

Abstract
7‐Carboxy‐7‐deazaguanine synthase, QueE, catalyzes the radical mediated ring contraction of 6‐carboxy‐5,6,7,8‐tetrahydropterin, forming the characteristic pyrrolopyrimidine core of all 7‐deazaguanine natural products. QueE is a member of the S‐adenosyl‐L‐methionine (AdoMet) radical enzyme superfamily, which harnesses the reactivity of radical intermediates to perform challenging chemical reactions. Members of the AdoMet radical enzyme superfamily utilize a canonical binding motif, a CX3CXϕC motif, to bind a [4Fe‐4S] cluster, and a partial (β/α)6 TIM barrel fold for the arrangement of AdoMet and substrates for catalysis. Although variations to both the cluster‐binding motif and the core fold have been observed, visualization of drastic variations in the structure of QueE from Burkholderia multivorans called into question whether a re‐haul of the defining characteristics of this superfamily was in order. Surprisingly, the structure of QueE from Bacillus subtilis revealed an architecture more reminiscent of the classical AdoMet radical enzyme. With these two QueE structures revealing varying degrees of alterations to the classical AdoMet fold, a new question arises: what is the purpose of these alterations? Here, we present the structure of a third QueE enzyme from Escherichia coli, which establishes the middle range of the spectrum of variation observed in these homologs. With these three homologs, we compare and contrast the structural architecture and make hypotheses about the role of these structural variations in binding and recognizing the biological reductant, flavodoxin.
Broader impact statement: We know more about how enzymes are tailored for catalytic activity than about how enzymes are tailored to react with a physiological reductant. Here, we consider structural differences between three 7‐carboxy‐7‐deazaguanine synthases and how these differences may be related to the interaction between these enzymes and their biological reductant, flavodoxin.
Keywords: AdoMet radical enzymes, flavodoxin, physiological reductant, iron–sulfur clusters, flavin mononucleotide
Short abstract
Introduction
Reduction by low potential electrons is required for the activity of a number of metalloenzymes, including the cobalamin‐dependent enzyme methionine synthase1 and proteins within the 100,000‐membered S‐adenosyl‐L‐methionine (AdoMet) radical enzyme superfamily.2 AdoMet radical enzymes utilize the reductive cleavage of a molecule of AdoMet ligated to a [4Fe‐4S] cluster to initiate radical chemistry [Fig. 1(A)].3 The highly reactive intermediate that is generated, 5′‐deoxyadenosyl radical or 5′‐dAdo•, can abstract a hydrogen‐atom (H‐atom) from diverse substrates, initiating a variety of chemically challenging and complex reactions.3, 4 This radical generation requires reduction of the AdoMet radical [4Fe‐4S] cluster from a resting oxidation state of +2 to +1. The biological reductant, flavodoxin, was first shown to be capable of this reduction in studies of pyruvate formate‐lyase activating enzyme.2 Subsequently, Escherichia coli flavodoxin (EcFldA) has been employed as a reductant for a number of AdoMet radical enzymes via an NADPH‐dependent flavodoxin reductase system5, 6, 7, 8, 9, 10, 11 [Fig. 1(B)].
Figure 1.
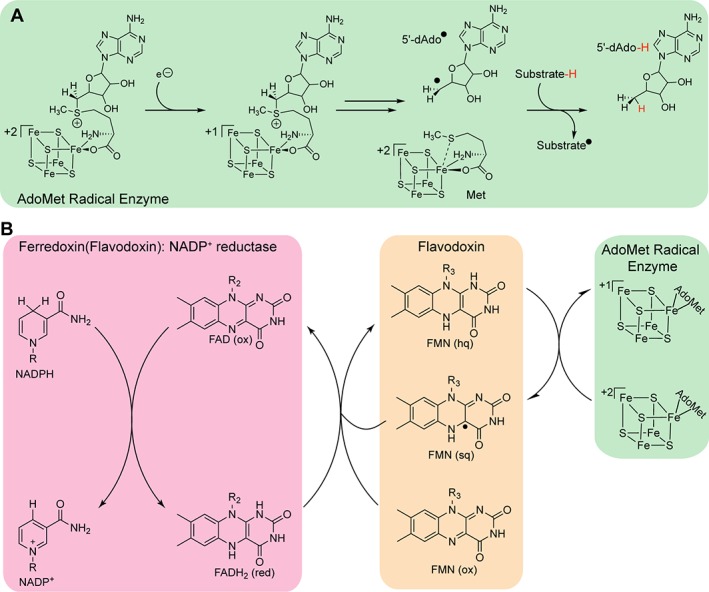
Flavodoxin reduces the AdoMet radical cluster. (A) To initiate radical chemistry through reductive cleavage of AdoMet, the AdoMet radical cluster needs to first be reduced from the resting +2 oxidation state to the +1 oxidation state. (B) Low potentials electrons from NADPH are transferred to the AdoMet radical cluster through the action of Ferredoxin (flavodoxin): NADP+ reductase/Flavodoxin system. Functional parts of NADPH, FAD and FMN are shown.
In addition to the biological EcFldA‐flavodoxin reductase system, dithionite can be used to provide reducing equivalents to AdoMet radical enzymes in vitro. In fact, in working with two anaerobic sulfatase enzymes, AtsB and anSME from Klebsiella pneumonia and Clostridium perfringens, respectively, Grove et al. noted that dithionite appeared to be the more robust reductant in that it increases activity 10–100 fold when compared to EcFldA‐flavodoxin reductase.12, 13 Similarly, more product is observed when dithionite instead of EcFldA is used to reduce 7‐carboxy‐7‐deazaguanine synthase (QueE) from Burkholdeira multivorans (BmQueE).14 Interestingly, the QueE from Bacillus subtilis (BsQueE) shows the opposite trend from BmQueE, with more product, 7‐carboxy‐7‐deazaguanine (CDG), observed upon incubation with the EcFldA‐flavodoxin reductase system compared to dithionite.15, 16 However, maximal production of CDG was observed for BsQueE in the presence of the native flavodoxins, YkuN and YkuP.15 Taken together these results underlie the need to understand the protein–protein interactions that occur between AdoMet radical enzymes and flavodoxins to begin to dissect the determinants for activation.
Here, we use the highly structurally divergent QueE enzyme family (Fig. 2) as our model system to investigate the hypothesis that the structures of AdoMet radical enzymes are tailored to make specific protein–protein interactions with particular flavodoxins. QueE enzymes are part of the biosynthetic pathway of 7‐deazapurine natural products (Fig. SI). They catalyze the radical‐mediated ring contraction of 6‐carboxy‐5,6,7,8‐tetrahydropterin (CPH4) (Fig. 2), forming the characteristic pyrrolopyrimidine core of all 7‐deazaguanine natural products, including the modified tRNA nucleoside queuosine17. Structures of two QueE homologs have been previously determined (BmQueE and BsQueE)14, 18 and here we present a third structure, that of QueE from E. coli (EcQueE) (Table S1). Interestingly, the structure of BmQueE revealed drastic deviations to the AdoMet radical core fold and the cluster‐binding motif: BmQueE folds into a pared‐down partial (β6/α3) TIM barrel, in comparison to the classic partial (β/α)6 TIM barrel, and contains a modified cluster‐binding motif, a CX14CXϕC motif,14 compared to the classic CX3CXϕC motif where ϕ is a conserved aromatic residue. In contrast, BsQueE adopts a partial (β6/α5) TIM barrel fold with minimal variations from the classic AdoMet radical fold and contains the standard CX3CXϕC cluster‐binding motif (Fig. S2).18 EcQueE, as described below, falls in between these two extremes. Given that the flavin mononucleotide (FMN) cofactor of flavodoxin must be within electron transfer distance from the AdoMet radical cluster for cluster reduction,19, 20, 21 we consider how these variations in protein folds observed in these QueE structures could explain the reductant specificity noted above for QueE enzymes.
