

Abstract
Chemoradiation therapy is the mainstay for treatment of locally advanced, borderline resectable pancreatic cancer. Pharmacological ascorbate (P-AscH-, i.e., intravenous infusions of ascorbic acid, vitamin C), but not oral ascorbate, produces high plasma concentrations capable of selective cytotoxicity to tumor cells. In doses achievable in humans, P-AscH- decreases the viability and proliferative capacity of pancreatic cancer via a hydrogen peroxide (H2O2)-mediated mechanism. In this study, we demonstrate that P-AscH- radiosensitizes pancreatic cancer cells but inhibits radiation-induced damage to normal cells. Specifically, radiation-induced decreases in clonogenic survival and double-stranded DNA breaks in tumor cells, but not in normal cells, were enhanced by P-AscH-, while radiation-induced intestinal damage, collagen deposition, and oxidative stress were also reduced with P-AscH- in normal tissue. We also report on our first-in-human phase 1 trial that infused P-AscH- during the radiation therapy ‘beam on.’ Specifically, treatment with P-AscH- increased median overall survival compared to our institutional average (21.7 vs. 12.7 mo, p = 0.08) and the E4201 trial (21.7 vs. 11.1 mo). Progression-free survival in P-AscH--treated subjects was also greater than our institutional average (13.7 vs. 4.6 mo, p < 0.05) and the E4201 trial (6.0 months). Results indicated that P-AscH- in combination with gemcitabine and radiation therapy for locally advanced pancreatic adenocarcinoma is safe and well tolerated with suggestions of efficacy. Because of the potential effect size and minimal toxicity, our findings suggest that investigation of P-AscH- efficacy is warranted in a phase 2 clinical trial.
Keywords: ascorbate, pancreatic cancer, clinical trial, radiation therapy, reactive oxygen species
Introduction
Over 55,000 patients are projected to be diagnosed with pancreatic adenocarcinoma in 2018, making it the 12th most common cancer in the United States.[1] Despite this modest incidence, it is the 4th deadliest cancer amongst both men and women (12.6 per 100,000) in the U.S. with 44,330 patients expected to succumb to the disease in 2018.[1] This lethality is likely due to a combination of late stage at diagnosis and poor survival at all stages. In fact, 29% of patients present with regional disease and 52% with metastatic disease with respective 5-year survival rates of 11.5% and 2.7%.[1, 2]
While there is a significant effort devoted to identify novel systemic agents for advanced disease, there is a tremendous clinical need to improve therapies for patients diagnosed with localized disease.2 One autopsy series demonstrated that approximately 30% of all stages of pancreatic cancer patients died as a result of local disease burden, irrespective of clinical stage at diagnosis, suggesting that patients with advanced disease may benefit from improved local disease treatment.[3] Surgery is one option for local therapy; however, the majority of patients with advanced disease are not amenable to resection. An alternative to surgery is radiation therapy in combination with chemotherapy but most patients will still experience local treatment failure and eventually succumb to their disease.[1, 4] Thus, it is clear that a significant segment of pancreatic cancer patients could benefit from improvements in the efficacy of radiation therapy. Chemoradiation therapy may also be initiated in this patient population as a front line therapy or after induction chemotherapy if there has been no metastatic progression.[4] The ECOG-E4201 study established 50.4 Gy radiation therapy delivered over 28 fractions plus gemcitabine as a superior regimen to gemcitabine alone for locally advanced disease with an increase in overall survival from 9.2 months to 11.1 months.[5]
The dose-limitation of radiation in pancreatic cancer is dictated by damage to surrounding tissue, in particular the small intestine.[6] The expected complication rate when the intestine receives a total dose of 50 Gy in 1.8 or 2 Gy fractions is 5% but rapidly increases to 50% when radiation doses reach a total dose of 60 Gy.[7] The maximum recommended conventional radiation dose for the treatment of pancreatic adenocarcinoma is 54 Gy, which poses a 9.4 – 24.4% complication rate.[6, 8] Unfortunately, efforts to find radio-protection agents that could potentially allow increased radiation dosing while avoiding concurrent tumor protection have been relatively unsuccessful.[9]
Vitamin C (ascorbic acid, ascorbate) is traditionally considered a donor antioxidant, yet is capable of behaving as a pro-oxidant at higher pharmacological doses (P-AscH-), producing H2O2 in the extravascular space.[10–12] Intravenous dosing is required to bypass the intestinal mechanisms that tightly control oral uptake (only micromolar concentrations achievable) and safely allows plasma concentrations to achieve levels 100-fold higher than oral administration (mM concentrations achievable).[13, 14] The H2O2 produced in the extravascular space at these levels is selectively cytotoxic to cancer cells.[15, 16] While the exact mechanism is still under study, H2O2 is thought to diffuse across the cell membrane and subsequently react with the relatively high levels of catalytic metals sequestered in cancer cells to produce toxic radical species.[15, 17] A recent phase 1 clinical trial combining P-AscH- with gemcitabine for advanced pancreatic cancer produced millimolar levels of plasma P-AscH-, demonstrated excellent tolerability, and suggested a synergistic affect with chemotherapy.[13, 18]
More recently, the radio-sensitizing effects of P-AscH- on cancer have been elucidated. Pre-clinical studies confirm that P-AscH- potentiates radiation-induced cytotoxicity via a H2O2-mediated mechanism, decreasing tumor growth and prolonging survival in an animal model.[19] A selective radiosensitizing agent with a protecting effect on surrounding tissue is novel and could potentially liberate patients from the current radiation dose limitations used, allowing more aggressive treatment of local disease. The goal of the present study is to determine the differential toxicity of P-AscH- in pancreatic cancer cells vs. normal cells and to evaluate the safety, tolerability, and potential efficacy of P-AscH- in a first-in-human phase 1 clinical trial combined with radiation and gemcitabine for locally advanced pancreatic cancer.
Materials and Methods
Cell Culture-
Human pancreatic cancer cell lines, MIA PaCa-2, PANC-1, and normal fetal human intestinal epithelial cell line, FHs 74 Int, were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and not further authenticated or tested. MIA PaCa-2 and PANC-1 were maintained as described.15, 16, 19 Patient-derived pancreatic cancer cell line, 403, from the Medical College of Wisconsin surgical oncology tissue bank [20, 21] was cultured in Dulbecco’s Modified Eagle’s Media Nutrient Mixture F-12 supplemented with 5% FBS, penicillin/streptomycin, human recombinant EGF, bovine pituitary extract, hydrocortisone, and human recombinant insulin. HUVEC cells were from Lonza (Basel, Switzerland) and cultured in Endothelial Basal Medium-Plus (EBM™ Medium) supplemented with Bovine Brain Extract (BBE), hydrocortisone, human Epidermal Growth Factor (hEGF), 2% FBS and Getamincin/Amphotericin-B. The immortalized normal pancreatic ductal epithelial cells H6c7 were cultured in keratinocyte-serum free medium (KSFM) with supplements (2.5 μg human recombinant EGF and 25 mg bovine pituitary extract).[22] All cells were maintained in a humidified atmosphere of 95% air/5% CO2 at 37 °C and passaged routinely for use until passage 20.
Reagents-
Stock solutions of ascorbate (pH 7.0) were made under argon and stored in screw-top sealed test tubes at 4 °C. Ascorbate concentration was verified using, ε265 = 14,500 M−1 cm−1.[23] The solution can be kept for several weeks without significant oxidation due to the lack of oxygen.
Clonogenic Survival Assay-
Clonogenic survival assays were performed as previously described.[19] Briefly, cells were plated and allowed to attach for 24 – 48 h. Cells were treated with P-AscH- (1 and 2 mM or 10 and 20 pmol cell−1, respectively) for 1 h in DMEM/10% FBS followed by irradiation at 0.5 – 2 Gy. Cells were immediately trypsinized, counted and seeded into 6-well plates at varying densities. FHs 74 Int and H6c7 cells were seeded on top of feeder cells that had been irradiated at 30 Gy. The dishes were maintained in a 37 °C, 4% O2, for 10–14 days to allow colony formation. The colonies were then fixed with 70% ethanol, stained with Coomassie™ Blue and counted (colonies containing > 50 cells were scored). Plating efficiency was determined by the formula: (number of colonies formed/number of cells inoculated) ×100.
Immunoblot Analysis-
Protein (20–40 μg) was electrophoresed in a 4% to 20% Bio-Rad ready gel and then electrotransferred to an Immobilon PVDF membrane (EMD Millipore). Membranes were blocked in 5% bovine serum albumin for 2 h and then treated with anti–γ-H2AX (Ser 139) antibody (1:1,000; EMD Millipore). Horseradish peroxidase (HRP)–conjugated goat anti-rabbit or goat anti-mouse (1:50,000; Chemicon International) was used as a secondary antibody. Anti-actin (1:4,000; Sigma-Aldrich, St. Louis, MO, USA) was used as a loading control. Blots were treated with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA, USA) and exposed to autoradiography film. To determine the role of oxidative stress in P-AscH--radiosensitization on both tumor and normal tissue, 4HNE was used a marker of lipid peroxidation and is a well-established marker of tissue oxidative stress.27 Mouse jejunal and tumor samples were excised and processed to determine 4-hydroxy-2-nonenal-(4HNE) modified proteins as previously described.19,26 Briefly, jejunal and tumor tissues were homogenized in 1.3 mM diethylenetriaminepentaacetic acid (DETAPAC), 10 mM butylated hydroxytoluene (BHT) and a complete mini-protease inhibitor (Roche Diagnostics) and assayed for protein concentration utilizing the Bradford assay. Protein (50 μg), in PBS, was blotted onto PVDF membranes. Blots were incubated with the primary antibody recognizing the Michael addition product of 4HNE-modified cellular proteins diluted 1:2,000 overnight at 4 °C.28 Secondary antibody incubation was performed for 1 h with an HRP-conjugated goat anti-rabbit polyclonal antibody (1:25,000). Immunoreactive protein was detected using chemiluminescence (ECL Plus Western Blotting Detection System) on X-ray film. Analysis was performed by comparing 4HNE immunoreactive protein normalized to protein loading determined by staining with Ponceau S stain. Normalized average integrated densities were determined with ImageJ software.
