

Abstract
Lactate monooxygenase (LMO) catalyzes the FMN‐dependent “coupled” oxidation of lactate and O2 to acetate, carbon dioxide, and water, involving pyruvate and hydrogen peroxide as enzyme‐bound intermediates. Other α‐hydroxy acid oxidase family members follow an “uncoupled pathway,” wherein the α‐keto acid product quickly dissociates before the reduced flavin reacts with oxygen. Here, we report the structures of Mycobacterium smegmatis wild‐type LMO and a wild‐type‐like C203A variant at 2.1 Å and 1.7 Å resolution, respectively. The overall LMO fold and active site organization, including a bound sulfate mimicking substrate, resemble those of other α‐hydroxy acid oxidases. Based on structural similarity, LMO is similarly distant from lactate oxidase, glycolate oxidase, mandelate dehydrogenase, and flavocytochrome b2 and is the first representative enzyme of its type. Comparisons with other α‐hydroxy acid oxidases reveal that LMO has a longer and more compact folded active site loop (Loop 4), which is known in related flavoenzymes to undergo order/disorder transitions to allow substrate/product binding and release. We propose that LMO's Loop 4 has an enhanced stability that is responsible for the slow product release requisite for the coupled pathway. We also note electrostatic features of the LMO active site that promote substrate binding. Whereas the physiological role of LMO remains unknown, we document what can currently be assessed of LMO's distribution in nature, including its unexpected occurrence, presumably through horizontal gene transfer, in halophilic archaea and in a limited group of fungi of the genus Beauveria.
Broad statement of impact
This first crystal structure of the FMN‐dependent α‐hydroxy acid oxidase family member lactate monooxygenase (LMO) reveals it has a uniquely large active site lid that we hypothesize is stable enough to explain the slow dissociation of pyruvate that leads to its “coupled” oxidation of lactate and O2 to produce acetate, carbon dioxide, and water. Also, the relatively widespread distribution of putative LMOs supports their importance and provides new motivation for their further study.
Keywords: flavoenzyme, lactate metabolism, enzyme mechanism, protein crystallography, electrostatics, horizontal gene transfer
Short abstract
Abbreviations and symbols
- LMO
lactate monooxygenase
- LOX
lactate oxidase
- GOX
glycolate oxidase
- MDH
mandelate dehydrogenase
- FCB2
flavocytochrome b2
- HGT
horizontal gene transfer
1. Introduction
An enzyme referred to as “lactate oxidase” was first discovered in Mycobacterium phlei in 1947,1 and it was proposed that this yellow protein was a flavoenzyme that was able to convert lactate and molecular oxygen to acetate and carbon dioxide via a pyruvate intermediate. Subsequent characterizations2 confirmed the enzyme to be FMN dependent3, 4 and to produce pyruvate as an enzyme‐bound intermediate.5 Although the name “lactate oxidase” persisted into the early 1990s, this acetate producing enzyme is now referred to as “lactate monooxygenase” (LMO), and “lactate oxidase” refers to a related flavoenzyme that converts lactate and molecular oxygen to pyruvate and hydrogen peroxide.6
LMO belongs to the α‐hydroxy acid oxidase family of flavoenzymes including lactate oxidase (LOX), glycolate oxidase (GOX), flavocytochrome b2 (FCB2), and mandelate dehydrogenase (MDH). The α‐hydroxy acid oxidases have a common reductive half reaction in which the α‐hydroxy acid is oxidized to an α‐keto‐acid and the enzyme‐bound flavin is reduced. After much debate, the consensus is that this reaction involves a hydride transfer.7, 8, 9 The oxidative half reaction (in which the flavin is reoxidized) can involve a reaction with molecular oxygen to produce hydrogen peroxide (e.g., for LMO, LOX, and GOX) or an electron transfer to a distant electron acceptor (e.g., for FCB2 and MDH). Among the family members reacting with oxygen, LMO is unique in that it proceeds along a “coupled pathway” [Fig. 1(A)], wherein rather than releasing the α‐keto‐acid as a product (in this case pyruvate), it remains in the active site, and after reaction with hydrogen peroxide undergoes an oxidative decarboxylation to produce acetate, carbon dioxide, and water.6 In contrast, LOX and GOX proceed through an “uncoupled pathway” wherein the α‐keto‐acid product dissociates from the active site before the reduced flavin reacts with oxygen. For example, in LOX, lactate and oxygen are converted to pyruvate and hydrogen peroxide [Fig. 1(A)].
Figure 1.
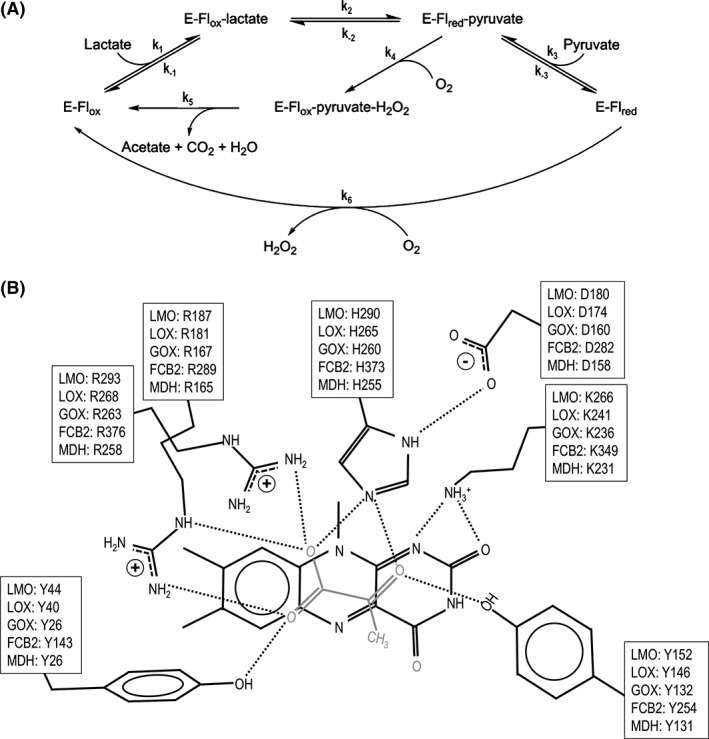
The LMO/LOX catalytic cycles and conserved active site of α‐hydroxy acid oxidases. (A) The inner loop represents the “coupled pathway” observed for LMO where pyruvate is an intermediate and acetate and CO2 are the final products. The outer loop represents the “uncoupled pathway” observed for LOX where pyruvate and H2O2 are produced by two uncoupled half‐reactions. E, Flox, and Flred represent the enzyme, oxidized flavin, and reduced flavin, respectively. Rate constants defined for each step are indicated. Adapted from Ref. 27. (B) Seven conserved residues in the active site of LMO, LOX, GOX, FCB2, and MDH are shown in front of the si‐face of the flavin along with pyruvate (grey) as it is seen bound to LOX in PDB entry 2E77. Typical hydrogen bonds (dotted lines) are also shown. The residue numbers are based on M. smegmatis LMO, and PDB entries 2E77 (LOX), 2RDU (GOX), 1KBI (FCB2), and 1P4C (MDH).
Working models for the active site of LMO have been based on the structures of LOX,6, 10 GOX,11, 12 FCB2,13 and MDH,14 all of which have a core TIM‐barrel of 8 α‐helices and 8 β‐strands, bind FMN at the C‐terminal end of the β‐barrel, and include an often disordered loop (Loop 4) that covers the active site. Sequence and structural comparisons along with chemical modification15, 16, 17, 18 and site‐directed mutagenesis studies19, 20, 21 have identified seven conserved key active site residues in the family members [Fig. 1(B)], implying that their modes of ligand binding and catalytic mechanisms are very similar.
Given the high active site similarity between LMO and the other α‐hydroxy acid oxidases, it was thought that its unique kinetics must somehow stem from subtle changes associated with differences in “second shell” residues near the LMO active site that could influence the flavin chemistry and substrate/product binding affinities. This made LMO an intriguing model for understanding and probing the fine‐tuning of flavoenzymes, and Vince Massey (to whom this article is dedicated) and his research group carried out a series of studies in the 1990s to discover the origin of its unique kinetic properties.6, 19, 20, 21, 22, 23 Some of these site‐directed mutagenesis studies were able to uncouple LMO, effectively converting its activity to that of LOX, but did so by greatly diminishing O2 reactivity and/or changing conserved active site residues21, 22, 23—neither of which can reflect what differentiates LMO from the other α‐hydroxy acid oxidases in nature. Indeed, LMO reacts readily with O2, with the free enzyme having a rate constant of ~9000 s−1, and the pyruvate complex being ~200‐fold faster, at 180,000 s−1.5 While slowing the O2 reactivity may uncouple the enzyme, this also has dramatic negative ramifications for catalysis and does not provide an understanding of the features differentiating LMO from LOX.