Figure 2.
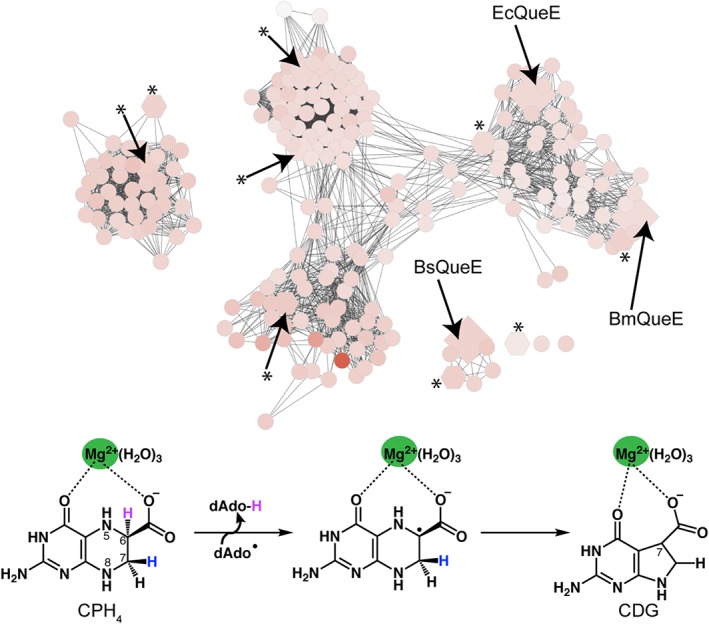
Sequence similarity network of the AdoMet radical enzyme subfamily, 7‐carboxy‐7‐deazaguanine synthases (QueE). The protein sequence similarity network38 for the QueE AdoMet radical subfamily was obtained from the Structure Function Linkage Database (http://sfld.rbvi.ucsf.edu/django) and visualized in Cytoscape.39 Each node represents sequences that share 50% identity or higher and node connections are filtered at a Blast Probability of 10−25. Nodes are colored based on increasing sequence length; White nodes denote the shortest sequences (149 amino acids) and the orange node denotes the longest sequence (509 amino acids). B. multivorans, B. subtilis and E. coli QueE sequences are shown as diamonds and sequences used in the sequence alignment (Fig. S2) are shown as hexagons and designated with an asterisk (*). QueE catalyzes the AdoMet and magnesium dependent rearrangement of 6‐carboxy‐5,6,7,8‐tetrahydropterin, CPH4, to 7‐carboxy‐7‐deazaguanine, CDG.
Results
EcQueE reveals an intermediary structure between BmQueE and BsQueE
The QueE homolog from E. coli (EcQueE) was produced in E. coli and its ability to convert CPH4 to CDG was confirmed in preliminary HPLC studies of the purified recombinant protein. The crystal structure of EcQueE was determined to 2.1‐Å resolution by multi‐wavelength anomalous dispersion (MAD) phasing and R work and R free of 0.205 and 0.238, respectively (Table S2). In the final structure, electron density was observed for most of the crystallization construct with the exception of the first nine residues of the N‐terminal hexahistidine tag (His6tag), residues 192–196 and the final 10 residues. EcQueE folds into a structural and functional head‐to‐tail homodimer, reminiscent of the published QueE structures from Burkholderia multivorans (BmQueE) and Bacillus subtilis (BsQueE)14, 18 [Fig. 3(A)]. The overall structure of EcQueE is similar to that of BmQueE (rmsd 1.8 Å) and BsQueE (rmsd 2.9 Å), with variations in the structure and orientation of the loops and α‐helices [Fig. 3(B)].
Figure 3.
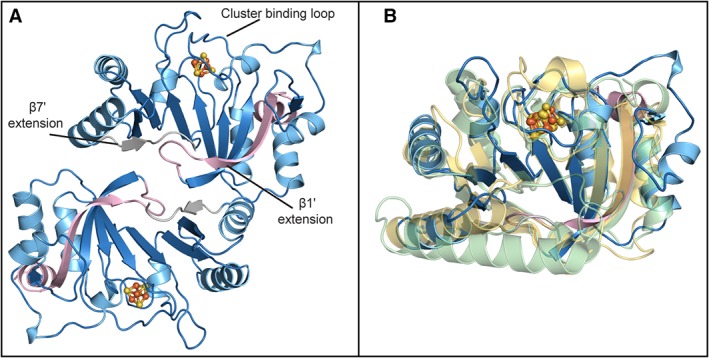
Structure of QueE from Escherichia coli. (A) Structure of EcQueE, shown as ribbons, folds into a head‐to‐tail functional dimer with the dimer interface composed of interactions between the N‐terminal (light pink) and C‐terminal (grey) extensions. The modified AdoMet core, a partial (β6/α5) TIM barrel, is shown in blue. (B) EcQueE (blue) monomer overlays well with the monomers of BsQueE, (PDB ID 5TH5), (translucent light green) and BmQueE, (PDB ID 4NJI), (translucent yellow). In both panels, [4Fe–4S] clusters are shown in a ball and stick representation, where iron is colored orange and sulfur is colored yellow.
All three QueE homologs fold into variants of the AdoMet radical core domain with extensions at the N‐ and C‐termini. The N‐terminal extensions of QueE structures comprise a single anti‐parallel β‐strand, β1′ [Fig. 4(A–C)], which is found adjacent to β1 of the AdoMet radical core. In EcQueE, the linker and the first residue of the His6tag are visible, forming an additional α‐helix, α1′, at the N‐terminus of the enzyme [Fig. 4(A)]. The C‐terminal regions of BmQueE and BsQueE fold into a β‐strand/α‐helix (β7′/α7′) pair, where the β7′ is found adjacent to β6 [Fig. 4(B) and (C)]. In the EcQueE structure, α7′ of the β7′/α7′ pair is not visible due to disorder of the final 10 amino acids of the protein [Fig. 4(A)]. The N‐ and C‐terminal extensions are important for both substrate binding and dimerization in BmQueE and BsQueE14, 18 and it is expected that they will serve the same function in EcQueE. Mutual interactions between the β1′‐loop‐β1 and β7′ of the adjacent QueE monomers create a dimeric interface such that the β‐strands of the N‐ and C‐terminal extensions not only extend the monomeric inner face, but also form an inter‐monomer 10‐stranded β‐sheet that is thought to resemble a crown [Fig. 4(A–C)].
Figure 4.