Animal Experiments-
Thirty-day-old athymic nude mice (Foxn1nu) were obtained from Envigo (Franklin Township, NJ, USA). The nude mice protocol was reviewed and approved by the Institutional Animal Care and Use Committee of The University of Iowa. The animals were housed four in a cage and were fed a sterile commercial stock diet and filtered tap water, ad libitum. Animals were allowed to acclimate in the unit for one week before any manipulations were performed. Mice were injected with with 3.0 × 106 MIA PaCa-2 pancreatic cancer cells and the tumors were allowed to grow to a size of 5–6 mm before treatments were initiated. Prior to radiation treatments, animals were anesthetized with 80 – 100 mg kg−1 ketamine and 10 mg kg−1 xylazine i.p. and shielded in a lead block such that only the lower body was irradiated, sparing the head and chest. Animals were euthanized by CO2 asphyxiation.
Mice were randomized to receive P-AscH- (4 g kg−1, IP, b.i.d.) or an equivalent volume of 1 M NaCl (4 g kg−1, IP, b.i.d.) for two days. On the third day, the animals received 10 Gy abdominal radiation (Pantak Therapax DXT 300, x-rays). The animals were shielded to protect the thoracic organs when the abdomen was irradiated. Dose rate for the abdominal radiation was 1.38 Gy/min. In the in vivo experiments, the mouse was in a lead shield resulting in just the tumor being irradiated. Dosimetry was used assuming the mouse leg is 1 cm thick and dose rate is to mid-leg while the mouse is in a body/leg container which is corrected for the depth of the container. The dose rate was 1.38 Gy/min. In separate groups of animals with tumor xenografts, mice received the same dosing schedule of P-AscH- or NaCl, but then had 5 Gy radiation delivered to the tumors. Treatment continued with either P-AscH- or NaCl on the fourth and fifth days; then all mice were sacrificed on day six. Jejunal tissue was obtained and evaluated with hematoxylin/eosin for tissue microstructure and trichrome blue for collagen deposition. A comparative pathologist blinded to the study assigned semi-quantitative scores to each jejunal section according to the grading system outlined in Supplemental Table 1. Mitochondrial damage was investigated using electron microscopy. Jejunal tissue was fixed in 2.5% glutaraldehyde in PBS overnight at 4 °C and then post fixed with 1% osmium tetroxide for 1 h. Following serial alcohol dehydration (50%, 75%, 95%, and 100%), the samples were embedded in Epon 12 (Ted Pella, Redding, CA). Ultramicrotomy was performed, and ultrathin sections (70 nm) were post stained with uranyl acetate and lead citrate. Samples were visualized with a JEOL 1230 transmission electron microscope (TEM) (Tokyo, Japan) at the Central Microscopy Research Facility of The University of Iowa. The number of total mitochondria visualized were counted as well as the number of damaged mitochondria.
Glutathione Assay-
The glutathione assay was kindly performed by the Antioxidant Enzyme Core Facility at The University of Iowa. Following incubation, jejunum slices were homogenized in 5% 5-sulfosalicylic acid (Sigma). The samples were then centrifuged to remove precipitating proteins and the supernatants assayed for total glutathione (GSH) content by the method of Griffith.[24] Glutathione disulfide (GSSG) was measured by adding 20 μl of a 1:1 mixture of 2-vinylpyridine and ethanol per 100 μl of sample and incubating for 2 h prior to assaying as described previously.[25] The SSA precipitated protein was resuspended in 0.1 N NaOH, and protein levels determined using the BCA Assay Kit (Thermo Scientific, Rockford, IL). Glutathione determinations were normalized to the protein content of whole homogenates.
Phase 1 Clinical Trial
Regulatory Requirements –
This clinical trial was conducted under an established investigator-sponsored IND. Compliant with ICMJE policy, the trial was listed on clinicaltrials.gov prior to enrollment (NCT01852890). Approval was obtained from The University of Iowa IRB-01 [Biomedical]. The trial was conducted under Good Clinical Practice consistent with International Council on Harmonization guidance document (ICH E6(R2)). Written or signed informed consent was obtained from all participating patients.
Study design and statistical analysis –
This was a single institution, open label, phase 1 study of P-AscH- administered concurrently with gemcitabine and radiation therapy. The P-AscH- dose was escalated via Storer’s Two-Stage Design BD for phase 1 trials.[26] Design B enrolls only a single participant per dose cohort until a dose-limiting toxicity is identified. The cohort was then expanded (Design D) to a 3-subject evaluation and continues with 3 subjects per cohort for subsequent cohorts. A minimum of 6 subjects must be evaluated at the recommended phase 2 dose with ≤ 1 dose-limiting toxicity occurring in those 6.
Participant Population –
Patients with histologically or cytologically diagnosed pancreatic adenocarcinoma and indication for concurrent gemcitabine and radiation therapy were invited to participate in this trial. Because P-AscH- infusions may cause red blood cell hemolysis in those deficient in the glucose-6-phosphate dehydrogenase (G6PD) enzyme, G6PD-deficient patients were excluded from the study (lower limit of normal 7 U g−1 hemoglobin, ARUP laboratories, Salt Lake, UT). Minimum Karnofsky performance status was 60. Required initial laboratory values included an absolute neutrophil count of ≥ 1,500/mm3, platelet count of ≥ 100,000/mm3, leukocytes > 3,000/mm3, creatinine of ≤ 1.5 mg/dL or creatinine clearance ≥ 60 mL/ min, total bilirubin ≤ 2 x upper limit of normal (ULN), transaminases ≤ 2.5 x ULN, and a prothrombin time and international normalized ratio (INR) within normal institutional limits. Key exclusion criteria included active comorbidities such as end stage congestive heart failure, unstable angina, or myocardial infarction within 6 months of enrollment. Consistent with Good Clinical Practice, subjects had to be capable of providing informed consent; legally authorized representatives and proxy consent were not allowed. The final eligibility criterion was tolerance of a test dose of 15 g pharmacological ascorbate (Mylan Pharmaceuticals, Rockford IL) in 250 mL sterile water for human use. Eligibility was independently confirmed by the Data and Safety Monitoring Committee of the Holden Comprehensive Cancer Center.
First-in-human –
This study is the first to actively infuse P-AscH- during the radiation ‘beam on’ time. P-AscH- infusion was initiated at least 20 min prior to radiation therapy delivery. Subjects were then transferred to the linear accelerator for therapy. After radiation, subjects were returned to the infusion area for the remainder of their infusion.
Treatment Plan –
The schema is provided (Figure 1). Subjects were assigned the P-AscH- dose as per open cohort slots (50 g in 750 mL, 75 g in 1000 mL, or 100 g in 1500 mL sterile water for human use). P-AscH- was infused daily. Gemcitabine was administered according to ECOG-E4201with an intravenous infusion at a dose of 600 mg/m2 over 30 min, once weekly for six weeks.[5] Intensity modulated radiation therapy (IMRT) was delivered with either Siemans Oncor or Elekta Versa HD treatment machines as either 50.4 Gy in 28 fractions or 50 Gy in 25 fractions as determined most appropriate by the treating radiation oncologist. 4-dimensional (4D) computed tomography–based simulation was performed for each subject. The 4D CT data sets included 10 respiratory phases: 20%, 40%, 60%, 80% and 100% inspiration phases, and 80%, 60%, 40%, 20% and 0% expiration phases. If target motion due to respiration was ≥ 1 cm, respiratory gating was used for treatment planning and delivery to limit target motion to ≤ 1 cm. In situations where the target motion was ≤ 1 cm, then an internal target volume (ITV) was generated using the 100% inspiration and 0% expiration phases. For subjects with residual disease at the time of simulation, the gross tumor volume (GTV) was defined as the primary tumor and/or any suspicious (≥ 1 cm) regional lymph nodes. The clinical target volume (CTV) was a 1.5 cm expansion on the GTV. The planning treatment volume (PTV) was defined as the CTV plus a 0.5 cm expansion in all directions. For subjects receiving adjuvant radiation therapy: a CTV volume was generated using the celiac axis, superior mesenteric artery, portal vein, pre-operative tumor volume, hepaticojejunostomy or pancreaticojejunostomy, aorta and surgical clips. A PTV was generated by expending the CTV by 0.5 cm. Surrounding normal structures including the liver, each kidney, spinal cord, duodenum, and bowel were contoured for dose-volume histogram generation and analysis of normal tissue radiation dose constraints. Subject positioning was confirmed prior to each treatment using either kilovoltage (kV) or megavoltage (MV) conebeam CT.
Figure 1. Schematic of phase I clinical treatment plan.