At that time, Vince Massey sought (in collaboration with our group) to gain insight through solving the LMO crystal structure, but while crystals were grown, no structure was solved.24 Since then, with no structural information to guide further studies, our knowledge of LMO has been stagnant. Some insight has been provided by two recent structure‐guided studies of LOX. Based on the authors’ observation that “product off‐rates appear to be dictated by partitioning of residues … from an active‐site lid loop into bulk solvent,”25 they mutated two “second shell” residues in the Loop 4 active site lid26, 27 and shifted its kinetic properties to be slightly more like LMO.
Here, we present the first direct structural information about any LMO, reporting both a 2.3 Å structure of M. smegmatis wild‐type LMO (WT) and a 1.7 Å structure of a variant in which Cys203 is mutated to Ala (C203A). Chemical modification studies had implicated Cys203 as an active site residue,28 but kinetic characterization showed this variant to “behave similarly to the wild‐type enzyme in all properties examined,”29 meaning it can be considered a reasonable surrogate for wild‐type LMO. These structures reveal that LMO has the expected structural similarity to other family members with good positional conservation of the seven conserved active site residues. In addition to providing a solid structural foundation for further studies of LMO, we observe structural features that lead us to propose that LMO's unique kinetics are not a consequence of specific residue changes that influence the flavin chemistry, but are due to a difference in its dynamic Loop 4 which we suggest folds more stably than those of other family members and slows pyruvate release. The distribution of putative LMOs in nature provides intriguing evidence that LMO can at least in some cases bring a selective advantage to the organisms in which it is found.
2. Results and Discussion
2.1. Structure determination and crystal packing
Crystals of wild‐type LMO were first grown and data, including the WT data set used here, were collected in the 1990s.24 At that time and still today, the most similar structurally known protein had only ~35% sequence identity, and no success was achieved in determining the structure using either molecular replacement or SeMet MAD phasing (unpublished). The advance allowing us to solve the structure now was the development of MR‐Rosetta30 as a more powerful molecular replacement approach. MR‐Rosetta was able to place four, well‐ordered chains (A, B, C, and D), but this left significant weak but unmodeled density in a large swath of the crystal (Fig. 2). Two additional, weaker chains (E and F) were placed with Phaser (Materials and Methods), resulting in six total chains in the asymmetric unit. While the two additional chains have much weaker density, both are reliably placed based on clear, unbiased density consistent with the expected positions of their active site FMN and bound sulfate (Supporting Information, Fig. S1).
Figure 2.
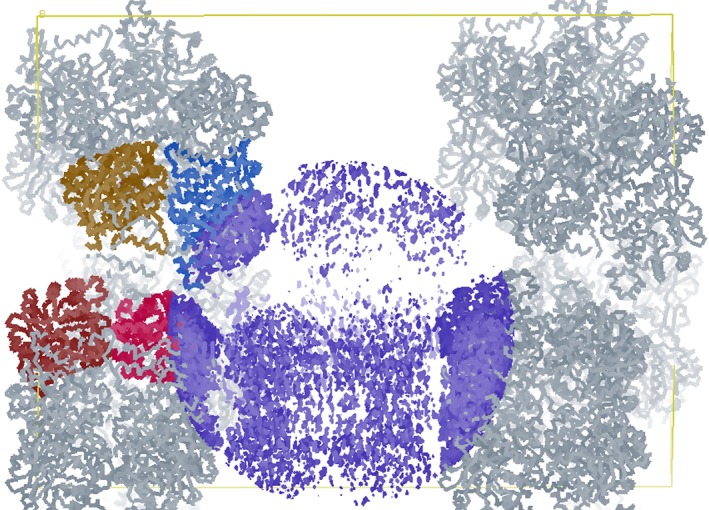
A swath of the LMO crystal has only very weak electron density. Shown are Cα‐traces of the four well‐ordered chains (mustard, blue, red, and burgundy) and their symmetry mates (grey) placed in the C203A unit cell (yellow box with the fourfold c‐axis lying horizontally). Shown also is a sphere of the 2F o–F c electron density map (blue; contoured at 1.3 ρ rms) centered in the large swath of the crystal in which no model was included when calculating this map. The chain E and F models (at 80% occupancy) later placed in the weak density have average B‐factors of ~110 Å2 and ~120 Å2, respectively, while chains A–D have average B‐factors of ~30, ~45, ~45, and ~65 Å2, respectively.
While we do not have a full understanding of this unusual crystal packing, we note that a fascinatingly similar phenomenon was reported for stefin B crystals.31 In that example, the lattice of the crystal was composed of five tetramer layers, four of which were well‐ordered and one of which had two alternative positions that could both be modeled. Even though we have only been able to model a single position for chains E and F, as the electron density in that part of the crystal is so weak, it could be that there are other alternate positions for the chains that are not well‐enough defined to model.
The refined model of LMO C203A at 1.70 Å resolution contains six chains in the asymmetric unit with R and R free values of 16.9% and 19.4%, respectively (Table 1). For five of the chains (A, B, C, D, and E), all 394 residues are sufficiently ordered to be modeled; for the sixth chain (F), the N‐terminal methionine (residue 0 according to canonical LMO numbering32) is not well‐ordered and only residues 1–393 are modeled. The refined model for WT at 2.30 Å resolution contains six chains in the asymmetric unit with R and R free values of 21.8% and 27.2%, respectively (Table 1). As in C203A, all 394 residues in chains A, B, C, and E are modeled and residues 1–393 are modeled in chain F. In WT, chain D is much less ordered and only residues 0–190, 203–216, and 261–393 are modeled.
Table 1.
Data Collection and Refinement Statistics for LMO Structures*
| WT | C203A | |
|---|---|---|
| Data statistics | ||
| Space group | P4212 | P4212 |
| Unit cell a, b, c axes (Å) | 148.4, 148.4, 272.6 | 149.6, 149.6, 274.3 |
| Resolution (Å) | 77.49–2.30 (2.38–2.30) | 30.90–1.70 (1.74–1.70) |
| Unique reflections | 123105 | 33380 (24764) |
| Multiplicity | 3.1 | 26.7 (27.5) |
| Average I/σ | 10.4 (2.6) | 15.5 (0.6) |
| R meas (%) | 10.2 (42) | 18.2 (690) |
| Completeness (%) | 91.0 (79.6) | 99.9 (100) |
| CC1/2 | n.d.† | 99.9 (22.7) |
| Refinement statistics | ||
| Amino acid residues | 2306 | 2363 |
| Solvent atoms | 1252 | 1464 |
| Non‐H atoms | 19112 | 20104 |
| RMS bonds (Å) | 0.015 | 0.014 |
| RMS angles (°) | 1.6 | 1.4 |
| ϕ, ψ favored (%) ‡ | 93.1 | 96.4 |
| ϕ, ψ outliers (%) ‡ | 1.0 | 0.6 |
| <B protein > (Å2) | 68 | 70 |
| <B solvent > (Å2) | 45 | 52 |
| R work (%) | 21.8 | 16.9 |
| R free (%) | 27.2 | 19.4 |
| PDB code | 6DVI | 6DVH |
Unless noted, all descriptions and analyses are based on the higher resolution structure of C203A. Given the stronger electron density of chains A–D, they are the only chains we consider reliable for defining details of the structures, so only they are described from here forward. The respectable R‐factors of this structure imply that information for these well‐ordered chains is not made less reliable by the presence of the less ordered chains. As chains A–D closely overlay with each other (Cα rmsd <0.4 Å), descriptions we provide based on chain A can be considered as true for all chains unless otherwise noted.
2.2. Structure of LMO
As expected for this family, the LMO monomer is organized into a β8/α8 TIM‐barrel fold [Fig. 3(A)]. Each chain contains an FMN with sulfate ion bound above the si face of the isoalloxazine ring where substrate would bind [Fig. 4(A)]. LMO is reported to assemble into an octamer in solution.32, 33 In the crystals, each of the LMO chains in the asymmetric unit is part of a tetramer created by the crystallographic fourfold symmetry. Pairs of the unique tetramers further assemble into an octamer, with octamers formed from chains A and C, chains B and D, and chains E and F [Fig. 3(B)]. In WT, the modeled chain D tetramer is rotated ~22° with respect to the chain B tetramer, generating a surprising single instance of an alternate octamer interface that has looser packing (Supporting Information, Fig. S2). We cannot rule out that this alternate octamer has physiological relevance, but given that it has substantially looser packing and is only seen in a minority of the crystallized octamers, we posit that it is an artifact of the non‐physiological crystallization conditions (at pH ~4.6 and high salt) that weakens the tetramer–tetramer interaction. As many of the related α‐hydroxy oxidases function as tetramers,34, 35, 36 it is to be expected that the tetramer is the stronger assembly, and the octamer would be more easily disrupted.