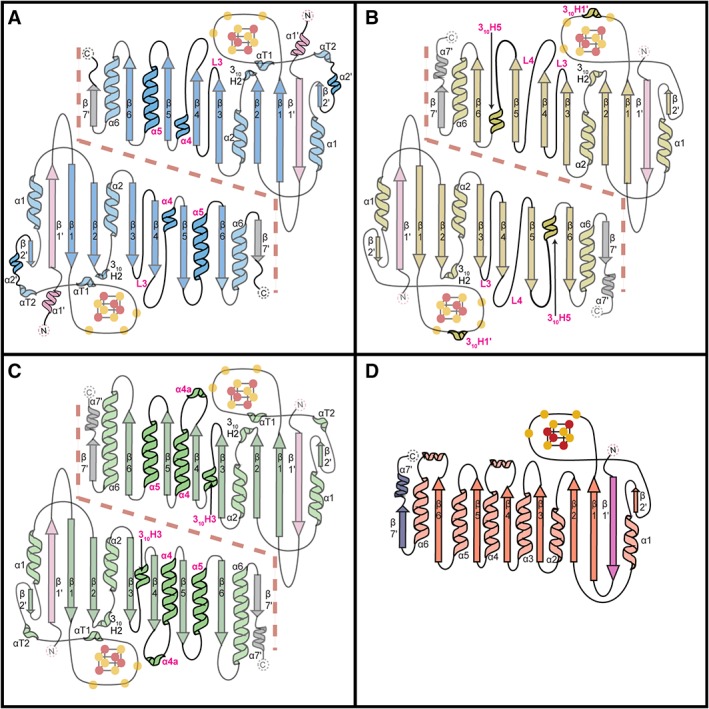
Topology diagrams for QueE homologs and PFL‐AE. (A) EcQueE, (B) BmQueE, (C) BsQueE and (D) PFL‐AE. The core AdoMet domains are colored blue for EcQueE, yellow for BmQueE and green for BsQueE whereas the N‐ and C‐terminal extensions are colored light pink and grey (respectively) for all three QueE structures. The differences between the QueE homologs structures are shown in bold and the corresponding secondary structure element denoted in magenta and the dashed line delineates the QueE dimer interface. The topology diagram of PFL‐AE is shown with the N‐ and C‐terminal extensions colored pink and slate respectively and AdoMet domain colored in coral. The iron atoms of the [4Fe–4S] clusters are colored orange and sulfur atoms are colored yellow. Cysteine ligands to the [4Fe–4S] cluster are shown as yellow circles. Structural elements outside the AdoMet radical core fold are labeled with a prime.
The core of the QueE homolog structures adopt three unique partial TIM barrel folds, where each variant differs in the number and type of α‐helices flanking the conserved parallel β‐sheet. The previously published structure of BmQueE shows the greatest variation of the three homologs and of the whole AdoMet radical superfamily characterized to date.20, 22 BmQueE sports a vastly pared down AdoMet radical fold, a β6/α3, where short loops, L3 and L4, replace α3 and α4 and a short 310‐helix, 310H5, replaces α5 [Fig. 4(B)]. The variations in the AdoMet radical core of BsQueE are the most conservative of the three homologs. BsQueE folds into a partial β6/α5 TIM barrel, which contains a non‐traditional short 310‐helix, 310H3 in place of α3 [Fig. 4(C)]. Similarly, the AdoMet radical domain of EcQueE folds into a partial β6/α5 TIM barrel with a variation at the α3 position, but this change is not as conservative as that seen in BsQueE [Fig. 4(A)]. In EcQueE, α3 is replaced by a short loop (L3), reminiscent of the α3 alternative in BmQueE, and also has a long loop connecting β4 to a very short α4.
The three QueE homologs, to date, are the smallest structurally characterized AdoMet radical enzymes, with BmQueE spanning only 210 amino acid residues, EcQueE, 223 amino acid residues and BsQueE 243 amino acid residues. The second smallest non‐QueE AdoMet radical enzyme structurally characterized, PFL‐AE (246 amino acid residues), shows surprising structural similarities to the QueE homologs, in particular BsQueE. PFL‐AE adopts a normal AdoMet radical core,21 a (β/α)6 TIM barrel, and contains N‐ and C‐terminal extensions, β1′ and β7′, which closely resemble those found in QueE [Fig. 4(D)]. Unlike QueE, PFL‐AE is a monomer, thus these terminal extensions do not play a role in oligomerization. However, similar to QueE, the N‐terminal extension is involved in substrate binding.21
In all three QueE structures, electron density was present for a [4Fe–4S] cluster bound by three cysteine ligands, leaving a site‐differentiated iron. Sequence analysis revealed an 11 amino acid insertion in the cluster‐binding loop of BmQueE, resulting in a CX14CXϕC sequence instead of the canonical CX3CXϕC cluster‐binding motif. Surprisingly, the insertion did not affect cysteine positioning and cluster binding and the cysteine ligands from the cluster binding loop superimposed well with other AdoMet radical enzymes [Fig. 3(B)]. Instead, the insertion folds into a short 310‐helix, 310H1, found on top of the AdoMet radical cluster [Fig. 4(B)] and further sequesters the cluster from solvent as well as increases the negative charge in that area. Following the cluster‐binding motif in BmQueE, the loop folds into a short β‐strand, β2′, before transitioning into α1, another structural addition outside of the AdoMet radical core. Sequence and structural analysis of BsQueE and EcQueE show a canonical cluster binding loop motif, CX3CXϕC, which positions the cluster at the top of the AdoMet radical barrel (Figs. 3 and 4). Akin to BmQueE, the transition in EcQueE and BsQueE from the cluster‐binding motif to α1 involves additional structural elements, a short α‐helix α2′ and/or helical turns αT1 and αT2, respectively, which precede β2′ and the subsequent α1 of the AdoMet radical core [Fig. 4(A) and (C)].
AdoMet binding motifs appear conserved in the QueE homologs
Structural analyses of AdoMet radical enzymes have revealed a number of structural motifs for securing AdoMet in position with respect to the [4Fe–4S] cluster for radical generation.20, 22 Structures of BmQueE with AdoMet bound (Table S1) revealed that alterations in the core fold and cluster‐binding motif, which were observed in that enzyme, did not lead to changes in the way that the enzyme bound AdoMet; AdoMet binding motifs were conserved [Figs. 5(A), S2, and S3].14 Likewise, a structure of BsQueE bound to an AdoMet‐derived adduct, 6‐carboxypterin‐5′‐deoxyadenosyl (6‐CP—dAdo) (Table S1), indicated conservation of AdoMet binding motifs [Figs. 5(B) and S2].18 Despite considerable effort, no structure of EcQueE has been obtained bound to either AdoMet or an AdoMet‐derived adduct, however, structural comparisons suggest that AdoMet‐binding residues are conserved (Figs. 5, S2, and S3). Interestingly, these residues in EcQueE are not pre‐organized for AdoMet binding. Modeling of AdoMet into the EcQueE active site [Figs. 5(C) and S3] indicates that side chain rearrangements will need to occur. No other QueE enzyme has been captured without a ligand bound (Table S1), thus the Ec structure is the first to show that the QueE active site is not pre‐organized to bind AdoMet.
Figure 5.
