

Abstract
Plasmon resonance biosensors provide ultimate sensitivity at the single-molecule level. This sensitivity is, however, associated with a nanometer-sized confined hotspot, and molecular transport toward the sensor relies on inefficient diffusion. Here, we combine a plasmonic nanoantenna with a solid-state nanopore and demonstrate that single DNA molecules can be efficiently delivered to the plasmonic hotspots and detected in a label-free manner at submillisecond acquisition rates by monitoring the backscattered light intensity from the plasmonic nanoantennas. Our method realizes a better than 200 μs temporal resolution together with a down to subsecond waiting time, which is orders of magnitude better than traditional single-molecule plasmonic resonance sensing methods. Furthermore, the electric field applied to the nanopore can actively drive biomolecules away from the hotspot, preventing molecules to permanently bind to the gold sensor surface and allowing efficient reuse of the sensor. Our plasmonic nanopore sensor thus significantly outperforms conventional plasmon resonance sensors and provides great opportunities for high-throughput optical single-molecule-sensing assays.
Keywords: Plasmon resonance sensing, single-molecule sensing, nanopore, plasmonic nanopore, single-particle scattering
Plasmon resonance sensing has been heralded as a high-throughput, high-speed, high-sensitivity, and label-free biosensing technique based on an optical readout.1 Biomolecule detection follows from a shift in the plasmon resonance of a plasmonic nanoantenna that results from changes in the refractive index of the local environment of the antenna induced by the presence of the analyte. This concept has been used extensively in bulk sensing2,3 and has been integrated into a variety of biological and chemical sensing devices.4 Recently, impressive developments in plasmonic sensing have pushed the sensitivity of these devices to the ultimate detection level of single molecules. This advancement has been achieved through engineering the hotspot, the nanoscale volume into which the plasmonic nanoantenna strongly concentrates the incident optical field.5−13
Despite substantial progress over the past years, single-molecule plasmon resonance sensing faces severe challenges. As the extreme increase in sensitivity requires the nanoscale electromagnetic (EM) hotspot to become exceedingly more confined, the probability that a biomolecule will be diffusing into the hotspot becomes vanishingly small for any practical analyte concentrations.14−16 Furthermore, molecules interact with the plasmonic sensor at many positions on the nanoantenna, not merely at its most sensitive region. Moreover, although surface interactions can be used to anchor and hence detect specific molecules to the plasmonic hotspot, the inability to release the molecules from the sensor’s surface quickly leads to saturation of the hotspot volume, and as a result, the sensor cannot be reused, limiting throughput. Hence, a technique that would be able to controllably deliver single molecules to a hotspot, position them there, as well as eject them from the spot again, would greatly enhance the sensing performance of plasmonic sensors. Multiple reports on the successful integration of such delivery systems to plasmonic sensors have been reported for bulk plasmonic sensing.17−19 However, the integration of such an active element at the single-molecule level is still lacking.
A solid-state nanopore is a biosensor that enables single biomolecules to be driven through a nanometer-sized aperture in a free-standing membrane. Acting as a gateway between two electrically biased reservoirs, the nanopore is the focus of a DC electric field that delivers and translocates charged biomolecules such as DNA or proteins across the membrane.20 The nanopore simultaneously can be used as a detector, as the passage of the molecules through the pore can be measured through a temporary blockade of the ionic current that is running through the pore by the same electric bias voltage. Although the ionic current-based nanopore sensing has been employed for single-molecule analysis with considerable success,21,22 particularly for DNA and protein sensing, the current-based readout has its own limitations. An optical readout would be beneficial for nanopore sensing,23−27 as it could be employed at a large measurement bandwidth, it could be operated irrespective of the chosen buffer conditions, and it does hold an excellent potential for high sensor integration densities.
Here we demonstrate label-free plasmonic resonance sensing of individual DNA molecules at a high turnover rate by integrating a nanopore with a plasmonic nanoantenna. The nanopore serves as an active element that electrophoretically drives biomolecules into the nanopore, thus forcing them exactly into the hotspot of the plasmonic sensor. We use top-down fabricated plasmonic nanostructures with sub-10 nm gaps to create highly localized and enhanced electromagnetic field hotspots that are excited by single-wavelength continuous-wave (CW) laser illumination of the nanoantenna. We show that the translocation of a single unlabeled DNA molecule through the nanopore in the gap can be monitored from a transient intensity change of the light that is elastically backscattered from the antenna (Figure 1a). We demonstrate that the optical signal originates from a shift in the resonance of the plasmonic antenna and show that there is an excellent correlation between the backscattered signals from the antenna and the ionic current signals as DNA molecules traverse the nanopore. By integrating a nanopore with a plasmonic nanoantenna, single molecules can thus be actively transported toward the sensor, be precisely positioned at will into the hotspot, and subsequently be read out optically in a label-free manner.
Figure 1.
