

Abstract
Many flavoenzymes catalyze hydroxylation of aromatic compounds especially phenolic compounds have been isolated and characterized. These enzymes can be classified as either single‐component or two‐component flavin‐dependent hydroxylases (monooxygenases). The hydroxylation reactions catalyzed by the enzymes in this group are useful for modifying the biological properties of phenolic compounds. This review aims to provide an in‐depth discussion of the current mechanistic understanding of representative flavin‐dependent monooxygenases including 3‐hydroxy‐benzoate 4‐hydroxylase (PHBH, a single‐component hydroxylase), 3‐hydroxyphenylacetate 4‐hydroxylase (HPAH, a two‐component hydroxylase), and other monooxygenases which catalyze reactions in addition to hydroxylation, including 2‐methyl‐3‐hydroxypyridine‐5‐carboxylate oxygenase (MHPCO, a single‐component enzyme that catalyzes aromatic‐ring cleavage), and HadA monooxygenase (a two‐component enzyme that catalyzes additional group elimination reaction). These enzymes have different unique structural features which dictate their reactivity toward various substrates and influence their ability to stabilize flavin intermediates such as C4a‐hydroperoxyflavin. Understanding the key catalytic residues and the active site environments important for governing enzyme reactivity will undoubtedly facilitate future work in enzyme engineering or enzyme redesign for the development of biocatalytic methods for the synthesis of valuable compounds.
Keywords: phenolic compounds, flavoenzyme‐catalyzed hydroxylation, flavin‐dependent hydroxylase, flavin‐dependent dehalogenases, flavin‐dependent catalyzed oxidative aromatic ring‐cleavage reactions
Abbreviations
- 3,4,5‐THCA
3,4,5‐trihydroxycinnamic acid
- 3,4,5‐THPA
3,4,5‐trihydroxyphenyl acetic acid
- 3,4‐DOHB
3,4‐dihydroxybenzoate
- 5HN
5‐hydroxynicotinic acid
- 5PAO
5‐pyridoxic acid oxygenase
- AAMS
α‐(N‐acetylaminomethylene) succinic acid
- C1
reductase component of p‐hydroxyphenylacetate 3‐hydroxylase from Acinetobacter baumannii
- C2
oxygenase component of p‐hydroxyphenylacetate 3‐hydroxylase from Acinetobacter baumannii
- DFT
density functional theory
- DHPA
3,4‐dihydroxyphenylacetate
- FADH−
reduced FAD
- FMNH−
reduced FMN
- HPA
4‐hydroxyphenylacetate
- HPAH
3‐hydroxyphenylacetate 4‐hydroxylase
- MHPC
2‐methyl‐3‐hydroxypyridine‐5‐carboxylate
- MHPCO
2‐methyl‐3‐hydroxypyridine‐5‐carboxylate oxygenase
- NMHN
N‐methyl‐5‐hydroxynicotinic acid
- P2O
pyranose 2‐oxidase
- PHBH
3‐hydroxy‐benzoate 4‐hydroxylase
- pOHB
4‐hydroxybenzoate
- QM/MM
quantum mechanical and molecular mechanical
- QSAR
quantitative structure and reactivity relationship
Introduction
Aromatic compounds in the phenolic acid family are some of the most abundant compounds found in nature. These compounds make up a significant part of lignocelluose, various natural products, and man‐made chemicals, and have a wide spectrum of applications in chemical, agricultural, food, and pharmaceutical industries.1, 2, 3 Many phenolic acids have useful biological activities such as antioxidant, anticancer, antimicrobial, antifungal, and anti‐inflammatory activities, and are widely used in the pharmaceutical and food industries.3 Hydroxylation of phenolic acid can generally increase the radical scavenging ability of the compound and may also increase other biological activities. For example, 3,4,5‐trihydroxycinnamic acid (3,4,5‐THCA), 3,4,5‐trihydroxyphenylacetic acid (3,4,5‐THPA), and gallic acid were reported to have greater antioxidant and anti‐inflammatory effects than their corresponding non‐hydroxylated phenolic acids which contain only the single hydroxyl group.4, 5, 6 Therefore, the hydroxylation of phenolic acids, especially via enzymatic methods is important for enhancing their biological activities.3, 7
Four enzyme types have been reported to catalyze aromatic hydroxylation, including the heme‐containing cytochrome P450, non‐heme iron, and pterin‐dependent and flavin‐dependent monooxygenases.8 This review will only cover flavin‐dependent systems and will aim to discuss in‐depth, the current mechanistic understanding of representative flavin‐dependent monooxygenases which catalyze only hydroxylation of aromatic compounds or hydroxylation plus additional reactions, and review the challenges facing their application. These flavin‐dependent enzymes discussed can be divided into two types: the first type binds flavin (oxidized FAD) as a prosthetic group (single‐component monooxygenase), while the second type does not bind tightly to oxidized flavin but rather, uses reduced flavin provided by a reductase as a substrate (two‐component monooxygenase). Representatives of both types will be discussed. We discuss monooxygenases which catalyze merely hydroxylation including 3‐hydroxy‐benzoate 4‐hydroxylase (PHBH, representative of single‐component hydroxylases) and 3‐hydroxyphenylacetate 4‐hydroxylase (HPAH, representative of two‐component hydroxylases), and also discuss monooxygenases catalyzing additional reactions in addition to hydroxylation including 2‐methyl‐3‐hydroxypyridine‐5‐carboxylate oxygenase (MHPCO, a representative single‐component enzyme that catalyzes aromatic‐ring cleavage), and HadA monooxygenase (HadA, a representative two‐component enzyme that catalyzes an additional group elimination reaction). Other flavin‐dependent monooxygenases are beyond the scope of this review, and information regarding a wide variety of reactions catalyzed by flavin‐dependent monooxygenases can be found in many excellent reviews.9, 10, 11, 12, 13, 14, 15, 16, 17, 18 In addition to mechanistic aspects, this review also discusses the potential applications of flavin‐dependent monooxygenases and the challenges currently encountered in these areas.
Overall catalytic reactions of flavin‐dependent monooxygenases
Single‐component flavin‐dependent monooxygenases
A typical reaction cycle of single‐component flavin‐dependent monooxygenases catalyzing aromatic hydroxylation can be divided into two parts – reductive and oxidative half‐reactions (Fig. 1). For the reductive half‐reaction, an aromatic substrate binds to the enzyme to form an active enzyme:substrate complex which can be reduced by NAD(P)H much faster than in the absence of the aromatic substrate (Steps 1 and 2). Single‐component monooxygenases use NAD(P)H directly as a reducing reagent to reduce their flavin cofactors, which to date, has only been reported to be FAD.10, 14, 19 For the oxidative half‐reaction, the reduced enzyme:substrate complex reacts with molecular oxygen to generate a C4a‐hydroperoxyflavin intermediate (Step 3), which further pursues hydroxylation of the aromatic substrate, resulting in hydroxylated product and C4a‐hydroxyflavin (Step 4). At the last step, the product is released from the active site and C4a‐hydroxyflavin dehydrates to regenerate oxidized flavin (Step 6). In the absence of substrate, or in the presence of compounds that can bind to the enzyme but cannot be hydroxylated, C4a‐hydroperoxyflavin only eliminates H2O2 to form the oxidized flavin (Step 7).
Figure 1.
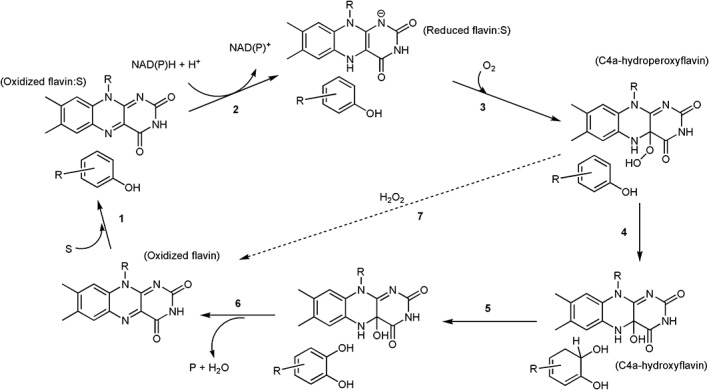
A typical catalytic cycle of phenolic hydroxylation catalyzed by single component flavin‐dependent monooxygenases (hydroxylases).
A reactive flavin species, C4a‐hydroperoxyflavin, is a key intermediate of all flavin‐dependent monooxygenases including sing‐component and two‐component types. This intermediate is the key reagent that inserts the hydroxyl group into aromatic substrates (C4a‐hydroperoxyflavin acts as an electrophile) (Fig. 2). In other types of flavin‐dependent monooxygenases which oxygenate different types of substrates such as Baeyer–Villiger monooxygenases, the reactive flavin species is instead, C4a‐peroxyflavin (C4a‐peroxyflavin acts as a nucleophile) (Fig. 2).14, 17 Monooxygenations by flavin‐dependent monooxygenases are involved in a wide variety of biological processes such as in the biodegradation of aromatic compounds in the environment, in the detoxification of drugs, and in the biosynthesis of antibiotics, hormones and vitamins.10, 14 Many of these enzymes also catalyze reactions with high regio‐ and/or stereo‐specificity, making them attractive for biocatalytic applications.3, 7, 10, 13, 14, 20, 21
Figure 2.
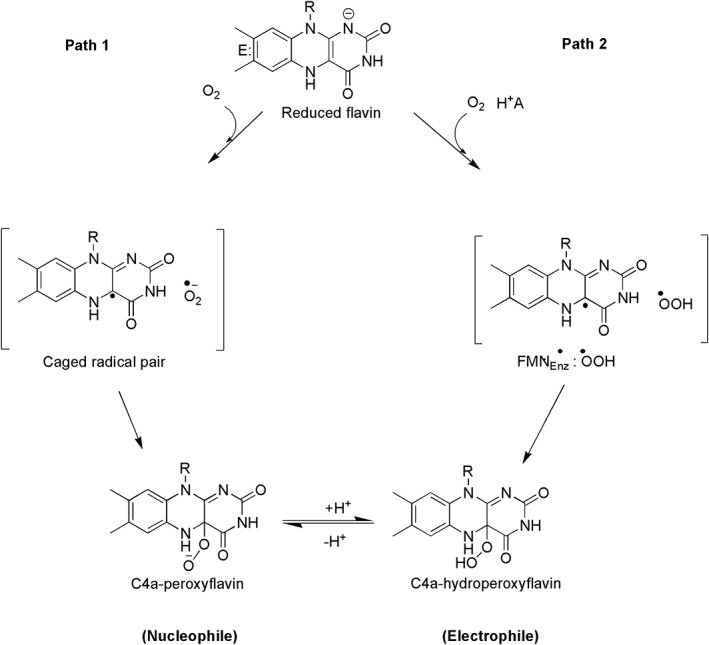
The reaction of reduced flavin with dioxygen to form C4a‐(hydro)peroxyflavin. The first step of the reaction is an electron transfer from reduced flavin to oxygen to generate a radical pair of flavin semiquionone and oxygen superoxide. Path 1, a model that has been traditionally proposed for reactions of flavin‐dependent monooxygenases, only involves electron transfer in the first step and combination of the radical pair to result in C4a‐peroxyflavin which can be further protonated in the following step. Using combined methods of theoretical chemistry and transient kinetic investigation of pyranose 2‐oxidase (P2O) and C2 monooxygenase, the electron transfer step was found to be synchronous with a proton transfer (proton‐coupled electron transfer) as in Path 2 to result in C4a‐hydroperoxyflavin.
As single‐component flavin‐dependent monooxygenases catalyze flavin reduction and substrate oxygenation in the same active site, the flavin cofactor remains bound to the protein throughout all steps of the catalytic cycle. Table 1 summarizes the biochemical and catalytic features of known single‐component monooxygenases. It is interesting to note that all of the single‐component flavin‐dependent monooxygenases use FAD as a cofactor, while FAD and FMN are both used in two‐component monooxygenases. This cofactor specificity must be due to the difference in structural motifs that interact with flavins. The overall folding of single‐component monooxygenases generally consists of three domains (Fig. 3): the FAD binding domain (gray), substrate binding domain (cyan), and dimer interface domain (purple). The FAD binding domain has a typical Rossmann fold which recognizes the ADP part of FAD, thus making FAD the only type of flavin typically found as a cofactor in single‐component monooxygenases.
Table 1.
Single Component Flavin‐Dependent Monooxygenase Catalyzed Hydroxylation of Aromatic Compounds
| Enzyme | Reaction | Crystal structure | References |
|---|---|---|---|
| 4‐Hydroxybenzoate 3‐hydroxylase (PHBH) from Pseudomonas fluorescens catalyzes the conversion of p‐hydroxybenzoate to 3,4‐dihydroxybenzoate. |
|
1PBE | 22 |
| 3‐Hydroxybenzoate‐6‐hydroxylase (3HB6H) from Rhodococcus jostii RHA1catalyzes the para‐hydroxylation of 3‐hydroxybenzoate to 2,5‐dihydroxybenzoate |
|
5HYM | 23 |
| 3‐Hydroxybenzoate 4‐hydroxylase (MHBH) from Comamonas testosteroni KH122‐3s catalyzes the conversion of 3‐hydroxybenzoate to 3,4‐dihydroxybenzoate |
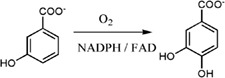
|
2DKH | 24 |
| Phenol hydroxylase (PHHY) from Trichosporon cutaneum catalyzes the ortho‐hydroxylation of phenol to catechol |
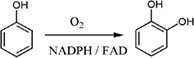
|
1FOH | 25 |
| Salicylate 1‐hydroxylase (SALH) from Pseudomonas putida S‐1 catalyzes the hydroxylation of salicylate to catechol with the release of CO2 by decarboxylation with a 1:1:1 stoichiometry |
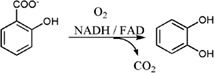
|
5EVY | 26 |
| RebC catalyzes hydroxylation in the biosynthesis of rebeccamycin in Lechevalieria aerocolonigenes |
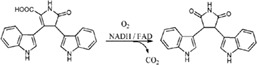
|
2R0P | 27 |
| PgaE catalyzes hydroxylation in angucycline biosynthesis in Streptomyces |
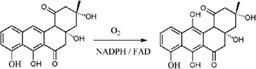
|
2QA1 | 28 |
| PhzS catalyzes hydroxylative decarboxylation in the biosynthesis of pyocyanin in Pseudomonas aeruginosa |
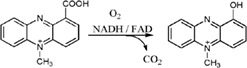
|
2RGJ | 29 |
| MHPCO catalyzes hydroxylation and ring‐opening of 2‐methyl‐3‐hydroxypyridine‐5‐carboxylic acid to form α‐(N‐acetylaminomethylene) succinic acid in Mesorhizobium loti |
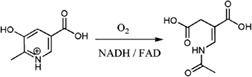
|
3GMC | 30 |
| 2,6‐Dihydroxypyridine‐3‐hydroxylase catalyzes hydroxylation of 2,6‐dihydroxypyridine to 2,3,6‐trihydroxypyridine in the nicotine degradation pathway in Arthrobacter nicotinovorans |
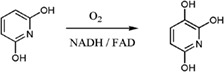
|
2VOU | 31 |
| Kynurenine 3‐monooxygenase catalyzes the hydroxylation of l‐kynurenine (l‐Kyn) to 3‐hydroxykynurenine (3OHKyn) in the pathway for tryptophan catabolism |
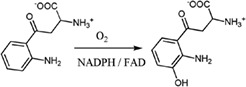
|
5Y7A | 32 |
| Aklavinone‐11‐hydroxylases catalyzes the hydroxylation of Aklavinone to ε‐rhodomycinone in the biosynthesis of anthracyclines in Streptomyces purpurascens |
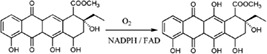
|
3IHG | 33 |
| 2‐Hydroxybiphenyl 3‐monooxygenase (HbpA) catalyzes the ortho‐hydroxylation of 2‐hydroxybiphenyl to 2,3‐dihydroxybiphenyl in the degradation of the fungicide 2‐hydroxybiphenyl |
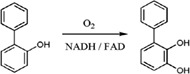
|
5BRT | 34 |
Figure 3.