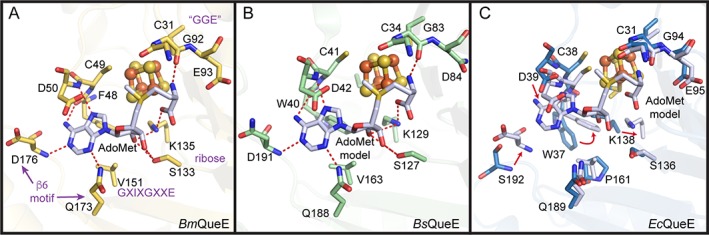
AdoMet binding pocket in QueE homologs. AdoMet binding within the AdoMet core (translucent ribbons) is shown for (A) BmQueE (PDB ID 4NJI), (B) BsQueE (PDB ID 5TH5) and (C) EcQueE. In (A), AdoMet binding motifs are labeled in magenta. See Fig. S3 for stereo views and further description of AdoMet binding. The binding pockets are composed of residues (sticks), which can provide hydrogen bonds (red) to AdoMet (white). The irons (orange) and the sulfurs (yellow) of the [4Fe‐4S] AdoMet radical cluster are shown as spheres. In (B), the intact AdoMet molecule is modeled using the adenosyl moiety of the 6‐carboxypterin‐5′‐deoxyadenosyl ester adduct (PDB ID 5TH5) and an intact AdoMet molecule (PDB ID 4NJI) as a guide. The AdoMet binding pocket of EcQueE (blue) is shown overlaid with the binding pocket from BmQueE (white) to highlight the changes that need to be made (red arrows) to allow binding of the modeled AdoMet (white) molecule.
Substrate binding motifs appear conserved among QueE homologs
Structures of BmQueE have been determined that depict the binding sites for substrate CPH4, product CDG, and the catalytically essential Mg2+ ion (Table S1). Given that analogous structures could not be obtained for the Bs and Ec enzymes, we used a BmQueE structure (PDB ID 4NJI) [Fig. 6(A) and (D)] to model substrate binding to BsQueE [Fig. 6(B) and (E)], and to EcQueE [Fig. 6(C) and (F)].14, 18 The QueE active site is found in the lateral opening of the partial TIM barrel and consists of residues from the AdoMet radical core and N‐ and C‐terminal extensions. The pterin ring of substrate is oriented in the active site through several interactions, including π‐π stacking with His and Phe residues in both BmQueE and BsQueE. In the EcQueE structure, T216 appears to be oriented to in place of these π–π stacking interactions with the substrate, but it is difficult to tell if this will remain true once substrate binds as the residues following T216, which includes H217, are disordered. In addition, the disordered C‐terminus does not allow for visualization of the C‐terminal plug in EcQueE, which is provided by the carboxylate moiety of the final residue of the protein, P210 in BmQueE or V243 in BsQueE. These C‐terminal residues provide interactions to the N2 exocyclic amino group, N3 and the C4 carbonyl group, whereas residues R27 and T90 in BmQueE, R30 and S81 in BsQueE, and possibly R27 and T92 in EcQueE position the C6 carboxyl group. Hydrogen bonds from the backbone of N‐terminal residues, G14 and L12 in BmQueE and EcQueE and G17 and Ile15 in BsQueE, further position substrate in the active site (Fig. 6). All QueE homologs tested require Mg2+ for catalysis,14 and the structure of BmQueE revealed its binding site [Fig. 6(A) and (D)]. Only one residue, T51 (BmQueE), directly interacts with the catalytic metal, Mg2+, and residues D50 and H204 (BmQueE) indirectly interact with the Mg2+ through water molecules [Fig. 6(D)]. Corresponding residues are S43, D42, and H223 in BsQueE [Fig. 6(E)] and with rearrangement upon ligand binding, T40, D39, and T216 in EcQueE [Fig. 6(F)]. Although Bs and Ec QueE structures do not have Mg2+ bound, water molecules are present in these structures that are already positioned for interaction with Mg2+.
Figure 6.
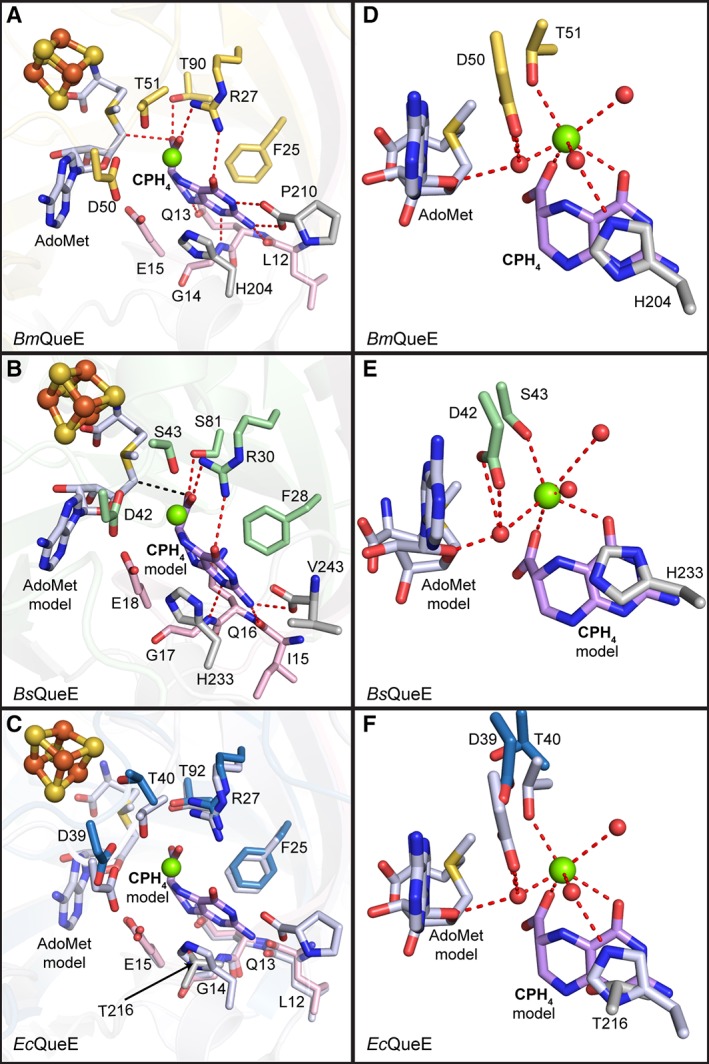
Substrate binding pocket. Residues (in sticks) that comprise the substrate‐binding pocket are shown for each of the QueE homologs. (A) CPH4 is bound to the active site by residues from the N‐terminal extension (pink), the AdoMet radical core fold (yellow) and the C‐terminal extension (grey) of BmQueE (PDB ID 4NJI). (B) In the modeled orientation, CPH4 appears to interact with the AdoMet radical domain (green) of BsQueE in addition to the N‐ and C‐terminal extensions, colored pink and grey, respectively. (C) CPH4 modeled into EcQueE. AdoMet radical domain in blue and N‐ and C‐terminal extensions in pink and grey, respectively, are shown overlaid with the active site of BmQueE (white). (D) CPH4 binding in BmQueE (PDB ID 4NJI) (yellow) creates a magnesium‐binding site. (E) CPH4 binding in BsQueE (PDB ID 5TH5) (green) is expected to create a magnesium‐binding site similar to that seen in BmQueE. (F) The putative magnesium‐binding site of EcQueE (blue) is shown overlaid with the CPH4 bound BmQueE (PDB ID 4NJI) (white). The substrate, CPH4, is shown in lilac, the catalytically essential magnesium is represented as a green sphere, the irons (orange) and the sulfurs (yellow) of the [4Fe–4S] AdoMet radical cluster are shown as spheres, AdoMet is shown in light blue and hydrogen bonds are shown as red dashes. Water molecules (red spheres) necessary for magnesium binding are shown.
EcQueE and EcFldA show surface charge–charge complementarity
Flavodoxins are small (~20 kDa) FMN‐containing proteins with limited sequence conservation (Fig. 7), but with a shared overall structural fold. They use a Rossmann‐like fold with a five‐stranded parallel β‐sheet that is surrounded by five helices (Figs. 7 and 8)23 to bind their cofactor FMN. Although no structure of a flavodoxin bound to an AdoMet radical enzyme has been determined, flavodoxin must make protein–protein contacts with the AdoMet radical enzyme in the vicinity of its [4Fe–4S] cluster to afford for facile electron transfer.24
Figure 7.