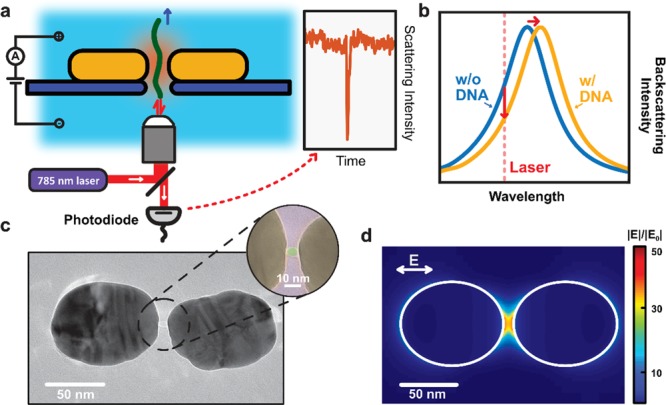
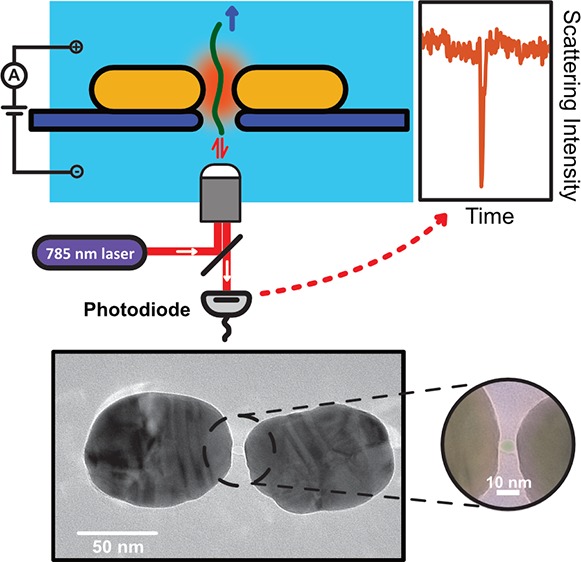
Plasmonic nanopores for single-molecule optical sensing. (a) Schematic side-view illustration of a DNA molecule that is electrophoretically driven through a plasmonic nanopore and detected by optical backscattering from the plasmonic antenna. (b) Illustration of the sensing principle. The temporary presence of the DNA in the hotspot region of the plasmonic antenna induces a shift of the resonance wavelength of the antenna, hence decreasing the scattering intensity that is detected at the excitation laser wavelength. (c) Typical TEM image of the plasmonic nanopore devices used in our experiments. The plasmonic nanopore consists of a gold dimer antenna with a ∼5 nm nanopore at the gap center. The inset shows a false colored TEM image of a zoom of the nanogap region, highlighting the nanopore. (d) Simulated electromagnetic field distribution of the plasmonic nanopore in longitudinal excitation (i.e., with a polarization of the E along the long axis of the structure, cf. image) with a wavelength of 785 nm, as used in our experiments. The simulation shows the extremely enhanced and confined electromagnetic field within the gap of the dimer antenna, which is required for label-free optical sensing of single molecules.
Plasmonic Nanopore Devices for Label-Free Single-Molecule Sensing
The principle for optical sensing with plasmonic nanopores relies on a refractive index change that is induced as an analyte molecule enters the hotspot of the plasmonic nanoantenna, i.e., the region where the electromagnetic field is most strongly localized. The DNA translocation will induce a redshift of the plasmon resonance of the entire nanostructure that can be observed through monitoring the backscattered light intensity from the antenna (Figure 1b). This shift can either be detected through tracking the plasmon resonance peak of the antenna,9 which is inevitably slow (tens of milliseconds), or by monitoring the scattered light intensity at a fixed excitation wavelength, which can be done at microsecond speeds. Note that, in the latter case, which is clearly advantageous for high-speed readouts, the presence of a biomolecule in the hotspot will produce either a decrease in intensity (if the excitation wavelength is shorter than the peak of the plasmon resonance, Figure 1b) or an increase (if the excitation wavelength is longer than the plasmon resonance peak). We adopted this detection principle by light excitation with a CW laser at 785 nm and collecting the elastically backscattered light from the plasmonic nanostructure with a balanced photodiode detector, which thus allows for fast monitoring of the hotspot region. Details on the experimental setup can be found in the Note S1 and Figure S1.
Our plasmonic nanopore devices are fabricated using a top-down approach based on two-step electron-beam lithography (EBL) to create an array of plasmonic nanoantennas combined with subsequent electron-beam sculpting to create a nanopore in the feed gap of a single antenna.28Figure 1c shows a TEM image of a typical plasmonic nanopore used in our experiments. Each nanoantenna consists of two elongated gold nanodiscs, positioned on a 20 nm thin free-standing silicon–nitride membrane, each with a 90 nm length and a 70 nm width, that are facing tip-to-tip and are separated by a ∼8 nm gap. A ∼5 nm diameter nanopore is drilled through the SiN membrane in the center of the nanogap (see inset Figure 1c). Details of the device fabrication can be found in text below and more TEM images of example devices can be found in Figure S2.
If the two elongated nanodiscs are separated by only a few-nanometer-sized gap, they do generate an extremely enhanced EM field when the gap-mode plasmon resonance is excited. We used finite difference time domain (FDTD) simulations to estimate the strength of the field enhancement as well as the spatial localization of the EM field in the nanostructure. Figure 1d shows the resulting simulated normalized electric field distribution for a nanostructure of two 70 nm × 90 nm × 30 nm (length × width × height) gold cylindroids with an 8 nm gap that is excited in longitudinal (i.e., along the longest direction of the nanostructure, see Figure 1d) polarization of the illumination light at 785 nm. The result shows that the magnitude of the electric field in the nanogap is strongly enhanced, over 50 times, and closely is localized to the nanometer-sized gap region. Figure S4 shows simulation results under transverse (i.e., along the shortest direction of the nanostructure) polarization, where no field enhancement occurs in the gap. Details of the FDTD simulations are provided in text below.
Before performing the single-molecule experiments, we characterize the devices and select a fitting nanoantenna for measurements. During the fabrication, we make an array of the nanoantennas with slightly different gap sizes, from which we chose one structure that is deemed most suitable to our experimental requirements, in which we drill a nanopore in its gap by use of a TEM. The device is then assembled as a separator between two compartments of a custom-made flow cell that are filled with an electrolyte (2 M LiCl buffered to pH 8 with tris-EDTA buffer), leaving the nanopore as the only connection between the two reservoirs. This flow cell is mounted on a piezo stage on an optical detection setup with a 60× 1.2 NA objective (see Supporting Information), and the membrane is scanned with a 100 μW longitudinally polarized 785 nm laser beam that is focused to a ∼0.5 μm spot. During scanning, both the backscattered light intensity and the ionic current are simultaneously recorded at each position. Figure 2 shows a typical backscattering and corresponding ionic current map resulting from such a scan. The scattering map (Figure 2a) shows an array of dots, where several antennas in the array scatter the focused laser beam significantly different than the background (Figure 2c and Figure S3). The ionic current map (Figure 2b) invariably shows only a single current maximum that is produced by local heating of the nanoantenna at the nanopore location.29 The heating from plasmonic nanostructures locally creates a temperature increase,30 and an associated increase in current will only be observed if the excited nanoantenna contains a nanopore. In this fashion, the plasmonic structure with a nanopore can easily be identified from the array and get aligned to the laser.