Figure 3.
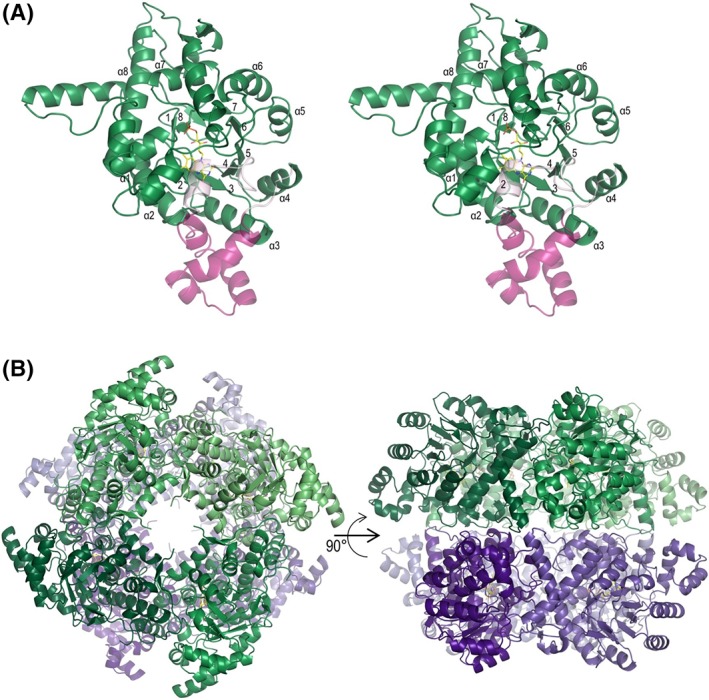
Tertiary and quaternary structures of LMO. (A) Stereoview of one subunit of LMO (C203A chain A) showing FMN (yellow carbons) and viewing into the barrel. The TIM‐barrel α‐helices (α1–α8) and β‐strands (1–8) are labeled. The segment connecting β4 and α4 is semitransparent with the αD conserved portion in light pink and the Loop 4 variable portion in dark pink. (B) Shown are views of the C203A LMO octamer looking down the crystallographic fourfold axis (left) and perpendicular to it (right). The two tetramers forming the octamer are based on chain A (shades of green) and chain C (shades of purple). See Supporting Information, Figure S2 for an alternate octamer observed for chains B and D in the wild‐type LMO crystals.
Figure 4.
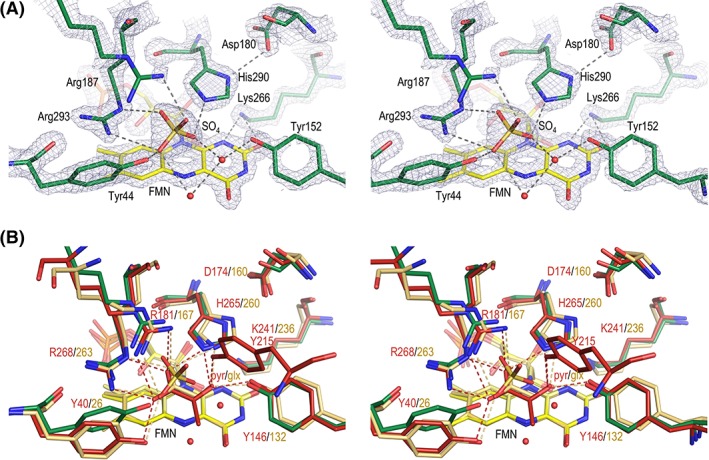
Active site of LMO. (A) Electron density map quality and active site of LMO. Stereoview of the LMO active site residues (green carbons), FMN (yellow carbons), sulfate, and ordered waters (red spheres) with 2F o–F c electron density (grey; contoured at 3.5ρ rms). Select hydrogen bonds (dashed line) are shown. Arg187 has an alternate conformation, but only the A conformation (50%) interacting with the sulfate is shown. The view roughly matches that shown in Figure 1(B) schematic. (B) Active site overlay of LMO with LOX and GOX. Stereoview of the LMO active site (colored as in Fig. 4) compared with LOX with bound pyruvate (red carbons; PDB entry 2E77) and GOX with bound glyoxylate (tan carbons; PDB entry 2RDU). Hydrogen bonds (dashed line) between active site side chains and the ligand in LOX (red) and GOX (beige) are shown.
In the active site of LMO, very clear electron density defines the positions of all seven residues conserved among the α‐hydroxy acid oxidases: Tyr44, Tyr152, Asp180, Arg187, Lys266, His290, and Arg293 [Fig. 4(A)]. Tyr44, Arg293, Arg187, and His290 hydrogen bond with the ordered sulfate. Asp180 hydrogen bonds with His290‐Nδ1 and is well‐positioned to stabilize the presumed active site base, His290, when it becomes protonated after abstracting a proton from the substrate.2, 6 Lys266 hydrogen bonds with FMN N1‐ and O2‐atoms and can stabilize the anionic semiquinone.2, 6, 19, 37 Two additional ordered waters form hydrogen bonds with the sulfate and Tyr152.
2.3. Relationships to other structurally known proteins
A structural similarity search performed using the DALI server38 shows that LMO is roughly equally similar to other members of the α‐hydroxy acid oxidase family including GOX (rmsd ~1.4–1.9 Å, 33%–36% sequence identity), MDH (rmsd 2.0 Å, 32% sequence identity), LOX (rmsd ~1.6–1.8 Å, 31%–33% sequence identity), and FCB2 (rmsd 1.7 Å, 31% sequence identity). Since LMO is as distant from each of the other α‐hydroxy acid oxidases as they are from each other, we conclude it not evolutionarily closer to LOX. Furthermore, this indicates that LMO truly is a distinct branch of the family and that even with all the structures that are known and have been solved from structural genomics projects, the structure we report here is still the first structure representing the LMO clade.
For carrying out a structure‐based sequence alignment (Fig. 5), using the DALI results, we selected a representative structure for each enzyme type with the greatest amount of its chain ordered. These were Aerococcus viridans LOX (2E77),10 human GOX (2RDU),12 Saccharomyces cerevisiae FCB2 (1KBI),13 and Pseudomonas putida MDH (1P4C).14 The resulting alignment shows good conservation of structural elements and active site residues across these enzymes. As we discuss further below, a notable feature is that there is substantial variation in both the length and structure of what has been called Loop 4, a segment between strand β4 and helix α4 that has been found to be disordered in many structures.
Figure 5.
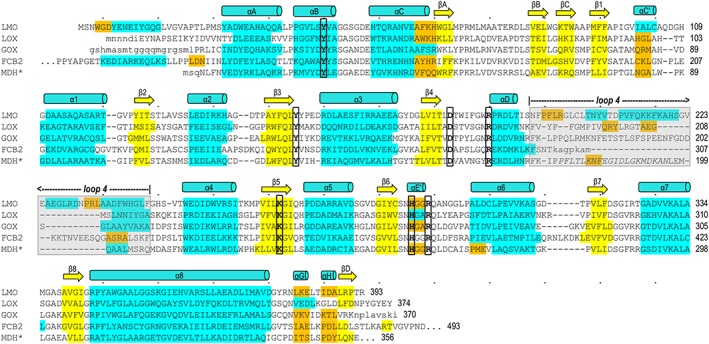
Structure‐based sequence alignment of LMO with representative α‐hydroxy acid oxidase family members. LMO (WT sequence, structure from C203A chain A) is shown first followed by LOX (PDB entry 2E77 chain B), GOX (2RDU chain A), FCB2 (1KBI chain A), and MDH* (MDH‐GOX chimera with chimeric region from spinach GOX italicized; 1P4C chain A). Conserved active site residues (bold, black outline) and residues in α‐helices (cyan), 310‐helices (orange), and β‐strands (yellow) are indicated. Secondary structural elements conserved among all family members are named in a manner consistent with naming conventions for other family members.6, 11, 39 Loop 4 (gray shading) is an active site lid that has been seen to undergo order/disorder transitions in other α‐hydroxy acid oxidases; when ordered, it covers the active site channel but is not structurally conserved between family members. Dots above the LMO sequence indicate every tenth character and a number is given for the residue at the end of each line. Residues in lower case letters are disordered in the respective structure.