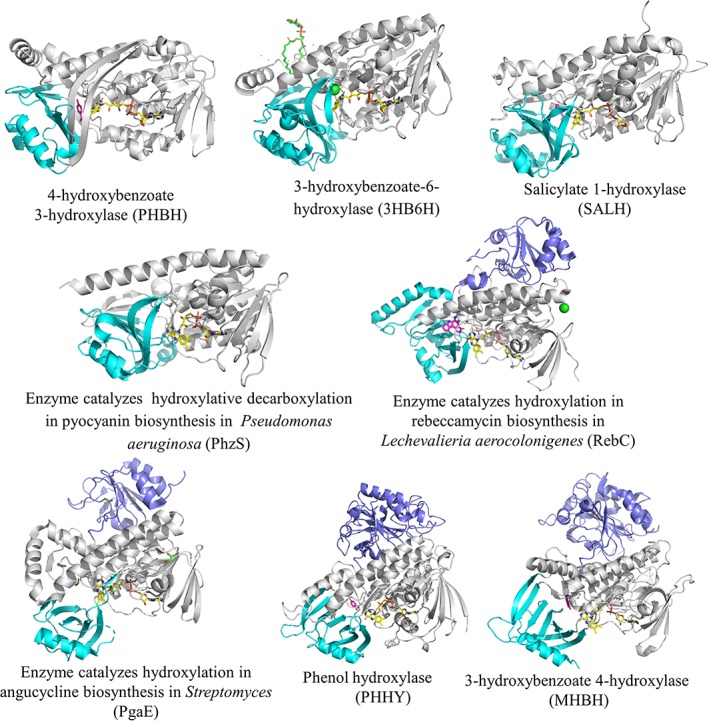
X‐ray structures of single component flavin‐dependent monooxygenases catalyze hydroxylation of aromatic compounds.
Two‐component flavin‐dependent monooxygenases
Unlike single‐component monooxygenases, two‐component flavin‐dependent monooxygenases require two proteins to carry out their reactions. The reductase is usually smaller than the oxygenase, ranging in size from 6 to 22 kDa for the reductase, and from 39 to 63 kDa for the monooxygenase (Table 2). The reductase component (C1) of p‐hydroxyphenylacetate 3‐hydroxylase (HPAH) from Acinetobacter baumannii is significantly larger (35 kDa) than those of other reductases41, 54 because C1 has an extra C‐terminal domain which serves as the binding site for the aromatic substrate which subsequently induces a conformational change in the enzyme to increase the rate of flavin reduction and release of reduced flavin.15, 55, 56
Table 2.
Two‐Component Flavin‐Dependent Monooxygenases Catalyze Hydroxylation of Aromatic Compounds
| Function | Reaction | Reductase component | Oxygenase component | References |
|---|---|---|---|---|
| Enzymes catalyze the hydroxylation of p‐hydroxyphenylacetate (HPA) yielding 3,4‐dihydroxyphenyl acetate (DHPA) in Escherichia coli,35 Pseudomonas aeruginosa, 36 Pseudomonas putida, 37 Klebsiella pneumonia, 38 Sulfolobus tokodaii, 39 Thermus thermophiles, 40 and Acinetobacter baumannii 41 |
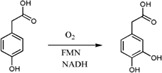
|
HpaA | HpaB | 35 |
| (17 kDa) | (59 kDa) | |||
| HpaC | HpaA | 36 | ||
| (19.4 kDa) | (58. kDa) | |||
| MhaB, | MhaA | 37 | ||
| (6 kDa) | (63 kDa) | |||
| HpaH | HpaA | 38 | ||
| (19 kDa) | (59 kDa) | |||
| HpaC | HpaB | 39 | ||
| (18 kDa) | ‐ | |||
| HpaC | HpaB | 40 | ||
| (16 kDa) | (54 kDa) | |||
| C1 | C2 | 41 | ||
| (35.5 kDa) | (47 kDa) | |||
| Enzymes catalyze hydroxylation in the first step in the degradation of phenol in Bacillus thermoglucosidasius A7 |
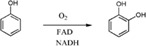
|
PheA2 | PheA1 | 42 |
| (18 kDa) | (57 kDa) | |||
| Enzymes catalyze hydroxylation involved in the antibiotic actinorhodin biosynthesis in Streptomyces coelicolor |
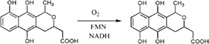
|
ActVB | ActVA | 43 |
| (18 kDa) | (39.7 kDa) | 44 | ||
| Enzymes catalyze hydroxylation of a carrier protein‐tethered substrate during the biosynthesis of the Enediyne antitumor antibiotic C‐1027 in Streptomyces globisporus |
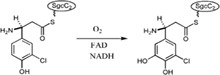
|
SgcE6 | SgcC | 45 |
| (19.5 kDa) | (57.9 kDa) | |||
| Enzymes catalyze hydroxylation of 2‐methyl‐6‐ethylaniline (MEA) in the downstream catabolic pathway of chloroacetanilide herbicides in Sphingomonads |
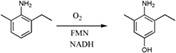
|
MeaY | MeaX | 46 |
| (21 kDa) | (46 kDa) | |||
| Enzymes catalyze hydroxylation to convert anthranilate to 3‐HAA in the initial reaction of the degradation pathway in Geobacillus thermodenitrificans NG80‐2 |
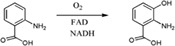
|
GTNG_3158 | GTNG_3160 | 47 |
| ‐ | ‐ | |||
| Enzymes catalyze the oxidation of 2,4,5‐trichlorophenoxyacetic acid (2,4,5‐TCP) to 2,5‐dichloro‐p‐benzoquinone and then to 2,5‐dichloro‐p‐hydroquinone (2,5‐DiCHQ). TftD then oxidizes the latter to 5‐chloro‐2‐hydroxy‐p‐benzoquinone in the degradation of 2,4,5‐TCP in Burkholderia cepacia AC1100 |
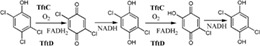
|
TftC | TftD | 48 |
| (22 kDa) | (58 kDa) | |||
| Enzymes catalyze reactions to convert 2,4,6‐trichlorophenol to 6‐chlorohydroxyquinone in the degradation of 2,4,6‐trichlorophenol in Cupriavidus necator JMP134 |
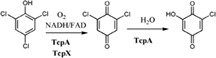
|
TcpX | TcpA | 49, 50 |
| (21 kDa) | (60.7 kDa) | |||
| Enzymes catalyze hydroxylation of 4‐nitrophenol to 4‐nitrocatechol in the 4‐NP degradation pathway in Rhodococcus sp. strain PN1 |
|
NphA2 | NphA1 | 51 |
| (20 kDa) | (53 kDa) | |||
| Enzymes catalyzes hydroxylation plus group elimination (halide) at position 4 of phenolic compounds with a para‐substituent in R. pickettii |
|
HadX | HadA | 52, 53 |
| (~19 kDa) | (~59 kDa) |
The flavin reduction in these enzymes takes place in the reductase component. These reductases typically bind FAD or FMN as cofactors or can be isolated in apoenzyme form.15, 41, 57, 58 They can be reduced directly by NAD(P)H without requiring the binding of aromatic compounds (Fig. 4, Step 1). The exception can be found in several reductases such as C1 in which aromatic compounds can bind and stimulate the flavin reduction (see more discussion later).
Figure 4.
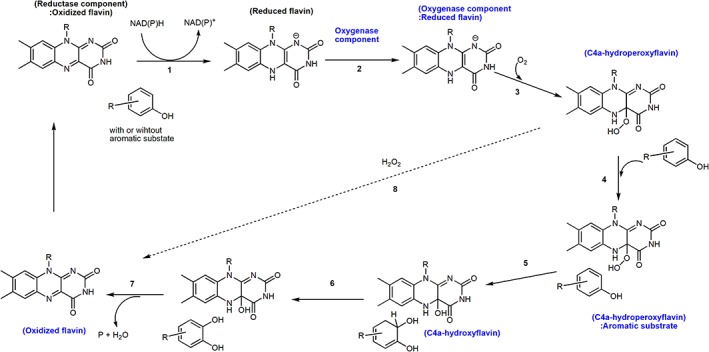
A typical catalytic cycle of phenolic hydroxylation catalyzed by two component flavin‐dependent monooxygenases (hydroxylases).
For the oxygenation reaction of two‐component enzymes, the enzyme‐bound reduced flavin reacts with oxygen to form C4a‐hydroperoxyflavin (Fig. 4, Step 3). All types of reduced flavins including FAD, FMN, and riboflavin can be used as substrates by two‐component enzymes, depending on their specific recognition features.15, 54, 57 The variety of flavin usage observed in two‐component monooxygenases is due to the flavin binding feature because only the isoalloxazine part is buried inside the active sites, while ribityl phosphate of FMN or ADP part of FAD generally protrudes outside59, 60 (Fig. 5).
Figure 5.
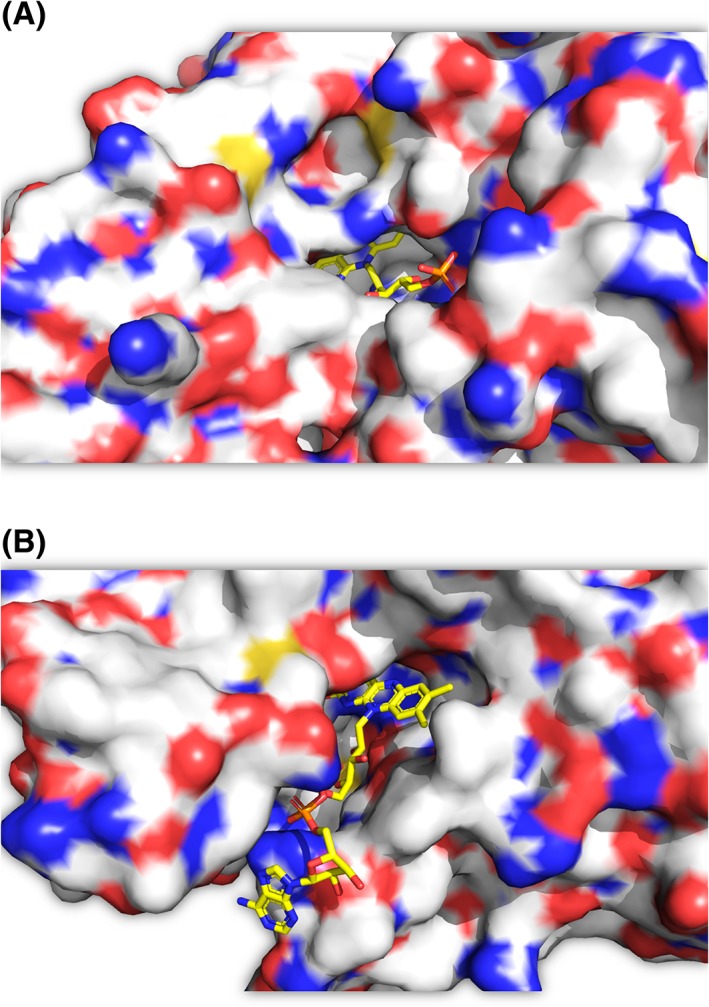
X‐ray structures of (A) C2 (pdb id: 2jbs) and (B) HpaB (pdb id: 2yyi), the isoalloxazine part is buried inside the active sites, while the ribityl phosphate of FMN or ADP part of FAD protrudes outside.
Aromatic substrates can be present or absent when the enzyme‐bound reduced flavin reacts with oxygen, depending on requirement of each enzyme. Although following the oxygenation step, the reaction of two‐component enzymes generally occurs similar to the reaction of single‐component enzymes, there are several distinctive features that are different between these two enzyme types. These include differences in the stability of C4a‐hydroperoxyflavin, the order of substrate binding, and the mode of reduced flavin transfer (Fig. 4, Step 2). Transfer of the reduced flavin is an essential part in the reaction of two‐component monooxygenases, but is not required for the single‐component monooxygenases (discussed more later in Section reaction mechanism of 4‐hydroxyphenylacetate 3‐hydroxylase).
Flavoenzymes catalyzing phenolic acid hydroxylation
Flavin‐dependent monooxygenases catalyzing hydroxylation of phenolic compounds are enzymes that are most extensively studied among the class. Here, the reactions of PHBH and HPAH are used as representative reactions of single‐component and two‐component enzymes, respectively.
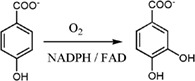
Reaction mechanism of 4‐hydroxybenzoate 3‐hydroxylase (PHBH) (single‐component monooxygenase)
PHBH represents one of the most extensively investigated single‐component hydroxylases. It catalyzes the hydroxylation of 4‐hydroxybenzoate (pOHB) to generate 3,4‐dihydroxybenzoate (3,4‐DOHB) using NADPH and O2 as co‐substrates [Fig. 6(A)].9, 19, 61, 62 The enzyme is classified as a member of Class A flavoenzyme monooxygenases.10 The reaction catalyzed by PHBH can be divided into two parts – the reductive half‐reaction and the oxidative half‐reaction, as described in Figure 1. The enzyme conformational change is important for controlling various steps in both half‐reactions.