Figure 2.
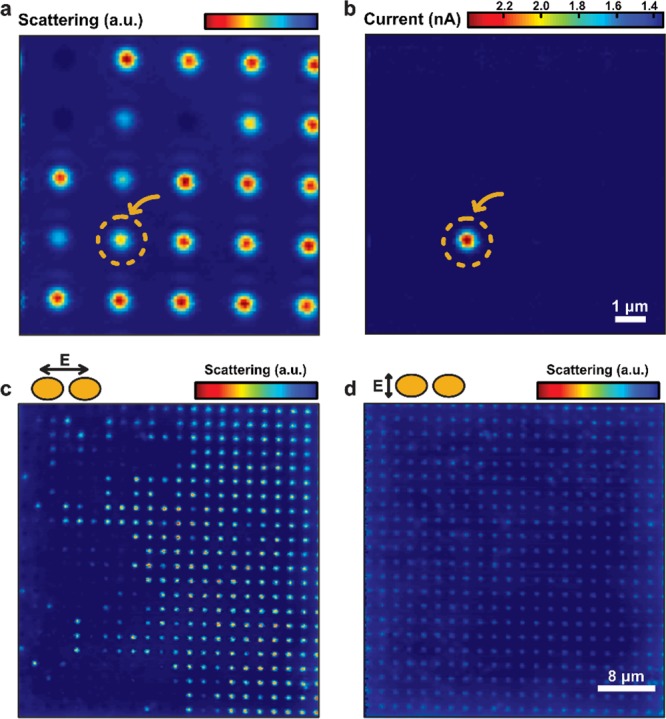
Backscattering and ionic current mapping from a plasmonic antenna array. (a) Backscattering intensity map from scanning a focused laser with longitudinal polarization across the plasmonic antenna array. Each of the nanoantennas in this image has a slightly different gap size, leading to varying scattering intensities. The one structure that had a nanopore drilled in the gap is marked with the yellow arrow/dashed circle. (b) Ionic current map to the scan corresponding to panel a. A clear current increase can be observed when the longitudinally polarized laser hit the plasmonic structure with a nanopore in the gap. The structure with a nanopore drilled in the gap is marked with the yellow arrow/dashed circle. (c and d) Backscattering intensities across an array of plasmonic nanoantennas with different gap sizes under longitudinal (c) and transverse (d) polarizations, respectively. Only under longitudinal polarization, we observe backscattering that is strong and sensitive to the gap size of the antenna, while the scattering in transverse excitation is weak and almost invariant to the gap size of the nanostructures.
The typical structures we choose for these experiments are aimed to have a gap-mode resonance wavelength close to, but slightly longer than, the illumination laser wavelength (785 nm); see simulation results in Figure S5. As expected, the antenna containing the nanopore has an excellent (but not the highest) backscattering baseline signal, as illustrated by the scattering map of Figure 2a, indicating that the antenna has a plasmon resonance close to the excitation wavelength. Moreover, excitation close to resonance will lead to significant absorption and heat generation. Indeed, a substantial temperature increase of about 40 °C30,31 can be estimated from the ionic current increase of 50% at a mere 100 μW of excitation power in Figure 2b (see also Figure S7). Such a temperature elevation is acceptable for these experiments with double-stranded DNA. If desired, one may use alternative designs for plasmonic nanodevices that yield a much lower temperature increase32 and that recently were also implemented for optical transmission detection of DNA translocation.27Figure 2c,d illustrates the strong difference in scattering strength if a transverse polarization is used. The scattering signal is weak from all antennas, as expected since the transverse mode does not excite the gap mode and has a peak resonance far off from the excitation wavelength. The observations clearly support the fact that backscattering in the longitudinal polarization is dominated by the gap-mode resonance excitation.
Label-Free Optical Detection of DNA Molecule Translocations
Next, we show the label-free detection of DNA molecules as they translocate through the nanopores. After locating and aligning the plasmonic nanopore to the focused laser spot, 10 kbp dsDNA molecules are flushed into the cis-side of the membrane (the bare SiN side without the gold nanostructures). In all of the experiments presented here, DNA molecules were exclusively translocated from the SiN side to the gold structure side to ensure that all DNA molecules that enter the hotspot will have passed through the nanopore and to prevent undesired binding of DNA on the peripheral ends of the gold nanostructure where it is harder to remove DNA electrophoretically (see below). Subsequently, a bias voltage (300 mV) is applied across the membrane while the laser excites the plasmonic nanostructure, and both the ionic current and the backscattering optical intensity are recorded simultaneously. (Details of the single-molecule experiments are described in text below.) Typical trajectories of both channels are shown in Figure 3a. Numerous concurrent transients can be clearly discerned in both the ionic current and backscattered light intensity. These signals can be immediately recognized as single DNA molecules passing freely both the optical and electrical detection volumes of the plasmonic nanopore. We observe the archetypical electrical transients that are exemplary for DNA translocations, with signals exhibiting excellent signal-to-noise characteristics due to the small pore (5 nm) and the large electrolyte concentration used (2 M LiCl). More importantly, the signals in the optical channel also display good signal-to-noise ratio characteristics and they correlate excellently with the signals from the ionic current channel, confirming that these optical signals derive from single DNA translocation events. This demonstrates that, remarkably, our plasmonic nanoantenna can be used for high-throughput label-free optical detection of single DNA molecules that are electrically driven through the hotspot.
Figure 3.