Flavodoxins sequence alignment. Sequences include flavodoxins from Helicobacter pylori, Escherichia coli, Anacystis nidulans, Aquifex aeolicus, Desulfovibrio gigas, Clostridium beijerinckii, Streptococcus pneumonia TIGR4, Bacillus subtilis (YkuN), Bacteroides fragilis NCTC 9343, and Burkholderia multivorans. The sequence alignment is colored according to secondary structure, blue for β‐strands and red for α‐helices, and the insertion for long chain flavodoxins and the chain insertion in flavodoxins from B. multivorans and B. fragilis are denoted with a box.
Figure 8.
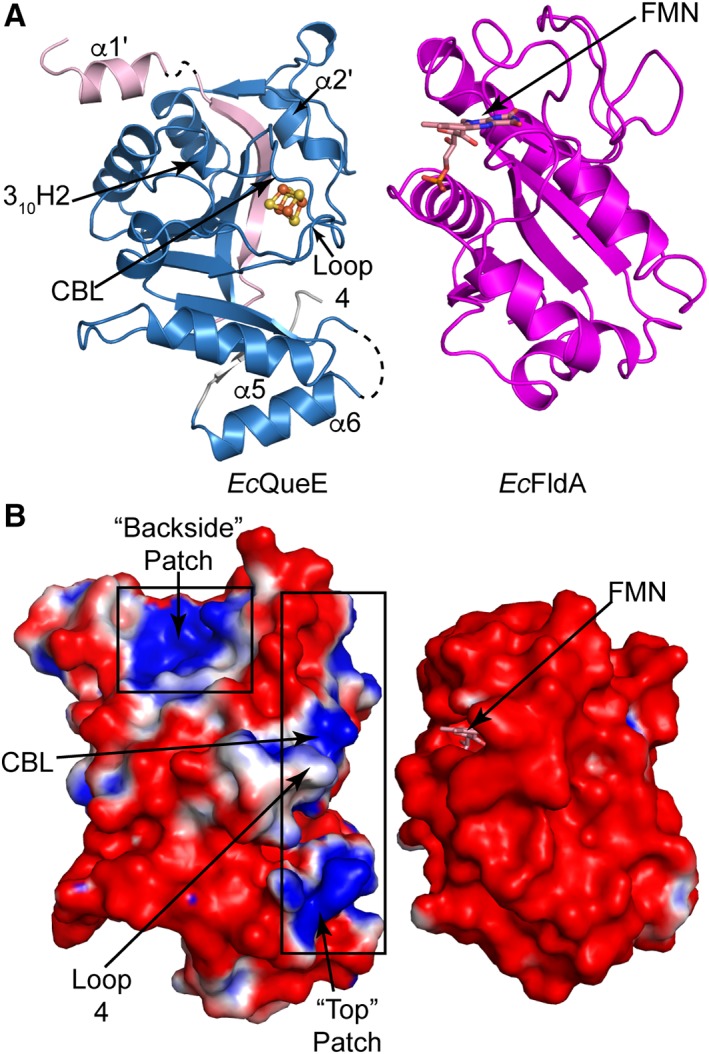
Electrostatic surface charge for EcQueE and the cognate Fld, EcfldA. (A) Ribbon drawing of monomer of EcQueE with the AdoMet radical core in blue and the N‐ and C‐terminals in light pink and grey, respectively, oriented such that the predicted binding sites are facing EcFldA. The structure of EcFldA (PDB ID 1AHN) is also shown in ribbon representation (magenta) with the FMN cofactor and the loops proposed to bind partner proteins facing EcQueE. (B) The solvent accessible electrostatic surface representations of EcQueE and EcFldA with FMN colored salmon are also displayed in the same orientation as in A. Electrostatic potentials are depicted on a colorimetric scale from red to blue for −1 to +1 kTe−1.
Electrostatics are a major driving force in protein–protein interactions, therefore we calculated the electrostatic surfaces for both our structure of EcQueE and the published structure of EcFldA.25 The surface of EcQueE is mainly negative with a positive strip running along the “top” of the partial TIM barrel, i.e. at C‐terminal ends of β‐strands where the cluster‐binding loop (CBL) and [4Fe–4S] cluster reside (Fig. 8). This “top positive patch” is made up of residues from α2′, the CBL, and the loop between β4 and the shortened α4 (Loop 4), the loop between β5 and α5 and the loop between β6 and α6 (Fig. 8). Another area with a positive electrostatic surface is found on the back side of the AdoMet radical barrel, made up of residues from the loop between α1′ and β1′ and the loop between β2 and α2.
The electrostatic surface of EcFldA is also largely negative (Fig. 8) with one major positive patch of electrostatic surface on the opposite side of the FMN binding pocket, corresponding to residues 20–30 and residues from the C‐terminal region. The electrostatic surface surrounding the FMN cofactor is negative and is therefore complementary to the large positive patch composed chiefly of residues from CBL and Loop 4 [Fig. 8(B)].
In addition to this charge complementary between the surface of the [4Fe–4S] cluster binding region of EcQueE and the surface of EcFldA, there is shape complementarity as well. The surface of EcQueE near the cluster, the “top” patch, appears to be a lock‐and‐key match with the surface of EcFldA that displays the FMN (Fig. 8).
QueE homologs display variable surfaces
The variations in fold between QueE homologs, from the replacement of helices with loops and the substitutions of long helices with shorter helices or with 310 turns, create a very different overall shape for these QueE enzymes (Fig. 9). BsQueE, which is the most traditional of the three QueEs in terms of the larger AdoMet radical enzyme family, has a monomeric unit whose overall shape is most barrel‐like and most spherical [Fig. 9(C)]. In contrast, the shorter helices or lack of helices in Ec and BmQueE homologs generate structures that are flatter by comparison with BsQueE (Fig. 9). Additionally, the electrostatic surfaces of these three QueE homologs are quite different (Fig. 9). The electrostatic surface of BmQueE is largely negatively charged with small positively charged patches [Fig. 9(A)]. In contrast, the electrostatic surfaces of the BsQueE [Fig. 9(C)] and EcQueE [Fig. 9(B)] contain considerably larger positive patches. Despite the difference in the sizes of the positive patches, the locations of these patches are similar. All QueEs have a “top” positive patch near the cluster binding loop (CBL), and a “backside” patch that corresponds to loops and α‐helices (α5 and α6, in particular) that flank the outside of the barrel (Fig. 9). The “top” patch, which is very large in BsQueE, is created by a number of secondary structural elements that surround the [4Fe–4S] cluster, including the N‐ and C‐terminal ends of the CBL, αT2, β2′, 310H2, loops following β3 (Loop 3) and β4 (Loop 4), the N‐terminal ends of β1′, α2, and α4 and the C‐terminal end of α5 [Fig. 9(C)]. In EcQueE, the corresponding “top” positive patch, which is intermediary in size between the Bm and Bs enzymes, is generated by residues of the CBL, αT1, α2′, the N‐terminus of the loop following β4 (Loop 4) and the N‐terminal ends of α5 and α6 [Fig. 9(B)]. BmQueE's smaller “top” patch is made up of β1′ and β2′, and N‐ and C‐termini of the CBL [Fig. 9(A)].