An overlay of LMO with sulfate bound onto LOX and GOX structures having pyruvate and oxalate, respectively, bound in the active sites shows that the FMN and side chain positions are quite similar [Fig. 4(B)]. Only one side chain, that of Tyr44 in LMO, has notable variation, being shifted ~2.5 Å compared to LOX Tyr40 to hydrogen bond with the bound sulfate. In addition to the seven key, conserved active site residues, LOX has an additional tyrosine (Tyr215) that contributes to the active site [Fig. 4(B)] and forms a hydrogen bond with the pyruvate product. This tyrosine is part of the variable Loop 4, and neither LMO nor GOX have a functionally or structurally equivalent residue.
Each of these nearly identical active sites is well set up to carry out the same chemistry: the FMN‐dependent oxidation of an α‐hydroxy acid substrate. In all structures, the putative catalytic base His (His290 in LMO, His265 in LOX, and His260 in GOX) is well‐oriented to abstract a proton from substrate, and an Asp (Asp180 in LMO, Asp174 in LOX, and Asp160 in GOX) is nearby to stabilize the protonated His. Likewise, a Lys (Lys266 in LMO, Lys241 in LOX, and Lys236 in GOX) is positioned to stabilize negative charge at the N1/O2 locus of the flavin during catalysis. The sulfate we see in the LMO active site appears to be a reasonable substrate mimic, as the hydrogen bonds with sulfate and ordered waters observed in LMO match well with interactions with ligand observed in LOX and GOX. Specifically, the carboxylate oxygens of the ligands bound in LOX and GOX overlay well with two of the oxygens of sulfate in LMO and form equivalent hydrogen bonds to those sulfate makes with Arg187 and Arg293 in LMO. Similarly, hydrogen bonds between O2 of pyruvate with residues equivalent to His290 and Tyr152 mimic the hydrogen bonds made with a sulfate oxygen and ordered water, respectively, in LMO. Given its high similarity with related structures, we do not see this structure as a source of further insight into the reaction chemistry, and so we do not comment further on that topic.
2.4. Major differences occur in Loop 4 that covers the active site
It has been noted that the α‐hydroxy acid oxidases have substantial sequence and structural divergence in a segment between β4 and α4, referred to as Loop 4 (Fig. 5).12, 39 In all structurally characterized family members including LMO, Loop 4 functions as a lid or flap, covering the active site pocket when folded (i.e., closed) and exposing the active site opening when unfolded (i.e., open) (Fig. 6 and Supporting Information, Fig. S3). Thus, movement of this loop is necessary for substrate binding and product release.10, 12, 25, 26, 27, 40 For LMO, LOX, and GOX, the opening of the loop not only makes the active site accessible, but also exposes a highly positive surface patch surrounding the active site opening that can promote catalysis by actively drawing the negatively charged α‐hydroxy acid substrate (e.g., lactate) into the active site [Fig. 6(A) and Supporting Information, Fig. S3]. Such an electrostatic guidance of substrate binding has been described for GOX,41 and one of the best‐studied examples of this phenomenon is copper superoxide dismutase.42 Interestingly, when Loop 4 is closed and blocks substrate binding, it also alters the electrostatic potential surface in this part of the protein, so substrate will not be attracted [Fig. 6(B) and Supporting Information, Fig. S3].
Figure 6.
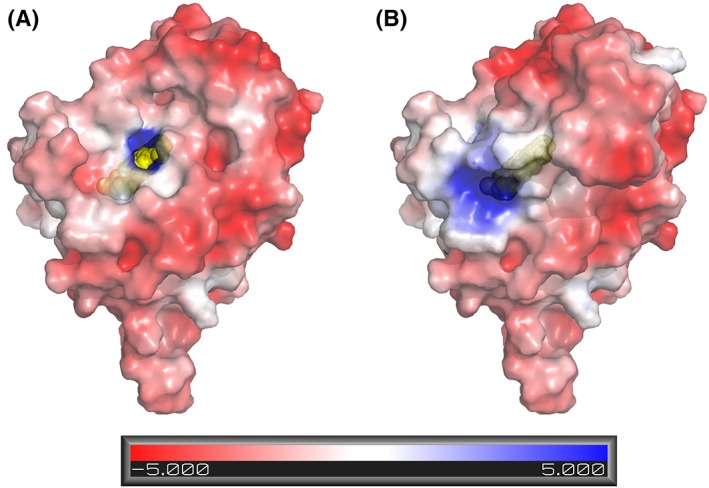
The open LMO active site electrostatically attracts substrate and Loop 4 folding blocks active site access. (A) Molecular surface and electrostatic potential of LMO with Loop 4 removed. FMN in the active site (yellow spheres) is visible through an opening. The surface electrostatic potential (semitransparent) is scaled from red to blue as shown. (B) Same as panel A, but with Loop 4 present.
For the other α‐hydroxy acid oxidases, partial or complete disordering of Loop 4 is seen in structures with no active site ligand and also in many structures containing a bound product or substrate/product analog.10, 11, 40 This implies that Loop 4 readily unfolds, and that it undergoes folding and unfolding transitions associated with substrate binding and product release. With the structure of LMO in hand, a notable feature is that, even though only a sulfate is bound at the active site, the Loop 4 of LMO is fully ordered and structured similarly in all chains, including the two less ordered chains (E and F). Also, compared to LOX and GOX, the LMO Loop 4 is both longer (Fig. 5) and folds quite differently [Fig. 7(A)]. In M. smegmatis LMO, Loop 4 is 49 residues long and adopts a compact association of four short α‐helices to create a small hydrophobic core [Fig. 7(B)]. A DALI search38 of the folded Loop 4 segment alone revealed no proteins that contain closely similar folds.
Figure 7.
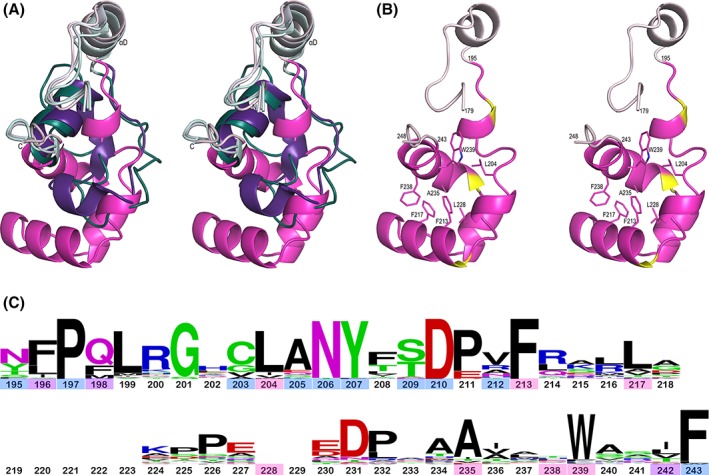
Comparison of Loop 4 in LMO, LOX, and GOX. (A) Stereoview of the chain between β4 and α4 of the LMO monomer (pink tones) overlaid with the LOX–pyruvate complex (purple tones; PDB entry 2E77) and the GOX–glyoxylate complex (teal tones; 2RDU). The N‐ and C‐termini are marked and segments between β4 and α4 are highlighted with the more spatially conserved parts at the beginning and end, including αD, in light colors and the varying Loop 4 portion in dark colors. The LOX and GOX Loop 4 segments follow similar paths to each other. (B) Stereoview of the LMO chain between β4 and α4 (orientation and ribbon coloring as in A, but with three helix‐capping Pro residues in yellow) with seven central core side chains shown as sticks and labeled. (C) WebLogo plot based on Loop 4 sequences from 48 representative LMOs using M. smegmatis LMO numbering. Residue numbers are colored based on the side chain role in packing: central core of Loop 4 (pink), interface between Loop 4 and the rest of LMO (blue), or both core and interface (purple). At 49 residues long, M. smegmatis LMO has the longest Loop 4 with the most common length being 42 residues; the 7 residues not present in most LMOs are blank spots in the WebLogo. The conservation of Gly201 which adopts a glycine‐preferred ϕ,ψ‐conformation and Pro197, Asp210/Pro211, and Asn231/Pro232 which form helix N‐capping motifs imply a strong conservation of these structural elements across all putative LMOs. The very conserved Asn206 and Tyr207 pack at the interface with the rest of LMO, with the Asn206 side chain hydrogen bonding to backbone atoms 135‐N and 129‐O, and Tyr207 packing between Phe163 and Loop 4 core residues.