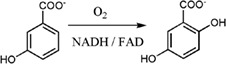
Figure 6.
Reactions catalyzed by flavin‐dependent monooxygenases in this review. (A) 3‐Hydroxy‐benzoate 4‐hydroxylase (PHBH), (B) 3‐hydroxyphenylacetate 4‐hydroxylase (HPAH), (C) 2‐methyl‐3‐hydroxypyridine‐5‐carboxylate oxygenase (MHPCO), and (D) HadA monooxygenase.
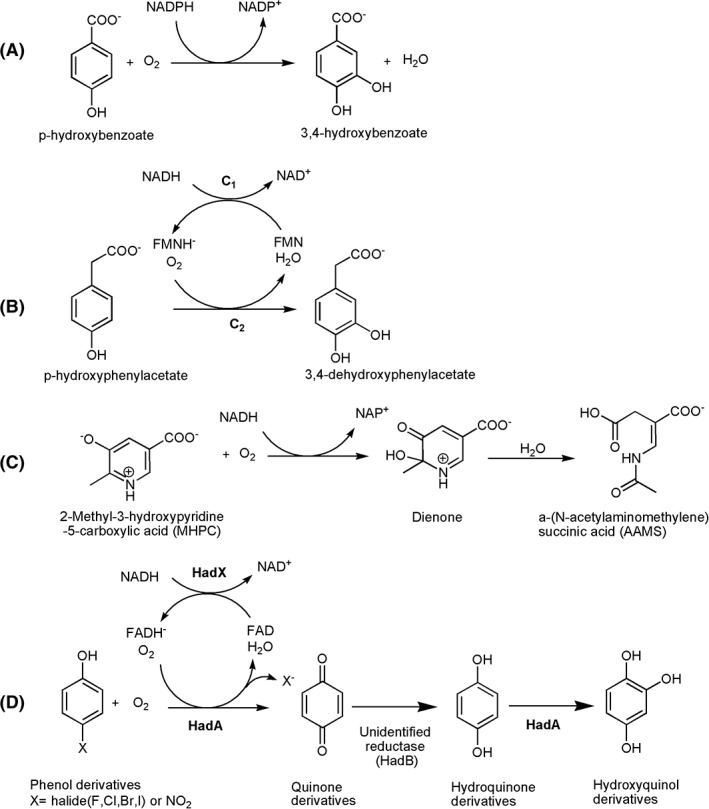
During the catalytic cycle, the isoalloxazine ring of the FAD moiety can change in position due to a change in the enzyme conformation to enhance catalysis. The first crystal structure of the enzyme in the form of a PHBH‐FAD‐pOHB complex shows that FAD is buried inside the active site.22 This enzyme form is designated as the “in” conformation, and this enzyme conformation does not facilitate the reduction of FAD by NADPH. However, in the presence of NADPH, the binding of pOHB allows the flavin ring to move to the “out” position. This out conformation was observed in the wild‐type enzyme in complex with 2,4‐dihydroxybenzoate.63 In this form, NADPH can bind to the re‐side of the isoalloxazine ring, and promote the flavin reduction, which is the transfer of a proR hydride of NADPH to the N5 of FAD.64 In comparison to the “in” conformation, the flavin ring in the “out” conformation is rotated around the ribityl side chain, resulting in movement of the N5 position of the isoalloxazine ring by about 7 Å (Fig. 7) closer to the protein surface where NADPH can bind closer to FAD.
Figure 7.
The “out”, “open,” and “in” conformations of the flavin ring were aligned and compared using pdb id: 1dod, 1k0j, and 1pbe, respectively.
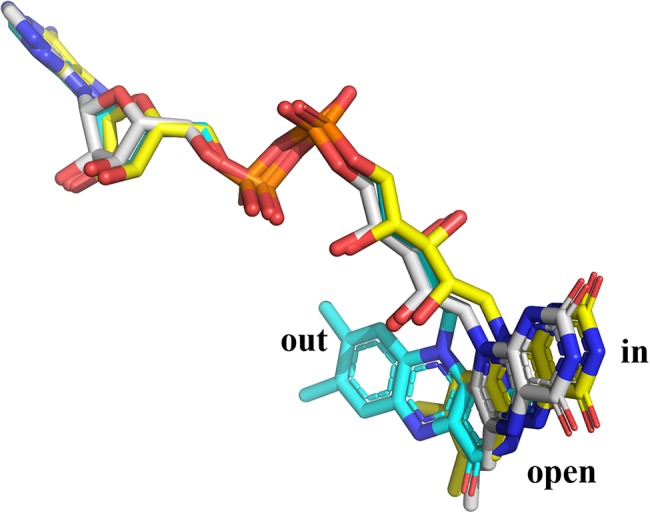
The crystal structure of PHBH with bound NADPH could only be observed with the structure of the Arg220Gln variant.65 However, the position of the flavin ring is different from what was previously designated as the “in” or “out” conformations (Fig. 7). In this protein complex, the isolloxazine ring swings out slightly from the active site to the protein surface in order to create space for the substrate and product to access the active site. NADPH binds in an elongated conformation along the cleft on the surface of PHBH. However, with this binding form, the nicotinamide ring is too far from FAD to allow flavin reduction. Thus, this conformation cannot facilitate the flavin reduction and was assigned as the “open” conformation. At this stage, the phenolic substrate (pOHB) can directly form hydrogen bonds with surface amino acids.65, 66 From the structure of the PHBH:pOHB complex, the substrate phenolic oxygen participates in a hydrogen bond network that extends about 13 Å and connects the backbone carbonyl of Pro293, Tyr201, Tyr385, a water molecule, and His72 (Fig. 8).22 These hydrogen‐bond networks can lower the pK a of the phenolic hydroxyl group by about 2 units.61 The deprotonated form of the substrate can trigger movement of the flavin ring to the “out” position in which NADPH can reduce FAD.67 Pre‐steady‐state kinetic data also support the importance of pOHB binding in promoting formation of the active enzyme conformation. The flavin reduction in the presence of aromatic substrate is 105 time faster than that of the reaction without aromatic substrate.61 After reduction, FADH− rotates back into the “in” conformation and may be stabilized by the positive electrostatic environment of the active site.68 Comparison of the structure of PHBH with other structures of enzymes in the same class such as phenol hydroxylase25 does not show similar H‐bond networks. Thus, the controlled reduction of flavin by the deprotonation of the substrate hydroxyl group may be unique to PHBH.
Figure 8.
The hydrogen bond network along Pro293, Try201, Try385, a water molecule, and His72 residues (pdb id: 1pbe) in PHBH.
During the oxidative half‐reaction, the isoalloxazin ring of the reduced enzyme complex with pOHB bound is in the “in” conformation in which it is buried and inaccessible to solvent. The pOHB hydroxyl group forms H‐bond networks with amino acid residues and is positioned close to the C4a position of the isoalloxazine ring. The active site of PHBH can facilitate the rapid reaction of oxygen with reduced flavin to form a C4a‐hydroperoxyflavin intermediate with a rate constant of 2.6 × 105 M−1 s−1.69 This reaction is about 103 time faster than the reaction of free reduced flavin with oxygen.69, 70 In the presence of pOBH, C4a‐hydroperoxyflavin hydroxylates the aromatic substrate via the electrophilic aromatic substitution mechanism and produces C4a‐hydroxyflavin and a non‐aromatic dienone product. The non‐aromatic dienone rapidly tautomerizes to generate the aromatic product (Fig. 1, Step 5).
After the hydroxylation, C4a‐hydroxyflavin dehydrates to eliminate water in order to regenerate the oxidized flavin. This step occurs synchronously with the release of aromatic product (3,4‐DOHB). At high concentrations of pOHB, the substrate inhibits the dehydration step of C4a‐hydroxyflavin, forming a trapped C4a‐hydroxyflavin:product complex which can slow down the overall turnover.71 The rate of product release is pH‐dependent and consistent with a pK a value of 7.1, suggesting that H‐bond networks that control substrate deprotonation with pK a of 7.1 are also involved with the process of product release. It was suggested that deprotonation of the para‐hydroxyl group of 3,4‐DOHB can result in the product dianionic form which can interact with Pro293 to induce the enzyme conformational change to promote positioning of the flavin in the “out” position, accelerating the dehydration process of C4a‐hydroxyflavin.22, 65, 72
Reaction mechanism of 4‐hydroxyphenylacetate 3‐hydroxylase (two‐component monooxygenase)
p‐Hydroxyphenylacetate 3‐hydroxylase (HPAH) is one of the most extensively studied two‐component flavin‐dependent monooxygenases in which the reaction mechanisms can be used as a model for understanding the reactions of other enzymes in this class. HPAH catalyzes the hydroxylation of 4‐hydroxyphenylacetate (HPA) to yield 3,4‐dihydroxyphenylacetate (DHPA) [Fig. 6(B)]. Although the net reactions of HPAH and PHBH are similar, as they both catalyze hydroxylation of phenolic acids, the modes of operation by these two enzymes are remarkably different. This is mainly due to the nature of one being a single‐component versus the other being a two‐component enzyme in which flavin reduction and oxygenation occur in a single or in separate active sites, respectively.
Many HPAHs from several bacteria species have been isolated and characterized as shown in Table 2. Among these, the reaction mechanism of HPAH from Acinetobacter baumannii is the most extensively investigated. This enzyme consists of a reductase component (C1) and an oxygenase component (C2).41, 54 The enzyme is part of the HPA degradation pathway in bacteria.73 C1 is a flavoprotein that binds FMN as a cofactor and catalyzes the reduction of oxidized FMN to generate reduced FMN using NADH as a reductant. Recently, the structure of C1 has been reported.74 C2 catalyzes the hydroxylation of HPA using oxygen and FMNH−, FADH− or reduced riboflavin as co‐substrates.54, 70
For two‐component flavin‐dependent monooxygenases, the reduced flavin generated by a reductase has to be transferred to a corresponding monooxygenase. The mode of reduced flavin transfer can occur via two modes – by free diffusion or through protein–protein interactions. Evidence from pre‐steady‐state kinetic experiments of several enzymatic systems suggests that the reduced FMN can be transferred from the reductase to the oxygenase efficiently without a requirement for protein–protein interaction.15, 52, 56, 75, 76 Investigation of the reduced flavin transfer mechanism from C1 to C2 indicates that the reduced flavin can be transferred via free diffusion and the rate‐limiting step of the process is the release of FMNH− from C1.15, 76 In HPAH, the substrate HPA acts as an effector to stimulate the reduction of FMN by changing the enzyme into a more active conformation.56, 74, 76 The rate constant of the FMN reduction increases by about 30 times in the presence of HPA. Recently, the X‐ray structure of C1 has been reported. Studies using small angle X‐ray diffraction analysis indicate that the presence of HPA induces an overall shape expansion of the enzyme.74 Presumably, this conformational change occurs to facilitate formation of a more active conformation of C1 in which it is able to interact more effectively with NADH. Interestingly, this effector enhancing NADH reduction of flavin is not present in all reductases involved with two‐component monooxygenase systems. Besides C1, experimental evidence of this property has only been observed for HPAH from Pseudomonas putida.77 However, based on the sequence analysis, several reductases such as the reductase of the nitrilotriacetate monooxygenase system have been proposed to have the ability to bind with their effectors and achieve rate enhancement of flavin reduction.54, 73, 74 Investigation on the control mechanism of the rate of flavin reduction indicates that the C‐terminal domain of C1 acts as an auto‐inhibitory domain in which the inhibition of NADH reduction is only alleviated upon HPA binding.56 In a recent report, the residues Tyr207 and Phe216 were identified based on site‐directed mutagenesis to be involved in HPA binding.74
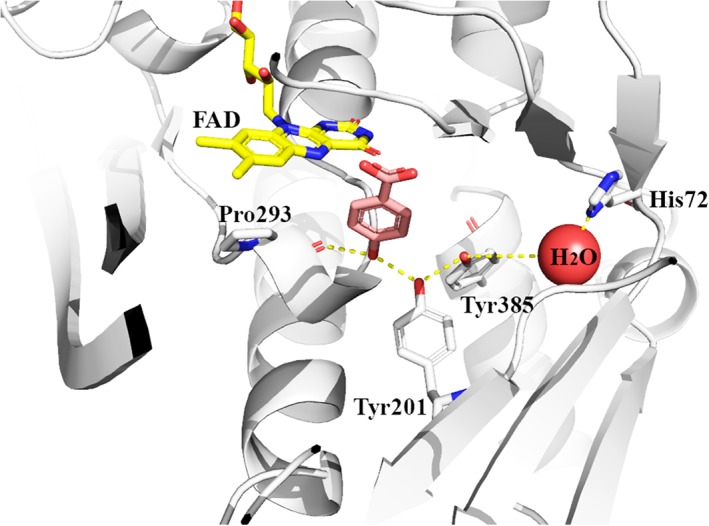
For the oxygenation which occurs on C2, FMNH− is the first substrate to bind to the enzyme. The binding of FMNH− to C2 occurs very rapidly with a second order rate constant of at least 107 M−1 s−1 and a K d value for the binding of ~1.2 μM.70 The C2 crystal structure shows that the isoalloxazine ring is buried inside the protein and is inaccessible to outside solvent.59 This binding arrangement is commonly found in other two‐component flavin‐dependent monooxygenases.60, 78, 79 The complex of C2:FMNH− reacts with O2 very quickly to form the C4a‐hydroperoxyflavin intermediate with a rate constant of 1.1 × 106 M−1 s−1. Molecular dynamics simulations indicate that the oxygen entrance into the active site of C2 is controlled by the gate‐keeping residue Phe266 (Fig. 9)80 in which the side chain of Phe266 is required to move away to allow for oxygen diffusion to the C4a‐position of flavin.
Figure 9.
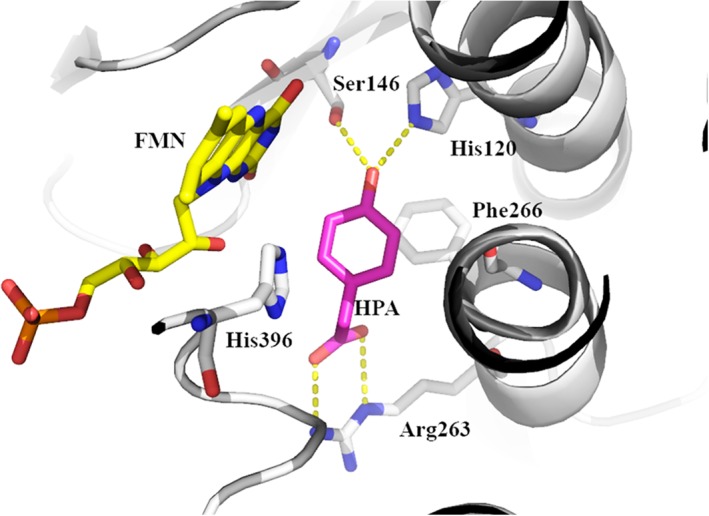
Important amino acid residues in the active site of C2 (pdb id: 2jbt).
