDX1 with peroxynitrite was performed according to the protocol reported in Reference 23 for PRDX2. Briefly, a molar excess of 5:1 peroxynitrite was added in a flux‐like addition to 100 μL of 130 μM disulfide PRDX1 (i.e., oxidized in the catalytic site but with C83 as thiol) in buffer A. To protect C83 before peroxynitrite treatment, disulfide PRDX1 was treated with 1 mM MMTS for 1 hour at room temperature, and then the mixture was passed through a Bio‐Spin column (BioRad) equilibrated with buffer A. Finally, peroxynitrite treatment was performed followed by protein reduction with DTT (the excess was removed by passing through a Bio‐Spin column). This sample was labeled C83‐SS‐R. As a control, disulfide PRDX1 was treated with 10 mM NaOH instead of peroxynitrite (nontreated sample, NT).
CD spectra
Spectra of PRDX1 were acquired at 25 °C using a Chirascan Q100 Spectropolarimeter (Applied Photophysics, Leatherhead, UK). Near‐UV measurements were carried out in 1 cm cells containing 50 μM PRDX1 and far‐UV with 5 μM PRDX1 in 0.1 cm cells. Protein was prepared in 10 mM sodium phosphate buffer, pH 7.4. DTT was removed in the reduced samples before measurements. A scan of the buffer was subtracted from the corresponding averaged sample spectra. Data for near and far UV spectra of PRDX2 were obtained from Reference 24.
Supporting information
Figure S1. Determination of second‐order rate constant for the reaction of HRP with peroxynitrite at pH 7.4 and 25 °C.
Figure S2. Time course and data fitting of PRDX1 oxidation by ONOOH. PRDX1 (0.25 μM) was reacted with 14 μM ONOOH (green line). The time course of the fluorescence (t > 2 ms) was fitted to a triple exponential function (black line). The residual shows the goodness of the fit.
Acknowledgments
Financial support was provided by Universidad de la República (CSIC C632‐348) to AD, Centro Argentino‐Brasileño de Biotecnología (CABBIO 2014‐05) to GFS and JS. JDR and LMR were partially supported by CAP Universidad de la República and PEDECIBA, Uruguay. The authors would like to thank Dr. Flavia Meotti and Dr. Moran Behar for kind donation of PRDX1 plasmid and Dr. Todd Lowther for PRDX2 plasmid.
REFERENCES
- 1. Wood ZA, Poole LB, Karplus PA (2003) Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 300:650–653. [DOI] [PubMed] [Google Scholar]
- 2. Wood ZA, Schroder E, Robin Harris J, Poole LB (2003) Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci 28:32–40. [DOI] [PubMed] [Google Scholar]
- 3. Rhee SG (2016) Overview on peroxiredoxin. Mol Cells 39:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sies H (2017) Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biol 11:613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fang J, Nakamura T, Cho DH, Gu Z, Lipton SA (2007) S‐nitrosylation of peroxiredoxin 2 promotes oxidative stress‐induced neuronal cell death in Parkinson's disease. Proc Natl Acad Sci USA 104:18742–18747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Engelman R, Weisman‐Shomer P, Ziv T, Xu J, Arner ES, Benhar M (2013) Multilevel regulation of 2‐Cys peroxiredoxin reaction cycle by S‐nitrosylation. J Biol Chem 288:11312–11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lowther WT, Haynes AC (2011) Reduction of cysteine sulfinic acid in eukaryotic, typical 2‐Cys peroxiredoxins by sulfiredoxin. Antiox Redox Signal 15:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rhee SG, Kang SW, Chang TS, Jeong W, Kim K (2001) Peroxiredoxin, a novel family of peroxidases. IUBMB Life 52:35–41. [DOI] [PubMed] [Google Scholar]
- 9. Nelson KJ, Knutson ST, Soito L, Klomsiri C, Poole LB, Fetrow JS (2011) Analysis of the peroxiredoxin family: using active‐site structure and sequence information for global classification and residue analysis. Proteins 79:947–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wood ZA, Poole LB, Hantgan RR, Karplus PA (2002) Dimers to doughnuts: redox‐sensitive oligomerization of 2‐cysteine peroxiredoxins. Biochemistry 41:5493–5504. [DOI] [PubMed] [Google Scholar]
- 11. Peskin AV, Low FM, Paton LN, Maghzal GJ, Hampton MB, Winterbourn CC (2007) The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. J Biol Chem 282:11885–11892. [DOI] [PubMed] [Google Scholar]
- 12. Park JW, Piszczek G, Rhee SG, Chock PB (2011) Glutathionylation of peroxiredoxin I induces decamer to dimers dissociation with concomitant loss of chaperone activity. Biochemistry 50:3204–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neumann CA, Krause DS, Carman CV, Das S, Dubey DP, Abraham JL, Bronson RT, Fujiwara Y, Orkin SH, Van Etten RA (2003) Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 424:561–565. [DOI] [PubMed] [Google Scholar]