Given that Loop 4 must unfold for product release, we asked if the Loop 4 subdomain of LMO might be more stably folded than those of LOX and GOX such that Loop 4 dynamics could dictate the speed of pyruvate release and LMO's kinetics. First, we asked if differences in loop lengths in the representative structures were characteristic, and found that LMOs do consistently have longer Loop 4s than LOXs and GOXs (Table 2). Then, we sought to assess if the longer LMO Loop 4 might also be more stably folded. There is a large body of literature indicating that buried surface area is correlated to stability in proteins,43, 44, 45, 46 so we calculated for each representative enzyme how much surface area is buried when going from an unfolded Loop 4 to the fully folded Loop 4 active site lid (Table 2). We found that folding of the LMO Loop 4 buries ~1500 and ~900 Å2 more surface than those of LOX and GOX, respectively (4750 vs. 3290 Å2 and 3870 Å2). This difference is due to the surface buried internal to Loop 4, consistent with the loop being more stably folded as a co‐dependent subdomain. Forming a central hydrophobic core of Loop 4 in LMO are Leu204, Phe213, Phe217, Leu228, Ala235, Phe238, and Trp239 [Fig. 7(B)]. Many of these core residues, along with residues forming the interface between Loop 4 and the rest of the protein, are relatively well conserved across representative LMOs [Fig. 7(C)], implying that the Loop 4 folding pattern and interaction with the rest of LMO are also well‐conserved.
Table 2.
Surface Areas (SA) Related to the Folding of Loop 4*
| LMO | LOX | GOX | |
|---|---|---|---|
| PDB entry analyzed | 6DVH | 2E77 | 2RDU |
| SA unfolded Loop 4 (Å2) | 6240 | 3630 | 4710 |
| SA buried internal to Loop 4 (Å2) | 2570 | 1140 | 1660 |
| SA Loop 4 interface with protein (Å2) | 1160 | 1190 | 1190 |
| SA protein interface with Loop 4 (Å2) | 1020 | 960 | 1020 |
| Total SA buried upon Loop 4 folding (Å2) † | 4750 | 3290 | 3870 |
| Range of Loop 4 lengths (residues) | 39–49 (49) | 27–29 (29) | 17–38 (38) |
See Materials and Methods for details of calculations.
Sum of the three previous entries.
Number in parenthesis is the loop length in this representative structure.
Taking these observations together, we hypothesize that the LMO Loop 4 has a greater propensity to be closed, especially when substrate or product is bound, and is the key factor that slows pyruvate release and leads LMO to follow the coupled reaction pathway.
2.5. Other studies involving Loop 4 residues support this hypothesis
An older pair of observations supporting this hypothesis is that treatment with fluoro‐dinitrobenzene (FDNB) blocked LMO activity through the modification of Cys104 and Cys203 and that these residues could be protected by inhibitor binding.28, 32 With the structure of LMO in hand, we now see that neither cysteine directly contributes to substrate binding or catalysis. Cys104 lines the flavin binding pocket where it is mostly buried behind the C7‐methyl of the flavin. Cys203 is in Loop 4 and is buried at the interface between Loop 4 and the rest of the protein. Importantly, both cysteines are positioned where Loop 4 folding and closure should protect them from modification, and also where a large modification (such as FDNB produces) would hinder enzyme activity by blocking proper folding of Loop 4. That inhibitor binding protected the residues from modification supports our hypothesis that Loop 4 is strongly closed in the presence of an active site ligand and could hinder product release. Also, the facile modification of the two Cys residues in the absence of an active site ligand implies the loop is dynamic enough in the absence of ligand to allow for rapid binding of substrate.
In newer work, as mentioned in the Introduction, there are two recent site‐directed mutagenesis studies that probed interactions involving Loop 4 residues in LOX. In both cases, the results provide experimental support for our hypothesis, because they directly link Loop 4 thermodynamics with the pyruvate dissociation rate. In LOX, Tyr215 from Loop 4 contributes to the active site (Figure 4B) and forms an additional hydrogen bond with the pyruvate product. In a Tyr215Phe mutant (Y215F), pyruvate release was decreased by approximately sevenfold.26 Similarly, mutating LOX Tyr191, another conserved Loop 4 residue, to Phe, Leu, or Ala slowed the pyruvate release step by ~5‐, 19‐, and 19‐fold, respectively.27 In both cases, it was concluded that the mutations shifted the Loop 4 dynamics to favor a closed conformation and that this hindered pyruvate release.26, 27 While these mutations slowed pyruvate release [k 3 in Fig. 1(A)], it did not become rate limiting, and no mutants had measurable flux through a coupled pathway. This is not surprising, because none of them came close to matching the ~3000‐fold slower k off observed in LMO.5
2.6. LMOs exist beyond bacteria
As far as we could find in the literature, LMO has exclusively been studied from mycobacteria since it was first identified in 1947.1, 2, 19, 33 However, the physiological role of LMO remains unknown. With an extensive number of genomes now having been sequenced, a database search can provide a more complete picture of the range of organisms that appear to have LMO (Fig. 8). Selecting all proteins with ~50% or higher sequence identity with M. smegmatis LMO yields a phylogenetic tree that groups all these putative LMOs more closely with each other than with LOX, GOX, FCB2, and MDH, confirming that with this cutoff the putative LMOs are at least much more similar to each other than to any of the other known α‐hydroxy acid oxidase enzymes.
Figure 8.
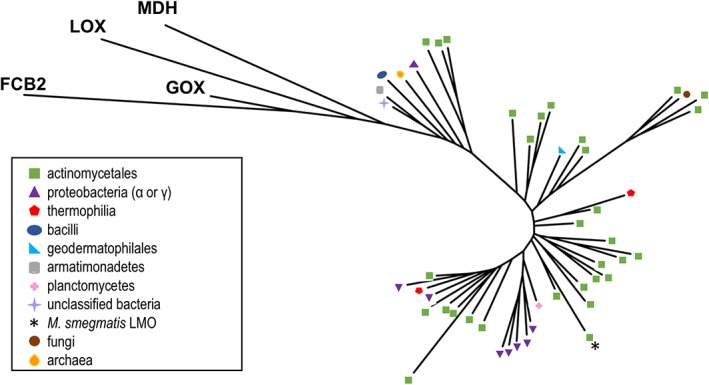
Relatedness tree and distribution of putative LMOs and representative α‐hydroxy acid oxidases. An unrooted tree is shown for the 48 representative putative LMO sequences along with proteins from representative PDB entries from LOX (PDB entry 2E77), GOX (2RDU), MDH (1P4C), and FCB2 (1KBI). The branches for the putative LMO sequences are annotated by symbols according to the organism where they are found, with a key in the figure indicating which symbols correspond to which organisms.
Using this criterion, putative LMOs are most abundantly represented in Actinomycetales, an order of Gram‐positive and generally aerobic bacteria including Mycobacterium, Nocardia, Streptomyces, Rhodococcus, and Frankia.47 LMO are also identified in bacteria from α‐ and γ‐proteobacteria, plantomycetes, pseudocardiales, thermophilia, geodermatophilales, and bacilli. Surprisingly, putative LMO sequences much like those in bacteria were found in a single fungal genus, Beauveria, and one archaeon, Halopiger salifodinae. As noted below, further investigation supports the conclusion that these sequences are reliably present in these nonbacterial sources and not due to contaminants, making it natural to suggest that they were derived through horizontal gene transfer (HGT).
In the case of Beauveria, a putative LMO gene that has a prokaryotic gene structure (i.e., lacks introns) is found in all sequenced strains of B. bassiana and its sister species, B. brongniartii (also known as Cordyceps brongniartii) (Supporting Information, Fig. S4), but is not found in other closely related entomopathogens such as Cordyceps (e.g., C. militaris and C. confragosa) and Metarhizium (e.g., Metarhizium album and Metarhizium robertsii). These Beauveria proteins group closely with those from the human pathogens, Mycobacterium abscessus, responsible for a multidrug‐resistant diseases in humans,48 and Tsukamurella pulmonis, commonly associated with infections in immunocompromised humans49 (Fig. 8 and Supporting Information, Fig. S4). While it may seem odd that these human pathogenic bacteria and insect pathogenic fungi would have closely related LMOs, T. pulmonis and many other mycobacteria have been isolated from insects,49, 50 and a variety of insects, including bedbugs and bloodsucking insects, have been implicated in the spread of mycobacteria.50 Thus, HGT between ancestors of these bacteria and of B. bassiana could have occurred in an insect carrier. In a potentially similar HGT scenario, the related entomopathogenic fungus Metarhizium anisopliae acquired a phosphoketolase from bacteria that is necessary for insect virulence and aids survival of the fungus in the insect haemocoel.51, 52 This raises the possibility that that LMO was acquired because it confers a selective advantage to Beauveria fungi.