In the absence of HPA, the C4a‐hydroperoxyflavin intermediate is quite stable because it slowly eliminates H2O2 with a rate constant of 0.003 s−1. Similar rate constants for reduced flavin reacting with oxygen and for the elimination of hydrogen peroxide were also observed in HPAH from Pseudomonas aeruginosa.36 This property is also unique to two‐component monooxygenases because similar stability of C4a‐hydroperoxyflavin in the absence of substrate can also be found in other two‐component monoxygenases such as bacterial luciferase,81 flavin‐dependent dehalogenase,53 styrene monooxygenase,82 and FMN‐dependent monooxygenase ActVA‐ActVB from Streptomyces coelicolor.83 Investigations using site‐directed mutagenesis and pre‐steady‐state kinetics suggest that a cavity located in front of the reduced FMN are perfectly fit for housing the peroxide moiety of the C4a‐hydroperoxyflavin intermediate13, 59 which can be stabilized by an H‐bond between the Oγ atom of Ser171 and the flavin N(5)‐H atom.84 Interruption of this H‐bond in a Ser171Ala variant increases the rate of H2O2 elimination by about 1400‐fold compared with that of the wild‐type enzyme.84
The His396 side chain located at the active site of C2 (Fig. 9) is important for C4a‐hydroperoxyflavin formation since when this residue was mutated to alanine, valine, and asparagine, the rate constant for the formation of intermediate decreased from 1.1 × 106 M−1 s−1 to 3.3–3.8 × 103 M−1 s‐1.84 The data imply that His396 may act as an instantaneous proton provider for the formation of C4a‐hydroperoxyflavin. This residue is also important for maintaining the protonation of C4a‐hydroperoxyflavin by acting as a hydrogen bond acceptor.85 Investigation using solvent kinetic isotope effects indicates that the proton transfer from His396 occurs rapidly before the formation of C4a‐hydroperoxyflavin and it is not the rate‐limiting step of the overall process of C4a‐hydroperoxyflavin formation.85 The information correlates well with results from density functional calculations in that the formation of C4a‐hydroperoxyflavin occurs via a proton‐coupled electron transfer mechanism in which molecular oxygen accepts an electron from reduced FMN simultaneously with a proton transfer from a protonated histidine (His396). The C2 active site is evolved to optimize this proton‐coupled electron transfer process so well because the energy associated with the C4a‐hydroperoxyflavin formation is nearly barrierless.86
Analysis by rapid‐quench flow experiments indicates that the C4a‐hydroperoxyflavin intermediate hydroxylates HPA to generate DHPA with a hydroxylation rate constant of 15–17 s−1.87 The hydroxylation rate constant is pH‐independent over the range of pH 6.2–9.9. Site‐directed mutagenesis, pH‐dependent, and pre‐steady‐state kinetics studies suggest that His120 (Fig. 9) may be positively charged throughout this pH range to facilitate formation of the deprotonated HPA, which is more prone to the oxygenation reaction via an electrophilic aromatic substitution mechanism.88 Site‐directed mutagenesis studies also show that Ser146 (Fig. 9) is important for the proper binding of substrate for efficient hydroxylation, as the rate of hydroxylation in the Ser146Ala variant was six‐fold slower than the wild‐type enzyme.
Flavin‐dependent monooxygenases that catalyze additional reactions after hydroxylation
Some of flavin‐dependent monooxygenases that can incorporate a hydroxyl group into aromatic or phenolic substrates can also further catalyze additional reactions. Here, we discuss two enzymatic systems in which one catalyzes breakage of an aromatic ring and another one catalyzes a group elimination reaction in addition to the initial hydroxylation reaction.
Flavin‐dependent monooxygenase that catalyzes oxidative aromatic ring‐cleavage reactions (single‐component monooxygenase)
Flavin‐dependent monooxygenases that catalyze aromatic ring‐cleavage reactions include 2‐methyl‐3‐hydroxypyridine‐5‐carboxylic acid oxygenase (MHPCO)89 and 5‐pyridoxic acid oxygenase (5PAO).90 These two flavoenzymes can be classified as members of Class A single‐component flavoprotein monooxygenases that mainly catalyze hydroxylation of aromatic substrates.10 MHPCO was successful overexpressed in Escherichia coli and its three‐dimensional structure is known. Much information regarding the MHPCO reaction and kinetic mechanisms is available. This review will highlight the current understanding of the MHPCO reaction as a representative flavoenzyme that can catalyze aromatic ring‐cleavage.
MHPCO is a single‐component flavoenzyme monooxygenase that catalyzes the ring‐opening reaction of a pyridine compound derivative, MHPC, to form an aliphatic product, α‐(N‐acetylaminomethylene) succinic acid (AAMS) [Fig. 6(C)]. This reaction is part of the vitamin B6 degradation pathway.91, 92, 93 Oxygen isotope labeling experiments showed that only one atom of molecular oxygen was incorporated into the AAMS product, while another atom of oxygen was from H2O.94 Similar to other flavoenzyme monooxygenases, the reaction of MHPCO can be divided into two parts – reductive and oxidative half‐reactions – similar to the reaction of PHBH.95
During the reductive half‐reaction, investigations using a substrate analog indicated that the tripolar ionic form of MHPC (Fig. 10) is the form that binds to the oxidized enzyme to form an active MHPCO–MHPC complex.96 The nitrogen atom of the substrate presumably remains protonated throughout the catalytic reaction. Site‐directed mutagenesis studies indicate that the protonation status of MHPC is probably stabilized by Tyr82 which can form an H‐bond with H2O and the nitrogen atom of the substrate as shown in Figure 11.30, 97 Tyr82 is crucial for the selective binding of the tripolar ionic form of the substrate. Upon changing Tyr82 to histidine and phenylalanine, the enzyme variants lost their selectivity for binding the tripolar ionic form of the substrate.97
Figure 10.
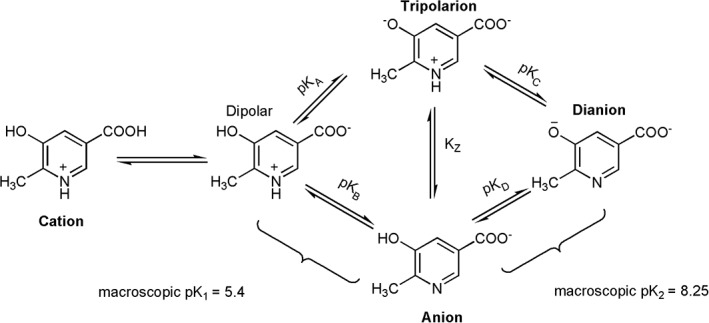
Protonation status of MHPC at various pHs.
MHPCO can also bind to the substrate analogues 5‐hydroxynicotinic acid (5HN) and N‐methyl‐5‐hydroxynicotinic acid (NMHN); the K d values for binding of MHPC, 5HN, and NMHN are 9.2 μM, 5.2 μM, and 55 μM, respectively.96 Similar to PHBH, the binding of MHPC or substrate analogues (5‐HN and NMHN) to MHPCO can increase the reduction rate of FAD by NADH. In the presence of MHPC, the flavin reduction rate is about 1600 time faster than the reaction without MHPC.96 Based on X‐ray structures, site‐directed mutagenesis and steady state kinetics, the data suggest that Tyr270 (Fig. 11), which can form an H‐bond with a carboxylic group of the substrate through a water molecule, is crucial for maintaining the optimal conformation for reduction of FAD by NADH.98
Figure 11.
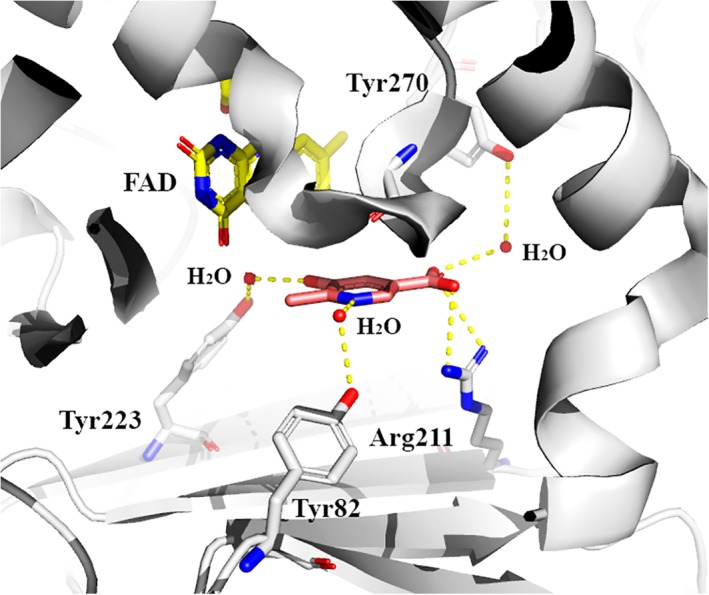
Important amino acid residues in the active site of MHPCO (pdb id: 3gmc).
During the oxidative half‐reaction, the complex of reduced enzyme with MHPC reacts with molecular oxygen to form the C4a‐hydroperoxyflavin intermediate with a bi‐molecular rate constant of 5.5×104 M−1 s−1.94 This reactive intermediate can oxygenate an aromatic substrate via an electrophilic aromatic substitution mechanism and generates the hydroxylated product and C4a‐hydroxyflavin. 5HN and NMNH can be used as efficient substrates with hydroxylation efficiencies of 92% and 70%, respectively, compared with the efficiency of MHPC of 91%.94, 96, 99 5HN is a useful substrate for investigating the reaction mechanism of MHPCO because the compound serves as a good substrate for MHPCO,96 and unlike MHPC, 5HN is commercially available. Comparison of the crystal structures of the MHPCO–5HN and MHPCO–MHPC complexes indicates that the binding mode of 5HN is almost the same as that of MHPC.98 Moreover, the rate constants of the MHPCO reaction using 5HN as a substrate are in the same range or slightly higher than the reaction using MHPC.94, 96
After hydroxylation, the process by which the hydroxylated product undergoes a ring opening reaction is still unclear and remains a mechanistic challenge for this enzyme. Three mechanisms have been proposed as shown in Figure 12. Mechanisms A and B were proposed by Chaiyen et al. and McMulloch et al., respectively. In both of these reactions, the ring‐cleavage reaction starts from the protonated hydroxy‐MHPC (I) (Fig. 12). In Chaiyen's pathway,94, 95, 96 the protonated hydroxy‐MHPC is attacked by water at the C3‐keto moiety group and is followed by rearrangements to yield AAMS as the product. In McMulloch's pathway,30 the protonated hydroxy‐MHPC undergoes an electrocyclic ring‐opening to form a ketene intermediate prior to attack by a water molecule. Recently, the Eriksson group performed density functional theory (DFT) calculations on a small model containing only C4a‐hydroperoxyflavin and substrate (using 5HN as substrate).100 The results suggest that both proposed mechanisms previously mentioned are less likely to occur since the activation energy for both of them are high (ΔΔG# gas = 38.5 kcal mol−1 and 41.9 kcal mol−1 for the Chaiyen pathway and McCulloch pathway, respectively). However, these values are not consistent with rate constants measured experimentally. The DFT calculations also suggest that the ring‐opening process must occur in the protein active site because in aqueous solution, the free hydroxylated 5HN can undergo re‐aromatization to form the hydroxylated aromatic product spontaneously.100 Based on DFT calculations, after hydroxylation, the proton of the protonated hydroxy‐MHPC (I) is proposed to be transferred to C4a‐hydroxyflavin intermediate. The deprotonated hydroxy‐MHPC (II) can proceed with the ring‐cleavage reaction with a much lower activation energy. Two pathways were proposed for the ring‐cleavage process, starting from the deprotonated hydroxy‐MHPC (II) (Fig. 12, pathway C). The energies associated with the two pathways are reasonably low (ΔΔG# gas = 14.1 kcal mol−1 and 11.1 kcal mol−1), allowing the reaction to proceed.100, 101 However, no experimental support is available to support results from the DFT calculations.
Figure 12.
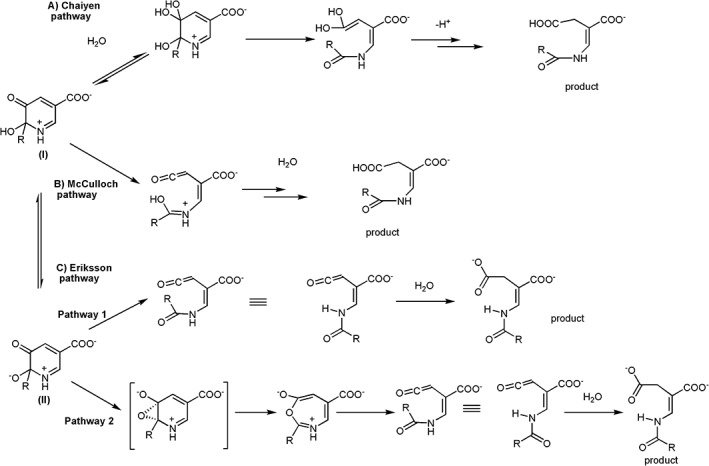
Proposed reaction mechanisms by Chaiyen et al., McCulloch et al., and Eriksson et al.