Figure 9.
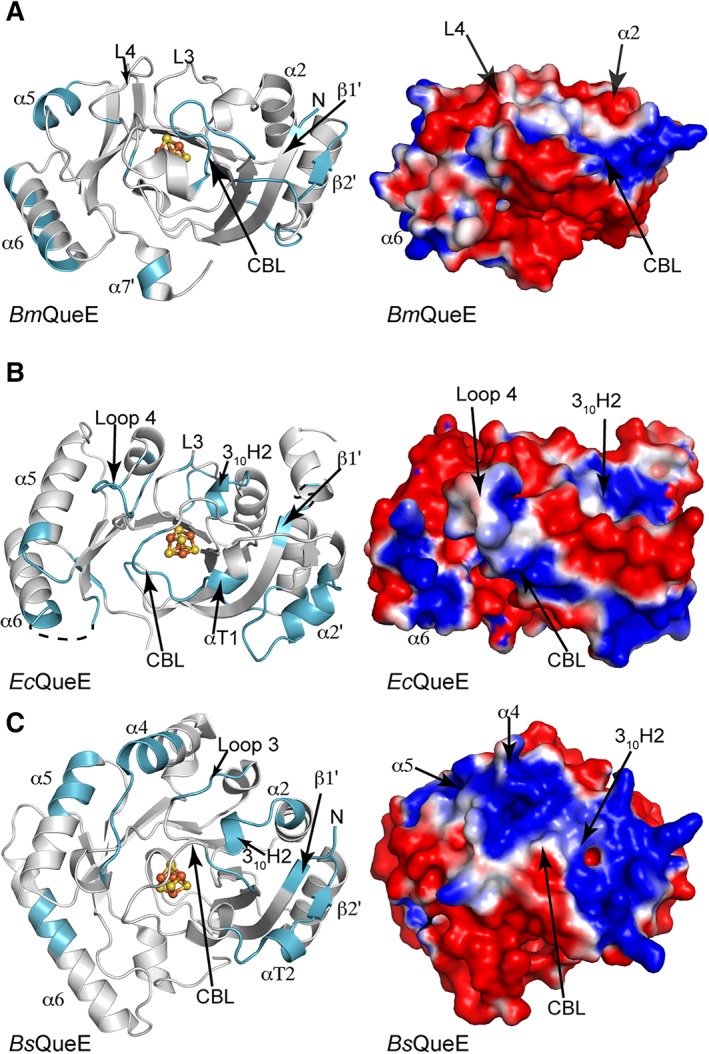
QueE orthologs show differential electrostatic surfaces. Ribbon drawing of QueE orthologs, shown as monomers in grey, with the structural elements contributing to the positive electrostatic surface highlighted in cyan. The electrostatic surface potential is shown colored from red to blue for −1 to +1 kTe−1 on the right of each panel for the corresponding orientation of the QueE orthologs. (A) BmQueE, (B) EcQueE, and (C) BsQueE.
Discussion
Here, we present the structure of EcQueE in the absence of substrate and compare this structure with previous QueE structures from Bm and Bs. Interestingly, these three QueEs, which all catalyze the exact same reaction, are farther apart in sequence space than are other AdoMet radical enzymes that catalyze completely different reactions.14 BmQueE is an outlier in an AdoMet radical enzyme superfamily with a minimal AdoMet core fold of (β6/α3) instead of the classic (β/α)6 partial TIM barrel fold, but these architectural differences are not required for 7‐carboxy‐7‐deazaguanine synthesis as the enzyme variant from Bs has a much more traditional (β6/α5) fold.18 It does not appear that the minimalist BmQueE is a one‐off outlier either. If this were the case, we would expect that all other QueE enzymes would look like BsQueE and they do not. This third QueE structure shows that the Ec enzyme has a fold that is intermediate between the other two QueEs. Further, the QueE sequence similarity network (SSN) (Fig. 2) suggests that even more variation is likely, with unexplored sequence space appearing to represent QueEs that will be even more distantly related than Bs, Ec, and Bm enzymes are to each other. Interestingly, there are QueEs of various lengths in all of the sequence clusters (Fig. 2), indicating that sequence length and sequence conservation are not highly correlated. What is the purpose of this QueE structural diversity? With the structural data that we now have in hand, we evaluated the relationship between fold variation and AdoMet binding, substrate binding, Mg2+ ion binding, and flavodoxin binding, and propose that the QueE structural variation is most likely in response to flavodoxin variations for the reasons outlined below.
Although we were not able to obtain a structure of Ec or BsQueEs with AdoMet, structural comparisons suggest that AdoMet binding residues are conserved. The Ec structure indicates that residues are not pre‐organized to bind AdoMet, but with modest side chain rearrangements, the binding pocket for the AdoMet cofactor is expected to be analogous to that visualized in the BmQueE structure. Similarly, side chain rearrangements are required for CPH4 binding in EcQueE, but sequence conservation and structural conservation near the active site predict an identical substrate binding mode in Ec and in BmQueE. BsQueE is also expected to bind substrate in an analogous fashion.18 Finally, the binding site for the required Mg2+ ion appears to be conserved. Thus, there is no indication that the sequence and structural diversity displayed by the QueE enzyme family is related to cofactor or substrate binding or to any variation in the enzyme mechanism used for 7‐carboxy‐7‐deazaguanine synthesis.
In contrast to the observations about AdoMet, substrate and Mg2+ binding, the QueE structures do appear to vary in the surface regions around the [4Fe–4S] cluster where flavodoxin must bind to deliver an electron to initiate radical generation. With the structure of EcFldA known,25 the determination of the QueE structure from E. coli provides the opportunity to evaluate the interaction surfaces for a physiological AdoMet radical enzyme–flavodoxin pair. EcFldA is a small, highly negatively charged protein that contains a partially exposed FMN cofactor, and here we find that EcQueE has a complementary positively charged patch surrounding the [4Fe–4S] cluster binding region (Fig. 9). Most of the rest of the surface of EcQueE is negatively charged, which should restrict non‐productive binding events. Additionally, the shape complementarity of the “top” ([4Fe–4S]‐cluster binding region) of EcQueE and the FMN‐exposed side of EcFldA is remarkable (Fig. 8). Protrusions of the EcFldA surface are matched with indentations in the EcQueE surface. The bringing together of these structurally and electrostatically complementary surfaces will juxtapose the FMN of EcFldA and the [4Fe–4S] cluster of EcQueE, facilitating electron transfer for this physiological redox protein pair.
