

Abstract
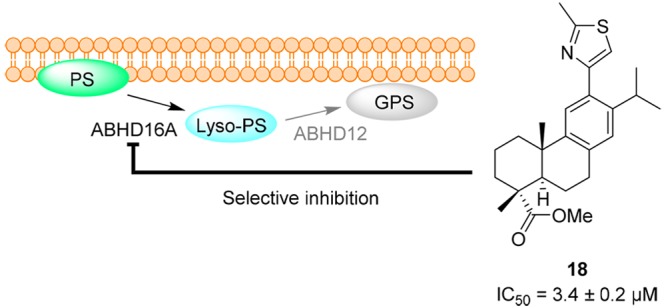
Screening of an in-house library of compounds identified 12-thiazole abietanes as a new class of reversible inhibitors of the human metabolic serine hydrolase. Further optimization of the first hit compound lead to the 2-methylthiazole derivative 18, with an IC50 value of 3.4 ± 0.2 μM and promising selectivity. ABHD16A has been highlighted as a new target for inflammation-mediated pain, although selective inhibitors of hABHD16A (human ABHD16A) have not yet been reported. Our study presents abietane-type diterpenoids as an attractive starting point for the design of selective ABHD16A inhibitors, which will contribute toward understanding the significance of hABHD16A inhibition in vivo.
Keywords: hABHD16A inhibitor, metabolic serine hydrolase, lysophosphatidylserine, dehydroabietic acid
Inflammation of tissue within the peripheral and central nervous system, known as neuroinflammation, has been implicated in the development of chronic pain via sensitization of nociceptive neurons.1,2 Therefore, identifying and targeting the processes and molecules involved in neuroinflammation is regarded as an effective strategy for innovative chronic pain treatments. In this regard, the metabolic serine hydrolase ABHD16A, also known as BAT5, belonging to the ABHD (α,β-hydrolase domain) enzyme family, is a potentially novel key target in inflammation-mediated pain.3,4 This enzyme is mostly expressed in the brain, muscle, heart, and testis, where its activity is closely coupled with that of ABHD12 to regulate the levels of pro-inflammatory lysophosphatidylserine (lyso-PS).3,5 More specifically, ABHD16A converts phosphatidylserine (PS) to lyso-PS, which is either recycled back to PS or further hydrolyzed by ABHD12 to glycerophosphoserine. Accordingly, deletion of ABHD12 results in high levels of lyso-PS in the brain and microglial activation, whereas pharmacological or genetic perturbation of ABHD16A decreases the levels of lyso-PS and ameliorates inflammation. It is therefore desirable to block the activity of ABHD16A without concomitant inhibition of ABHD12.
ABHD16A is a membrane-bound enzyme and in the absence of a crystal structure, the search for inhibitors has been based on competitive activity-based protein profiling (ABPP).3,6 Palmostatin B, tetrahydrolipstatin (THL), 1,3,4-oxadiazol-2(3H)-ones, and KC01 have all been reported to inhibit ABHD16A, some with nanomolar potency, however, the selectivity is still poor (Figure 1).3,6,7
Figure 1.

Examples of known ABHD16A inhibitors and their reported selectivity. aRP = Relative potency ratio related to THL expressed as IC50 (THL)/IC50 (compound), under the same assay conditions.
Previously, we have shown that pentacyclic triterpenoids including maslinic acid bear unprecedented selectivity for inhibition of ABHD12 over other serine hydrolases, as well as over cannabinoid receptors.8 A ligand-based pharmacophore model highlighted the planar hydrophobic hydrocarbon core among the relevant pharmacophoric features. Compounds in this class of triterpenoids, however, have high molecular weights and poor aqueous solubility, which limits their use in biological assays and further development into drug leads. Smaller molecular weight diterpenoids of the abietane family are much more interesting from this perspective especially because a few, including methyl carnosate and miltirone, cross the blood–brain barrier and bear analgesic properties.9
When we screened an in-house library of 50 abietanes for their ability to inhibit hydrolase activity in lysates of HEK293 cells transiently overexpressing human ABHD16A,6 at 10 μM, ruling out concomitant inhibition of human ABHD12,7 the 2-aminothiazole 5 (Scheme 1) was the only compound showing moderate inhibition of hABHD16A without interfering with hABHD12 (Table 1). Its synthesis proceeded from methyl dehydroabietate 2, which gave 3 by Fridel–Crafts acylation (Scheme 1),10 followed by bromination of the carbon alpha to the carbonyl group with CuBr2, in refluxing MeOH.11 The reaction resulted in a mixture of mono- and dibromo compounds 4a and 4b, which was used without further purification in the reaction with thiourea to give 5, in 40% yield, over two steps.