H. salifodinae is a halophilic archaeon and its putative LMO groups closely with that of the halophilic proteobacterium Salincola salarius (Fig. 8). A BLAST search using the H. salifodinae protein as query retrieves an extensive set of putative LMOs from a range of halophilic archaea (Supporting Information, Fig. S5). It has been observed that anaerobic and thermophilic bacteria share an unusually large number of genes with archaea and that HGT is especially rampant among archaea and bacteria found in similar environments,53, 54 making it plausible that the presence of LMO in these organisms presents another example of this evolutionary phenomenon.
2.7. Outlook
This long overdue structure determination of an LMO provides a piece that has been missing in studies of the α‐hydroxy acid oxidase family. It lays a solid foundation for reinvigorating structure–function studies on this enzyme, as well as providing a specific, testable hypothesis that a more stable folding of Loop 4 is responsible for LMO's unique coupled reaction that directly produces acetate and carbon dioxide and does not release pyruvate as a product. Further, our analysis of the distribution of putative LMOs in nature, including the varieties of bacteria in which it is found and its surprising presence in a fungus and in halophilic archaea, provides new motivation to discover the physiological role it plays in these organisms.
3. Materials and Methods
3.1. Crystallography and structure determination
Recombinant LMO (WT and C203A) from M. smegmatis was provided by Dr Vince Massey (University of Michigan). The protein sample was shipped at 4°C as precipitated protein in 1.0 M acetate, pH 5.4. Before crystallization, the protein was transferred to 5 mM HEPES pH 7.0, 10 μM PMSF and concentrated to 20 mg/mL. Aliquots were frozen and stored at −80°C.
The proteins were crystallized at 4°C in hanging drops with a reservoir solution of 0.1 M sodium citrate pH 4.6, 0.4 M lithium sulfate, and 0.4 M ammonium sulfate. For WT, crystals were transferred into a series of artificial mother liquors containing 0%, 5%, 15%, and 25% glycerol and plunged into liquid nitrogen. Data were collected in 1996 at CHESS station A1 using Δφ = 0.5° rotation and processed using DENZO.55
For C203A, crystals were transferred to an artificial mother liquor containing 25% glycerol and plunged into liquid nitrogen. Data were collected at beamline 5.0.2 at the Advanced Light Source using λ = 1.0 Å, Δφ = 0.25° rotation, 0.25 s exposure, collecting 360° total. Data were processed and handled using XDS56 and the CCP4 program suite.57 A CC1/2 cutoff criteria58 of ~0.2 after merging was used to define the resolution cutoff of 1.70 Å, and a random 5% of reflections in resolution bins were marked for cross‐validation.
Largely due to the unusual observed crystal packing, these structures proved challenging for molecular replacement and were tackled in an atypical, multistep manner. The phase problem was initially solved by molecular replacement using MR‐Rosetta with default settings30 using the 2.3 Å WT dataset and a homology search model generated by HHpred.59 The initial solution contained four chains with 1687 residues built and R/R free of 0.34/0.38. All manual modeling building and refinement was done in Coot60, 61 and Phenix,62 respectively.
To have plausible crystal packing, it was clear that further chains had to be placed even though there was only very weak density in a swath of the crystal along the c‐axis (Fig. 2). Attempts to place these chains using the WT data were not successful, but we were able to place the chains using the higher resolution data from the C203A crystal and a combination of MR‐Rosetta, Autobuild, and Phaser. After obtaining a higher resolution C203A dataset, the existing WT model was used as a placed model for model‐building using MR‐Rosetta with the 1.7 Å C203A dataset. The resulting solution from MR‐Rosetta contained four chains with 1522 residues built and R/R free of 0.26/0.27. This solution was used to seed Autobuild,63 which led to no discernable changes in the models (and we presume was not a necessary step). After some additional refinement, the four chains were fed into Phaser64 as an already placed model, and molecular replacement using C203A Chain A—without its FMN and bound sulfate or waters—as a search model and with the packing criteria turned off gave a solution that contained two additional chains. These chains were in the weaker electron density region of the crystal, and a confirmation of their correctness was provided by a difference map that had strong positive peaks in both chains corresponding to where FMN and sulfate should be found (Supporting Information, Fig. S1).
This model containing the four well‐ordered chains (A, B, C, and D) and two less ordered chains (E and F) was further refined. Because the electron density is so weak for chains E and F, we suspect there may be alternate ways for these chains to pack that support crystal formation. For this reason, we allowed the occupancies of these chains to refine, and set the occupancy for both chains at 80% based on the average occupancy of their atoms. As modeled, chains E and F each closely overlay with the well‐ordered chains, with both having a Cα rmsd of <0.4 Å compared to chain A. Water molecules were manually placed based on having electron density ≥ 4.0 ρ rms in F o–F c maps and ≥ 0.9 ρ rms in 2F o–F c maps and reasonable hydrogen bonding interactions. Further refinement of the C203A structure, with riding hydrogens and TLS using one group per chain (i.e., six total TLS groups), led to the final model (statistics in Table 1).
The LMO WT structure was resolved by molecular replacement using a nearly final C203A structure as the search model, following the same methods as above for modeling and refinement. As chain D was clearly different in the WT structure (Supporting Information, Fig. S2), before refinement progressed, that chain was replaced by the model for chain D that was obtained from the initial WT MR‐Rosetta solution, which was correctly positioned.
3.2. Phylogenetic studies
We carried out a BLAST search in March 2018 using the WT LMO sequence as the query and generated 1000 putative LMO sequences. The sequences all had e‐values above ~8 × 10−112 and sequence identity of ~50% or higher. To generate a smaller set of representative sequences, the 1000 sequences were clustered into 48 groups using CD‐Hit65 with sequence identity cutoff of 70%. The representative 48 sequences output by CD‐Hit and the four sequences of representative PDB structures of GOX, LOX, MDH, and FCB2 were aligned. To assess the reliability of putative LMO sequences in Beauveria and archaea, we carried out additional BLAST searches using the B. bassiana LMO sequence and H. salifodinae LMO sequence as query, respectively. In the B. bassiana search, all fungal sequences included had ~95% sequence identity or higher and the most similar bacterial sequence had ~70% sequence identity. This set was aligned with Beauveria sequences provided by Dr Claudio Valero (University of Wageningen), WT LMO, and LMO sequences from Mycobacterium abscessus and Tsukamurella pulmonis. From the H. salifodinae search, we generated a set of archaeal sequences that included all archaeal sequences that were more similar to H. salifodinae LMO than the most similar bacterial sequence; this included sequences of ~60% sequence identity and higher. They were aligned with WT LMO. In all cases, sequences were aligned with MUSCLE,66 and trees were generated using PhyML67 and Interactive Tree of Life.68
3.3. Electrostatic potential and surface area calculations
Electrostatic potentials were calculated using the PDB2PQR server69 with the AMBER force field and Adaptive Poisson–Boltzmann Solver (APBS) server.70, 71 Parameters for FMN were generated using PRODRG72 with atomic partial charges from Rohr et al.73 Electrostatic potentials were rendered using Pymol74 in conjunction with the APBS plugin.71
The solvent‐accessible surface areas of folded proteins, both with and without Loop 4, were calculated using Areaimol.75, 76 The solvent‐accessible surface areas of the unfolded Loop 4 were calculated using the upper and lower bound model proposed by Creamer et al.,77, 78, 79 with the values reported in Table 2 being the average between the calculated upper and lower limits. For these calculations, the residues defining the beginning and end of Loop 4 were set, respectively, as the equivalent to LMO residues 195 and 243 based on the residues for which representative structures for GOX, LOX, FCB2, and MDH deviated in a structural overlay. The range of Loop 4 lengths in the LMO family was determined based on the same 48 sequences used for the phylogenetic tree. For the LOX and GOX families, the ranges were determined using COBALT alignments generated from a BLAST search using the representative enzyme (2E77 for LOX; 2RDU for GOX) and the top 100 (down to ~60% sequence identity) or 250 sequences (down to ~70% sequence identity), respectively. In the case of GOX, two sequences were removed, one of which was annotated as “low‐quality protein” and one of which was an incomplete sequence.
The WebLogo80 plot for LMO was generated using the same 48 sequences used for the phylogenetic tree. Residues in the core and/or interface of Loop 4 were determined based on both visual assessment and by comparing the residue‐level solvent exposed surface reported by DSSP81 in the absence and presence of the rest of the protein.