Interestingly, structural comparisons of the three QueE homologs show substantial differences in shape and charge (Fig. 9). The replacement of helices with loops and variations in helical lengths observed for these QueE enzymes has the net effect of changing the overall shape of the monomeric unit. These structural differences along with sequence variations alter the electrostatic charge of the resulting surfaces. These structural observations are consistent with the report that EcFldA does not promote the CDG synthesis activity of all three QueEs uniformly. EcFldA works to some degree with BsQueE; it is more effective than chemical dithionite in promoting CDG synthesis, but falls short of the activity observed with the B. subtilis partner proteins. EcFldA, on the other hand, is less effective than chemical dithionite in promoting activity of BmQueE.14, 15 In addition to lower turnover numbers when a non‐physiological FldA is used, the ratio of AdoMet abortive cleavage events to turnover events also increases.15 Given the shape differential between EcQueE and BsQueE (Fig. 9), it is a bit surprising that EcFldA is able to work as well as it does. It is likely that the large positively charged patches on BsQueE compensate for imperfect shape complementarity. BmQueE, in contrast, lacks shape complementarity, lacks the large surface positive charge (Fig. 9) and is not activated to a significant extent by EcFldA. BmQueE is the most negatively charged QueE, and given that flavodoxins tend to be negatively charged, it is tempting to speculate that the cognate Bm flavodoxin may be atypical. No structures are known of any of the three flavodoxins from Bm, but a sequence of one of the Bm flavodoxins that is shown in Figure 7 does suggest that this BmFld will be unusual. In particular, it has two inserts that are not present in the EcFldA or in most other flavodoxins (Fig. 7). Taken together, these data are in agreement with the idea that structural variations observed in QueEs may be matched with changes to their cognate flavodoxins or other biological reductant. In short, structures of EcQueE and EcFldA help us understand why these partner proteins work well together, and QueE structural comparisons provide explanations of why turnover is lower and abortive cleavage is higher when EcFldA is paired with BmQueE or BsQueE. Structures of flavodoxins from Bm will provide further validation of this idea and will allow us to understand how the unusual BmQueE is activated for catalysis in vivo.
Conclusion
Although variations to the AdoMet radical core have been observed before outside of the QueE system, these changes have been attributed to tailoring of the enzyme to the chemistry performed and/or substrate binding. Structural analysis of three QueE homologs, which perform identical chemistry on the same substrate, revealed both structural and electrostatic differences. We believe these variations serve to dictate binding to their cognate biological reductant. Charge–charge complementarity could serve as a hard discriminant, preventing flavodoxins with incompatibly charged surfaces from binding to QueE. Surface complementarity (dictated by the structure) can further fine‐tune these interactions, allowing for activation of the enzyme. It is only when there is both charge and surface complementarity that full activation of the enzyme occurs. Thus, we expect some sort of co‐evolution of flavodoxins–ligand pairs, to allow for complementarity needed for optimal activation.
Materials and Methods
Preparation of EcQueE
The gene corresponding to QueE was cloned from E. coli W3110 into the NdeI and HindIII sites of pET28a for expression of His6tagged protein. Expression, purification, reconstitution, and activity assays were carried out as described previously for BsQueE.16
Crystallization and data collection of EcQueE
Crystallization conditions for His6tagged EcQueE were initially identified by sparse matrix screening within a room temperature MBraun anaerobic chamber using a TTP Mosquito pipetting robot and optimized by sitting drop vapor diffusion within a Coy scientific anaerobic chamber. Data quality crystals were obtained by equilibrating drops containing 1.5 μL of protein (10 mg/mL in 50 mM Tris•HCl pH 8.0 and 10 mM dithiothreitol) and 0.5 μL of reservoir (175–200 mM magnesium chloride, 25–30% PEG 400 and 100 mM Tris•HCl pH 8.5) over a reservoir of 500 μL. Brown 200–300 μm × 30 μm rod‐like crystals were obtained after 24 h. Crystals were harvested from the mother liquor with no further cryoprotecting and cryo‐cooled in liquid nitrogen within the Coy anaerobic chamber.
Diffraction data were collected at the Advanced Photon Source (Argonne, IL) at beamline 24‐ID‐C, using a Pilatus 6 M pixel detector at 100 K. Data were collected on the same crystal at two different wavelengths. An Fe‐peak data set was collected in six 35° wedges using an inverse beam method (Friedel mates were measured consecutively, rotating the crystal 180° every 120 frames with 0.3° oscillation steps and an exposure time of 0.3 s) at a wavelength of 1.7384 Å to 2.6‐Å resolution. The remote data set was collected at a wavelength of 0.9792 Å to 2.1‐Å resolution, using the continuous vector scan method (the crystal was continuously translated along its major crystallographic axis during data collection). All data were processed in HKL200026 in the space group P212121.
Structure determination and refinement
The structure of EcQueE with two molecules in the asymmetric unit was solved using Fe multi‐wavelength anomalous dispersion (MAD) phasing. Two Fe sites were identified with occupancies above 0.9 using the remote and peak data sets trimmed to 4‐Å resolution in ShelxD/E27 in HKL2MAP.28 Heavy atom site refinement, experimental map generation, automated model building and density modification were performed in SOLVE and RESOLVE in Phenix AutoSol.29 The figure of merit‐weighted electron density map (FOM = 0.64 to 4‐Å resolution) obtained was sufficient for tracing protein secondary structure elements manually in Coot. The automated model was extensively rebuilt to produce a model for one monomer in the asymmetric unit. The second monomer was placed in the asymmetric unit using Phenix AutoBuild30 and the resulting model was subjected to iterative rounds of refinement and density modification using Resolve,30 and phenix.refine,31 respectively, and the resolution was extended to the full‐length of the data, 2.1‐Å resolution. The resulting R‐factors were 25.6% and 29.9% working and free R‐factors, respectively.
Iterative rounds of model building in Coot32 and refinement in Phenix31 using atomic coordinates, atomic displacement parameters (B‐factors), and non‐crystallographic symmetry (NCS) restraints, without sigma cutoffs, completed the model. In advanced stages of refinement, water molecules were manually added in Coot32 and in final stages, NCS restraints were released and refinement included translation, libration, screw (TLS) parameterization with one TLS group per monomer. The model was validated using simulated annealing composite omit maps calculated in Phenix. Analysis of geometry using MolProbity33 indicates that 96.45%, 3.55%, and 0.0% of residues were in the favored, allowed, and disallowed regions of the Ramachandran plot, respectively. The final structure of EcQueE contains 224 residues (out of 243) and a [4Fe‐4S] cluster in chain A and 229 residues (out of 243) and a [4Fe–4S] cluster in chain B. In both chains, the His6tag linker region containing the Tobacco Etch Virus (TEV) protease cleavage site is visible as well as a His residue from the His6tag. Crystallography software packages were compiled by SBGrid.34
Manual docking of AdoMet and substrates
Docking of AdoMet, and CPH4 molecules into EcQueE was performed manually in Coot32 using BmQueE (PDB ID 4NJI) as a guide. In BsQueE (PDB ID 5TH5), 6‐CP‐dAdo binding foretold the binding interactions of AdoMet and substrate, therefore, the adduct was used, in addition to BmQueE (PDB ID 4NJI), to configure intact AdoMet and CPH4 in the active site of EcQueE.
Preparation of Figures and electrostatic surfaces
All crystallographic figures were created with PyMOL Software and electrostatic surface potentials were calculated using the Adaptive Poisson–Boltzmann Solver plugin implemented in PyMOL, using default parameters.35, 36, 37
Supporting information
Appendix S1: Supporting Information
Acknowledgments
This research was supported by National Institutes of General Medical Sciences of the National Institutes of Health grants R35 GM12682 (awarded to C.L.D.) and R01 GM72623 (awarded to V.B.) with Administrative Supplement GM72623 S01 to V.B. for the collaboration between V.B. and C.L.D, R35 GM126982 (awarded to C.L.D.), a National Science Foundation Graduate Research Fellowship under Grant no. 1122374 (awarded to T.A.J.G.), a Helen Hay Whitney Fellowship (awarded to C.N), and funding from the MIT UROP office (B.B.). C.L.D. is a Howard Hughes Medical Institute Investigator. The work presented here is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Pilatus 6 M detector on 24‐ID‐C beamline is funded by a NIH‐ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE‐AC02‐06CH11357.