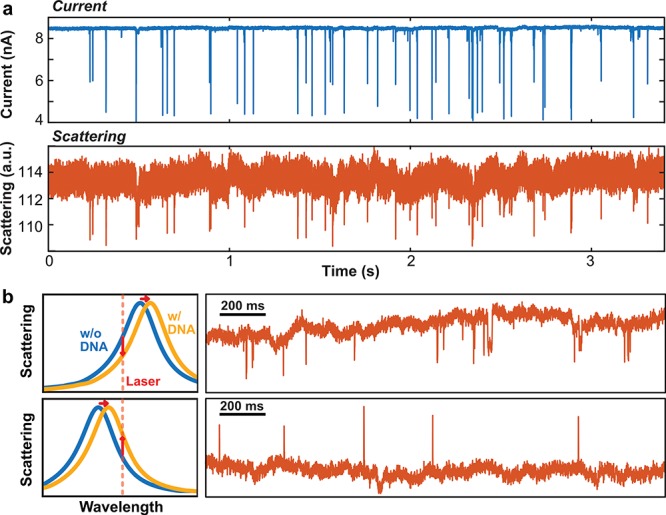
Optical detection of single DNA translocations through plasmonic nanopores. (a) Example of corresponding ionic current (blue) and backscattering (orange) time traces during a DNA translocation experiment. Single-molecule events appear as blockades in both the ionic current, where the ion flow is blocked by DNA, and the optical signals, where transient decreases in the backscattering intensity of nanoantenna are induced by DNA molecules that traverse the hotspot region. Traces are filtered with a 5 kHz low-pass filter. (b) Examples of single-molecule optical events that, remarkably, display different signal polarities. The top and bottom traces are obtained for two different devices with a different plasmonic resonance peak wavelength. The schematics in the left panel illustrate the mechanism that explains the decrease or increase of the scattering intensity induced by single DNA molecules. As the DNA molecules always induce a red shift of the antenna’s resonance wavelength, a decrease will be observed if the resonance wavelength is longer than the 785 nm excitation laser wavelength, while an increase will be observed if the resonance wavelength is shorter than the excitation wavelength.
The assertion that the optical signals arise from a shift of the plasmon resonance is corroborated by Figure 3b, where the backscattering signals for DNA translocations are displayed for two different devices with slightly different geometries. The top trace of Figure 3b shows that the scattering intensity reduces during DNA translocation events, whereas the bottom trace, from a different device, shows increases in the scattering intensity for translocation events. The different signal polarities are caused by the different plasmon resonance peak wavelength in both devices with respect to the excitation wavelength. The TEM images of these two devices are shown in Figure 1 and Figure S2c, respectively. Since the refractive index of DNA is larger than water at optical frequencies,33 insertion of a DNA molecule into the hotspot will induce a redshift of the plasmon resonance of the gold antenna.34 Subsequently, as illustrated on the left of Figure 3b, the backscattering intensity reduces when the resonance peak wavelength is longer than the wavelength of excitation, but increases when it is shorter. The observation of these different signal polarities serves as a clear hallmark of the plasmonic resonance origin of the DNA signals.
Microscopic Insight of the DNA Translocation through a Plasmonic Nanopore
Figure 4a shows a closer inspection of the typical signals from both the optical and electrical channels. The ionic current signals (blue traces) display the characteristic levels that are typical for DNA translocations: before translocation, an open pore current runs through the pore, which is subsequently partially reduced when a molecule is inserted into the nanopore. The DNA molecule can traverse the pore in a linear head-to-tail fashion (where it enters the pore with one of its ends and only one double strand of DNA resides in the nanopore during the translocation), or in a folded mode35 (where it enters the pore in a folded fashion with at first two double strands of DNA residing in the nanopore) that is distinctive for electrophoretically driven DNA translocations through a solid-state nanopore. Surprisingly, however, in our plasmonic devices, we observe that the ionic current after translocation does not immediately recover to the baseline value, but remains at a very shallow current blockade level that lasts for a few milliseconds. This signal strength is too shallow to be attributed to DNA that is inserted in the nanopore. Instead, it indicates that the DNA remains near the nanopore without inserting exactly in it; i.e., it suggests that part of the nearby DNA molecule contributes to an additional access resistance that slightly lowers the current.36 The simultaneously acquired signals in the optical channel further illuminate these translocation events (Figure 4a, orange traces). Initially a strong reduction of the backscattered light intensity from the baseline can be observed, which correlates well with the translocation of the DNA molecule through the nanopore, albeit that the optical signal does not seem to distinguish strongly between linear and folded translocation modes. Remarkably, the signal strength after DNA translocation through the pore reduces only slightly (if at all), indicating that the DNA molecule remains present in the hotspot region. The duration of the extended signal matches well with the shallow level observed in the current channel, implying a close proximity of the DNA molecule near the nanopore while it resides in the hotspot.
Figure 4.