3.4. Accession numbers
Coordinates and structure factors for LMO WT and C203A have been deposited in the Protein Data Bank with the accession numbers 6DVI and 6DVH, respectively.
Supporting information
Figure S1 Evidence for the correct placement of the two less ordered LMO chains, E and F
Figure S2. Alternate octamer observed in wild‐type LMO crystals
Figure S3. Electrostatic potential surface of LOX and GOX without Loop 4 channels substrate into the active site
Figure S4. Relatedness tree for putative lactate monooxygenases in the entomopathogenic fungi of the genus Beauveria
Figure S5. Relatedness tree for putative lactate monooxygenases in archaea
Acknowledgments
The authors thank Zhan Deng for growing the original crystals and collecting the data used here to solve the wild‐type structure. They also thank Dale Tronrud for his assistance in tackling this challenging molecular replacement, and Claudio Valero and Jan van Kan (University of Wageningen) for their guidance, insights, and unpublished sequences related to the LMO genes found in Beauveria, and also Michael Freitag and Joey Spatafora (Oregon State University) for helpful conversations.
Dedication: Dedicated to Vincent Massey.
References
- 1. Edson NL (1947) The oxidation of lactic acid by Mycobacterium phlei . Biochem J 41:145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ghisla S, Massey V. L‐Lactate oxidase In: Müller F, Ed., 1991. Chemistry and biochemistry of flavoenzymes. Boca Raton, FL: CRC Press. [Google Scholar]
- 3. Sutton WB (1954) Isolation and properties of a lactic oxidative decarboxylase from Mycobacterium phlei . J Biol Chem 210:309–320. [PubMed] [Google Scholar]
- 4. Sullivan PA (1968) Crystallization and properties of L‐lactate oxidase from Mycobacterium smegmatis . Biochem J 110:363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lockridge O, Massey V, Sullivan PA (1972) Mechanism of action of the flavoenzyme lactate oxidase. J Biol Chem 247:8097–8106. [PubMed] [Google Scholar]
- 6. Maeda‐Yorita K, Aki K, Sagai H, Misaki H, Massey V (1995) L‐lactate oxidase and L‐lactate monooxygenase: mechanistic variations on a common structural theme. Biochimie 77:631–642. [DOI] [PubMed] [Google Scholar]
- 7. Fitzpatrick PF (2015) Combining solvent isotope effects with substrate isotope effects in mechanistic studies of alcohol and amine oxidation by enzymes. Biochim Biophys Acta 1854:1746–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sobrado P, Fitzpatrick PF (2003) Solvent and primary deuterium isotope effects show that lactate CH and OH bond cleavages are concerted in Y254F flavocytochrome b2, consistent with a hydride transfer mechanism. Biochemistry 42:15208–15214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tabacchi G, Zucchini D, Caprini G, Gamba A, Lederer F, Vanoni MA, Fois E (2009) L‐lactate dehydrogenation in flavocytochrome b2: a first principles molecular dynamics study. FEBS J 276:2368–2380. [DOI] [PubMed] [Google Scholar]
- 10. Li SJ, Umena Y, Yorita K, Matsuoka T, Kita A, Fukui K, Morimoto Y (2007) Crystallographic study on the interaction of L‐lactate oxidase with pyruvate at 1.9 Angstrom resolution. Biochem Biophys Res Commun 358:1002–1007. [DOI] [PubMed] [Google Scholar]
- 11. Lindqvist Y (1989) Refined structure of spinach glycolate oxidase at 2 Å resolution. J Mol Biol 209:151–166. [DOI] [PubMed] [Google Scholar]
- 12. Murray MS, Holmes RP, Lowther WT (2008) Active site and loop 4 movements within human glycolate oxidase: implications for substrate specificity and drug design. Biochemistry 47:2439–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cunane LM, Barton JD, Chen ZW, Welsh FE, Chapman SK, Reid GA, Mathews FS (2002) Crystallographic study of the recombinant flavin‐binding domain of Baker's yeast flavocytochrome b(2): comparison with the intact wild‐type enzyme. Biochemistry 41:4264–4272. [PubMed] [Google Scholar]
- 14. Sukumar N, Dewanti AR, Mitra B, Mathews FS (2004) High resolution structures of an oxidized and reduced flavoprotein. The water switch in a soluble form of (S)‐mandelate dehydrogenase. J Biol Chem 279:3749–3757. [DOI] [PubMed] [Google Scholar]
- 15. Murphy CJ, Shepherd MG, Sullivan PA (1983) Chemical modification of L‐lactate 2‐monooxygenase with fluorodinitrobenzene: evidence for two essential histidine residues. Biochemistry 22:1665–1669. [DOI] [PubMed] [Google Scholar]
- 16. Soon CY, Shepherd MG, Sullivan PA (1978) Inactivation and modification of lactate oxidase with fluorodinitrobenzene. Biochem J 173:255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Soon CY, Shepherd MG, Sullivan PA (1977) Modification of lactate oxidase with diethyl pyrocarbonate. Evidence for an active‐site histidine residue. Biochem J 165:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peters RG, Jones WC, Cromartie TH (1981) Inactivation of L‐lactate monooxygenase with 2,3‐butanedione and phenylglyoxal. Biochemistry 20:2564–2571. [DOI] [PubMed] [Google Scholar]
- 19. Muh U, Massey V, Williams CH Jr (1994) Lactate monooxygenase. I. Expression of the mycobacterial gene in Escherichia coli and site‐directed mutagenesis of lysine 266. J Biol Chem 269:7982–7988. [PubMed] [Google Scholar]
- 20. Muh U, Williams CH Jr, Massey V (1994) Lactate monooxygenase. II. Site‐directed mutagenesis of the postulated active site base histidine 290. J Biol Chem 269:7989–7993. [PubMed] [Google Scholar]
- 21. Muh U, Williams CH, Jr., Massey V (1994) Lactate monooxygenase. III. Additive contributions of active site residues to catalytic efficiency and stabilization of an anionic transition state. J Biol Chem 269:7994–8000. [PubMed] [Google Scholar]
- 22. Sun W, Williams CH Jr, Massey V (1997) The role of glycine 99 in L‐lactate monooxygenase from Mycobacterium smegmatis . J Biol Chem 272:27065–27076. [DOI] [PubMed] [Google Scholar]
- 23. Sun W, Williams CH Jr, Massey V (1996) Site‐directed mutagenesis of glycine 99 to alanine in L‐lactate monooxygenase from Mycobacterium smegmatis . J Biol Chem 271:17226–17233. [DOI] [PubMed] [Google Scholar]
- 24. Deng Z. Structural studies of the active site and NADP+ binding of plant‐type ferredoxin:NADP+ reductase. Ithaca, NY: Cornell University, 1999. [Google Scholar]
- 25. Stoisser T, Rainer D, Leitgeb S, Wilson DK, Nidetzky B (2015) The Ala95‐to‐Gly substitution in Aerococcus viridans l‐lactate oxidase revisited ‐ structural consequences at the catalytic site and effect on reactivity with O2 and other electron acceptors. FEBS J 282:562–578. [DOI] [PubMed] [Google Scholar]
- 26. Stoisser T, Brunsteiner M, Wilson DK, Nidetzky B (2016) Conformational flexibility related to enzyme activity: evidence for a dynamic active‐site gatekeeper function of Tyr(215) in Aerococcus viridans lactate oxidase. Sci Rep 6:27892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stoisser T, Klimacek M, Wilson DK, Nidetzky B (2015) Speeding up the product release: a second‐sphere contribution from Tyr191 to the reactivity of L‐lactate oxidase revealed in crystallographic and kinetic studies of site‐directed variants. FEBS J 282:4130–4140. [DOI] [PubMed] [Google Scholar]
- 28. Giegel DA, Massey V, Williams CH (1987) L‐lactate‐2‐monooxygenase. Sequence of peptides containing residues modified by 1‐fluoro‐2,4‐dinitrobenzene. J Biol Chem 262:5705–5710. [PubMed] [Google Scholar]
- 29. Muh U, Williams CH, Massey V. Lactate monooxygenase: studies of active site mutations In: Yagi K, Ed., 1994. Flavins and flavoproteins. Berlin: Walter de Gruyter; p. 201–210. [Google Scholar]
- 30. Terwilliger TC, Dimaio F, Read RJ, Baker D, Bunkoczi G, Adams PD, Grosse‐Kunstleve RW, Afonine PV, Echols N (2012) phenix.mr_rosetta: molecular replacement and model rebuilding with Phenix and Rosetta . J Struct Funct Genom 13:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Renko M, Taler‐Vercic A, Mihelic M, Zerovnik E, Turk D (2014) Partial rotational lattice order‐disorder in stefin B crystals. Acta Cryst D70:1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Giegel DA, Williams CH, Jr , Massey V (1990) L‐lactate 2‐monooxygenase from Mycobacterium smegmatis. Cloning, nucleotide sequence, and primary structure homology within an enzyme family. J Biol Chem 265:6626–6632. [PubMed] [Google Scholar]