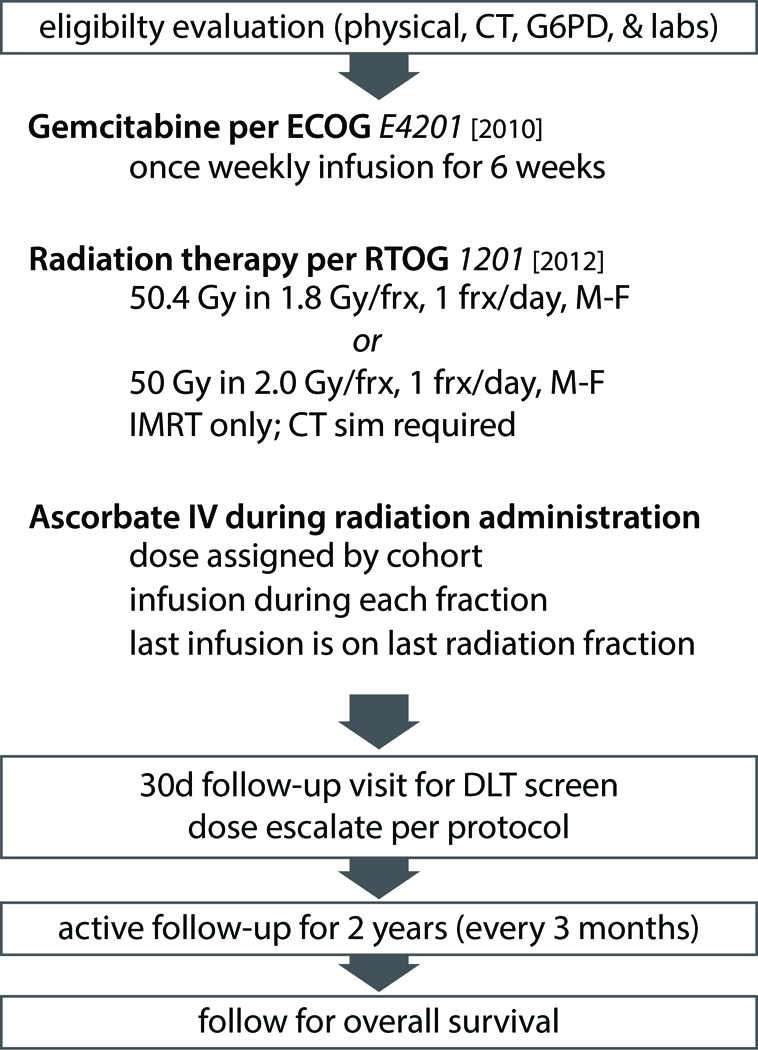
Patients were evaluated for G6PD deficiency, as they are pre-disposed to hemolysis with P-AscH-. Gemcitabine and radiation therapy were given per published protocol.[5] After tolerance of a 15-g test dose, patients were escalated to treatment dose (50, 75, or 100 g) and followed for dose-limiting toxicities.
Dose-limiting toxicities were defined as the following events, regardless of attribution to the P-AscH-: vomiting resulting in hypokalemia that did not respond to treatment; vomiting resulting in hypotension that did not respond to treatment; grade 4 febrile neutropenia, grade 4 intra-abdominal hemorrhage; or grade 4 weight loss. Also, any serious adverse event (21 CFR§312.32) with a causality to the P-AscH- infusion was deemed a dose-limiting toxicity. Subjects continued P-AscH- treatment throughout combination therapy. Blood samples were drawn weekly, pre- and post- infusion (≤ 15 min). Blood samples were stored on ice in a cooler and transferred to a dark refrigerator until prepared for assay. No P-AscH- degradation was expected to occur during processing and storage conditions.
Response and toxicity criteria –
Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events 4.03 (CTCAE). Consistent with recent FDA guidance on risk based monitoring and safety assessments,[27, 28] assessment to P-AscH- was required for all serious adverse events or adverse events ≥ grade 3. Lower-graded events were assumed to be related to tumor burden, side effects of antineoplastic therapy, and activities of daily life. Causality of P-AscH- to the adverse event was determined by the sponsor-investigator (JC) as per Federal Code.
Sample preparation for plasma and RBCs –
Whole blood (NaHeparin 75 USP units, BD Vacutainer® green top blood collection tube, 4 mL) was collected from clinical trial participants. Samples were centrifuged at 2000 RCF for 15 min, and then RBC-free platelet depleted plasma was collected and stored frozen at −80 °C in multiple aliquots for subsequent P-AscH- or F2-isoprostane measurement and other biochemical analyses. Ascorbate levels in subject plasma samples were determined with a published plate reader-based assay as described in Vislisel et al with minor modifications to detect ascorbate concentrations at nutritional (μM) and post-intravenous P-AscH- infusion levels (mM).[29] Minor modifications to this protocol have been described and implemented in this trial.[13, 29]
Measurement of Isoprostanes –
Isoprostanes are arachidonic acid derivatives, formed from non-enzymatic, free radical peroxidation. F2-isoprostanes are chemically stable, and found in all biological fluids, making them the most ideal current measure of systemic oxidative stress.[30] Plasma samples were collected prior to initiating chemoradiotherapy, three weeks into chemoradiotherapy, and at the completion of chemoradiotherapy for F2-isoprostane comparison. To quantify F2-isoprostane levels, gas chromatography-mass spectrometry was utilized (Eicosanoid Core Laboratory, Vanderbilt University, Nashville, TN). This assay was chosen because it has been shown to be capable of detecting changes indicative of in vivo oxidative stress in plasma samples and is verifiable.[30–32]
Institutional Comparators
There were 19 subjects enrolled in the comparator arm of the study. These subjects met the same inclusion criteria as subjects receiving P-AscH-. All subjects received standard regimens of concurrent gemcitabine and radiation therapy with curative intent. Additional clinical data for the comparator subjects is available in supplemental information, Table 2.Blood was obtained in four subjects undergoing concurrent chemoradiation to determine F2-isoprostane levels. Approval was obtained from The University of Iowa IRB-01 (IRB 201508733, Cullen PI).
Results
Differential effects of P-AscH- on radiation-induced clonogenic cell death –
MIA PaCa-2, 403, H6c7, and FHs 74 Int cells were treated with P-AscH- (1 mM, 10 pmol cell−1) and radiation (1 Gy). Individually, P-AscH- (1 mM) and radiation (1 Gy) induced clonogenic cell death in the MIA PaCa-2 pancreatic cancer cell line but not in the 403 pancreatic cancer cell line. The combination treatment, however, did induce clonogenic cell death in both cell lines (Figure 2A and2B). These data are consistent with previous studies of P-AscH- combined with radiation on pancreatic cancer cell lines [19].To determine whether H2O2 was responsible for the cytotoxic effects of P-AscH- and radiation, the 403 cell lines was treated with bovine catalase (100 U/mL) and clonogenic survival was determined. In addition, P-AscH- and P-AscH- combined with radiation decreased clonogenic survival which was reversed with catalase pretreatment (Supplementary Figure 1). In contrast, the H6c7 non-tumorigenic pancreatic ductal epithelial cell lines did not demonstrate significant cytotoxicity with a combination treatment of P-AscH- and radiation (Figure 2C). Furthermore, the FHs 74 Int cell line experienced a recovery in clonogenic survival capability when radiated cells were treated with P-AscH-, suggesting a possible radioprotective effect in this normal cell line (Figure 2D).
Figure 2. P-AscH- exacerbates radiation-induced cell death in cancer cells by increasing radiation-induced double-stranded DNA breaks, but protects normal cells.
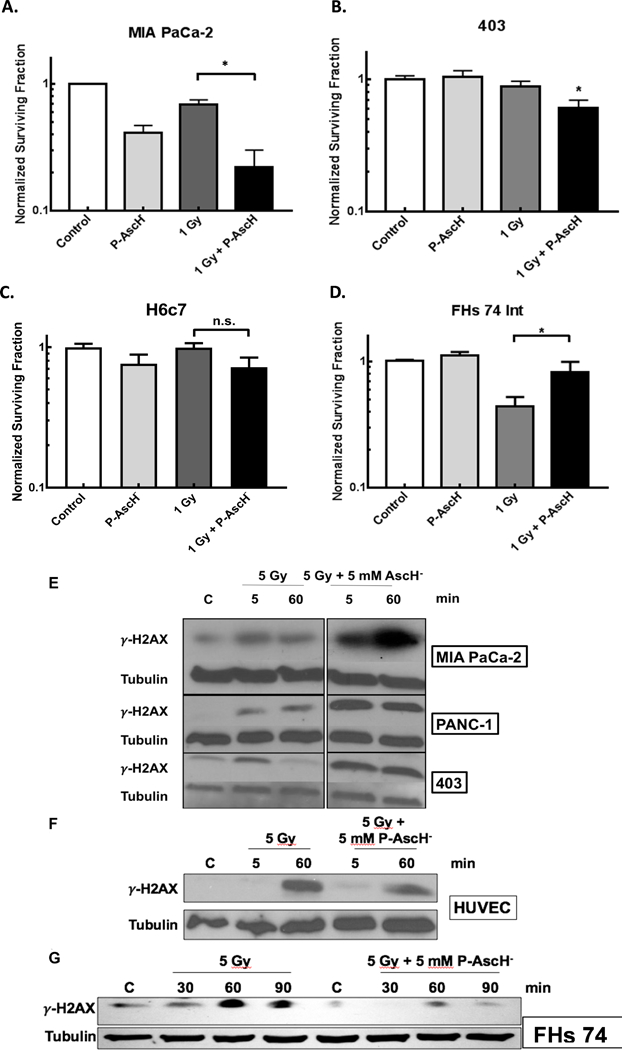
A. MIA-PaCa-2 cells demonstrated significantly decreased clonogenic survival when treated with 1 mM (10 pmol cell−1) P-AscH- or 1 Gy radiation alone, with additive effect when radiation and P-AscH- are given in combination. (* p < 0.05)
B. Significant decreases in clonogenic survival in the 403 cell line with the combination of 1 mM (10 pmol cell−1) P-AscH- and 1 Gy radiation compared to control (* p < 0.05).