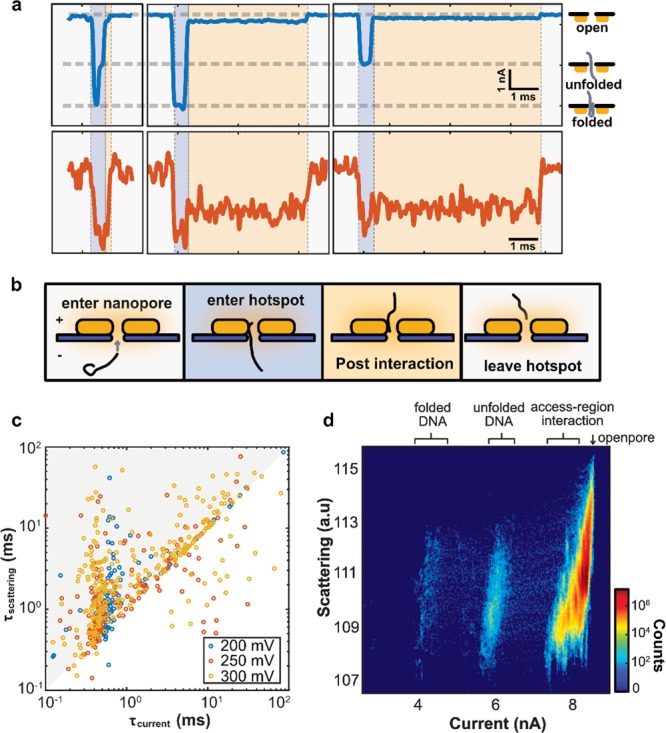
Comparison of optical and current blockade single-molecule signals. (a) Examples of single-molecule DNA translocations. Top traces (blue) are ionic current signals; bottom traces (orange) are optical backscattering signals. The ionic current reports on different folding conformations during translocation (cf. insets on the right). Interestingly, a clear post-translocation optical signal and shallow current blockade signal can be observed, indicating the presence of the molecule in the hotspot of the plasmonic antenna on the exit access region of the nanopore. (b) Cartoons for illustrating the different phases of DNA translocation of plasmonic nanopores (background colors correspond to panel a). A DNA molecule enters the nanopore, inducing a blockade of the ionic current. It then moves virtually instantaneously into the hotspot, resulting in optical detection of the molecule. After the DNA molecule has translocated, post-translocation DNA–gold interactions maintain an extended presence of the DNA molecule in the hotspot that is located in the exit access region of the nanopore, leading to a pronounced optical and weak ionic current signal. Finally, the molecule is unloaded from the nanogap. (c) Comparison of the signal duration τ (the time taken between two baseline crossings) of the optical and electrical signals under 200, 250, and 300 mV bias voltages. (d) Comparison of ionic current and scattering intensity for all data points at 300 mV bias. Events with anomalously long (integral of current signal > 20 nA ms) sticking of the DNA are ruled out from this analysis.
The data lead to a physical picture of the translocation process as sketched in Figure 4b. Once a DNA molecule diffuses into the capture region of the nanopore, the negatively charged molecule is driven toward the nanopore prior to translocation. Next, the single DNA molecule enters the nanopore in either a linear or folded fashion, blocking part of the ion flow and producing a distinctive blockade current signal. Directly after the DNA molecule traverses through the 20 nm short nanopore, it enters the hotspot region of the gold nanoantenna, incurring a clear change in the scattered light intensity. The DNA polymer continues to be reeled through the pore and eventually exits the nanopore, but at that point, it remains present in the optical hotspot (as well in the electrical access region) by virtue of the interactions between the DNA molecule and the surface of gold nanoantenna. Finally, the DNA molecule also escapes from this region.
This picture is further supported by an in-depth analysis of the signals. Figure 4c provides a log–log scatter plot that compares the signals durations τ from both channels for all translocation events at different driving voltages. The events along the diagonal have an identical optical and electrical dwell time (as in the examples in Figure 4a). A large number of events is, however, observed above the diagonal in the diagram, representing events with a longer signal duration in the optical channel than in the electrical channel. Since the post-translocation interaction cannot always be discerned in the ionic current, as the blockade is shallow and its strength depends on the position of the molecule in the electrical access region,36 optical signals typically last longer than the associated electrical ones. The signal strengths of both the electrical and optical channels are compared in Figure 4d. This all-points heatmap of the current and scattering intensities for all translocation events shows a strong clustering of data points around the current blockade levels corresponding to the folded and linear translocation events (near 6 and 4 nA, respectively), consistent with the ionic current examples shown in Figure 4a. However, most of the data points cluster at the access-region contribution to the electrical signals (near 8 nA), since the molecules typically spend a much longer time in the hotspot region than in the nanopore (see Figure S8). In the optical channel, no tight clustering is observed, which can, as discussed in Note S3, be attributed to inhomogeneities in the EM field distribution along the gold nanoantenna gap.
Our plasmonic nanopores significantly outperform previous single-molecule plasmonic biosensors in several aspects. Table 1 provides a detailed comparison between our results and those from pioneering methods such as photothermal microscopy, dark-field spectroscopy, single-nanorod scattering, and double-nanohole plasmonic trapping. The integration of a nanopore in the hotspot provides the plasmonic nanostructure with the capability of actively attracting biomolecules into the sensing region as well as releasing the molecules after acquisition of the sensor signals. Even though some other sensing strategies have, as indicated in Table 1, demonstrated great selectivity through biochemical modification of the sensor surface, our work provides clear advantages on time resolution, waiting time, and maximum number of events for each nanoantenna over these previously reported plasmonic single-molecule sensing approaches. The limited signal-to-noise ratio of the previous methods typically requires a long integration time for obtaining distinguishable signals. In our method, the detection bandwidth can easily be set to 5 kHz or higher, an improvement of more than a factor of 500, while further improvements can be achieved by optimizing the nanoantenna geometry. The number of molecules that can be sensed is also dramatically enhanced in our approach. In our nanopore approach, molecules are actively captured and delivered into the sensing region, which reduces the waiting time between events from hundreds of seconds down to the millisecond regime, an improvement of 3 orders of magnitude from the conventional diffusion-limited techniques. Finally, previous methods can at most detect ∼10 molecules per antenna in total because of a saturation of the sensing region. In our plasmonic nanopore approach, the applied electrophoretic force actively releases the analyzed molecules from the sensing region, and hence the hotspot of the nanoantenna is not consumed, removing any limit to the maximum number of molecules that can be detected by a single nanoantenna.