Flavin‐dependent dehalogenase that catalyzes additional dehalogenation or denitration of aromatic compounds (two‐component monooxygenase)
Halogenated compound waste is found wide spread in the biosphere as a consequence of their heavy usage as solvents, pharmaceuticals102 and agrochemicals such as pesticides, herbicides, insecticides, and fungicides. The toxic nature of halogenated compounds and their potential long‐term persistence in the environment raises a serious threat to the environment, and to the health of humans and animals. Oxidative dehalogenation involves the replacement of a halogen with a hydroxyl group from molecular oxygen or hydrogen peroxide which typically can be catalyzed by flavin‐dependent oxygenases, cytochrome P450 oxidases, and dehaloperoxidases.17, 103, 104, 105
Several aerobic microbes possess dehalogenases with the promising ability to degrade halogenated compounds to much less toxic compounds such as benzoquinone.52, 53, 106 Degradation of chlorophenols can be carried out by various enzymatic systems such as tft clusters from Burkholderia cepacia AC1100,48 tcp clusters from Ralstonia eutropha JMP134,107 had clusters from Ralstonia pickettii DTP0602,53, 108 and pcp clusters from Sphingobium chlorophenolicum.109 Many enzymes that catalyze oxidative dehalogenation can use a broad range of substrates.110, 111 A well‐studied single‐component dechlorinase is PcpB from S. chlorophenolicum. This enzyme has FAD as a prosthetic group and uses NADPH and molecular oxygen as co‐substrates for the conversion of pentachlorophenol to 2,3,5,6‐tetrachlorobenzoquinone. The quinone product can be chemically reduced to 2,3,5,6‐tetrachloro‐p‐hydoquinone.109, 112
Many oxidative dechlorination reactions are also catalyzed by two‐component flavin‐dependent enzymes. Of these enzymes, the reaction mechanism of HadA from R. pickettii is the most extensively studied [Fig. 6(D)].52, 53, 106 Transient kinetics based on stopped‐flow and rapid‐quench flow experiments have shown that HadA binds to reduced flavin as the first substrate to form a binary complex which can further react with oxygen to generate C4a‐hydroperoxyflavin (Fig. 13).53 Previous investigations of TftD also detected a species which was speculated to be a C4a‐flavin adduct (C4a‐hydroperoxyflavin or C4a‐peroxyflavin), but transient kinetic experiments have never been carried out with the TftD system.113 Studies of PcpB also demonstrated the existence of C4a‐flavin adducts.109, 112 4‐Chlorophenol binds to the HadA enzyme after the reactive intermediate C4a‐hydroperoxyflavin forms.53 The ternary complex then bifurcates into the coupling path of hydroxylation plus Cl− elimination and the uncoupling path of H2O2 elimination (Fig. 13).53
Figure 13.
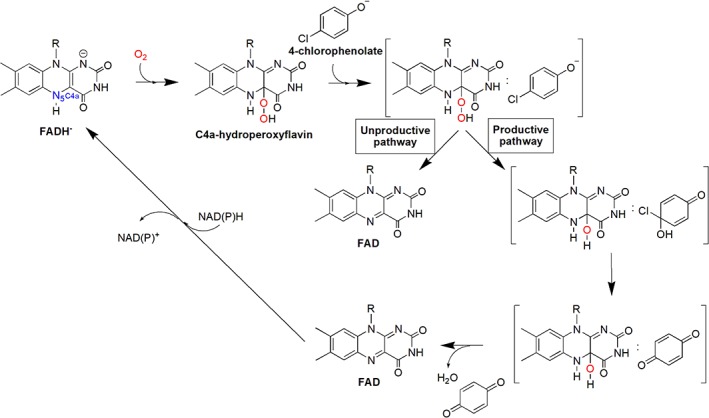
Kinetic mechanisms of the HadA reaction with 4‐chlorophenol.
Recently, studies using various substrates and density functional analysis to investigate the reaction mechanism of HadA indicate that HadA has dual activities of oxidative dehalogenation (hydroxylation plus halide elimination) and denitration (hydroxylation plus nitro elimination). Quantitative structure and reactivity relationship (QSAR) analysis was performed in order to correlate the rate constants associated with the individual steps of the HadA reactions with various physio‐chemical parameters. This analysis indicated that the deprotonation of the phenolic substrate is the step with the highest activation energy amongst all of the steps involved in product formation because the deprotonation of substrates controls the overall reaction of product formation. Phenolic substrates with a lower pK a give higher rate constants of hydroxylation than substrates with higher pK a values, indicating that substrate deprotonation has a higher energy barrier than the other steps such as –OH transfer, and thus controls the hydroxylation step (Fig. 14). These results contradict previous TftD computational calculations. Results from quantum mechanical and molecular mechanical (QM/MM) calculations of TftD showed that the –OH transfer has the highest energy barrier and did not give a high energy barrier of substrate deprotonation, thus explaining why the proton and –OH transfer is a concerted process.114
Figure 14.
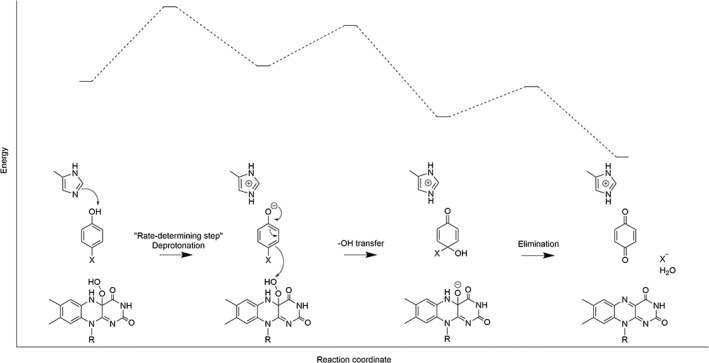
The proposed model and energy diagram for oxidative dehalogenation and denitration of HadA.
The substrate deprotonation mechanism of HadA and other two‐component monooxygenases are likely different from that of single‐component monooxygenases. The substrate deprotonation mechanism of PHBH is mediated through an H‐bond network on the enzyme surface through the active site.9 The deprotonation of pOHB is important for the –OH transfer from C4a‐hydroperoxyflavin to the substrate via an electrophilic aromatic substitution mechanism which is the key step that controls the hydroxylation.115, 116, 117, 118 QM/MM studies of phenol hydroxylase suggest that binding of the phenol substrate to Asp54 facilitates substrate deprotonation to the phenolate form.119 For MHPCO, the hydroxypyridine binds to the enzyme in the deprotonated form95 and the enzyme active site environment ensures substrate deprotonation.97 Whereas for HadA, TftD, and TcpA, an active site His was proposed to be the key catalytic base for initiation of proton abstraction.48, 120 However, it still remains unclear how the environment of the HadA active site can promote additional group elimination in addition to hydroxylation.
Current mechanistic understanding and challenging aspects of flavin‐dependent monooxygenases that hydroxylate aromatic compounds
Hydroxylation mechanism
Studies of flavin‐dependent monooxygenases have been instrumental in understanding the mechanisms of oxygen activation in many flavoenzymes.13, 17, 86, 121, 122 There is no specific binding site for oxygen in flavoenzymes that use oxygen as a substrate. The combined approaches of molecular dynamics simulations, site‐directed mutagenesis and transient kinetics indicate that oxygen diffuses through specific pathways in these enzymes to enter the area close to the C4a‐position of the flavin of C2,80 alditol oxidase from Streptomyces coelicolor A3,80 aryl alcohol oxidase,123 etc. The reaction of reduced enzyme with molecular oxygen was proposed to occur via a single electron transfer from reduced flavin to molecular oxygen to form a radical pair of flavin semiquinone and superoxide anion before the radical pair collapses to form a C4a‐peroxyflavin intermediate prior to its protonation to form C4a‐hydroperoxyflavin (Fig. 2, Path 1).11, 124, 125, 126 Recently, DFT calculations of the oxygenase component (C2) of p‐hydroxyphenylacetate 3‐hydroxylase (HPAH) indicate that the formation of C4a‐hydroperoxyflavin occurs via a proton‐coupled electron transfer mechanism86 in which the single electron transfer process to generate the radical pair is tightly coupled with proton transfer (Fig. 2, Path 2). This mechanism is also consistent with the mechanism of C4a‐hydroperoxyflavin formation in pyranose 2‐oxidase (P2O).122 P2O is an oxidase that can form C4a‐hydroperoxyflavin as an intermediate before elimination of the typical H2O2 product for flavoenzyme oxidases. Although all of the flavin‐depdendent monooxygenases can form C4a‐flavin oxygen adducts as intermediates, different monooxygeases facilitate different protonation states of the adduct (either to form C4a‐hydroperoxyflavin or C4a‐peroxyflavin), depending on its requirement to act electrophile or nucleophile.10, 14, 17 It still remains unclear at this stage how the environment inside these proteins can control different protonation states so that the enzymes can be fine‐tuned to perform the expected tasks.
Based on the currently available information, the mechanisms of –OH group transfer from C4a‐hydroperoxyflavin to aromatic substrates in flavin‐dependent monooxygenases are mostly consistent with electrophilic aromatic substitution in which the –OH moiety of C4a‐hydroperoxyflavin acts as an electrophile (Fig. 15). In PHBH and MHPCO, the QSAR correlation using flavin analogues with different substituents at the 8‐postion indicates that the rate constants of hydroxylation increase with more electron withdrawing substituents due to the stabilization of the flavin C4a‐alkoxide leaving group after hydroxylation (Fig. 15). These data suggest that an OH‐moiety of C4a‐hydroperoxyflavin is transferred as an electrophile to aromatic substrate.118, 127 The QSAR analysis for the correlation of k cat values with the E LUMO–E HOMO energy of –OH transfer in a series of fluorinated p‐hydroxybenzoate in PHBH128 and halogenated phenol for phenol hydroxylase119 indicate that this energy gap controls the hydroxylation because the k cat values of PHBH and phenol hydroxylase are higher when the molecular orbital energy gaps of C4a‐hydroperoxyflavin and the substrates are lower. The results suggest that the –OH transfer is the rate‐limiting step for PHBH and phenol hydroxylase. In the HadA reaction discussed previously, the –OH transfer is not the rate‐limiting step in the reaction, but rather, the deprotonation process is the step with the highest energy.106 Therefore, within the same –OH incorporation via electrophilic aromatic substitution mechanism, the energy landscape that controls these reaction steps are varied. It would be interesting to learn more about the energy landscapes associated with other flavin‐dependent monooxygenases. Although most of the mechanisms are consistent with the electrophilic aromatic substitution, it remains unclear for the mode of substrate activation to facilitate this –OH group transfer. As the deprotonated form of the substrate would promote the incorporation of +OH into the aromatic ring, the active site features that facilitate the process are undoubtedly important for this catalysis. In the reaction catalyzed by PHBH, it was clearly demonstrated that protein and FAD conformational changes coupled with interactions through H‐bond networks control the process of substrate deprotonation. Studies using QM/MM methods for PHBH,129 phenol hydroxylase119 salicylate hydroxylase130 suggest that the deprotonation of the aromatic substrate increases the rate of hydroxylation by decreasing the energy barrier. In PHBH, the hydrogen bond network between Pro293, Tyr201, Tyr385, a water molecule, and His72 (Fig. 8) promotes formation of the deprotonated from of the hydroxyl group of pOHB by decreasing the pK a of pOHB by about 2 units.22 Interruption of this H‐bond network by site‐directed mutagenesis resulted in a decrease in the hydroxylation rate constant.131, 132 In other systems, although candidate residues for interacting with a substrate hydroxyl group have been proposed (previously discussed), detailed explanation regarding their control of substrate deprotonation are less clear and remain to be investigated.
Figure 15.
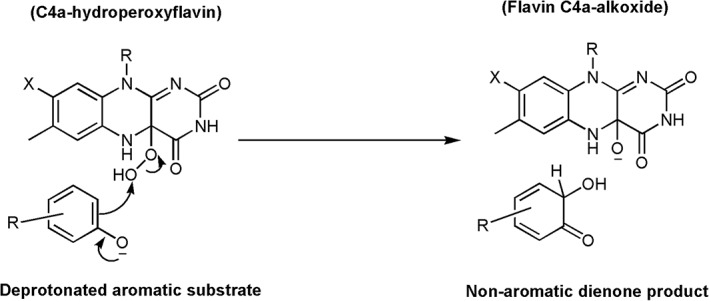
Electrophilic aromatic substitution mechanism.
Mechanisms related to additional reactions
A major challenge regarding the reaction mechanism of MHPCO is to understand how the enzyme active site facilitates additional ring‐cleavage after hydroxylation. It is not clear how the active site promotes attack of the dienone hydroxylated MHPC by water and why this water addition does not occur in other aromatic hydroxylases. The ring‐breaking mechanisms proposed by DFT calculations remain to be investigated to for consistency with experimental results. An understanding of the active site architecture that is crucial for this C–C bond breaking will be valuable for future applications in biocatalysis or enzyme redesign to construct an organic‐biomimetic catalyst capable of converting aromatic to aliphatic compounds without involvement of metal.
Although the hydroxylation of HadA likely proceeds via an electrophilic aromatic substitution mechanism in which the substrate deprotonation controls the overall process, it is still unclear why the active site of HadA promotes group elimination in addition to hydroxylation. The next step for investigating the HadA reaction requires an X‐ray structure of the HadA:substrate complex so that the residues involved in the group elimination reaction can be identified and further analyzed by site‐directed mutagenesis. An understanding of the group elimination reaction catalyzed by HadA should be valuable for biocatalysis applications, as it would allow the incorporation of the group elimination activity of HadA in other flavin‐dependent hydroxylases through enzyme engineering, allowing these enzymes to perform tasks that are useful for detoxification or for the synthesis of valuable chemicals.
Potential use in biocatalysis
Regio‐ and stereo‐specific oxygen insertions by oxygenase enzymes are useful for industrial applications for production of pharmaceutical ingredients and fine chemicals.133, 134, 135, 136 Expansion of enzymatic specificity is generally accomplished by enzyme engineering through a random, semi‐random, or rational approach.137, 138 Various aromatic hydroxylases that are useful in biocatalysis have been documented.21 The HPAH system can efficiently synthesize antioxidants 3,4,5‐THPA and 3,4,5‐THCA from HPA and p‐coumaric acid, respectively. Rational engineering of HPAH resulted in the production of the Tyr398Ser variant that was shown to be more effective than the wild‐type enzyme for preparation of 3,4,5‐THCA.139, 140 HPAH possesses several properties that are suitable for biocatalytic applications, including the ability to obtain a high yield of protein overexpression during production, stability over a pH range of 6–1087 and high thermostability.139 Engineering of HPAH to use an aniline instead of a phenolic compound was achieved in the Ser146Ala variant which can convert 4‐aminophenylacetic acid to 3‐hydroxy‐4‐aminophenylacetic acid.140 This reaction should be useful for the synthesis of many pharmaceutical compounds.141 Recently, the reaction of HPAH has been demonstrated to be applicable to conversion of agricultural waste such as phenolic compounds from palm oil mill effluent to antioxidants such as THCA.142
Dehalogenation and denitration by the HadA reaction should be useful for biodetoxification of halogenated phenols and nitrophenols accumulated in environment. Potential applications in this aspect should be further explored by engineering cells to contain clusters of enzymes capable of degrading these toxic compounds.105, 143, 144 In addition, low‐cost detoxification via biodegradation also offers a means for converting toxic waste into value‐added products, and have the potential to contribute to waste biorefining and the development of a circular economy. The resulting quinone and hydroquinone resulting from the HadA reaction can be further metabolized by other enzymes in the had operon to yield common metabolites in cells. Some of these metabolites such as di‐acid and hydroxyl‐acid can be used as commodity chemicals such as precursors in bioplastics.7
In order to successfully apply these monooxygenases in industrial applications, further developments regarding all engineering aspects need to be achieved. Comprehensive activity profiling and enzyme engineering are necessary for understanding the value of each enzyme in biocatalysis. This can be achieved by rational design, directed‐evolution or by the use of ancestral reconstruction and characterization of evolutionary pathways between distinct functional families.145, 146, 147 Enzyme engineering is necessary to overcome protein stability issues in some hydroxylases such as HadA. In order to accomplish economically viable biocatalysis, process optimization to design efficient scale‐up is necessary for industrial implementation. Recent improvements in enzyme immobilization methods should also be helpful in providing enzyme reusability, recyclability, and operational stability, thus leading to reduced cost of production. The techniques employed for hydroxylase immobilization in the past included carrier bonding, cross‐linking, and entrapment.148, 149, 150
Conclusions
Many flavoenzymes catalyze hydroxylation of phenolic compounds and hydroxylation plus additional reactions have been isolated and characterization in both single component and two component flavin‐dependent hydroxylase. Despite some common features in the overall catalytic reactions, single‐component and two‐component aromatic hydroxylase are different in most catalytic respects. MHPCO and HadA are examples of one‐component and two‐component monooxygenases that can catalyze additional reactions beside hydroxylation. It is not known how the key active site features in these enzymes facilitate these additional reactions. Many outstanding questions regarding the mechanistic details of these monooxygenases remain to be investigated. Further developments in engineering aspects are necessary for the successful implementation of these enzymes in large‐scale or industrial applications.