C. H6c7 cells demonstrate no significant cytotoxic effect of radiation in combination with P-AscH-.
D. FHs 74 Int cells, derived from fetal human small intestinal epithelium, demonstrated significant decreased clonogenic survival when treated with 1 Gy radiation alone, but this effect is significantly reversed when co-treated with P-AscH-, 1 mM (10 pmol cell−1) (* p < 0.05).
E. Pancreatic cancer cell lines (MIA PaCa-2, PANC-1, and 403) demonstrate increased γ-H2AX immunoreactivity with 5 Gy Radiation at 5 and 60 min. This effect is increased and prolonged when cells are co-treated with 5 mM P-AscH-.
F. HUVEC cells, derived from normal umbilical vein endothelium, demonstrate increased γ-H2AX immunoreactivity 60 min after 5 Gy radiation. This effect is ameliorated when cells are co-treated with 5 mM P-AscH-.
G. FHs 74 Int cells demonstrate increased γ-H2AX immunoreactivity 60 and 90 min after radiation. Again, this effect is ameliorated when cells are co-treated with 5 mM P-AscH-.
Differential effects of P-AscH- on radiation-induced DNA damage –
All cell lines were treated with P-AscH- (5 mM, 5 pmol cell−1) for 1 hr and radiated (5 Gy). Protein was collected at 5 min and 60 min after radiation to determine γ-H2AX peak. In the pancreatic cancer cell lines, co-treatment with 1 hr of P-AscH- enhanced γ-H2AX immunoreactivity at 5 min and 60 min (Figure 2E). In contrast, γ-H2AX immunoreactivity was decreased at these time points when the non-tumorigenic cell lines (HUVEC and FHs 74 Int) were treated with IR and P-AscH- (Figures 2F and2G). To quantify and further clarify the DNA damage response, a quantitative PCR analysis of DNA damage was determined using a recently described and published method from our group.[16] Using quantitative PCR method, the formation and removal of DNA damage were measured in cells. In MIA PaCa-2 cells, P-AscH- (5 mM) plus radiation (5 Gy) increased the frequency of DNA lesions (0.31 ± 0.05 lesions/10 kb) (kb = kilobase pairs, Means ± SEM, n = 3) compared to cells irradiated with 5 Gy only (0.09 ± 0.01 lesions/10 kb). In contrast, FHs 74 Int cells had a decrease in the frequency of DNA lesions from (0.17 ± 0.05 lesions/10 kb, Means ± SEM, n = 3, P < 0.05 vs. MIA PaCa-2) in cells irradiated alone, to undetectable levels in cells treated with P-AscH- (5 mM) plus 5 Gy. These data are consistent with the Western blots for γ-H2AX immunoreactivity in non-tumor and tumor cell lines treated with P-AscH- and radiation.
P-AscH- limits radiation-induced intestinal damage and collagen deposition –
The clinical effect of radiation damage on the small intestine is demonstrated by villous blunting, loss of crypt cells, and collagen deposition. Semi-quantitative analysis was conducted by a comparative pathologist (KGC), blinded to the study, according to a standard scoring system (Supplemental Information, Table 1). The hematoxylin/eosin staining demonstrated radiotoxic effects of mouse jejunum following 10 Gy radiation, including extensive villous blunting and loss of crypt architecture (Figure 3A). This effect was partially inhibited when radiated mice were treated with P-AscH- (Figure 3A and3B). Similarly, collagen deposition and permeation into the submucosa and lamina propria was significantly increased in mice treated with radiation alone (Figure 3C). This effect was also partially ameliorated in mice treated with P-AscH- (Figure 3C and3D). The mitochondrial structure of jejunal epithelial cells from mice radiated with or without P-AscH- was analyzed utilizing electron microscopy. Radiation alone resulted in 91% damaged mitochondria characterized by swelling, vacuolization, loss of cristae structure, and membrane rupture (Figure 3E). P-AscH- once again partially inhibited the radiation-induced mitochondrial damage (Figure 3E and3F). To determine the role of oxidative stress as a mechanism whereby P-AscH- ameliorates radiation-induced damage to normal tissue, a glutathione assay was performed on irradiated jejunum (Figure 3G). The ratios of reduced glutathione (GSH) to oxidized glutathione disulfide (GSSG) were compared in mice with abdominal radiation alone and abdominal radiation with P-AscH-. Jejunum from mice treated with radiation and P-AscH- displayed lower levels of GSSG suggesting that P-AscH- may be acting as a systemic antioxidant to normal tissue during radiation. In addition, 4HNE expression demonstrated that irradiated jejunal tissue treated with P-AscH- expressed lower levels of 4HNE than irradiated tumor tissue treated with P-AscH- (Figure 3H). Taken together, these data suggest that P-AscH- radiosensitizes pancreatic tumors and acts as a pro-oxidant in irradiated tumor tissue, but may act as an antioxidant in radiated normal tissue.
Figure 3. P-AscH- reduces radiation-induced jejunal mucosal damage, intestinal collagen deposition, normal tissue mitochondrial damage, and oxidative stress in vivo.
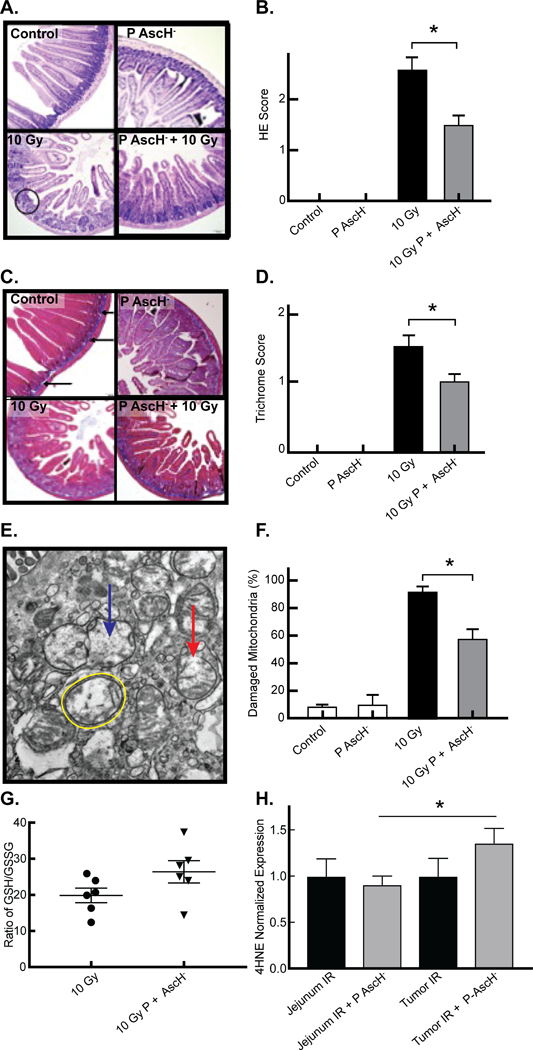
A. Light microscopy of representative samples of Hematoxylin/Eosin stained mouse jejunal samples demonstrates villous blunting and loss of fusion (circle) and crypt architecture in mice radiated with 10 Gy radiation, and reversal of this effect in mice treated with P-AscH- 2 days prior and 2 days after radiation (4 g kg−1, twice daily).
B. Semi-quantitation, performed by a comparative pathologist blinded to the study, confirms increased jejunal damage in radiated mice. Radiation damage is significantly decreased in mice treated with P-AscH- (Means ± SEM, n = 3, * p < 0.05)
C. Light microscopy of trichrome-blue stained mouse jejunal samples. Blue staining identifies collagen with normal deposition limited to the submucosa of controls (arrows). There is increased collagen thickness with penetration into the lamina propria in mice radiated with 10 Gy radiation, and inhibition of this effect in mice treated with P-AscH- 2 days prior and 2 days after radiation (4 g kg−1, twice daily).
D. Semi-quantitation, performed by a comparative pathologist blinded to the study, confirms increased collagen deposition in radiated mice which is significantly decreased in mice treated with P-AscH- (Means ± SEM, n = 3, * p < 0.05)
E. Electron microscopy of representative samples of mouse jejunal epithelial cells samples demonstrates increased mitochondrial damage characterized by mitochondrial swelling (yellow circle), loss of cristae structure (blue arrow), and membrane rupture (red arrow) in mice radiated with 10 Gy radiation, and partial inhibition of this effect in mice treated with P-AscH- 2 days prior and 2 days after radiation (4 g kg−1, twice daily).
F. Percentage of damaged mitochondria in control, ascorbate, 10 Gy and 10 Gy + P-AscH- treated mice (Means ± SEM, n = 3, * p < 0.05)
G. Ratio of GSH to GSSG in normal mouse jejunum exposed to 10 Gy vs. 10 Gy + P-AscH- (Means ± SEM, n = 5 – 6, *p < 0.05)
H. 4HNE expression in mouse jejunum exposed to radiation and radiation + P-AscH- in comparison to 4HNE expression in tumor xenografts exposed to either radiation and radiation + P-AscH- (Means ± SEM, n = 4 – 5, * p < 0.05).