Scheme 1. Synthesis of 12-Thiazole Abietanes 5–8 and 11–14 from Dehydroabietic Acid 1.

Reagents and conditions: (a) MeI, K2CO3, DMF, rt, 2.5 h; (b) MeCOCl, AlCl3, CH2Cl2, 0 °C → rt, 3.5 h; (c) CuBr2, MeOH, 65 °C, 16 h; (d) thiourea, Et3N, dry EtOH, reflux, 1 h 45 min, or 120 °C with microwaves, 30 min; (e) thioformamide, dry 1,4-dioxane, 100 °C with microwaves, 10 min; (f) LiAlH4, THF, 0 °C → rt; (g) KOH, ethylene glycol, H2O, 130 °C; (h) HOBt, EDC-HCl, aq NH3, DMF, 0 °C → rt, 19 h.
Table 1. Remaining Hydrolase Activity for hABHD12 and hABHD16A (% Control) at 10 μM Concentration.
| compd | hABHD16A (% control) mean ± SD, n = 2–3 | hABHD12 (% control) mean ± SD, n = 2–3 |
|---|---|---|
| 5 | 58.2 ± 18.1 | 102.6 ± 2.3 |
| 6 | 50.3 ± 4.6 | 91.5 ± 0.4 |
| 7 | 74.6 ± 7.5 | 87.8 ± 0.8 |
| 8 | 65.9 ± 2.8 | 96.3 ± 1.0 |
| 11 | 80.0 ± 5.6 | 96.9 ± 0.8 |
| 12 | 82.1 ± 4.3 | 98.7 ± 0.2 |
| 13 | 91.5 ± 2.8 | 98.4 ± 0.2 |
| 14 | 62.9 ± 3.4 | 77.8 ± 1.0 |
| 15 | 73.8 ± 4.7 | 89.8 ± 0.1 |
| 16 | 86.6 ± 0.8 | 100.9 ± 4.1 |
| 17 | 99.3 ± 2.4 | 96.6 ± 8.8 |
| 18 | 23.0 ± 16.3 | 100.1 ± 1.1 |
| 19 | 48.7 ± 0.5 | 73.8 ± 3.2 |
| 20 | 42.8 ± 4.1 | 94.3 ± 0.7 |
| 21 | 66.3 ± 5.9 | 98.7 ± 2.2 |
| 22 | 98.4 ± 0.8 | 101.9 ± 0.0 |
| 23 | 80.6 ± 1.8 | 99.6 ± 1.0 |
| 24 | 82.7 ± 4.2 | 92.8 ± 1.2 |
| 25 | 81.5 ± 4.9 | 91.1 ± 1.7 |
| 26 | 80.1 ± 1.6 | 91.1 ± 2.9 |
In the light of the well-known canonical esterase mechanism for α/β-hydrolase fold enzymes,12 we reasoned that the ring A ester in 5 would likely account for the observed activity. In addition, aminothiazoles are generally known to be promiscuous assay-interfering compounds,13 so it was important to determine whether the 2-amino substituent was relevant for the observed activity. The ester was first modified by reduction14 to alcohol 7 or replaced with the amide 11, as depicted on Scheme 1. A thiazole derivative 6, devoid of a 2-amino substituent, was prepared by reacting 4 and thioformamide under microwave conditions, in 53% yield.
Modification of the ring A ester resulted in modest inhibition of hABHD16A in 7 and 11, whereas 6 inhibited the enzyme slightly more efficiently than 5. A new round of modifications of the ring A ester in 6 gave the amide 12 via a synthetic route similar to that of 11, and the carboxylic acid 13, from the alkaline hydrolysis of 6 (Scheme 1). Enzyme inhibition was almost abrogated with the amide 12 and the carboxylic acid 13. However, it was retained by the alcohol 8, even if less pronounced when compared to the ester 6 (Table 1).