- 14. Lee TH, Kim SU, Yu SL, Kim SH, Park DS, Moon HB, Dho SH, Kwon KS, Kwon HJ, Han YH, Jeong S, Kang SW, Shin HS, Lee KK, Rhee SG, Yu DY (2003) Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood 101:5033–5038. [DOI] [PubMed] [Google Scholar]
- 15. Parsonage D, Nelson KJ, Ferrer‐Sueta G, Alley S, Karplus PA, Furdui CM, Poole LB (2015) Dissecting peroxiredoxin catalysis: separating binding, peroxidation, and resolution for a bacterial AhpC. Biochemistry 54:1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carvalho LAC, Truzzi DR, Fallani TS, Alves SV, Toledo JC Jr, Augusto O, Netto LES, Meotti FC (2017) Urate hydroperoxide oxidizes human peroxiredoxin 1 and peroxiredoxin 2. J Biol Chem 292:8705–8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Portillo‐Ledesma S, Randall LM, Parsonage D, Dalla Rizza J, Karplus PA, Poole LB, Denicola A, Ferrer‐Sueta G (2018) Differential kinetics of two‐cysteine peroxiredoxin disulfide formation reveal a novel model for peroxide sensing. Biochemistry 57:3416–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Manta B, Hugo M, Ortiz C, Ferrer‐Sueta G, Trujillo M, Denicola A (2009) The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Arch Biochem Biophys 484:146–154. [DOI] [PubMed] [Google Scholar]
- 19. Cox AG, Pearson AG, Pullar JM, Jonsson TJ, Lowther WT, Winterbourn CC, Hampton MB (2009) Mitochondrial peroxiredoxin 3 is more resilient to hyperoxidation than cytoplasmic peroxiredoxins. Biochem J 421:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Floris R, Piersma SR, Yang G, Jones P, Wever R (1993) Interaction of myeloperoxidase with peroxynitrite. A comparison with lactoperoxidase, horseradish peroxidase and catalase. Eur J Biochem 215:767–775. [DOI] [PubMed] [Google Scholar]
- 21. Ogusucu R, Rettori D, Munhoz DC, Netto LE, Augusto O (2007) Reactions of yeast thioredoxin peroxidases I and II with hydrogen peroxide and peroxynitrite: rate constants by competitive kinetics. Free Rad Biol Med 42:326–334. [DOI] [PubMed] [Google Scholar]
- 22. Reyes AM, Vazquez DS, Zeida A, Hugo M, Pineyro MD, De Armas MI, Estrin D, Radi R, Santos J, Trujillo M (2016) PrxQ B from Mycobacterium tuberculosis is a monomeric, thioredoxin‐dependent and highly efficient fatty acid hydroperoxide reductase. Free Rad Biol Med 101:249–260. [DOI] [PubMed] [Google Scholar]
- 23. Randall LM, Manta B, Hugo M, Gil M, Batthyany C, Trujillo M, Poole LB, Denicola A (2014) Nitration transforms a sensitive peroxiredoxin 2 into a more active and robust peroxidase. J Biol Chem 289:15536–15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Randall L, Manta B, Nelson KJ, Santos J, Poole LB, Denicola A (2016) Structural changes upon peroxynitrite‐mediated nitration of peroxiredoxin 2; nitrated Prx2 resembles its disulfide‐oxidized form. Arch Biochem Biophys 590:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peskin AV, Dickerhof N, Poynton RA, Paton LN, Pace PE, Hampton MB, Winterbourn CC (2013) Hyperoxidation of peroxiredoxins 2 and 3: rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J Biol Chem 288:14170–14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bolduc JA, Nelson KJ, Haynes AC, Lee J, Reisz JA, Graff AH, Clodfelter JE, Parsonage D, Poole LB, Furdui CM, Lowther WT (2018) Novel hyperoxidation resistance motifs in 2‐Cys peroxiredoxins. J Biol Chem 293:11901–11912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trindade DF, Cerchiaro G, Augusto O (2006) A role for peroxymonocarbonate in the stimulation of biothiol peroxidation by the bicarbonate/carbon dioxide pair. Chem Res Toxicol 19:1475–1482. [DOI] [PubMed] [Google Scholar]
- 28. Ferrer‐Sueta G, Manta B, Botti H, Radi R, Trujillo M, Denicola A (2011) Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem Res Toxicol 24:434–450. [DOI] [PubMed] [Google Scholar]
- 29. Trujillo M, Radi R (2002) Peroxynitrite reaction with the reduced and the oxidized forms of lipoic acid: new insights into the reaction of peroxynitrite with thiols. Arch Biochem Biophys 397:91–98. [DOI] [PubMed] [Google Scholar]
- 30. Lee W, Choi KS, Riddell J, Ip C, Ghosh D, Park JH, Park YM (2007) Human peroxiredoxin 1 and 2 are not duplicate proteins: the unique presence of CYS83 in Prx1 underscores the structural and functional differences between Prx1 and Prx2. J Biol Chem 282:22011–22022. [DOI] [PubMed] [Google Scholar]