Phase 1 Clinical Trial
Participant demographics –
Table 1 provides a clinical summary of the subjects enrolled in the clinical trial and the comparator group. A total of sixteen subjects were enrolled for P-AscH- treatment from June 2014 to November 2016. Of the sixteen subjects treated with P-AscH-, one subject withdrew due to chronic back pain exacerbation resulting from sitting during the infusion. Another subject withdrew after a grade 3 transient blood pressure elevations due to hypervolemia. The remaining fourteen completed all protocol therapy, making them evaluable for statistical purposes. Adverse events were reviewed monthly at investigator meetings and reviewed against protocol requirements for DLTs. Average treatment duration was 5.7 weeks. No subjects were lost to follow-up.
Table 1.
Subject characteristics prior to treatment
| Characteristic | P-AscH- | 50 g | 75 g | 100 g | Comparator |
|---|---|---|---|---|---|
| Sex | |||||
| Male | 6 | 1 | 3 | 2 | 13 |
| Female | 8 | 2 | 3 | 3 | 6 |
| Age (years) | |||||
| Mean | 59 | 54 | 62 | 59 | 63 |
| SD | 7 | 8 | 5 | 6.0 | 10 |
| Median | 62 | 50 | 62 | 58 | 62 |
| Range | 48–69 | 48–65 | 53–69 | 52–68 | 46–80 |
| Karnofsky | |||||
| 70 | 2 | 0 | 0 | 2 | 3 |
| 80 | 3 | 1 | 2 | 0 | 5 |
| 90 | 8 | 2 | 4 | 3 | 9 |
| 100 | 1 | 0 | 0 | 1 | 2 |
| Smoking History | |||||
| Never | 7 | 2 | 4 | 1 | 9 |
| Former | 2 | 0 | 0 | 2 | 4 |
| Current | 5 | 1 | 2 | 2 | 6 |
| Clinical Stage | |||||
| I | 1 | 0 | 0 | 1 | 1 |
| II | 9 | 2 | 3 | 4 | 12 |
| III | 3 | 1 | 2 | 0 | 5 |
| IV | 1 | 0 | 1 | 0 | 1 |
| T Stage | |||||
| T2 | 2 | 0 | 0 | 2 | 3 |
| T3 | 8 | 2 | 3 | 3 | 12 |
| T4 | 4 | 1 | 3 | 0 | 4 |
| N Stage | |||||
| N0 | 7 | 3 | 4 | 2 | 10 |
| N1 | 7 | 0 | 2 | 3 | 9 |
| M Stage | |||||
| M0 | 13 | 0 | 5 | 5 | 18 |
| M1 | 1 | 0 | 1 | 0 | 1 |
| Ca 19–9 | |||||
| <40 | 8 | 1 | 4 | 3 | 2 |
| >40 | 6 | 2 | 2 | 2 | 16 |
| n/a | - | - | - | - | 1 |
| Prior Chemo | |||||
| FOL | 12 | 3 | 4 | 5 | 1 |
| G/A | 3 | 0 | 2 | 1 | 0 |
| Gem | 3 | 0 | 1 | 2 | 1 |
Safety and Toxicity –
Average Karnofsky Performance Score both prior to chemo-radiotherapy and post-chemo-radiotherapy was 86 ± 2 (SEM). One subject experienced a decrease in Karnofsky score, two subjects showed an increase, and eleven showed no change. No adverse events occurred during the concurrent P-AscH- infusion with combination therapy administration or immediately thereafter. In general, adverse events were consistent with those previously described (Table 2). Three adverse events were considered attributable to P-AscH-: dry mouth, thirst, and transient blood pressure elevation. For the purposes of this clinical trial, transient blood pressure elevation is described as a blood pressure elevation (systolic pressure, diastolic pressure, or both) causing the post-infusion blood pressure grade to be higher than pre-infusion grade. Grading was harmonized to CTCAE hypertension grading. Transient blood pressure elevation was considered a separate event from hypertension because the blood pressure elevation was typically not sustained beyond 30 min. Five subjects experienced a grade 3 transient blood pressure elevation at least once. Dry mouth and thirst were transient symptoms alleviated the same day as infusion. Only one dose limiting toxicity occurred (subject 3, 75 g dose). Within 2 h after P-AscH- infusion, subject 3 reported to a routine medical oncology visit. During the exam, subject described left-sided chest heaviness followed by sharp stabbing pain lasing for about 20 min. Subject 3 was admitted for cardiac workup and observation and ultimately diagnosed with esophageal spasm (CTCAE non-cardiac chest pain, grade 3); the event was filed with FDA through the MedWatch 3500A mechanism due to the contemporaneousness of the event to the P-AscH- infusion. Subject 3 was re-challenged without incident and continued on the trial. The cohorts were then expanded from Storer’s Design B to Design D as per protocol. Table 3 provides the summary of maximum grade for toxicities for all treatments combined. Grade 3 and 4 hematologic toxicities included anemia (n = 1), white blood cell count decreased (n = 8), lymphocyte count decreased (n = 13), absolute neutrophil count decreased (n = 7), and platelet count decreased (n = 2). Hematologic toxicities were consistent with gemcitabine and radiation therapy.
Table 2.
Toxicity Summary
| Grade | |||||
|---|---|---|---|---|---|
| CTCAE term | 1 | 2 | 3 | 4 | 5 |
| Anemia | 2 | 5 | 1 | - | - |
| White blood cell decreased | 1 | 4 | 7 | 1 | - |
| Lymphocyte count decreased | - | - | 6 | 7 | - |
| Neutrophil count decreased | 2 | 3 | 7 | - | - |
| Platelet count decreased | 6 | 4 | 2 | - | - |
| Alkaline phosphatase increased | 2 | - | - | - | - |
| Alanine aminotransferase increased | 3 | - | - | - | - |
| Aspartate aminotransferase increased | 1 | - | - | - | - |
| GGT increased | 2 | - | - | - | - |
| Hyperglycemia | 2 | 3 | - | - | - |
| Hypoalbuminemia | 1 | 2 | - | - | - |
| Hypocalcemia | 2 | - | - | - | - |
| Hypoglycemia | 1 | - | - | - | - |
| Hypokalemia | 3 | - | 3 | 1 | - |
| Hypophosphatemia | 1 | 1 | - | - | - |
| Abdominal bruising | 1 | - | - | - | - |
| Abdominal pain | 8 | - | - | - | - |
| Anal pain | - | 1 | - | - | - |
| Anorexia | 2 | - | - | - | - |
| Atrial fibrillation | - | - | 1 | - | - |
| Bleeding | 1 | - | - | - | - |
| Cardiac disorder - other: irregular heartrate | 1 | - | - | - | - |
| Confusion | 1 | - | - | - | - |
| Cough | 1 | - | - | - | - |
| Chills | 11 | 1 | - | - | - |
| Constipation | 2 | - | - | - | - |
| Dehydration | 2 | 1 | - | - | - |
| Dermatitis radiation | 1 | - | - | - | - |
| Diarrhea | 3 | - | - | - | - |
| Dry mouth | 9 | 1 | - | - | - |
| Dry skin | 1 | - | - | - | - |
| Dysgeusia | 2 | - | - | - | - |
| Epigastric pain | 1 | - | - | - | - |
| Eye disorders - Other, vision decrement | 1 | - | - | - | - |
| Fatigue | 5 | 5 | - | - | - |
| Fever | 1 | - | - | - | - |
| Flushing (malar flushing) | 1 | - | - | - | - |
| Gastroesophageal reflux | 1 | 1 | - | - | - |
| Gastrointestinal disorders - Other, early satiety | 1 | - | - | - | - |
| Headache | 5 | - | - | - | - |
| Hemorrhoids | - | 1 | - | - | - |
| Hypertension | - | 2 | - | - | - |
| Hypotension | 1 | - | - | - | - |
| Infections and infestations - Other, common cold | 1 | - | - | - | - |
| Lower extremity swelling | 1 | - | - | - | - |
| Nausea | 6 | 4 | - | - | - |
| Noncardiac chest pain | - | - | 1 | - | - |
| Pain | 1 | - | - | - | - |
| Sinus bradycardia | - | 1 | - | - | - |
| Sinusitis | - | 1 | - | - | - |
| Skin and subcutaneous tissue disorders - Other, increased inguinal sweating | 1 | - | - | - | - |
| Thirst | 2 | - | - | - | - |
| Vascular disorders – Other, transient blood pressure elevation | - | 7 | 5 | 1 | - |
| Vomiting | 9 | - | - | - | - |
| Watering eyes | 1 | - | - | - | - |
| Worst Degree | - | - | 4 | 10 | - |
Maximum Tolerated Dose –
All three dose cohorts were evaluated. Due to the single dose limiting toxicity, six subjects total were evaluated in the 75 g dose cohort prior to escalating to 100 g. Of the five subjects treated at the 100 g dose, one subject’s blood pressure elevated to 201/102 mmHg but resolved to 127/88 mmHg within 30 min. A second subject, who did not have a history of baseline hypertension, developed post-infusion elevated blood pressures that did not resolve as quickly, remaining in a systolic range of 140–150 mmHg. The fluid volume for a 100 g dose is 1500 mL and is less than the typical hydration volume used for cisplatin. For this reason, these adverse events were attributed as possible to the P-AscH- and were considered dose limiting. Both subject’s doses were reduced to 75 g for the remainder of therapy. Given the increased blood pressure events, the increased infusion time (120 min vs. 180 min), and the diminished return on the average plasma ascorbate level, 100 g was determined to be the maximum tolerated dose and 75 g was selected as a recommended phase 2 dose.