Acknowledgments
The authors acknowledge research support from The Thailand Research Fund MRG6180156 (to P. Chenprakhon), RTA5980001 (to P. Chaiyen), MRG6080234 (to T. Wongnate), Mahidol University (to P. Chenprakhon), and VISTEC (to P. Chaiyen and T. Wongnate).
References
- 1. Balasundram N, Sundram K, Samman S (2006) Phenolic compounds in plants and agri‐industrial by‐products: antioxidant activity, occurrence, and potential uses. Food Chem 99:191–203. [Google Scholar]
- 2. Acosta‐Estrada BA, Gutiérrez‐Uribe JA, Serna‐Saldívar SO (2014) Bound phenolics in foods, a review. Food Chem 152:46–55. [DOI] [PubMed] [Google Scholar]
- 3. Maenpuen S, Tinikul R, Chenprakhon P, Chaiyen P, Production of valuable phenolic compounds from lignin by biocatalysis: state‐of‐the‐art perspective. In: Chang HN, Ed. (2018) Emerging areas in bioengineering. Weinheim: Wiley‐VCH Verlag GmbH & Co. KGaA, pp 105–123.
- 4. Siquet C, Paiva‐Martins F, Lima JL, Reis S, Borges F (2006) Antioxidant profile of dihydroxy‐and trihydroxyphenolic acids—a structure–activity relationship study. Free Radical Res 40:433–442. [DOI] [PubMed] [Google Scholar]
- 5. Esteves M, Siquet C, Gaspar A, Rio V, Sousa JB, Reis S, Marques MPM, Borges F (2008) Antioxidant versus cytotoxic properties of hydroxycinnamic acid derivatives—a new paradigm in phenolic research. Arch Pharm Chem Life Sci 341:64–173. [DOI] [PubMed] [Google Scholar]
- 6. Badhani B, Sharma N, Kakkar R (2015) Gallic acid: a versatile antioxidant with promising therapeutic and industrial applications. RSC Adv 5:27540–27557. [Google Scholar]
- 7. Tinikul R, Chenprakhon P, Maenpuen S, Chaiyen P (2018) Biotransformation of plant derived phenolic acids. Biotechnol J 13:1700632. [DOI] [PubMed] [Google Scholar]
- 8. Mitchell KH, Rogge CE, Gierahn T, Fox BG (2003) Insight into the mechanism of aromatic hydroxylation by toluene 4‐monooxygenase by use of specifically deuterated toluene and p‐xylene. Proc Natl Acad Sci USA 100:3784–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ballou DP, Entsch B, Cole LJ (2005) Dynamics involved in catalysis by single‐component and two‐component flavin‐dependent aromatic hydroxylases. Biochem Biophys Res Commun 338:590–598. [DOI] [PubMed] [Google Scholar]
- 10. van Berkel WJH, Kamerbeek NM, Fraaije MW (2006) Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J Biotechnol 124:670–689. [DOI] [PubMed] [Google Scholar]
- 11. Mattevi A (2006) To be or not to be an oxidase: challenging the oxygen reactivity of flavoenzymes. Trends Biochem Sci 31:276–283. [DOI] [PubMed] [Google Scholar]
- 12. Joosten V, van Berkel WJ (2007) Flavoenzymes. Curr Opin Chem Biol 11:195–202. [DOI] [PubMed] [Google Scholar]
- 13. Chaiyen P, Fraaije MW, Mattevi A (2012) The enigmatic reaction of flavins with oxygen. Trends Biochem Sci 37:373–380. [DOI] [PubMed] [Google Scholar]
- 14. Huijbers MM, Montersino S, Westphal AH, Tischler D, van Berkel WJ (2014) Flavin dependent monooxygenases. Arch Biochem Biophys 544:2–17. [DOI] [PubMed] [Google Scholar]
- 15. Sucharitakul J, Tinikul R, Chaiyen P (2014) Mechanisms of reduced flavin transfer in the two‐component flavin‐dependent monooxygenases. Arch Biochem Biophys 555:33–46. [DOI] [PubMed] [Google Scholar]
- 16. Piano V, Palfey BA, Mattevi A (2017) Flavins as covalent catalysts: new mechanisms emerge. Trends Biochem Sci 42:457–469. [DOI] [PubMed] [Google Scholar]
- 17. Romero E, Gómez Castellanos JR, Gadda G, Fraaije MW, Mattevi A (2018) Same substrate, many reactions: oxygen activation in flavoenzymes. Chem Rev 118:1742–1769. [DOI] [PubMed] [Google Scholar]
- 18. Heine T, van Berkel W, Gassner G, van Pée KH, Tischler D (2018) Two‐component FAD‐dependent monooxygenases: current knowledge and biotechnological opportunities. Biology 7:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Palfey BA, McDonald CA (2010) Control of catalysis in flavin‐dependent monooxygenases. Arch Biochem Biophys 493:26–36. [DOI] [PubMed] [Google Scholar]
- 20. Pazmino DT, Winkler M, Glieder A, Fraaije MW (2010) Monooxygenases as biocatalysts: classification, mechanistic aspects and biotechnological applications. J Biotechnol 146:9–24. [DOI] [PubMed] [Google Scholar]
- 21. Montersino S, Tischler D, Gassner GT, van Berkel WJ (2011) Catalytic and structural features of flavoprotein hydroxylases and epoxidases. Adv Synth Catal 353:2301–2319. [Google Scholar]
- 22. Schreuder HA, Prick PA, Wierenga RK, Vriend G, Wilson KS, Hol WG, Drenth J (1989) Crystal structure of the p‐hydroxybenzoate hydroxylase‐substrate complex refined at 1.9 Å resolution: analysis of the enzyme‐substrate and enzyme‐product complexes. J Mol Biol 208:679–696. [DOI] [PubMed] [Google Scholar]
- 23. Montersino S, Te Poele E, Orru R, Westphal AH, Barendregt A, Heck AJ, van Berkel WJ (2017) 3‐Hydroxybenzoate 6‐hydroxylase from Rhodococcus jostii RHA1 contains a phosphatidylinositol cofactor. Front Microbiol 8:1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hiromoto T, Fujiwara S, Hosokawa K, Yamaguchi H (2006) Crystal structure of 3‐hydroxybenzoate hydroxylase from Comamonas testosteroni has a large tunnel for substrate and oxygen access to the active site. J Mol Biol 364:878–896. [DOI] [PubMed] [Google Scholar]
- 25. Enroth C, Neujahr H, Schneider G, Lindqvist Y (1998) The crystal structure of phenol hydroxylase in complex with FAD and phenol provides evidence for a concerted conformational change in the enzyme and its cofactor during catalysis. Structure 6:605–617. [DOI] [PubMed] [Google Scholar]
- 26. Uemura T, Kita A, Watanabe Y, Adachi M, Kuroki R, Morimoto Y (2016) The catalytic mechanism of decarboxylative hydroxylation of salicylate hydroxylase revealed by crystal structure analysis at 2.5 Å resolution. Biochem Biophys Res Commun 469:158–163. [DOI] [PubMed] [Google Scholar]
- 27. Ryan KS, Howard‐Jones AR, Hamill MJ, Elliott SJ, Walsh CT, Drennan CL (2007) Crystallographic trapping in the rebeccamycin biosynthetic enzyme RebC. Proc Natl Acad Sci USA 104:15311–15316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Koskiniemi H, Metsä‐Ketelä M, Dobritzsch D, Kallio P, Korhonen H, Mäntsälä P, Niemi J (2007) Crystal structures of two aromatic hydroxylases involved in the early tailoring steps of angucycline biosynthesis. J Mol Biol 372:633–648. [DOI] [PubMed] [Google Scholar]
- 29. Greenhagen BT, Shi K, Robinson H, Gamage S, Bera AK, Ladner JE, Parsons JF (2008) Crystal structure of the pyocyanin biosynthetic protein PhzS. Biochemistry 47:5281–5289. [DOI] [PubMed] [Google Scholar]
- 30. McCulloch KM, Mukherjee T, Begley TP, Ealick SE (2009) Structure of the PLP degradative enzyme 2‐methyl‐3‐hydroxypyridine‐5‐carboxylic acid oxygenase from Mesorhizobium loti MAFF303099 and its mechanistic implications. Biochemistry 48:4139–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Treiber N, Schulz GE (2008) Structure of 2,6‐dihydroxypyridine 3‐hydroxylase from a nicotine‐degrading pathway. J Mol Biol 379:94–104. [DOI] [PubMed] [Google Scholar]
- 32. Gao J, Yao L, Xia T, Liao X, Zhu D, Xiang Y (2018) Biochemistry and structural studies of kynurenine 3‐monooxygenase reveal allosteric inhibition by Ro 61‐8048. FASEB J 32:2036–2045. [DOI] [PubMed] [Google Scholar]
- 33. Lindqvist Y, Koskiniemi H, Jansson A, Sandalova T, Schnell R, Liu Z, Mantsala P, Miemi J, Schneider G (2009) Structural basis for substrate recognition and specificity in aklavinone‐11‐hydroxylase from rhodomycin biosynthesis. J Mol Biol 393:966–977. [DOI] [PubMed] [Google Scholar]
- 34. Kanteev M, Bregman‐Cohen A, Deri B, Shahar A, Adir N, Fishman A (2015) A crystal structure of 2‐hydroxybiphenyl 3‐monooxygenase with bound substrate provides insights into the enzymatic mechanism. Biochim Biophys Acta 1854:1906–1913. [DOI] [PubMed] [Google Scholar]
- 35. Prieto MA, Garcia JL (1994) Molecular characterization of 4‐hydroxyphenylacetate 3‐hydroxylase of Escherichia coli. A two‐protein component enzyme. J Biol Chem 269:22823–22829. [PubMed] [Google Scholar]
- 36. Chakraborty S, Ortiz‐Maldonado M, Entsch B, Ballou DP (2010) Studies on the mechanism of p‐hydroxyphenylacetate 3‐hydroxylase from Pseudomonas aeruginosa: a system composed of a small flavin reductase and a large flavin‐dependent oxygenase. Biochemistry 49:372–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Arias‐Barrau E, Sandoval A, Naharro G, Olivera ER, Luengo JM (2005) A two‐component hydroxylase involved in the assimilation of 3‐hydroxyphenyl acetate in Pseudomonas putida . J Biol Chem 280:26435–26447. [DOI] [PubMed] [Google Scholar]
- 38. Gibello A, Suarez M, Allende JL (1997) Molecular cloning and analysis of the genes encoding the 4‐hydroxyphenylacetate hydroxylase from Klebsiella pneumonia . Arch Microbiol 167:160–166. [PubMed] [Google Scholar]
- 39. Okai M, Kudo N, Lee WC, Kamo M, Nagata K, Tanokura M (2006) Crystal structures of the short‐chain flavin reductase HpaC from Sulfolobus tokodaii strain 7 in its three states: NAD (P)+‐free, NAD+‐bound, and NADP+‐bound. Biochemistry 45:5103–5110. [DOI] [PubMed] [Google Scholar]
- 40. Kim SH, Hisano T, Iwasaki W, Ebihara A, Miki K (2008) Crystal structure of the flavin reductase component (HpaC) of 4‐hydroxyphenylacetate 3‐monooxygenase from Thermus thermophilus HB8: structural basis for the flavin affinity. Proteins: Struct Funct Bioinf 70:718–730. [DOI] [PubMed] [Google Scholar]
- 41. Chaiyen P, Suadee C, Wilairat P (2001) A novel two‐protein component flavoprotein hydroxylase. Eur J Biochem 268:5550–5561. [DOI] [PubMed] [Google Scholar]
- 42. Kirchner U, Westphal AH, Müller R, van Berkel WJ (2003) Phenol hydroxylase from Bacillus thermoglucosidasius A7, a two‐protein component monooxygenase with a dual role for FAD. J Biol Chem 278(48):47545–47553. [DOI] [PubMed] [Google Scholar]
- 43. Valton J, Fontecave M, Douki T, Kendrew SG, Nivière V (2005) An aromatic hydroxylation reaction catalyzed by a two‐component FMN‐dependent monooxygenase: the ActVA‐ActVB system from Streptomyces coelicolor . J Biol Chem 281:27–35. [DOI] [PubMed] [Google Scholar]
- 44. Filisetti L, Fontecave M, Nivière V (2003) Mechanism and substrate specificity of the flavin reductase ActVB from Streptomyces coelicolor . J Biol Chem 278:296–303. [DOI] [PubMed] [Google Scholar]
- 45. Chang CY, Lohman JR, Cao H, Tan K, Rudolf JD, Ma M, Xu W, Bingman CA, Yennamalli RM, Bigelow L, Babnigg G, Yan X, Joachimiak A, Phillips GN Jr, Shen B (2016) Crystal structures of SgcE6 and SgcC, the two‐component monooxygenase that catalyzes hydroxylation of a carrier protein‐tethered substrate during the biosynthesis of the enediyne antitumor antibiotic C‐1027 in Streptomyces globisporus . Biochemistry 55:5142–5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cheng M, Meng Q, Yang Y, Chu C, Chen Q, Li Y, Cheng D, Hong Q, Yan X, He J (2017) The two‐component monooxygenase MeaXY initiates the downstream pathway of chloroacetanilide herbicide catabolism in sphingomonads. Appl Environ Microbiol 83(7):e03241‐16 10.1128/AEM.03241-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu X, Dong Y, Li X, Ren Y, Li Y, Wang W, Wang L, Feng L (2010) Characterization of the anthranilate degradation pathway in Geobacillus thermodenitrificans NG80‐2. Microbiology 156:589–595. [DOI] [PubMed] [Google Scholar]