- 33. Sullivan PA, Soon CY, Schreurs WJ, Cutfield JF, Shepherd MG (1977) The structure of L‐lactate oxidase from Mycobacterium smegmatis . Biochem J 165:375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Umena Y, Yorita K, Matsuoka T, Kita A, Fukui K, Morimoto Y (2006) The crystal structure of L‐lactate oxidase from Aerococcus viridans at 2.1A resolution reveals the mechanism of strict substrate recognition. Biochem Biophys Res Commun 350:249–256. [DOI] [PubMed] [Google Scholar]
- 35. Sukumar N, Xu Y, Gatti DL, Mitra B, Mathews FS (2001) Structure of an active soluble mutant of the membrane‐associated (S)‐mandelate dehydrogenase. Biochemistry 40:9870–9878. [DOI] [PubMed] [Google Scholar]
- 36. Xia ZX, Shamala N, Bethge PH, Lim LW, Bellamy HD, Xuong NH, Lederer F, Mathews FS (1987) Three‐dimensional structure of flavocytochrome b2 from baker's yeast at 3.0‐A resolution. Proc Natl Acad Sci U S A 84:2629–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Massey V, Muller F, Feldberg R, Schuman M, Sullivan PA, Howell LG, Mayhew SG, Matthews RG, Foust GP (1969) The reactivity of flavoproteins with sulfite. Possible relevance to the problem of oxygen reactivity. J Biol Chem 244:3999–4006. [PubMed] [Google Scholar]
- 38. Holm L, Rosenstrom P (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cunane LM, Barton JD, Chen ZW, Le KH, Amar D, Lederer F, Mathews FS (2005) Crystal structure analysis of recombinant rat kidney long chain hydroxy acid oxidase. Biochemistry 44:1521–1531. [DOI] [PubMed] [Google Scholar]
- 40. Furuichi M, Suzuki N, Dhakshnamoorhty B, Minagawa H, Yamagishi R, Watanabe Y, Goto Y, Kaneko H, Yoshida Y, Yagi H, Waga I, Kumar PKR, Mizuno H (2008) X‐ray structures of Aerococcus viridans lactate oxidase and its complex with D‐lactate at pH 4.5 show an alpha‐hydroxyacid oxidation mechanism. J Mol Biol 378:436–446. [DOI] [PubMed] [Google Scholar]
- 41. Raychaudhuri S, Younas F, Karplus PA, Faerman CH, Ripoll DR (1997) Backbone makes a significant contribution to the electrostatics of alpha/beta‐barrel proteins. Protein Sci 6:1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Getzoff ED, Cabelli DE, Fisher CL, Parge HE, Viezzoli MS, Banci L, Hallewell RA (1992) Faster superoxide dismutase mutants designed by enhancing electrostatic guidance. Nature 358:347–351. [DOI] [PubMed] [Google Scholar]
- 43. Miller S, Lesk AM, Janin J, Chothia C (1987) The accessible surface area and stability of oligomeric proteins. Nature 328:834–836. [DOI] [PubMed] [Google Scholar]
- 44. Chothia C (1984) Principles that determine the structure of proteins. Annu Rev Biochem 53:537–572. [DOI] [PubMed] [Google Scholar]
- 45. Lesk AM, Chothia C (1980) Solvent accessibility, protein surfaces, and protein folding. Biophys J 32:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou H, Zhou Y (2004) Quantifying the effect of burial of amino acid residues on protein stability. Proteins 54:315–322. [DOI] [PubMed] [Google Scholar]
- 47. Barka EA, Vatsa P, Sanchez L, Gaveau‐Vaillant N, Jacquard C, Meier‐Kolthoff JP, Klenk HP, Clement C, Ouhdouch Y, van Wezel GP (2016) Taxonomy, physiology, and natural products of actinobacteria. Microbiol Mol Biol Rev 80:1–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee MR, Sheng WH, Hung CC, Yu CJ, Lee LN, Hsueh PR (2015) Mycobacterium abscessus complex infections in humans. Emerg Infect Dis 21:1638–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Safaei S, Fatahi‐Bafghi M, Pouresmaeil O (2018) Role of Tsukamurella species in human infections: first literature review. New Microbes New Infect 22:6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kazda J, Pavlik I, Falkinham JO, Hruska K. The ecology of Mycobacteria: impact on animal's and human's health. Netherlands: Springer, 2009. [Google Scholar]
- 51. Fitzpatrick DA (2012) Horizontal gene transfer in fungi. FEMS Microbiol Lett 329:1–8. [DOI] [PubMed] [Google Scholar]
- 52. Duan Z, Shang Y, Gao Q, Zheng P, Wang C (2009) A phosphoketolase Mpk1 of bacterial origin is adaptively required for full virulence in the insect‐pathogenic fungus Metarhizium anisopliae . Environ Microbiol 11:2351–2360. [DOI] [PubMed] [Google Scholar]
- 53. Papke RT, Corral P, Ram‐Mohan N, de la Haba RR, Sanchez‐Porro C, Makkay A, Ventosa A (2015) Horizontal gene transfer, dispersal and haloarchaeal speciation. Life 5:1405–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fuchsman CA, Collins RE, Rocap G, Brazelton WJ (2017) Effect of the environment on horizontal gene transfer between bacteria and archaea. PeerJ 5:e3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- 56. Kabsch W (2010) Xds. Acta Cryst D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Collaborative Computational Project N (1994) The CCP4 suite: programs for protein crystallography. Acta Cryst D50:760–763. [DOI] [PubMed] [Google Scholar]
- 58. Karplus PA, Diederichs K (2012) Linking crystallographic model and data quality. Science 336:1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Soding J, Biegert A, Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–W248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 61. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Cryst D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L‐W, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarity NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Cryst D66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Terwilliger TC, Grosse‐Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Cryst D64:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Huang Y, Niu BF, Gao Y, Fu LM, Li WZ (2010) CD‐HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26:680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum‐likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. [DOI] [PubMed] [Google Scholar]
- 68. Letunic I, Bork P (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44:W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA (2004) PDB2PQR: an automated pipeline for the setup of Poisson‐Boltzmann electrostatics calculations. Nucleic Acids Res 32:W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, Brookes DH, Wilson L, Chen JH, Liles K and others (2018) Improvements to the APBS biomolecular solvation software suite. Protein Sci 27:112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schuttelkopf AW, van Aalten DM (2004) PRODRG: a tool for high‐throughput crystallography of protein‐ligand complexes. Acta Cryst D60:1355–1363. [DOI] [PubMed] [Google Scholar]
- 73. Rohr AK, Hersleth HP, Andersson KK (2010) Tracking flavin conformations in protein crystal structures with Raman spectroscopy and QM/MM calculations. Angew Chem Int Ed Engl 49:2324–2327. [DOI] [PubMed] [Google Scholar]
- 74. The PyMOL molecular graphics system. Version 1.3. San Carlos, CA: Schrödinger, LLC, 2010. [Google Scholar]
- 75. Lee B, Richards FM (1971) The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55:379–400. [DOI] [PubMed] [Google Scholar]
- 76. Saff EB, Kuijlaars ABJ (1997) Distributing many points on a sphere. Math Intell 19:5–11. [Google Scholar]
- 77. Creamer TP, Srinivasan R, Rose GD (1995) Modeling unfolded states of peptides and proteins. Biochemistry 34:16245–16250. [DOI] [PubMed] [Google Scholar]
- 78. Creamer TP, Srinivasan R, Rose GD (1997) Modeling unfolded states of proteins and peptides. II. Backbone solvent accessibility. Biochemistry 36:2832–2835. [DOI] [PubMed] [Google Scholar]
- 79. Estimate of Unfolded state Accessible Surface Area . <http://folding.chemistry.msstate.edu/utils/unfolded.html>. Apr 22, 2018.
- 80. Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers 22:2577–2637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Evidence for the correct placement of the two less ordered LMO chains, E and F
Figure S2. Alternate octamer observed in wild‐type LMO crystals
Figure S3. Electrostatic potential surface of LOX and GOX without Loop 4 channels substrate into the active site
Figure S4. Relatedness tree for putative lactate monooxygenases in the entomopathogenic fungi of the genus Beauveria
Figure S5. Relatedness tree for putative lactate monooxygenases in archaea