References
- 1. Fujii K, Huennekens F (1974) Activation of methionine synthetase by a reduced triphosphopyridine nucleotide‐dependent flavoprotein system. J Biol Chem 249:6745–6753. [PubMed] [Google Scholar]
- 2. Knappe J, Schacht J, Möckel W, Höpner T, Vetter H Jr, Edenharder R (1969) Pyruvate formate‐lyase reaction in Escherichia coli: The enzymatic system converting and inactive form of the lyase into the catalytically active enzyme. Eur J Biochem 11:316–327. [DOI] [PubMed] [Google Scholar]
- 3. Broderick JB, Duffus BR, Duschene KS, Shepard EM (2014) Radical S‐adenosylmethionine enzymes. Chem Rev 114:4229–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jarrett JT (2003) The generation of 5′‐deoxyadenosyl radicals by adenosylmethionine‐dependent radical enzymes. Curr Opin Chem Biol 7:174–182. [DOI] [PubMed] [Google Scholar]
- 5. Harder J, Eliasson R, Pontis E, Ballinger MD, Reichard P (1992) Activation of the anaerobic ribonucleotide reductase from Escherichia coli by S‐Adenosylmethionine. J Biol Chem 267:25548–25552. [PubMed] [Google Scholar]
- 6. Bianchi V, Reichard P, Eliasson R, Pontis E, Krook M, Jörnvall H, Haggård‐Ljungquist E (1993) Escherichia coli ferredoxin NADP+ reductase: activation of E. coli anaerobic ribonucleotide reduction, cloning of the gene (fpr), and overexpression of the protein. J Bacteriol 175:1590–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ifuku O, Koga N, Haze S‐I, Kishimoto J, Wachi Y (1994) Flavodoxin is required for conversion of dethiobiotin to biotin in Escherichia coli . Eur J Biochem 224:173–178. [DOI] [PubMed] [Google Scholar]
- 8. Bianchi V, Eliasson R, Fontecave M, Mulliez E, Hoover DM, Matthews RG, Reichard P (1993) Flavodoxin is required for the activation of the anaerobic ribonucleotide reductase. Biochem Biophys Res Commun 197:792–797. [DOI] [PubMed] [Google Scholar]
- 9. Brazeau BJ, Gort SJ, Jessen HJ, Andrew AJ, Liao HH (2006) Enzymatic activation of lysine 2,3‐aminomutase from Porphyromonas gingivalis . Appl Environ Microbiol 72:6402–6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cicchillo RM, Iwig DF, Jones AD, Nesbitt NM, Baleanu‐Gogonea C, Souder MG, Tu L, Booker SJ (2004) Lipoyl synthase requires two equivalents of S‐adenosyl‐L‐methionine to synthesize one equivalent of lipoic acid. Biochemistry 43:6378–6386. [DOI] [PubMed] [Google Scholar]
- 11. Layer G, Verfurth K, Mahlitz E, Jahn D (2002) Oxygen‐independent coproporphyrinogen‐III oxidase HemN from Escherichia coli . J Biol Chem 277:34136–34142. [DOI] [PubMed] [Google Scholar]
- 12. Grove TL, Ahlum JH, Qin RM, Lanz ND, Radle MI, Krebs C, Booker SJ (2013) Further characterization of Cys‐type and Ser‐type anaerobic sulfatase maturating enzymes suggests a commonality in the mechanism of catalysis. Biochemistry 52:2874–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grove TL, Lee K‐H, St Clair J, Krebs C, Booker SJ (2008) In vitro characterization of AtsB, a radical SAM formylglycine‐generating enzyme that contains three [4Fe‐4S] clusters. Biochemistry 47:7523–7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dowling DP, Bruender NA, Young AP, McCarty RM, Bandarian V, Drennan CL (2014) Radical SAM enzyme QueE defines a new minimal core fold and metal‐dependent mechanism. Nat Chem Biol 10:106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bruender NA, Young AP, Bandarian V (2015) Chemical and biological reduction of the radical SAM enzyme CPH4 synthase. Biochemistry 54:2903–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McCarty RM, Krebs C, Bandarian V (2012) Spectroscopic, steady‐state kinetic, and mechanistic characterization of the radical SAM enzyme QueE, which catalyzes a complex cyclization reaction in the biosynthesis of 7‐deazapurines. Biochemistry 52:188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCarty RM, Somogyi Á, Lin G, Jacobsen NE, Bandarian V (2009) The deazapurine biosynthetic pathway revealed: in vitro enzymatic synthesis of preQ0 from guanosine 5′‐triphosphate in four steps. Biochemistry 48:3847–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bruender NA, Grell TA, Dowling DP, McCarty RM, Drennan CL, Bandarian V (2017) 7‐Carboxy‐7‐deazaguanine synthase: a radical S‐adenosyl‐L‐methionine enzyme with polar tendencies. J Am Chem Soc 139:1912–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Layer G, Moser J, Heinz DW, Jahn D, Schubert W‐D (2003) Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of radical SAM enzymes. EMBO J 22:6214–6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vey JL, Drennan CL (2011) Structural insights into radical generation by the radical SAM superfamily. Chem Rev 111:2487–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vey JL, Yang J, Li M, Broderick WE, Broderick JB, Drennan CL (2008) Structural basis for glycyl radical formation by pyruvate formate‐lyase activating enzyme. Proc Natl Acad Sci USA 105:16137–16141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dowling DP, Vey JL, Croft AK, Drennan CL (2012) Structural diversity in the AdoMet radical enzyme superfamily. Biochim Biophys Acta 1824:1178–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sancho J (2006) Flavodoxins: sequence, folding, binding, function and beyond. Cell Mol Life Sci 63:855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Page CC, Moser CC, Chen X, Dutton PL (1999) Natural engineering principles of electron tunnelling in biological oxidation–reduction. Nature 402:47–52. [DOI] [PubMed] [Google Scholar]
- 25. Hoover DM, Ludwig ML (1997) A flavodoxin that is required for enzyme activation: the structure of oxidized flavodoxin from Escherichia coli at 1.8 Å resolution. Protein Sci 6:2525–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Minor W, Otwinowski Z. HKL2000 (Denzo‐SMN) software package. Processing of X‐ray diffraction data collected in oscillation mode, 1997. Methods in Enzymology, Macromolecular Crystallography. New York: Academic Press. [DOI] [PubMed] [Google Scholar]
- 27. Sheldrick GM (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Cryst D66:479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pape T, Schneider TR (2004) HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J Appl Cryst 37:843–844. [Google Scholar]
- 29. Terwilliger TC, Adams PD, Read RJ, McCoy AJ, Moriarty NW, Grosse‐Kunstleve RW, Afonine PV, Zwart PH, Hung L‐W (2009) Decision‐making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Cryst D65:582–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Terwilliger TC, Grosse‐Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung L‐W, Read RJ, Adams PD (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Cryst D 64:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L‐W, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarity NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Cryst D 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 33. Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Cryst D 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, Sliz P (2013) Cutting edge: collaboration gets the most out of software. elife 2:e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schrödinger L (2010) PyMOL The PyMOL Molecular Graphics System. Version.
- 36. DeLano WL (2002) The PyMOL molecular graphics system. http://pymol.org.
- 37. Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, Brookes DH, Wilson L, Chen J, Liles K (2018) Improvements to the APBS biomolecular solvation software suite. Protein Sci 27:112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Atkinson HJ, Morris JH, Ferrin TE, Babbitt PC (2009) Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PloS one 4:e4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27:431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information