- 48. Webb BN, Ballinger JW, Kim E, Belchik SM, Lam KS, Youn B, Nissen MS, Xun L, Kang C (2010) Characterization of chlorophenol 4‐monooxygenase (TftD) and NADH:FAD oxidoreductase (TftC) of Burkholderia cepacia AC1100. J Biol Chem 285:2014–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Louie TM, Webster CM, Xun L (2002) Genetic and biochemical characterization of a 2, 4, 6‐trichlorophenol degradation pathway in Ralstonia eutropha JMP134. J Bacteriol 184:3492–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Belchik SM, Xun L (2008) Functions of flavin reductase and quinone reductase in 2, 4, 6‐trichlorophenol degradation by Cupriavidus necator JMP134. J Bacteriol 190:1615–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Takeo M, Murakami M, Niihara S, Yamamoto K, Nishimura M, Kato DI, Negoro S (2008) Mechanism of 4‐nitrophenol oxidation in Rhodococcus sp. strain PN1: characterization of the two‐component 4‐nitrophenol hydroxylase and regulation of its expression. J Bacteriol 190:7367–7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pimviriyakul P, Chaiyen P (2018) A complete bioconversion cascade for dehalogenation and denitration by flavin‐dependent enzymes. J Biol Chem. 10.1074/jbc.RA118.005538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pimviriyakul P, Thotsaporn K, Sucharitakul J, Chaiyen P (2017) Kinetic mechanism of the dechlorinating flavin‐dependent monooxygenase HadA. J Biol Chem 292:4818–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thotsaporn K, Sucharitakul J, Wongratana J, Suadee C, Chaiyen P (2004) Cloning and expression of p‐hydroxyphenylacetate 3‐dydroxylase from Acenetobacter baumannii: evidence of the divergence of enzyme in the class of two‐protein component aromatic hydroxylase. Biochim Biophys Acta Gene Struct Expression 1680:60–66. [DOI] [PubMed] [Google Scholar]
- 55. Sucharitakul J, Chaiyen P, Entsch B, Ballou DP (2005) The reductase of p‐hydroxyphenylacetate 3‐hydroxylase from Acinetobacter baumannii requires p‐hydroxyphenylacetate for effective catalysis. Biochemistry 44:10434–10442. [DOI] [PubMed] [Google Scholar]
- 56. Phongsak T, Sucharitakul J, Thotsaporn K, Oonanant W, Yuvaniyama J, Svasti J, Ballou DP, Chaiyen P (2012) The C‐terminal domain of 4‐hydroxyphenylacetate 3‐hydroxylase from Acinetobacter baumannii is an autoinhibitory domain. J Biol Chem 287:26213–26222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ellis HR (2010) The FMN‐dependent two‐component monooxygenase systems. Arch Biochem Biophys 497:1–12. [DOI] [PubMed] [Google Scholar]
- 58. Nijvipakul S, Wongratana J, Suadee C, Entsch B, Ballou DP, Chaiyen P (2008) LuxG is a functioning flavin reductase for bacterial luminescence. J Bacteriol 190:1531–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Alfieri A, Fersini F, Ruangchan N, Prongjit M, Chaiyen P, Mattevi A (2007) Structure of the monooxygenase component of a two‐component flavoprotein monooxygenase. Proc Nat Acad Sci USA 104:1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim SH, Hisano T, Takeda K, Iwasaki W, Ebihara A, Miki K (2007) Crystal structure of the oxygenase component (HpaB) of the 4‐hydroxyphenylacetate 3‐monooxygenase from Thermus thermophilus HB8. J Biol Chem 282:33107–33117. [DOI] [PubMed] [Google Scholar]
- 61. Husain M, Massey V (1979) Kinetic studies on the reaction of p‐hydroxybenzoate hydroxylase. Agreement of steady state and rapid reaction data. J Biol Chem 254:6657–6666. [PubMed] [Google Scholar]
- 62. Entsch B, van Berkel WJ (1995) Structure and mechanism of para‐hydroxybenzoate hydroxylase. FASEB J 9:476–483. [DOI] [PubMed] [Google Scholar]
- 63. Gatti DL, Palfey BA, Lah MS, Entsch B, Massey V, Ballou DP, Ludwig ML (1994) The mobile flavin of 4‐OH benzoate hydroxylase. Science 266:10–114. [DOI] [PubMed] [Google Scholar]
- 64. Manstein DJ, Pai EF, Schopfer LM, Massey V (1986) Absolute stereochemistry of flavins in enzyme‐catalyzed reactions. Biochemistry 25:6807–6816. [DOI] [PubMed] [Google Scholar]
- 65. Wang J, Ortiz‐Maldonado M, Entsch B, Massey V, Ballou D, Gatti DL (2002) Protein and ligand dynamics in 4‐hydroxybenzoate hydroxylase. Proc Nat Acad Sci USA 99:608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cole LJ, Entsch B, Ortiz‐Maldonado M, Ballou DP (2005) Properties of p‐hydroxybenzoate hydroxylase when stabilized in its open conformation. Biochemistry 44:14807–14817. [DOI] [PubMed] [Google Scholar]
- 67. Palfey BA, Basu R, Frederick KK, Entsch B, Ballou DP (2002) Role of protein flexibility in the catalytic cycle of p‐hydroxybenzoate hydroxylase elucidated by the Pro293Ser mutant. Biochemistry 41:8438–8446. [DOI] [PubMed] [Google Scholar]
- 68. Moran GR, Entsch B, Palfey BA, Ballou DP (1997) Electrostatic effects on substrate activation in parahydroxybenzoate hydroxylase: studies of the mutant lysine 297 methionine. Biochemistry 36:7548–7556. [DOI] [PubMed] [Google Scholar]
- 69. Entsch B, Ballou DP, Massey V (1976) Flavin‐oxygen derivatives involved in hydroxylation by p‐hydroxybenzoate hydroxylase. J Biol Chem 251:2550–2563. [PubMed] [Google Scholar]
- 70. Sucharitakul J, Chaiyen P, Entsch B, Ballou DP (2006) Kenetic mechanisms of the oxygenase from a two‐component enzyme, p‐hydroxyphenylacetate 3‐hydroxylase from Acinetobacter baumannii . J Biol Chem 281:17044–17053. [DOI] [PubMed] [Google Scholar]
- 71. Entsch B, Ballou DP, Husain M, Massey V (1976) Catalytic mechanism of p‐hydroxybenzoate hydroxylase with p‐mercaptobenzoate as substrate. J Biol Chem 251:7367–7379. [PubMed] [Google Scholar]
- 72. Palfey BA, Moran GR, Entsch B, Ballou DP, Massey V (1999) Substrate recognition by “Password” in p‐hydroxybenzoate hydroxylase. Biochemistry 38:1153–1158. [DOI] [PubMed] [Google Scholar]
- 73. Thotsaporn K, Tinikul R, Maenpuen S, Phonbuppha J, Watthaisong P, Chenprakhon P, Chaiyen P (2016) Enzymes in the p‐hydroxyphenylacetate degradation pathway of Acinetobacter baumannii . J Mol Catal B Enzym 134:353–366. [Google Scholar]
- 74. Yuenyao A, Petchyam N, Kamonsutthipaijit N, Chaiyen P, Pakotiprapha D (2018) Crystal structure of the flavin reductase of Acinetobacter baumannii p‐hydroxyphenylacetate 3‐hydroxylase (HPAH) and identification of amino acid residues underlying its regulation by aromatic ligands. Arch Biochem Biophys 653:24–38. [DOI] [PubMed] [Google Scholar]
- 75. Tinikul R, Pitsawong W, Scharitakul J, Nijvipakul S, Ballou DP, Chaiyen P (2013) The transfer of reduced flavin mononucleotide from LuxG oxidoreductase to luciferase occurs via free diffusion. Biochemistry 52:6834–6843. [DOI] [PubMed] [Google Scholar]
- 76. Sucharitakul J, Phongsak T, Entsch B, Svasti J, Chaiyen P, Ballou DP (2007) Kinetic of a two‐component p‐hydroxyphenylacetate hydroxylase explain how reduced flavin is transferred from the reductase to the oxygenase. Biochemistry 46:8611–8623. [DOI] [PubMed] [Google Scholar]
- 77. Arunachalam U, Massey V, Miller SM (1994) Mechaniam of p‐hydroxyphenylacetate 3‐hydroxylase: a two‐protein enzyme. J Biol Chem 269:150–155. [PubMed] [Google Scholar]
- 78. Li L, Liu X, Yang W, Xu F, Wang W, Feng L, Bartlam M, Wang L, Rao Z (2008) Crystal structure of long‐chain alkane monooxygenase (LadA) in complex with coenzyme FMN: unveiling the long‐chain alkane hydroxylase. J Mol Biol 376:453–465. [DOI] [PubMed] [Google Scholar]
- 79. Hino T, Hamamoto H, Suzuki H, Yagi H, Ohshiro T, Nagano S (2017) Crystal structures of TdsC, a dibenzothiophene monooxygenase from the thermophile Paenibacillus sp. A11‐2, reveal potential for expanding its substrate selectivity. J Biol Chem 292:15804–15813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Baron R, Riley C, Chenprakhon P, Thotsaporn K, Winter RT, Alfieri A, Forneris F, van Berkel WJH, Chaiyen P, Fraaije MW, Mattevi A (2009) Multiple pathways guide oxygen diffusion into flavoenzyme active sites. Proc Natl Acad Sci USA 106:10603–10605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Suadee C, Nijvipakul S, Svasti J, Entsch B, Ballou DP, Chaiyen P (2007) Luciferase from Vibrio campbellii is more thermostable and binds reduced FMN better than its homologues. J Biochem 142:539–552. [DOI] [PubMed] [Google Scholar]
- 82. Kantz A, Chin F, Nallamothu N, Nguyen T, Gassner GT (2005) Mechanism of flavin transfer and oxygen activation by the two‐component flavoenzyme styrene monooxygenase. Arch Biochem Biophys 442:102–116. [DOI] [PubMed] [Google Scholar]
- 83. Valton J, Mathevon C, Fontecave M, Nivière V, Ballou DP (2008) Mechanism and regulation of the two‐component FMN‐dependent monooxygenase ActVA‐ActVB from Streptomyces coelicolor . J Biol Chem 283:10287–10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Thotsaporn K, Chenprakhon P, Sucharitakul J, Mattevi A, Chaiyen P (2011) Stabilization of C4a‐hydroperoxyflavin in a two‐component flavin‐dependent monooxygenase is achieved through interactions at flavin N5 and C4a atoms. J Biol Chem 286:28170–28180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chenprakhon P, Trisrivirat D, Thotsaporn K, Sucharitakul J, Chaiyen P (2014) Control of C4a‐hydroperoxyflavin protonation in the oxygenase component of p‐hydroxyphenylacetate‐3‐hydroxylase. Biochemistry 53(25):4084–4086. [DOI] [PubMed] [Google Scholar]
- 86. Visitsatthawong S, Chenprakhon P, Chaiyen P, Surawatanawong P (2015) Mechanism of oxygen activation in a flavin‐dependent monooxygenase: a nearly barrierless formation of C4a‐hydroperoxyflavin via proton‐coupled electron transfer. J Am Chem Soc 137:9363–9374. [DOI] [PubMed] [Google Scholar]
- 87. Ruangchan N, Tongsook C, Sucharitakul J, Chaiyen P (2011) pH‐dependent studies reveal an efficient hydroxylation mechanism of the oxygenase component of p‐hydroxyphenylacetate 3‐hydroxylase. J Biol Chem 286:223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tongsook C, Sucharitakul J, Thotsaporn K, Chaiyen P (2011) Interactions with the substrate phenolic group are essential for hydroxylation by the oxygenase component of p‐hydroxyphenylacetate 3‐hydroxylase. J Biol Chem 286:44491–44502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chaiyen P, Ballou DP, Massey V (1997) Genecloning, sequence analysis, and expression of 2‐methyl‐3‐hydroxypyridine‐5‐carboxylic acid oxygenase. Proc Natl Acad Sci USA 94:7233–7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nelson MJ, Snell EE (1986) Enzymes of vitamin B6 degradation. Purification and properties of 5‐pyridoxic‐acid oxygenase from Arthrobacter sp. J Biol Chem 261:5115–15120. [PubMed] [Google Scholar]
- 91. Snell EE (1993) From bacterial nutrition to enzyme structure: a personal odyssey. Annu Rev Biochem 62:1–26. [DOI] [PubMed] [Google Scholar]
- 92. Yuan B, Yokochi N, Yoshikane Y, Ohnishi K, Yagi T (2006) Molecular cloning, identification and characterization of 2‐methyl‐3‐hydroxypyridine‐5‐carboxylic‐acid‐dioxygenase‐coding gene from the nitrogen‐fixing symbiotic bacterium Mesorhizobium loti. J Biosci Bioeng 102:504–510. [DOI] [PubMed] [Google Scholar]
- 93. Mukherjee T, Hanes J, Tews I, Ealick SE, Begley TP (2011) Pyridoxal phosphate: biosynthesis and catabolism. Biochim Biophys Acta Proteins Proteomics 1814:1585–1596. [DOI] [PubMed] [Google Scholar]
- 94. Chaiyen P, Brissette P, Ballou DP, Massey V (1997) Unusual mechanism of oxygen atom transfer and product rearrangement in the catalytic reaction of 2‐methyl‐3‐hydroxypyridine‐5‐carboxylic acid oxygenase. Biochemistry 2960:8060–8070. [DOI] [PubMed] [Google Scholar]
- 95. Chaiyen P (2010) Flavoenzymes catalyzing oxidative aromatic ring‐cleavage reactions. Arch Biochem Biophys 493:62–70. [DOI] [PubMed] [Google Scholar]
- 96. Chaiyen P, Brissette P, Ballou DP, Massey V (1997) Reaction of 2‐Methyl‐3‐hydroxypyridine‐5‐carboxylic acid (MHPC) oxygenase with N‐methyl‐5‐hydroxynicotinic acid: studies on the mode of binding, and protonation status of the substrate. Biochemistry 36:13856–13864. [DOI] [PubMed] [Google Scholar]
- 97. Luanloet T, Sucharitakul J, Chaiyen P (2015) Selectivity of substrate binding and ionization of 2‐methyl‐3‐hydroxypyridine‐5‐carboxylic acid oxygenase. FEBS J 282:3107–3125. [DOI] [PubMed] [Google Scholar]