- 31. Chae HZ, Oubrahim H, Park JW, Rhee SG, Chock PB (2012) Protein glutathionylation in the regulation of peroxiredoxins: a family of thiol‐specific peroxidases that function as antioxidants, molecular chaperones, and signal modulators. Antiox Redox Signal 16:506–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Randall LM, Ferrer‐Sueta G, Denicola A (2013) Peroxiredoxins as preferential targets in H2O2‐induced signaling. Methods Enzymol 527:41–63. [DOI] [PubMed] [Google Scholar]
- 33. Benfeitas R, Selvaggio G, Antunes F, Coelho PM, Salvador A (2014) Hydrogen peroxide metabolism and sensing in human erythrocytes: a validated kinetic model and reappraisal of the role of peroxiredoxin II. Free Rad Biol Med 74:35–49. [DOI] [PubMed] [Google Scholar]
- 34. Jarvis RM, Hughes SM, Ledgerwood EC (2012) Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Rad Biol Med 53:1522–1530. [DOI] [PubMed] [Google Scholar]
- 35. Sobotta MC, Liou W, Stocker S, Talwar D, Oehler M, Ruppert T, Scharf AN, Dick TP (2015) Peroxiredoxin‐2 and STAT3 form a redox relay for H2O2 signaling. Nat Chem Biol 11:64–70. [DOI] [PubMed] [Google Scholar]
- 36. Romero N, Radi R, Linares E, Augusto O, Detweiler CD, Mason RP, Denicola A (2003) Reaction of human hemoglobin with peroxynitrite. Isomerization to nitrate and secondary formation of protein radicals. J Biol Chem 278:44049–44057. [DOI] [PubMed] [Google Scholar]
- 37. Cao Z, Bhella D, Lindsay JG (2007) Reconstitution of the mitochondrial PrxIII antioxidant defence pathway: general properties and factors affecting PrxIII activity and oligomeric state. J Mol Biol 372:1022–1033. [DOI] [PubMed] [Google Scholar]
- 38. Haynes AC, Qian J, Reisz JA, Furdui CM, Lowther WT (2013) Molecular basis for the resistance of human mitochondrial 2‐Cys peroxiredoxin 3 to hyperoxidation. J Biol Chem 288:29714–29723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Santos J, Marino‐Buslje C, Kleinman C, Ermacora MR, Delfino JM (2007) Consolidation of the thioredoxin fold by peptide recognition: interaction between E. coli thioredoxin fragments 1‐93 and 94‐108. Biochemistry 46:5148–5159. [DOI] [PubMed] [Google Scholar]
- 40. Mulrooney SB (1997) Application of a single‐plasmid vector for mutagenesis and high‐level expression of thioredoxin reductase and its use to examine flavin cofactor incorporation. Prot Express Purif 9:372–378. [DOI] [PubMed] [Google Scholar]
- 41. Nelson DP, Kiesow LA (1972) Enthalpy of decomposition of hydrogen peroxide by catalase at 25 degrees C (with molar extinction coefficients of H 2 O 2 solutions in the UV). Analyt Biochem 49:474–478. [DOI] [PubMed] [Google Scholar]
- 42. Radi R, Beckman JS, Bush KM, Freeman BA (1991) Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem 266:4244–4250. [PubMed] [Google Scholar]
- 43. Wang PF, Veine DM, Ahn SH, Williams CH Jr (1996) A stable mixed disulfide between thioredoxin reductase and its substrate, thioredoxin: preparation and characterization. Biochemistry 35:4812–4819. [DOI] [PubMed] [Google Scholar]
- 44. Trujillo M, Ferrer‐Sueta G, Radi R (2008) Kinetic studies on peroxynitrite reduction by peroxiredoxins. Methods Enzymol 441:173–196. [DOI] [PubMed] [Google Scholar]
- 45. Hayashi Y, Yamazaki I (1979) The oxidation‐reduction potentials of compound I/compound II and compound II/ferric couples of horseradish peroxidases A2 and C. J Biol Chem 254:9101–9106. [PubMed] [Google Scholar]
- 46. Staudacher V, Trujillo M, Diederichs T, Dick TP, Radi R, Morgan B, Deponte M (2018) Redox‐sensitive GFP fusions for monitoring the catalytic mechanism and inactivation of peroxiredoxins in living cells. Redox Biol 14:549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grassetti DR, Murray JF Jr (1967) Determination of sulfhydryl groups with 2,2′‐ or 4,4′‐dithiodipyridine. Arch Biochem Biophys 119:41–49. [DOI] [PubMed] [Google Scholar]
- 48. Chae HZ, Chung SJ, Rhee SG (1994) Thioredoxin‐dependent peroxide reductase from yeast. J Biol Chem 269:27670–27678. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Determination of second‐order rate constant for the reaction of HRP with peroxynitrite at pH 7.4 and 25 °C.
Figure S2. Time course and data fitting of PRDX1 oxidation by ONOOH. PRDX1 (0.25 μM) was reacted with 14 μM ONOOH (green line). The time course of the fluorescence (t > 2 ms) was fitted to a triple exponential function (black line). The residual shows the goodness of the fit.