Interestingly, 14, which was the main product obtained in the hydrolysis reaction of 6, showed inhibition of hABHD16A comparable to that of the alcohol 8, however, with weaker selectivity, as approximately 20% inhibition of ABHD12 was observed. Overall, these results suggested that both the thiazole ring and the substituent at ring A were relevant for the activity with the 2-amino substituent in the heterocycle not being an essential feature, and ring A substitution being flexible between an ester and an alcohol, the ester performing better in terms of potency and selectivity.
We proceeded to study what substituents were tolerated at position 2 of the thiazole ring. We synthesized the bulky phenyl and chlorophenyl derivatives 15–17 with the methyl ester at C18, according to Scheme 2. The chlorophenyl compounds 16 and 17 did not inhibit hABHD16A, and 15 was a modest inhibitor.
Scheme 2. Variation of the Position 2 of the Thiazole Ring.

Reagents and conditions: (a) respective thioamide, dry EtOH, 120 °C with microwaves; (b) 7 M NH3 in MeOH, rt, 2 d; (c) Et3N, TFAA, THF, 0 °C → rt, 3 h; (d) LiAlH4, THF, 0 °C → rt, 2 h; (e) KOH, ethylene glycol, H2O, 130 °C, 1 d; (f) HOBt, EDC-HCl, aq NH3, DMF, 0 °C → rt, 22 h.
Introduction of the less bulky methyl group at position 2 of the thiazole ring resulted in 18, the most active compound in the set with approximately 80% inhibition of the activity of hABHD16A, and no inhibition of hABHD12 (Scheme 2). We again ruled out the effect of having the methyl ester on ring A by synthesizing the carboxylic acid and amide derivatives 22 and 23, respectively. Compound 23 was synthesized following activation of 22 with hydroxybenzotriazole (HOBt) and N-(3-(dimethylamino)propyl-N′-ethylcarbodiimide hydrochloride (EDC-HCl), and further reaction with ammonia, in 77% yield. As before, inhibition of hABH16A was almost abrogated in the carboxylic acid and amide derivatives, whereas the alcohol 21 was half as potent as the parent ester (Table 1). The synthesis of 20, where the methyl substituent was replaced by an electron-withdrawing nitrile group, proceeded from 4 and ethyl thiooxamate and continued with the hydrolysis of 19, with NH3 in MeOH to give an intermediate amide, which was further converted to the nitrile 20 using trifluoroacetic anhydride and triethylamine in tetrahydrofuran, in 46% yield.15 Both 19 and 20 inhibited the activity of hABDH16A by approximately 60%, with 20 being selective. Altogether, we concluded that the bulkiness of the substituent at position 2 of the thiazole ring seems to be more relevant for the activity than its electron donating or withdrawing ability.
The synthesis of the thiazolium salts 24–26, where the ring nitrogen is no longer available for hydrogen bonding and is in addition more prone to nucleophilic attack, was attempted next (Scheme 3). Compound 18 and iodomethane or iodoethane, respectively, were refluxed in acetone to give 24 and 25, in 73% and 11% yield, respectively.16 Compound 26 was obtained similarly from 6 and iodomethane in 65% yield and none of the thiazolium derivatives inhibited the activity of hABHD16A significantly.
Scheme 3. Synthesis of the Thiazolium Salts 24–26.

Reagents and conditions: (a) MeI or EtI, acetone, 56 °C.
We characterized in more detail the two most promising hABHD16A inhibitors of the series, 18 and 20. The dose–responses were determined using four concentrations (0.1–100 μM) of the compounds (Figure 2A). For reference purposes, the potent hABHD16A inhibitor palmostatin B was tested at 0.01–10 μM concentrations and in the present experiments it inhibited hABHD16A with an IC50 value of 99 ± 12 nM (mean ± SD, n = 2) in close agreement with our previous findings.6 For 18 and 20, we determined the IC50 values (mean ± SD, n = 2) of 3.4 ± 0.2 and 5.2 ± 0.3 μM, respectively. It is noteworthy that in contrast to palmostatin B which comprehensively inhibited hABHD16A in these assays, 18 and 20 could not fully inhibit hABHD16A activity but 12 ± 1% and 25 ± 1% residual activity remained at 100 μM concentration, respectively.