Plasma Ascorbate Levels –
Mean plasma ascorbate concentrations immediately upon completion of the IV infusion were 15 mM for 50 g (n = 17, 95% CI 13 – 17 mM), 20 mM for 75 g (n = 37, 95% CI 19–21 mM), and 20 mM for 100 g (n = 32, 95% CI 19–22 mM), respectively. Plasma ascorbate levels in the 50-g cohort were significantly lower than the 75-g and 100-g cohorts (One-way ANOVA with Post-hoc Tukey analysis, n = 86, p < 0.05, Figure 4A). Comparison of plasma ascorbate levels to subject metrics revealed the most predictable dosing on a per kg basis (Figure 4B). Body mass index, body surface area (BSA), and height, all correlated significantly with plasma ascorbate levels, (Supplemental Figure 2).
Figure 4. P-AscH- produces high plasma ascorbate concentrations, stabilizes hemoglobin concentration during chemoradiotherapy, and decreases plasma F2-Isoprostane levels during chemoradiotherapy.
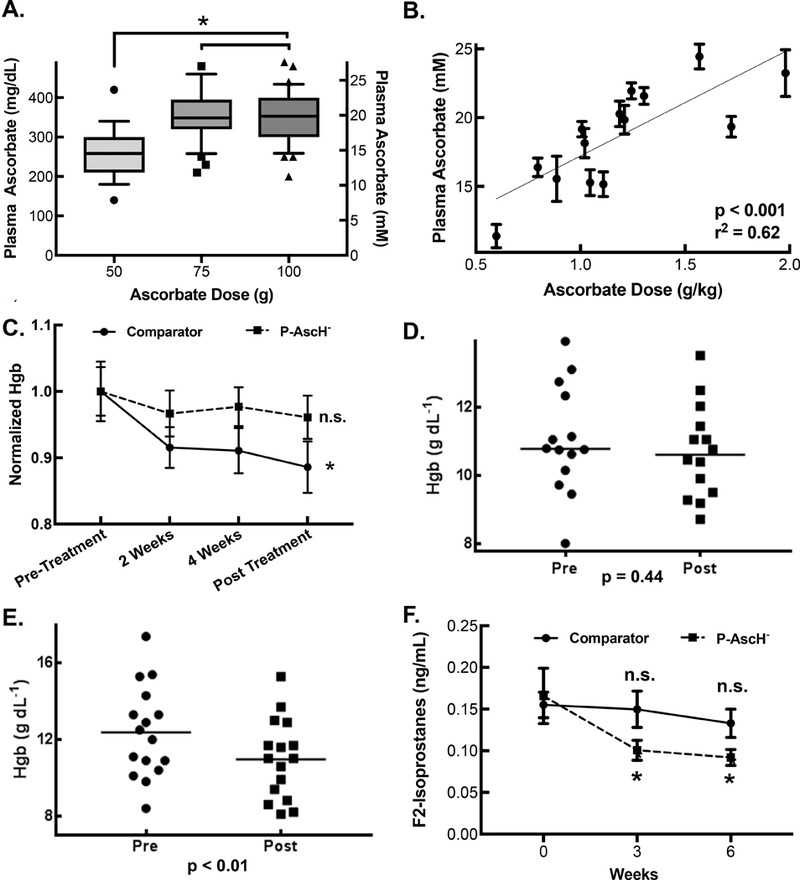
A. Plasma ascorbate concentrations. Box represents interquartile range with ranges representing 10th and 90th percentiles. Mean plasma ascorbate concentrations for 50-, 75-, and 100-g doses are 265 mg dL-1 (15 mM, n = 17, 95% CI 232 – 299 mg dL-1), 355 mg dL-1 (20 mM, n = 37, 95% CI 333 – 376 mg dL-1), and 356 mg dL-1 (20 mM, n = 32, 95% CI 331 – 380 mg dL-1), respectively (Means ± SEM, n = 17 – 32, * p < 0.05)
B. Plasma ascorbate concentration as a function of gram per kilogram dosing. The gram per kilogram dose was calculated as an average weight of the patient over the course of treatment. Linear Regression demonstrates significant non-zero slope and R2 = 0.62.
C. A one way repeated measure ANOVA was performed on Hgb (Hgb, g dL−1) values over the course of the chemoradiation therapy cycle for comparator patients and patients receiving P-AscH-. Comparator patients experienced a change in Hgb over this time period (Means ± SEM, n = 13, * p < 0.01) while patients receiving P-AscH- did not (Means ± SEM, n = 14, p = 0.47).
D. P-AscH- treated subject hemoglobin pre- and post-chemoradiotherapy were determined by linear regression from all Hgb measurement within each subject to minimized expected variation. Mean change in Hgb −0.34 ± 0.43 g dL−1 (SEM, n = 14, *p < 0.01).
E. Hemoglobin levels in subjects not treated with P-AscH- before and after completion of chemoradiotherapy as calculated from linear regression of all subject hemoglobin measurements collected to minimized expected variation. Mean change in hemoglobin −1.47 ± 0.34 g dL−1 (n = 13, p = 0.44).
F. Plasma was collected prior to therapy (week 0), after the 3rd week of therapy and at the finish of 6 weeks of chemoradiotherapy. Two-way ANOVA analysis was performed. There was no significant difference between week 0 plasma F2-Isoprostane levels compared to week 3 of treatment (Means ± SEM, n = 4, p = 0.99) or immediately following completion of chemoradiotherapy (Means ± SEM, n = 4, p = 0.88) in control subjects. There was a significant difference between week 0 plasma F2-Isoprostane levels compared to week 3 of treatment (Means ± SEM, n = 14, * p < 0.05) and immediately following completion of chemoradiotherapy (Means ± SEM, n = 14, * p < 0.05) in subjects treated with P-AscH-.
Anemia –
Despite the potential of hemolysis by P-AscH-, as determined from in vitro experiments,[33] which could negatively impact the tumoricidal effects, the incidence of anemia is consistent with published literature.[5] Hemoglobin levels collected were collected periodically over the six week chemoradiation cycle (Supplemental Figure 3). This data was analyzed by one way repeated measure ANOVA for both P-AscH- treated patients and comparator patients (Figure 4C). There was no change in Hgb level over time when P-AscH- was added to combination therapy treated patients (p = 0.44). However, a change in Hgb level over the course of combination therapy was detected in comparator patients (p < 0.01). Paired t-tests did not detect any change in pre vs post combination therapy Hgb values (Mean Δ Hgb −0.34 g dL−1, 95% CI −1.27 to 0.59 mg dL−1, p > 0.05) (Figure 4D). However, a statistically significant decrease in Hgb was found in the thirteen of nineteen comparator patients with Hgb data available for analysis (Mean Δ Hgb −1.47 g dL−1, 95% CI −2.21 to 0.73 mg dL−1 p < 0.01, Figure 4E). One of fourteen subjects (7%) treated with P-AscH- suffered grade 3 anemia as defined by hemoglobin concentration. This also compared favorably to historical controls, where six of thirty-four subjects (18%) receiving combination therapy alone suffered grade 3 or 4 anemia as defined by hemoglobin concentration.[5]
Blood Pressure –
Despite concerns that P-AscH- may have acute hypotensive effects when given in dosages over 30 g,[34] hypotension was not a realized adverse event (Table 3). To the contrary, transient elevation in blood pressure was documented (grade 2: n=7; grade 3, n=5; grade 4, n=1). These elevations typically resolved within 30 min upon completion of infusion. Two subjects had shifts in hypertension (as defined by CTCAE, from grade 1 to grade 2) during active therapy. One returned to baseline post-treatment blood pressure levels, the other did not.
F2-Isoprostanes –
Plasma F2-isprostanes are a reliable marker of systemic lipid peroxidation and are used as markers of oxidative injury.[35] F2-isoprostane levels from P-AscH- treated subjects drawn prior to beginning combination therapy, during combination therapy (after the 3rd week), and after completion of all six weeks of combination therapy, showed a significant decrease over time from 0.17 ± 0.01 ng mL−1 prior to treatment initiation to 0.09 ± 0.01 ng mL−1 at the end of the study (p = 0.02, Means ± SEM, n = 14, Figure 4F). In contrast, F2-Isoprostane levels did not change over the course of treatment for comparator subjects treated with combination therapy alone starting at a level of 0.16 ± 0.02 ng mL−1 prior to treatment initiation to 0.13 ± 0.02 ng mL−1 at the conclusion of the trial (p = 0.88, Means ± SEM, n = 4) (Figure 4F).
Treatment compliance –
Eight of fourteen subjects (57%) received all six gemcitabine doses, five received five doses and one received four doses. Two subjects had a dose delay of 1 week (week 6) as absolute neutrophil count decreased and platelet count decreased. Eight subjects had gemcitabine dose reductions. All subjects completed radiation therapy. The average dose was 50.4 Gy. One subject received 50 Gy in 25 doses and the remaining thirteen subjects received 50.4 Gy in 28 doses. Eight of fourteen subjects (57%) received all doses of P-AscH-. In six subjects, at least one dose of P-AscH- was withheld. Subjects received an average of 97% ± 5% of the prescribed ascorbate. Of the 364 doses of ascorbate prescribed, 354 were infused.