Table 1. Comparison of Different Single-Molecule Plasmon Resonance Sensing Methods.
| method | time resolution (ms) | waiting time | max no. of events | specificity | ref |
|---|---|---|---|---|---|
| photothermal microscopy | 100 | tens to hundreds of seconds (100 nM protein) | <10 (streptavidin) | yes | (10) |
| dark-field spectroscopy | 24 | 50 s (average, 1.25 μg/mL fibronectin) | ∼10–20 (fibronectin) | yes | (9) |
| single-nanorod scattering | 6 | ∼1–100 s (25–2.5 nM antibody) | ∼10b (antibody) | yes | (11) |
| double-nanohole plasmonic trapping | <1 | seconds to hoursa | not determined (no binding site consumed) | no | (5) and (6) |
| plasmonic nanopores | <200 μs | ∼100 ms (1 μg/mL dsDNA) | >500 (no binding site consumed) | no | this work |
Waiting times in these devices vary widely depending on slight differences in sample preparation and poorly understood surface repulsion.6
On each nanorod.
In conclusion, we have introduced a new method to optically detect single DNA molecules in solution. Plasmonic nanoantennas are used to create a well-defined and highly EM field-enhanced plasmonic hotspot, into which molecules are precisely delivered via a solid-state nanopore. The backscattered light from the antenna enables the detection of, in a label-free manner, the presence of single DNA molecules in the hotspot. The nanopore in the plasmonic gap actively captures analyte molecules from the bulk and ejects them after signal acquisition so that the most sensitive hotspot region can be reused for probing the next molecules. The plasmonic nanopores provide orders of magnitude improvements on the time resolution, waiting time, and maximum number of events over previously reported plasmon resonance-based single-molecule sensing methods. Improved antenna design and lower noise optical detectors can yield yet higher sensitivities and detection bandwidth than reported in these first proof-of-principle experiments. We anticipate plentiful applications and extensions of the technique. For example, additional selectivity can be provided to the plasmonic nanopore sensor through modification of the gold surface, akin to previous plasmonic resonance sensing schemes. Moreover, the plasmonic resonance nanopore sensing approach is easily applicable to other biomolecules such as proteins, as the refractive index sensing mechanism will apply generally to any analyte. Finally, these plasmonic nanopores can be fabricated in massively parallel arrays, where each nanoantenna is simultaneously read out, which will allow the development of high-throughput single-molecule optical assays for sensing a variety of analytes.
Fabrication of Plasmonic Nanopores
The plasmonic nanopore devices were fabricated using an electron-beam-lithography (EBL)-based top-down approach, as we reported before.28 In brief, an array of plasmonic bowtie nanoantennas, each consisting of two elongated nanodiscs with a length of 90 nm and width of 70 nm, was defined using EBL in two steps. The pattern from each step contains a dimer half, such that alignment of the two patterns will provide a variety of gap spacings between adjacent dimers. For each EBL step, ∼100 nm PMMA resist layer (950 K MW, 3% in anisole) was spin coated on the SiN membranes and then exposed using a Raith EBPG 5200 EBL system, at an accelerating voltage of 100 kV, with pressure below 5 × 10–7 mbar, and with e-beam doses ranging from 2000 to 2500 μC cm–2. The EBL-defined patterns were developed in a methyl isobutyl ketone (MIBK) and isopropanol (IPA) mixture (a volume ratio 1:3, MIBK/IPA) for 60 s. Then, a 30 nm gold layer with 1 nm titanium as the adhesion layer was deposited using electron-beam evaporation, and the lift-off was performed by immersing the samples in 80 °C PRS-3000 solution overnight. The nanodiscs arrays defined in the first e-beam step were manually aligned to the center of the free-standing membranes, and the second-step patterns were aligned by an automatic marker search routine on markers defined in the first step. Finally, a single nanopore was drilled using a TEM (FEI Tecnai 200S, 200 kV) in the gap of a single nanoantenna on each free-standing membrane.
FDTD Simulation
FDTD solutions (Lumerical Solutions, Inc., Canada) were used to model the electric field distribution around the plasmonic nanopore. The elongated gold disc dimer was modeled as two 70 nm × 90 nm × 30 nm (width × length × thickness) cylindroids separated by an 8 nm gap on a 20 nm thin SiN membrane (refractive index, RI = 2) with a 1 nm Ti layer under the gold. A 6 nm in diameter nanopore through the SiNx membrane was placed at the gap center. The RI of the surrounding medium was set to 1.33. Symmetry on the boundaries was used to reduce the computational time. The modeled antenna was excited by a total-field scattered-field source propagating along the axis perpendicular to the membrane. Figures S4–6 show additional FDTD simulation results.
Single-Molecule Experiments
The plasmonic nanopore devices were oxygen-plasma cleaned before all of the experiments. Before the experiments, a nanopore device was placed in a flow cell (design reported in ref (37)) containing a 2 M LiCl electrolyte solution with 20 mM Tris (tris(hydroxymethyl)-aminomethane) and 2 mM EDTA (ethylenediaminetetraacetic) at pH 8. The flow cell was installed and fixed in the optical detection setup described in Note 1. A 785 nm continuous wave laser was focused on the device and moved to the free-standing SiN membrane with the piezo stage. The position of the laser focus spot and the location of the membrane could be devised from the camera in the optical setup. A pair of Ag/AgCl electrodes was immersed into the flow cell, one in each side of the reservoir, and connected to a patch clamp amplifier (Axon Axopatch 200B, Molecular Devices). A DC voltage was applied across the membrane, and the ionic current through the nanopore was recorded by the amplifier. The backscattered light is detected by a balanced photodiode detector. The output of both the amplifier and the photodiode detector was simultaneously sampled with a DAC board (USB-6251, National Instruments) and transferred to a computer. The laser was precisely focused onto the plasmonic nanopore device by moving the piezo stage and maximizing both the ionic current and the backscattering intensities. Next, double-stranded DNA (10 kbp, 5 ng μL–1) was added to the compartment of flow cell facing the etch pit of the chip (SiN side of the chip). Addition of DNA to the compartment facing the gold nanostructures resulted in an unsteady baseline, and translocation events could not be resolved, presumably due to irreversible unspecific binding of DNA to the nanostructure. The DNA molecules were then electrophoretically driven through the nanopore by an applied bias voltage. The ionic current and the backscattering signals were recorded using a custom-designed LabVIEW program.