Figure 2.

Biological characterization of 18 and 20 in comparison with palmostatin B. (A) Dose–response curves for palmostatin B, 18 and 20 to inhibit 1-LG hydrolysis in lysates of hABHD16A-HEK cells. Lysates were pretreated for 30 min with DMSO (control) or with the indicated concentrations of the inhibitors before adding the substrate (25 μM final concentration). Glycerol liberated from 1-LG hydrolysis was determined as previously described.6 Note that in contrast to 10 μM palmostatin B, which comprehensively blocks 1-LG hydrolysis, ∼20% and ∼30% residual activity remains at 100 μM concentration of 18 and 20, respectively. Values are mean ± SD of duplicate wells from two independent experiments. (B–D) Reversible nature of hABHD16A inhibition by 18 and 20. Fast 40-fold dilution of inhibitor-treated hABHD16A-HEK293 lysate preparation in the reversibility assay results in notable drop of the diterpene inhibitor potency, as compared to potency values obtained using the routine assay. In contrast, the potency for the β-lactone palmostatin B remains similar during the time-course of this study. Data are mean ± SD from two independent experiments. (E) Competitive ABPP using rat cerebellar membrane proteome reveals ABHD16A as the sole serine hydrolase targeted by 18 and 20. THL (10 μM), palmostatin B (10 μM), and compound 44 (1 μM) were used as positive controls to identify ABHD16A and in line with our previous study,6 all three blocked TAMRA-FP labeling of a band migrating at ∼63 kDa, corresponding to ABHD16A. Compound 18 dose-dependently inhibits TAMRA-FP labeling of ABHD16A, whereas 20 was marginally effective only at the higher concentration. Note that in contrast to THL and palmostatin B, no additional targets are evident for 18 or 20 among the metabolic serine hydrolases. The gel is representative of two independent ABPP runs with similar outcome. FAAH, fatty acid amide hydrolase; LYPLA1/2, lysophospholipase A1/A2.
To test whether 18 and 20 reversibly inhibit hABHD16A, we compared inhibitor potency obtained in the routine assays to that obtained following rapid, 40-fold dilution of the enzyme–inhibitor complex (Figure 2B–D).8 As the compounds showed micromolar potency values in the routine assay, we extended the inhibitor concentration range up to 1 mM concentration in the dilution assays. A notable drop in inhibitor potency was evident for 18 (11.4-fold) and 20 (6.4-fold), obviously due to dissociation of the enzyme–inhibitor complex following the dilution step. In contrast, the IC50 values for the reference compound palmostatin B remained similar (1.8-fold change) between the two assay formats, indicating that within the time frame of these studies, the β-lactone irreversibly inhibited hABHD16A.
Next we probed selectivity of 18 and 20 among the serine hydrolase in native rat brain membrane proteome. Competitive ABPP was used to evaluate the hABHD16A selectivity of 18 and 20 among the serine hydrolases in rat cerebellar membrane proteomes. We chose this proteome for these studies, as a recent study indicated that in the rodent brain, hABHD16A activity as revealed by the ABPP approach is higher in the cerebellum as compared to cortex and hippocampus.3 In competitive ABPP, binding of an inhibitor masks the enzyme’s active site, thereby inhibiting subsequent labeling with the active-site targeting probe. For reference purposes, the previously characterized hABHD16A inhibitors THL, palmostatin B, and compound 44(6) were included.