Survival –
Although the trial was not prospectively powered to determine differences in survival, treatment with P-AscH- appears to be no worse than combination therapy when compared to the University of Iowa’s median OS of 12.7 mo (range: 3.1 – 54.6 mo; Log-Rank test p = 0.08) and the historical median OS of 11.1 from ECOG-E4201.[5] Similarly, median progression free survival (PFS) was 13.7 mo (range: 2.4 – 43.8 mo) compared to The University of Iowa’s 4.6 mo median (range: 1.9 – 26.5 mo; p = 0.02) (Figure 5). Two subjects with borderline resectable disease became eligible for pancreaticoduodenectomy following trial completion. Subject #1 was initially staged with cT2N1M0 disease and considered borderline resectable due to impingement on the first jejunal branch of the gastroduodenal artery. Following initial FOLFIRINOX treatment, there was no longer evidence of lymphadenopathy but there was disease progression at the interface between the tumor and the superior mesenteric artery and superior mesenteric vein. The subject enrolled in the clinical trial with P-AscH- and chemoradiation. After completing the trial, restaging imaging demonstrated disease regression. The subject was determined suitable for surgery and a pancreaticoduodenectomy was performed. Pathology demonstrated marked treatment effect and seventeen lymph nodes negative for disease. Subject #4 was initially staged with cT3N1M0 disease and determined to be borderline resectable based on suspected invasion of the portal vein, abutment of the common hepatic artery and regional lymph node adenopathy. The subject initially underwent two cycles of FOLFIRINOX and three cycles of gemcitabine and nab-paclitaxel. Restaging CT scan demonstrated stable disease. The subject was enrolled in the clinical trial and received P-AscH- in combination with chemoradiation. After completion of the trial, restaging imaging showed disease regression with resolution of regional lymphadenopathy making the subject eligible for surgery. A pancreaticoduodenectomy was performed and surgical pathology revealed no residual viable invasive adenocarcinoma; seventeen lymph nodes were negative for disease. Both subjects are currently alive and are 44 mo and 35 mo from diagnosis, respectively, without evidence of disease recurrence. At the time of submission of this manuscript, five of fourteen subjects remain alive and three subjects are without evidence of disease recurrence.
Figure 5. Patient Survival.
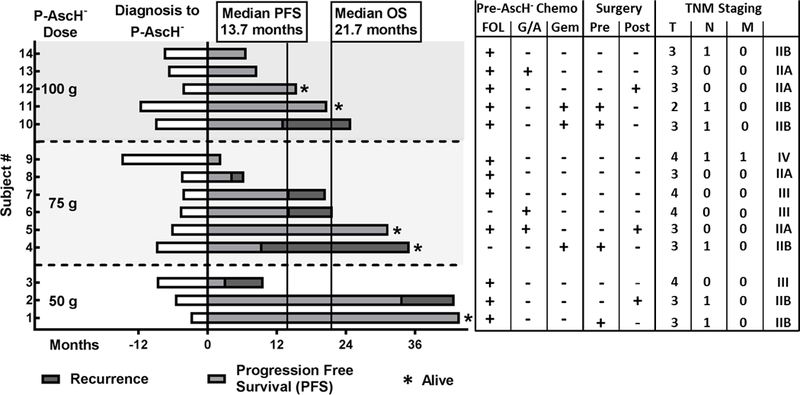
Waterfall plot by dosing cohort demonstrating median progression free survival (13.7 mo) and overall survival (21.7 mo) of all subjects treated with P-AscH- plus combination therapy. Of fourteen subjects enrolled in the trial, five remain alive, three without disease progression. Pre-P-AscH- treatments are outlined indicating which chemotherapy regimens patients received prior to starting the clinical trial. Patients who received pancreas resections are indicated as well. TNM staging data is also available. FOL = FOLFIRINOX, G/A = Gemcitabine + Abraxane, Gem = Gemcitabine alone, Pre = pancreas resection prior to P-AscH- + chemoradiation, Post = pancreas resection following P-AscH- + chemoradiation
Discussion
Our study demonstrates that P-AscH- selectively radioprotects normal cells and radiosensitizes tumor cells. P-AscH- reverses radiation-induced clonogenic cell death in non-tumorigenic cells but radiosensitized a variety of human pancreatic cancer cell lines. A potential mechanism is the differential oxidative effect of radiation-induced DNA damage in cancer vs. normal cells which the present study demonstrates in vitro and in vivo. Radiation damage to the intestine is demonstrated by villous blunting, loss of crypt cells, collagen deposition, mitochondrial damage, and increased GSSG which were all partially ameliorated with P-AscH-. Results from our first-in-human phase 1 clinical trial suggest that adding daily P-AscH- to combination therapy is safe for locally advanced pancreatic cancer. While not statistically powered to demonstrate efficacy, PFS and OS are increased compared to similar patients treated at our institution as well as ECOG-E4201.
There are limitations to the present study. The mechanism underlying the opposing effects of P-AscH- radioprotecting normal cells while radiosensitizing tumor cells is an active area of investigation. A possible explanation is that the H2O2 produced by P-AscH- has a much greater detrimental effect on cancer cells than normal cells. Cancer cells, which have elevated levels of reactive metals, are likely to have increased formation of hydroxyl radical, causing DNA damage.[15, 17] Normal cells, which have relatively lower fluxes of O2●-, have lower levels of labile metals, and more abundant catalase to detoxify H2O2, leaving P-AscH- to act as an antioxidant alone.[12, 16] Others have demonstrated that the NF-κB transcription factor RelB can determine the differential effects of P-AscH- in normal vs. cancer cells.[36] In addition, this phase 1 trial was not powered to make any conclusive statements regarding the efficacy of P-AscH- for locally advanced pancreatic cancer. Additionally, the retrospective comparator patients are not ideal controls for a variety of reasons including differences in chemotherapy regimens prior to chemoradiation, differing starting CA19–9 levels, and the retrospective nature of their data collection. Due to the retrospective nature of the data in the comparator patients, our study can only evaluate the reduced normal tissues toxicity in the in vitro and in vivo studies and not in human subjects at this time. However, we do believe this comparator group adds clinical context for which to interpret the potential of P-AscH- treatments for locally advanced disease in both historical controls and our institutional comparator subjects. The clinical data and pathologic findings in the resected surgical specimens from P-AscH- treated patients are also encouraging.
There is growing evidence that P-AscH- is safe with suggestion of efficacy in a variety of cancer types.[17, 37, 38] The safety of P-AscH- in combination with gemcitabine in metastatic pancreatic adenocarcinoma has been previously established.[13] We establish here the safety and tolerability of P-AscH- administered concomitantly with radiation therapy and gemcitabine for locally advanced pancreatic adenocarcinoma. Fourteen subjects received the combined therapy in this clinical trial. Of these subjects, 57% received gemcitabine as prescribed (6 cycles) and 100% received the prescribed radiation dose, a major improvement from the published data whereby 29% completed all planned cycles of chemotherapy and 24% of subjects received less than 45 Gy of radiation therapy.[5] The improved ability to remain adherent to chemo-radiation therapy regimens may be partially explained by decreased systemic oxidative stress. Plasma F2-isoprostanes significantly decreased over the course of therapy in subjects treated with P-AscH- but not in comparator subjects. This phenomenon has been observed before in previous studies of patients receiving P-AscH- and gemcitabine.[13] However, this trial evaluated F2-isoprostane levels in subjects treated with P-AscH-, gemcitabine and radiation therapy, and furthermore compared these plasma levels to control subjects receiving gemcitabine and radiation therapy alone. These results taken together with the current in vitro and in vivo data suggest a potential systemic antioxidant effect in spite of local pro-oxidative treatments. In translational studies of glioblastoma, P-AscH- has been shown to synergize with radiation in vitro, but protect tumor cells from radiation killing in vivo.[39] These differences from our present study could be due to different ascorbate metabolism in the central nervous system, single vs. multiple doses of radiation, tumor microenvironment, and different tumor types.
The effect size suggested by this first-in-man trial is promising. This trial demonstrates an increase in median overall survival compared to our institutional median (21.7 vs. 12.7 mo, p = 0.08) and the ECOG-E4201 trial (21.7 vs. 11.1 mo).[5, 40–43] Progression free survival in P-AscH--treated subjects was also greater than our institutional median (13.7 vs. 4.6 mo, p = 0.02) and the ECOG-E4201 trial (6.0 months).[5]
In conclusion, P-AscH- enhances pancreatic tumor cell radiation cytotoxicity in addition to offering potential protection from radiation damage in normal surrounding tissue, making it an optimal agent for potentially improving locally advanced pancreatic adenocarcinoma. This is the first human study to not only infuse P-AscH- in combination with chemoradiation but to infuse while the radiation beam is on. These clinical trial results suggest adding P-AscH- daily to radiation therapy is safe with minimal additional toxicity and a promising therapeutic effect. A phase 2 trial powered to determine efficacy of P-AscH- at a dose of 75 g, when added to current standard of care combination therapy for pancreatic adenocarcinoma is indicated.
Supplementary Material
Statement of Significance.
Findings demonstrate that pharmacological ascorbate enhances pancreatic tumor cell radiation cytotoxicity in addition to offering potential protection from radiation damage in normal surrounding tissue, making it an optimal agent for improving treatment of locally advanced pancreatic adenocarcinoma.