Acknowledgments
We acknowledge Meng-Yue Wu for TEM drilling of nanopores and Prof. Aleksei Aksimentiev, Sergii Pud, Wayne Yang, and Nils Klughammer for discussions. This work was supported by the National Human Genome Research Institute of the National Institutes of Health (1R01HG007406-01), by the ERC Advanced grant SynDiv (669598), and by The Netherlands Organization for Scientific Research (NWO/OCW), as part of the Frontiers of Nanoscience program. X.S. thanks Prof. Yi-Tao Long and acknowledges support from the China Scholarship Council (CSC201606740021)
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.8b04146.
Additional discussions about the experimental setup, backscattering mapping of dimer antenna array, and spatial field distribution of the electromagnetic hotspot in the plasmonic gaps, schematic of the experimental setup, more examples of plasmonic nanopore devices, additional scattering and ionic current mappings, additional FDTD simulation results, IV curves of typical plasmonic nanopore, and the signal durations of corresponding ionic current and backscattering events (PDF)
Author Contributions
C.D. directed the project. X.S, D.V.V., and C.D. designed the experiments. X.S and D.V.V. fabricated the plasmonic devices. X.S. performed the single-molecule experiments. X.S. and D.V.V. analyzed data. All authors discussed the results, contributed to writing, and approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Willets K. A.; Van Duyne R. P. Localized Surface Plasmon Resonance Spectroscopy and Sensing. Annu. Rev. Phys. Chem. 2007, 58, 267–297. 10.1146/annurev.physchem.58.032806.104607. [DOI] [PubMed] [Google Scholar]
- Xin H.; Namgung B.; Lee L. P. Nanoplasmonic Optical Antennas For life Sciences and Medicine. Nature Reviews Materials 2018, 3, 228–243. 10.1038/s41578-018-0033-8. [DOI] [Google Scholar]
- Mayer K. M.; Hafner J. H. Localized Surface Plasmon Resonance Sensors. Chem. Rev. 2011, 111, 3828–3857. 10.1021/cr100313v. [DOI] [PubMed] [Google Scholar]
- Brolo A. G. Plasmonics for Future Biosensors. Nat. Photonics 2012, 6, 709. 10.1038/nphoton.2012.266. [DOI] [Google Scholar]
- Pang Y.; Gordon R. Optical Trapping of a Single Protein. Nano Lett. 2012, 12, 402–406. 10.1021/nl203719v. [DOI] [PubMed] [Google Scholar]
- Gordon R. Biosensing with Nanoaperture Optical Tweezers. Opt. Laser Technol. 2019, 109, 328–335. 10.1016/j.optlastec.2018.07.019. [DOI] [Google Scholar]
- Juan M. L.; Gordon R.; Pang Y.; Eftekhari F.; Quidant R. Self-Induced Back-Action Optical Trapping of Dielectric Nanoparticles. Nat. Phys. 2009, 5, 915. 10.1038/nphys1422. [DOI] [Google Scholar]
- Al Balushi A. A.; Gordon R. Label-Free Free-Solution Single-Molecule Protein–Small Molecule Interaction Observed by Double-Nanohole Plasmonic Trapping. ACS Photonics 2014, 1, 389–393. 10.1021/ph5000314. [DOI] [Google Scholar]
- Ament I.; Prasad J.; Henkel A.; Schmachtel S.; Sönnichsen C. Single Unlabeled Protein Detection on Individual Plasmonic Nanoparticles. Nano Lett. 2012, 12, 1092–1095. 10.1021/nl204496g. [DOI] [PubMed] [Google Scholar]
- Zijlstra P.; Paulo P. M.; Orrit M. Optical Detection of Single Non-Absorbing Molecules Using the Surface Plasmon Resonance of a Gold Nanorod. Nat. Nanotechnol. 2012, 7, 379. 10.1038/nnano.2012.51. [DOI] [PubMed] [Google Scholar]
- Beuwer M. A.; Prins M. W.; Zijlstra P. Stochastic Protein Interactions Monitored by Hundreds of Single-Molecule Plasmonic Biosensors. Nano Lett. 2015, 15, 3507–3511. 10.1021/acs.nanolett.5b00872. [DOI] [PubMed] [Google Scholar]
- Ertsgaard C. T.; Wittenberg N. J.; Klemme D. J.; Barik A.; Shih W.-C.; Oh S.-H. Integrated Nanogap Platform for Sub-Volt Dielectrophoretic Trapping and Real-Time Raman Imaging of Biological Nanoparticles. Nano Lett. 2018, 18, 5946–5953. 10.1021/acs.nanolett.8b02654. [DOI] [PubMed] [Google Scholar]
- Chen X.; Lindquist N. C.; Klemme D. J.; Nagpal P.; Norris D. J.; Oh S.-H. Split-Wedge Antennas with Sub-5 Nm Gaps for Plasmonic Nanofocusing. Nano Lett. 2016, 16, 7849–7856. 10.1021/acs.nanolett.6b04113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A. B.; Zijlstra P. Single-Molecule Plasmon Sensing: Current Status and Future Prospects. ACS Sensors 2017, 2, 1103–1122. 10.1021/acssensors.7b00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackie E. J.; Le Ru E. C.; Etchegoin P. G. Single-Molecule Surface-Enhanced Raman Spectroscopy of Nonresonant Molecules. J. Am. Chem. Soc. 2009, 131, 14466–14472. 10.1021/ja905319w. [DOI] [PubMed] [Google Scholar]
- Le Ru E. C.; Etchegoin P. G. Single-Molecule Surface-Enhanced Raman Spectroscopy. Annu. Rev. Phys. Chem. 2012, 63, 65–87. 10.1146/annurev-physchem-032511-143757. [DOI] [PubMed] [Google Scholar]