In line with previous findings,6 these experiments indicated that the reference inhibitors totally blocked activity probe binding to hABHD16A at the used concentrations (Figure 2E). As previously shown, palmostatin B also inhibited probe labeling of several additional serine hydrolases, most notably LYPLA1/2 in this proteome. As 18 and 20 showed reversible mode of inhibition in the glycerol-based hydrolase assays, these compounds were tested at 20 and 200 μM concentrations mimicking the approach that was successfully used to reveal serine hydrolase targets for reversible inhibitors of ABHD12.8 Interestingly, hABHD16A was the only apparent target of 18 as probe labeling of this particular serine hydrolase was dose-dependently inhibited by the two concentrations of 18. In line with less potent behavior in substrate-based activity assays, 20 targeted hABHD16A less efficiently, with clearly visible effect only at 200 μM concentration. Interestingly, incomplete inhibition of ABHD16A was also evident in ABPP experiments using hABHD16A-HEK lysates (Supporting Information, Figure S1), raising the possibility that the observed incomplete inhibitory activity could derive from an allosteric rather than active site mechanism of binding for these abietanes.
In summary, herein we have identified 12-thiazole abietanes as a new class of reversible inhibitors of hABHD16A in vitro and detailed key structure–activity relationships for enzyme inhibition. The good selectivity of 18 for hABHD16A among other serine hydrolyses warrants further investigations into this class of compounds in search of more potent and equally selective derivatives that will surely greatly contribute toward translating the observed in vitro effects to an in vivo context, thereby establishing the significance of inhibiting this enzyme in neuroinflammation and inflammatory-mediated pain.
Acknowledgments
We acknowledge Taina Vihavainen and Juha Niskanen (University of Eastern Finland) for conducting the hydrolase assays and Adjunct Prof. Mikko Airavaara (Institute of Biotechnology, HiLife Unit, University of Helsinki) for revising the manuscript.
Glossary
Abbreviations Used
- ABHD
α,β-hydrolase domain
- hABHD16A
human ABHD16A
- hABHD12
human ABHD12
- PS
phosphatidylserine
- ABPP
activity-based protein profiling
- THL
tetrahydrolipstatin
- 1-LG
1-linoleylglycerol
- FAAH
fatty acid amide hydrolase
- LYPL
lysophospholipase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00442.
Synthetic procedures, Figure S1, analytical data, assay protocols (PDF)
Author Contributions
T.J.A. designed, synthesized, and characterized the compounds with the support of V.M.M. and J.Y.-K. J.R.S. and J.T.L. performed the biological evaluation. The manuscript was written through contributions of all authors.
The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no 602919. J.T.L. acknowledges the Academy of Finland (decision 278212) for funding support.
The authors declare no competing financial interest.
Supplementary Material
References
- Gao Y.-J.; Ji R.-R. Chemokines, Neuronal–glial Interactions, and Central Processing of Neuropathic Pain. Pharmacol. Ther. 2010, 126 (1), 56–68. 10.1016/j.pharmthera.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y.; Zhang L.; Cheng J.-K.; Ji R.-R. Cytokine Mechanisms of Central Sensitization: Distinct and Overlapping Role of Interleukin-1β, Interleukin-6, and Tumor Necrosis Factor-α in Regulating Synaptic and Neuronal Activity in the Superficial Spinal Cord. J. Neurosci. 2008, 28, 5189–5194. 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat S. S.; Camara K.; Parsons W. H.; Chen D. H.; Dix M. M.; Bird T. D.; Howell A. R.; Cravatt B. F. Immunomodulatory Lysophosphatidylserines Are Regulated by ABHD16A and ABHD12 Interplay. Nat. Chem. Biol. 2015, 11 (2), 164–171. 10.1038/nchembio.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. Y. Phospholipids: A Neuroinflammation Emerging Target. Nat. Chem. Biol. 2015, 11 (2), 99–100. 10.1038/nchembio.1740. [DOI] [PubMed] [Google Scholar]