Acknowledgments
We acknowledge the staff at the University of Iowa College of Medicine and Holden Comprehensive Cancer Center Radiation and Free Radical Research (RFRR) Core for radiation services. We acknowledge the invaluable support of the ESR Facility. Supported by NIH grants P01 CA217797 (J. Cullen, and D. Spitz), P30CA086862, CA184051 (J. Cullen), CA148062 (M. Alexander and J. Wilkes), CA169046 (G. Buettner), CA086862 (D. Spitz), and a grant from Gateway for Cancer Research (J. Cullen).
Footnotes
Conflict of interest statement: The authors have no conflicts of interest to disclose.
References
- [1].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: A Cancer Journal for Clinicians 2018;68(1):7–30. [DOI] [PubMed] [Google Scholar]
- [2].Ryerson AB, Eheman CR, Altekruse SF, Ward JW, Jemal A, Sherman RL, et al. Annual Report to the Nation on the Status of Cancer, 1975–2012, featuring the increasing incidence of liver cancer. Cancer 2016;122(9):1312–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2009;27(11):1806–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Balaban EP, Mangu PB, Khorana AA, Shah MA, Mukherjee S, Crane CH, et al. Locally Advanced, Unresectable Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline. Journal of Clinical Oncology 2016;34(22):2654–68. [DOI] [PubMed] [Google Scholar]
- [5].Loehrer PJ Sr., Feng Y, Cardenes H, Wagner L, Brell JM, Cella D, et al. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an Eastern Cooperative Oncology Group trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29(31):4105–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bahl A, Bhattacharyya T, Kapoor R, Singh OA, Parsee T, Sharma SC. Postoperative radiotherapy in periampullary cancers: a brief review. J Gastrointest Cancer 2013;44(1):111–4. [DOI] [PubMed] [Google Scholar]
- [7].Perrakis N, Athanassiou E, Vamvakopoulou D, Kyriazi M, Kappos H, Vamvakopoulos NC, et al. Practical approaches to effective management of intestinal radiation injury: benefit of resectional surgery. World journal of gastroenterology 2011;17(35):4013–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Huguet F, Goodman KA, Azria D, Racadot S, Abrams RA. Radiotherapy technical considerations in the management of locally advanced pancreatic cancer: American-French consensus recommendations. International journal of radiation oncology, biology, physics 2012;83(5):1355–64. [DOI] [PubMed] [Google Scholar]
- [9].Moding EJ, Kastan MB, Kirsch DG. Strategies for optimizing the response of cancer and normal tissues to radiation. Nat Rev Drug Discov 2013;12(7):526–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A 2005;102(38):13604–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen Q, Espey MG, Sun AY, Lee JH, Krishna MC, Shacter E, et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci U S A 2007;104(21):8749–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Buettner GR, Jurkiewicz BA. Catalytic metals, ascorbate and free radicals: combinations to avoid. Radiation research 1996;145(5):532–41. [PubMed] [Google Scholar]
- [13].Welsh JL, Wagner BA, van’t Erve TJ, Zehr PS, Berg DJ, Halfdanarson TR, et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): results from a phase I clinical trial. Cancer Chemother Pharmacol 2013;71(3):765–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Levine M, Conry-Cantilena C, Wang Y, Welch RW, Washko PW, Dhariwal KR, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci U S A 1996;93(8):3704–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Du J, Wagner BA, Buettner GR, Cullen JJ. Role of labile iron in the toxicity of pharmacological ascorbate. Free radical biology & medicine 2015;84:289–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Doskey CM, Buranasudja V, Wagner BA, Wilkes JG, Du J, Cullen JJ, et al. Tumor cells have decreased ability to metabolize H2O2: Implications for pharmacological ascorbate in cancer therapy. Redox Biol 2016;10:274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schoenfeld JD, Sibenaller ZA, Mapuskar KA, Wagner BA, Cramer-Morales KL, Furqan M, et al. O2- and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer cell 2017;31(4):487–500 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Espey MG, Chen P, Chalmers B, Drisko J, Sun AY, Levine M, et al. Pharmacologic ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free radical biology & medicine 2011;50(11):1610–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Du J, Cieslak JA 3rd, Welsh JL, Sibenaller ZA, Allen BG, Wagner BA, et al. Pharmacological Ascorbate Radiosensitizes Pancreatic Cancer. Cancer research 2015;75(16):3314–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Roy I, Zimmerman NP, Mackinnon AC, Tsai S, Evans DB, Dwinell MB. CXCL12 chemokine expression suppresses human pancreatic cancer growth and metastasis. PloS one 2014;9(3):e90400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, Gallick GE. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc 2009;4(11):1670–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Qian J, Niu J, Li M, Chiao PJ, Tsao MS. In vitro modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis. Cancer research 2005;65(12):5045–53. [DOI] [PubMed] [Google Scholar]
- [23].Buettner GR. In the absence of catalytic metals ascorbate does not autoxidize at pH 7: ascorbate as a test for catalytic metals. Journal of biochemical and biophysical methods 1988;16(1):27–40. [DOI] [PubMed] [Google Scholar]
- [24].Tietze F Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: Applications to mammalian blood and other tissues. Analytical biochemistry 1969;27(3):502–22. [DOI] [PubMed] [Google Scholar]
- [25].Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Analytical biochemistry 1980;106(1):207–12. [DOI] [PubMed] [Google Scholar]
- [26].Storer BE. Design and analysis of phase I clinical trials. Biometrics 1989;45(3):925–37. [PubMed] [Google Scholar]
- [27].Administration USDoHaHSFaD. Safety assessment for IND safety reporting: guidance for industry., http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM477584.pdf. ; 2017. [accessed October 3rd.2017].
- [28].Administration USDoHaHSFaD. Guidance for Industry Oversight of Clinical Investigations — A Risk-Based Approach to Monitoring, https://www.fda.gov/downloads/Drugs/Guidances/UCM269919.pdf; [accessed October 3rd.2017].
- [29].Vislisel JM, Schafer FQ, Buettner GR. A simple and sensitive assay for ascorbate using a plate reader. Analytical Biochemistry 2007;365(1):31–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ 2nd, . A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci U S A 1990;87(23):9383–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kadiiska MB, Gladen BC, Baird DD, Germolec D, Graham LB, Parker CE, et al. Biomarkers of oxidative stress study II: are oxidation products of lipids, proteins, and DNA markers of CCl4 poisoning? Free radical biology & medicine 2005;38(6):698–710. [DOI] [PubMed] [Google Scholar]
- [32].Kadiiska MB, Gladen BC, Baird DD, Graham LB, Parker CE, Ames BN, et al. Biomarkers of oxidative stress study III. Effects of the nonsteroidal anti-inflammatory agents indomethacin and meclofenamic acid on measurements of oxidative products of lipids in CCl4 poisoning. Free radical biology & medicine 2005;38(6):711–8. [DOI] [PubMed] [Google Scholar]
- [33].Zhang ZZ, Lee EE, Sudderth J, Yue Y, Zia A, Glass D, et al. Glutathione Depletion, Pentose Phosphate Pathway Activation, and Hemolysis in Erythrocytes Protecting Cancer Cells from Vitamin C-induced Oxidative Stress. The Journal of biological chemistry 2016;291(44):22861–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ried K, Travica N, Sali A. The acute effect of high-dose intravenous vitamin C and other nutrients on blood pressure: a cohort study. Blood Press Monit 2016;21(3):160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proceedings of the National Academy of Sciences of the United States of America 1990;87(23):9383–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wei X, Xu Y, Xu FF, Chaiswing L, Schnell D, Noel T, et al. RelB Expression Determines the Differential Effects of Ascorbic Acid in Normal and Cancer Cells. Cancer research 2017;77(6):1345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Monti DA, Mitchell E, Bazzan AJ, Littman S, Zabrecky G, Yeo CJ, et al. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PloS one 2012;7(1):e29794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, Chen Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci Transl Med 2014;6(222):222ra18. [DOI] [PubMed] [Google Scholar]
- [39].Grasso C, Fabre M-S, Collis SV, Castro ML, Field CS, Schleich N, et al. Pharmacological Doses of Daily Ascorbate Protect Tumors from Radiation Damage after a Single Dose of Radiation in an Intracranial Mouse Glioma Model. Frontiers in oncology 2014;4:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhang X, Huang HJ, Feng D, Yang DJ, Wang CM, Cai QP. Is concomitant radiotherapy necessary with gemcitabine-based chemotherapy in pancreatic cancer? World journal of gastroenterology 2014;20(46):17648–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ben-Josef E, Schipper M, Francis IR, Hadley S, Ten-Haken R, Lawrence T, et al. A phase I/II trial of intensity modulated radiation (IMRT) dose escalation with concurrent fixed-dose rate gemcitabine (FDR-G) in patients with unresectable pancreatic cancer. International journal of radiation oncology, biology, physics 2012;84(5):1166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hammel P, Huguet F, van Laethem JL, Goldstein D, Glimelius B, Artru P, et al. Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients With Locally Advanced Pancreatic Cancer Controlled After 4 Months of Gemcitabine With or Without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA 2016;315(17):1844–53. [DOI] [PubMed] [Google Scholar]
- [43].Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364(19):1817–25. [DOI] [PubMed] [Google Scholar]
- 43].Du Bois D, Du Bois EF. A formula to estimate the approximate surface area if height and weight be known. 1916. Nutrition 1989;5(5):303–11; discussion 12–3. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.