- Eftekhari F.; Escobedo C.; Ferreira J.; Duan X.; Girotto E. M.; Brolo A. G.; Gordon R.; Sinton D. Nanoholes as Nanochannels: Flow-through Plasmonic Sensing. Anal. Chem. 2009, 81, 4308–4311. 10.1021/ac900221y. [DOI] [PubMed] [Google Scholar]
- Jonsson M. P.; Dahlin A. B.; Feuz L.; Petronis S.; Höök F. Locally Functionalized Short-Range Ordered Nanoplasmonic Pores for Bioanalytical Sensing. Anal. Chem. 2010, 82, 2087–2094. 10.1021/ac902925e. [DOI] [PubMed] [Google Scholar]
- Yanik A. A.; Huang M.; Artar A.; Chang T.-Y.; Altug H. Integrated Nanoplasmonic-Nanofluidic Biosensors with Targeted Delivery of Analytes. Appl. Phys. Lett. 2010, 96, 021101 10.1063/1.3290633. [DOI] [Google Scholar]
- Van Dorp S.; Keyser U. F.; Dekker N. H.; Dekker C.; Lemay S. G. Origin of the Electrophoretic Force on DNA in Solid-State Nanopores. Nat. Phys. 2009, 5, 347. 10.1038/nphys1230. [DOI] [Google Scholar]
- Chen K.; Juhasz M.; Gularek F.; Weinhold E.; Tian Y.; Keyser U. F.; Bell N. A. Ionic Current-Based Mapping of Short Sequence Motifs in Single DNA Molecules Using Solid-State Nanopores. Nano Lett. 2017, 17, 5199–5205. 10.1021/acs.nanolett.7b01009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waduge P.; Hu R.; Bandarkar P.; Yamazaki H.; Cressiot B.; Zhao Q.; Whitford P. C.; Wanunu M. Nanopore-Based Measurements of Protein Size, Fluctuations, and Conformational Changes. ACS Nano 2017, 11, 5706–5716. 10.1021/acsnano.7b01212. [DOI] [PubMed] [Google Scholar]
- Assad O. N.; Gilboa T.; Spitzberg J.; Juhasz M.; Weinhold E.; Meller A. Light-Enhancing Plasmonic-Nanopore Biosensor for Superior Single-Molecule Detection. Adv. Mater. 2017, 29, 1605442. 10.1002/adma.201605442. [DOI] [PubMed] [Google Scholar]
- Gilboa T.; Meller A. Optical Sensing and Analyte Manipulation in Solid-State Nanopores. Analyst 2015, 140, 4733–4747. 10.1039/C4AN02388A. [DOI] [PubMed] [Google Scholar]
- Ivankin A.; Henley R. Y.; Larkin J.; Carson S.; Toscano M. L.; Wanunu M. Label-Free Optical Detection of Biomolecular Translocation through Nanopore Arrays. ACS Nano 2014, 8, 10774–10781. 10.1021/nn504551d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulizzi F. Building a Better Nanopore. Nat. Nanotechnol. 2016, 11, 105–105. 10.1038/nnano.2016.15. [DOI] [PubMed] [Google Scholar]
- Verschueren D.; Pud S.; Shi X.; De Angelis L.; Kuipers K.; Dekker C.. Label-Free Optical Detection of DNA Translocations through Plasmonic Nanopores, in progress. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X.; Verschueren D.; Pud S.; Dekker C. Integrating Sub-3 Nm Plasmonic Gaps into Solid-State Nanopores. Small 2018, 14, 1703307. 10.1002/smll.201703307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson M. P.; Dekker C. Plasmonic Nanopore for Electrical Profiling of Optical Intensity Landscapes. Nano Lett. 2013, 13, 1029–1033. 10.1021/nl304213s. [DOI] [PubMed] [Google Scholar]
- Nicoli F.; Verschueren D.; Klein M.; Dekker C.; Jonsson M. P. DNA Translocations through Solid-State Plasmonic Nanopores. Nano Lett. 2014, 14, 6917–6925. 10.1021/nl503034j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschueren D. V.; Jonsson M. P.; Dekker C. Temperature Dependence of DNA Translocations through Solid-State Nanopores. Nanotechnology 2015, 26, 234004. 10.1088/0957-4484/26/23/234004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkin M.; Chao S.-H.; Jonsson M. P.; Dekker C.; Aksimentiev A. Plasmonic Nanopores for Trapping, Controlling Displacement, and Sequencing of DNA. ACS Nano 2015, 9, 10598–10611. 10.1021/acsnano.5b04173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikkaraddy R.; Turek V.; Kongsuwan N.; Benz F.; Carnegie C.; Van De Goor T.; de Nijs B.; Demetriadou A.; Hess O.; Keyser U. F.; et al. Mapping Nanoscale Hotspots with Single-Molecule Emitters Assembled into Plasmonic Nanocavities Using DNA Origami. Nano Lett. 2018, 18, 405–411. 10.1021/acs.nanolett.7b04283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Giessen H.; Lalanne P. Simple Analytical Expression for the Peak-Frequency Shifts of Plasmonic Resonances for Sensing. Nano Lett. 2015, 15, 3439–3444. 10.1021/acs.nanolett.5b00771. [DOI] [PubMed] [Google Scholar]
- Storm A. J.; Chen J.; Zandbergen H.; Dekker C. Translocation of Double-Strand DNA through a Silicon Oxide Nanopore. Phys. Rev. E 2005, 71, 051903 10.1103/PhysRevE.71.051903. [DOI] [PubMed] [Google Scholar]
- Carlsen A. T.; Zahid O. K.; Ruzicka J.; Taylor E. W.; Hall A. R. Interpreting the Conductance Blockades of DNA Translocations through Solid-State Nanopores. ACS Nano 2014, 8, 4754–4760. 10.1021/nn501694n. [DOI] [PubMed] [Google Scholar]
- Keyser U.; Van der Does J.; Dekker C.; Dekker N. Optical Tweezers for Force Measurements on DNA in Nanopores. Rev. Sci. Instrum. 2006, 77, 105105. 10.1063/1.2358705. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.