- Blankman J. L.; Long J. Z.; Trauger S. A.; Siuzdak G.; Cravatt B. F. ABHD12 Controls Brain Lysophosphatidylserine Pathways That Are Deregulated in a Murine Model of the Neurodegenerative Disease PHARC. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 1500–1505. 10.1073/pnas.1217121110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savinainen J. R.; Patel J. Z.; Parkkari T.; Navia-Paldanius D.; Marjamaa J. J. T.; Laitinen T.; Nevalainen T.; Laitinen J. T. Biochemical and Pharmacological Characterization of the Human Lymphocyte Antigen B-Associated Transcript 5 (BAT5/ABHD16A). PLoS One 2014, 9, e109869. 10.1371/journal.pone.0109869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navia-Paldanius D.; Savinainen J. R.; Laitinen J. T. Biochemical and Pharmacological Characterization of Human α/β-Hydrolase Domain Containing 6 (ABHD6) and 12 (ABHD12). J. Lipid Res. 2012, 53 (11), 2413–2424. 10.1194/jlr.M030411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkkari T.; Haavikko R.; Laitinen T.; Navia-Paldanius D.; Rytilahti R.; Vaara M.; Lehtonen M.; Alakurtti S.; Yli-Kauhaluoma J.; Nevalainen T.; Savinainen J. R.; Laitinen J. T. Discovery of Triterpenoids as Reversible Inhibitors of α/β-Hydrolase Domain Containing 12 (ABHD12). PLoS One 2014, 9 (5), e98286 10.1371/journal.pone.0098286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hügel H. M.; Jackson N. Danshen Diversity Defeating Dementia. Bioorg. Med. Chem. Lett. 2014, 24 (3), 708–716. 10.1016/j.bmcl.2013.12.042. [DOI] [PubMed] [Google Scholar]
- Kolsi L. E.; Krogerus S.; Brito V.; Rüffer T.; Lang H.; Yli-Kauhaluoma J.; Silvestre S. M.; Moreira V. M. Regioselective Benzylic Oxidation of Aromatic Abietanes: Application to the Semisynthesis of the Naturally Occurring Picealactones A, B and C. ChemistrySelect 2017, 2 (24), 7008–7012. 10.1002/slct.201701477. [DOI] [Google Scholar]
- Burkhart J. P.; Gates C. A.; Laughlin M. E.; Resvick M. E.; Peet N. P. Inhibition of Steroid C17(20)Lyase with C-17-Heteroaryl Steroids. Bioorg. Med. Chem. 1996, 4 (9), 1411–1420. 10.1016/0968-0896(96)00135-6. [DOI] [PubMed] [Google Scholar]
- Rauwerdink A.; Kazlauskas R. J. How the Same Core Catalytic Machinery Catalyzes 17 Different Reactions: The Serine-Histidine-Aspartate Catalytic Triad of α/β-Hydrolase Fold Enzymes. ACS Catal. 2015, 5 (10), 6153–6176. 10.1021/acscatal.5b01539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine S. M.; Mulcair M. D.; Debono C. O.; Leung E. W. W.; Nissink J. W. M.; Lim S. S.; Chandrashekaran I. R.; Vazirani M.; Mohanty B.; Simpson J. S.; Baell J. B.; Scammells P. J.; Norton R. S.; Scanlon M. J. Promiscuous 2-Aminothiazoles (PrATs): A Frequent Hitting Scaffold. J. Med. Chem. 2015, 58 (3), 1205–1214. 10.1021/jm501402x. [DOI] [PubMed] [Google Scholar]
- González M. A.; Correa-Royero J.; Agudelo L.; Mesa A.; Betancur-Galvis L. Synthesis and Biological Evaluation of Abietic Acid Derivatives. Eur. J. Med. Chem. 2009, 44 (6), 2468–2472. 10.1016/j.ejmech.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Dang Q.; Kasibhatla S. R.; Jiang T.; Fan K.; Liu Y.; Taplin F.; Schulz W.; Cashion D. K.; Reddy K. R.; van Poelje P. D.; Fujitaki J. M.; Potter S. C.; Erion M. D. Discovery of Phosphonic Diamide Prodrugs and Their Use for the Oral Delivery of a Series of Fructose 1,6-Bisphosphatase Inhibitors. J. Med. Chem. 2008, 51 (14), 4331–4339. 10.1021/jm8001235. [DOI] [PubMed] [Google Scholar]
- Hojo M.; Ichi T.; Masuda R.; Kobayashi M.; Shibano H. Isolation and Reaction of New Aromatic Dications. 4-Thioniapyridinium Dications: 2-aryl-(or 2-alkyl-)4-methyl-3-oxo-2H-1,4-thiazin-2-ylium Tetrafluoroborates. J. Org. Chem. 1988, 53 (10), 2209–2213. 10.1021/jo00245a016. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.