- 98. Kobayashi J, Yoshida H, Yagi T, Kamitori S, Hayashi H, Mizutani K, Takahashi N, Mikami B (2017) Role of the Tyr270 residue in 2‐methyl‐3‐hydroxypyridine‐5‐carboxylic acid oxygenase from Mesorhizobium loti . J Biosci Bioeng 123:154–162. [DOI] [PubMed] [Google Scholar]
- 99. Chaiyen P, Brissette P, Ballou DP, Massey V (1997) Thermodynamics and reduction kinetics properties of 2‐methyl‐3‐ hydroxypyridine‐5‐carboxylic acid oxygenase. Biochemistry 36:2612–2621. [DOI] [PubMed] [Google Scholar]
- 100. Tian B, Tu Y, Strid Å, Eriksson LA (2010) Hydroxylation and ring‐opening mechanism of an unusual flavoprotein monooxygenase, 2‐methyl‐3‐hydroxypyridine‐5‐carboxylic acid oxygenase: a theoretical study. Chem Eur J 16:2557–2566. [DOI] [PubMed] [Google Scholar]
- 101. Tian B, Tu Y, Strid Å, Eriksson LA (2011) Catalytic roles of active‐site residues in 2‐methyl‐3‐hydroxypyridine‐5‐carboxylic acid oxygenase: an ONIOM/DFT study. J Phys Chem B 115:1918–1926. [DOI] [PubMed] [Google Scholar]
- 102. Gribble GW (2003) The diversity of naturally produced organohalogens. Chemosphere 52:289–297. [DOI] [PubMed] [Google Scholar]
- 103. Isin EM, Guengerich FP (2007) Complex reactions catalyzed by cytochrome P450 enzymes. Biochim Biophys Acta Gen Subj 1770:314–329. [DOI] [PubMed] [Google Scholar]
- 104. Borja J, Taleon DM, Auresenia J, Gallardo S (2005) Polychlorinated biphenyls and their biodegradation. Process Biochem 40:1999–2013. [Google Scholar]
- 105. Arora PK, Srivastava A, Singh VP (2014) Bacterial degradation of nitrophenols and their derivatives. J Hazard Mater 266:42–59. [DOI] [PubMed] [Google Scholar]
- 106. Pimviriyakul P, Surawatanawong P, Chaiyen P (2018) Oxidative dehalogenation and denitration by a flavin‐dependent monooxygenase is controlled by substrate deprotonation. Chem Sci 9:7468–7482. 10.1039/c8sc01482e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Xun L, Webster CM (2004) A monooxygenase catalyzes sequential dechlorinations of 2,4,6‐trichlorophenol by oxidative and hydrolytic reactions. J Biol Chem 279:6696–6700. [DOI] [PubMed] [Google Scholar]
- 108. Hatta T, Fujii E, Takizawa N (2012) Analysis of two gene clusters involved in 2,4,6‐trichlorophenol degradation by Ralstonia pickettii DTP0602. Biosci Biotechnol Biochem 76:892–899. [DOI] [PubMed] [Google Scholar]
- 109. Rudolph J, Erbse AH, Behlen LS, Copley SD (2014) A radical intermediate in the conversion of pentachlorophenol to tetrachlorohydroquinone by Sphingobium chlorophenolicum. Biochemistry 53:6539–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Valverde C, Orozco A, Becerra A, Jeziorski MC, Villalobos P, Solis JC (2004) Halometabolites and cellular dehalogenase systems: an evolutionary perspective. Int Rev Cytol 234:143–199. [DOI] [PubMed] [Google Scholar]
- 111. Franzen S, Thompson MK, Ghiladi RA (2012) The dehaloperoxidase paradox. Biochim Biophys Acta Proteins Proteomics 1824:578–588. [DOI] [PubMed] [Google Scholar]
- 112. Hlouchova K, Rudolph J, Pietari JM, Behlen LS, Copley SD (2012) Pentachlorophenol hydroxylase, a poorly functioning enzyme required for degradation of pentachlorophenol by Sphingobium chlorophenoum . Biochemistry 51:3848–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Gisi MR, Xun L (2003) Characterization of chlorophenol 4‐monooxygenase (TftD) and NADH:flavin adenine dinucleotide oxidoreductase (TftC) of Burkholderia cepacia AC1100. J Bacteriol 185:2786–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Li YW, Zhang RM, Du LK, Zhang QZ, Wang WX (2015) Insights into the catalytic mechanism of chlorophenol 4‐monooxygenase: a quantum mechanics/molecular mechanics study. RSC Adv 5:13871–13877. [Google Scholar]
- 115. Ridder L, Harvey JN, Rietjens IM, Vervoort J, Mulholland AJ (2003) Ab initio QM/MM modeling of the hydroxylation step in p‐hydroxybenzoate hydroxylase. J Phys Chem B 107:2118–2126. [Google Scholar]
- 116. Tishchenko O, Pham‐Tran NN, Kryachko ES, Nguyen MT (2001) Protonation of gaseous halogenated phenols and anisoles and its interpretation using DFT‐based local reactivity indices. J Phys Chem A 105:8709–8717. [Google Scholar]
- 117. Matsui T, Shigeta Y, Morihashi K (2017) Assessment of methodology and chemical group dependences in the calculation of the pK a for several chemical groups. J Chem Theory Comput 13:4791–4803. [DOI] [PubMed] [Google Scholar]
- 118. Chaiyen P, Sucharitakul J, Svasti J, Entsch B, Massey V, Ballou DP (2004) Use of 8‐substituted‐FAD analogues to investigate the hydroxylation mechanism of the flavoprotein 2‐methyl‐3‐hydroxypyridine‐5‐carboxylic acid oxygenase. Biochemistry 43:3933–3943. [DOI] [PubMed] [Google Scholar]
- 119. Ridder L, Mulholland AJ, Rietjens IM, Vervoort J (2000) A quantum mechanical/molecular mechanical study of the hydroxylation of phenol and halogenated derivatives by phenol hydroxylase. J Am Chem Soc 122:8728–8738. [Google Scholar]
- 120. Hayes RP, Webb BN, Subramanian AK, Nissen M, Popchock A, Xun L, Kang C (2012) Structural and catalytic differences between two FADH(2)‐dependent monooxygenases: 2,4,5‐TCP 4‐monooxygenase (TftD) from Burkholderia cepacia AC1100 and 2,4,6‐TCP 4‐monooxygenase (TcpA) from Cupriavidus necator JMP134. Int J Mol Sci 13:9769–9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hernández‐Ortega A, Quesne MG, Bui S, Heyes DJ, Steiner RA, Scrutton NS, de Visser SP (2015) Catalytic mechanism of cofactor‐free dioxygenases and how they circumvent spin‐forbidden oxygenation of their substrates. J Am Chem Soc 137:7474–7487. [DOI] [PubMed] [Google Scholar]
- 122. Wongnate T, Surawatanawong P, Visitsatthawong S, Sucharitakul J, Scrutton NS, Chaiyen P (2013) Proton‐coupled electron transfer and adduct configuration are important for C4a‐hydroperoxyflavin formation and stabilization in a flavoenzyme. J Am Chem Soc 136:241–253. [DOI] [PubMed] [Google Scholar]
- 123. Hernández‐Ortega A, Borrelli K, Ferreira P, Medina M, Martínez AT, Guallar V (2011) Substrate diffusion and oxidation in GMC oxidoreductases: an experimental and computational study on fungal aryl‐alcohol oxidase. Biochem J 436:341–350. [DOI] [PubMed] [Google Scholar]
- 124. Eberlein G, Bruice TC (1983) The chemistry of a 1,5‐diblocked flavin. 2. Proton and electron transfer steps in the reaction of dihydroflavins with oxygen. J Am Chem Soc 105:6685–6697. [Google Scholar]
- 125. Bruice TC (1984) Oxygen‐flavin chemistry. Isr J Chem 24:54–61. [Google Scholar]
- 126. Massey V (1994) Activation of molecular oxygen by flavins and flavoproteins. J Biol Chem 269:22459–22462. [PubMed] [Google Scholar]
- 127. Ortiz‐Maldonado M, Ballou DP, Massey V (1999) Use of free energy relationships to probe the individual steps of hydroxylation of p‐hydroxybenzoate hydroxylase: studies with a series of 8‐substituted flavins. Biochemistry 38:8124–8137. [DOI] [PubMed] [Google Scholar]
- 128. Ridder L, Palfey BA, Vervoort J, Rietjens IM (2000) Modelling flavin and substrate substituent effects on the activation barrier and rate of oxygen transfer by p‐hydroxybenzoate hydroxylase. FEBS Lett 478:197–201. [DOI] [PubMed] [Google Scholar]
- 129. Ridder L, Mulholland AJ, Rietjens IM, Vervoort J (1999) Combined quantum mechanical and molecular mechanical reaction pathway calculation for aromatic hydroxylation by p‐hydroxybenzoate‐3‐hydroxylase. J Mol Graph Mod 17:163–175. [DOI] [PubMed] [Google Scholar]
- 130. Wang X, Hou Q, Liu Y (2018) Insights into the decarboxylative hydroxylation of salicylate catalyzed by the flavin‐dependent monooxygenase salicylate hydroxylase. Theor Chem Acc 137:89. [Google Scholar]
- 131. Entsch B, Palfey BA, Ballou DP, Massey V (1991) Catalytic function of tyrosine residues in para‐hydroxybenzoate hydroxylase as determined by the study of site‐directed mutants. J Biol Chem 266:17341–17349. [PubMed] [Google Scholar]
- 132. Ortiz‐Maldonado M, Entsch B, Ballou DP (2004) Oxygen reactions in p‐hydroxybenzoate hydroxylase utilize the H‐bond network during catalysis. Biochemistry 43:15246–15257. [DOI] [PubMed] [Google Scholar]
- 133. Leisch H, Morley K, Lau PC (2011) Baeyer–Villiger monooxygenases: more than just green chemistry. Chem Rev 111:4165–4222. [DOI] [PubMed] [Google Scholar]
- 134. Schmidt S, Scherkus C, Muschiol J, Menyes U, Winkler T, Hummel W, Groger H, Liese A, Herz HG, Bornscheuer UT (2015) An enzyme cascade synthesis of ε‐caprolactone and its oligomers. Angew Chem Int Ed 54:2784–2787. [DOI] [PubMed] [Google Scholar]
- 135. Zhu Y, Zhang Q, Li S, Lin Q, Fu P, Zhang G, Zhang H, Shi R, Zhu W, Zhang C (2013) Insights into caerulomycin A biosynthesis: a two‐component monooxygenase CrmH‐catalyzed oxime formation. J Am Chem Soc 135:18750–18753. [DOI] [PubMed] [Google Scholar]
- 136. Hollmann F, Hofstetter K, Habicher T, Hauer B, Schmid A (2005) Direct electrochemical regeneration of monooxygenase subunits for biocatalytic asymmetric epoxidation. J Am Chem Soc 127:6540–6541. [DOI] [PubMed] [Google Scholar]
- 137. Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K (2012) Engineering the third wave of biocatalysis. Nature 485:185–194. [DOI] [PubMed] [Google Scholar]
- 138. Bornscheuer UT (2013) From commercial enzymes to biocatalysts designed by protein engineering. Synlett 24:150–156. [Google Scholar]
- 139. Dhammaraj T, Phintha A, Pinthong C, Medhanavyn D, Tinikul R, Chenprakhon P, Sucharitakul J, Vardhanabhuti N, Jiarpinitnun C, Chaiyen P (2015) p‐Hydroxyphenylacetate 3‐hydroxylase as a biocatalyst for the synthesis of trihydroxyphenolic acids. ACS Catal 5:4492–4502. [Google Scholar]
- 140. Dhammaraj T, Pinthong C, Visitsatthawong S, Tongsook C, Surawatanawong P, Chaiyen P (2016) A single‐site mutation at ser146 expands the reactivity of the oxygenase component of p‐hydroxyphenylacetate 3‐hydroxylase. ACS Chem Biol 11:2889–2896. [DOI] [PubMed] [Google Scholar]
- 141. Muro F, Iimura S, Yoneda Y, Chiba J, Watanabe T, Setoguchi M, Takayama G, Yokoyama M, Takashi T, Nakayama A, Machinaga N (2009) A novel and potent VLA‐4 antagonist based on trans‐4‐substituted cyclohexanecarboxylic acid. Bioorg Med Chem 17:1232–1243. [DOI] [PubMed] [Google Scholar]
- 142. Pinthong C, Phoopraintra P, Chantiwas R, Pongtharangkul T, Chenprakhon P, Chaiyen P (2017) Green and sustainable biocatalytic production of 3,4,5‐trihydroxycinnamic acid from palm oil mill effluent. Process Biochem 63:122–129. [Google Scholar]
- 143. Arora PK, Bae H (2014) Bacterial degradation of chlorophenols and their derivatives. Microb Cell Fact 13:31–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Harms H, Schlosser D, Wick LY (2011) Untapped potential: exploiting fungi in bioremediation of hazardous chemicals. Nat Rev Microbiol 9:177–192. [DOI] [PubMed] [Google Scholar]
- 145. Dong J, Fernandez‐Fueyo E, Hollmann F, Paul CE, Pesic M, Schmidt S, Wang Y, Younes S, Zhang W (2018) Biocatalytic oxidation reactions: a chemist's perspective. Angew Chem Int Ed Engl 57:9238–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Lancaster L, Abdallah W, Banta S, Wheeldon I (2018) Engineering enzyme microenvironments for enhanced biocatalysis. Chem Soc Rev 47:5177–5186. [DOI] [PubMed] [Google Scholar]
- 147. Arnold FH (2018) Directed evolution: bringing new chemistry to life. Angew Chem Int Ed Engl 57:4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Ducharme J, Auclair K (2018) Use of bioconjugation with cytochrome P450 enzymes. Biochim Biophys Acta Proteins Proteomics 1866:32–51. [DOI] [PubMed] [Google Scholar]
- 149. Ba S, Vinoth Kumar V (2017) Recent developments in the use of tyrosinase and laccase in environmental applications. Crit Rev Biotechnol 37:819–832. [DOI] [PubMed] [Google Scholar]
- 150. Bucko M, Gemeine P, Schenkmayerova A, Krajcovic T, Rudroff F, Mihovilovic MD (2016) Baeyer‐Villiger oxidations: biotechnological approach. Appl Microbiol Biotechnol 100:6585–6599. [DOI] [PubMed] [Google Scholar]