

Abstract
The redox chemistry of flavoproteins is often gated by substrate and iodotyrosine deiodinase (IYD) has the additional ability to switch between reaction modes based on the substrate. Association of fluorotyrosine (F‐Tyr), an inert substrate analog, stabilizes single electron transfer reactions of IYD that are not observed in the absence of this ligand. The co‐crystal of F‐Tyr and a T239A variant of human IYD have now been characterized to provide a structural basis for control of its flavin reactivity. Coordination of F‐Tyr in the active site of this IYD closely mimics that of iodotyrosine and only minor perturbations are observed after replacement of an active site Thr with Ala. However, loss of the side chain hydroxyl group removes a key hydrogen bond from flavin and suppresses the formation of its semiquinone intermediate. Even substitution of Thr with Ser decreases the midpoint potential of human IYD between its oxidized and semiquinone forms of flavin by almost 80 mV. This decrease does not adversely affect the kinetics of reductive dehalogenation although an analogous Ala variant exhibits a 6.7‐fold decrease in its k cat/K m. Active site ligands lacking the zwitterion of halotyrosine are not able to induce closure of the active site lid that is necessary for promoting single electron transfer and dehalogenation. Under these conditions, a basal two‐electron process dominates catalysis as indicated by preferential reduction of nitrophenol rather than deiodination of iodophenol.
Keywords: Iodotyrosine, halotyrosine, deiodinase, dehalogenase, flavoprotein, redox control, nitroreduction
Short abstract
PDB Code(s): 5yak
Abbreviations
- AQS
anthraquinone‐2‐sulfonate
- Flox
oxidized flavin
- Flsq
semiquinone (one electron reduced) flavin
- Flhq
hydroquinone (two‐electron reduced) flavin
- F‐Tyr
3‐fluoro‐L‐tyrosine
- I2‐Tyr
3,5‐diiodo‐L‐tyrosine
- I‐Tyr
3‐iodo‐L‐tyrosine
- HhIYD
Haliscomenobacter hydrossis IYD
- HsIYD
human iodotyrosine deiodinase
- PSF
phenosafranine
- safranin O
SFO
Introduction
Iodotyrosine deiodinase (IYD) was first discovered by its ability to salvage iodide from iodotyrosine. This activity is crucial for iodide homeostasis in vertebrates and helps to sustain formation of thyroid hormone. IYD also represents a rare example of a reductive dehalogenase that acts under aerobic conditions and requires a flavin cofactor, FMN (Fig. 1).1, 2 Surprisingly, IYD is not limited to organisms that produce thyroid hormone and instead is found in most all metazoa and even certain bacteria and archaea.3, 4 Its broader role in biology has not yet been defined and may alternatively relate to its ability to debrominate and dechlorinate the corresponding halotyrosines5, 6 since a requirement for iodide has not been widely identified beyond vertebrates. A preference for halotyrosine versus other halogenated compounds, however, remains constant for all enzyme homologs characterized to date ranging from human (HsIYD) to bacteria (Haliscomenobacter hydrossis, HhIYD) IYD.7 Additionally, human and Drosophila melanogaster IYD are equally active with both bromo‐ and iodotyrosine6, 8 and, as discovered recently, either one of these substrates may have a role in Drosophila fertility.9
Figure 1.
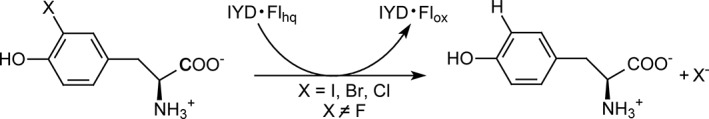
Iodotyrosine deiodinase (IYD) promotes reductive dehalogenation of halotyrosines while oxidizing flavin hydroquinone (Flhq) to flavin (Flox).
IYD represents one subgroup within the very large and ancient nitroreductase superfamily (>20,000 sequences).4 This enzyme is also likely ancient since homologs in mammals, bacteria and archaea all share a similar sequence identity of ~40%.2 Only a very limited number of proteins within this superfamily have yet been characterized and most studied to date rely on their bound flavin cofactor (Fl) to promote hydride transfer, a two‐electron process (Fig. 2). Moreover, the flavin semiquinone intermediate (Flsq) generated by one electron transfer is significantly destabilized in enzymes catalyzing the namesake reaction of this superfamily, reduction of nitro groups.10 In contrast, all mechanistic evidence to date suggests that dehalogenation is catalyzed by a process involving sequential single electron transfer.2 Although redox titration of human and bacteria IYD does not generate observable quantities of Flsq in the absence of an active site ligand, significant concentrations of this species are evident when redox titrations are repeated in the presence of fluorotyrosine (F‐Tyr), an inert substrate analog.7, 11 F‐Tyr is proposed to co‐ordinate Fl and induce closure of an active site lid in analogy to that observed in co‐crystals of I‐Tyr and IYD containing Flox.7, 11, 12, 13 In these structures, the aromatic portion of I‐Tyr stacks over Flox and its zwitterion is chelated by the pyrimidine ring of Flox and a conserved set of active site residues, Tyr, Glu, and Lys. This association stabilizes closure of an active site lid and dramatically changes the environment surrounding Flox. 2‐Iodophenol can also stack over Flox but lacks the functional groups to establish the remaining interactions.7 Consequently, 2‐iodophenol does not induce lid closure nor stabilize a Flsq intermediate and its efficiency of dehalogenation is suppressed by 4 orders of magnitude.7
Figure 2.
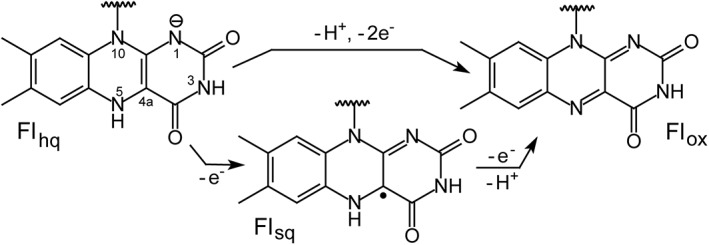
Redox states of flavin (Fl).
Crystal structures of human and mouse IYD suggest that a conserved Thr shifts into a hydrogen bonding range of the Fl N5 in response to binding of a halotyrosine and this change may be responsible for stabilizing Flsq.11, 12 Hydrogen bonding through a side chain hydroxyl group is prevalent in enzymes of the nitroreductase superfamily that appear to stabilize Flsq intermediates. In contrast, most members of this superfamily contain a backbone derived hydrogen bond to Fl N5 and suppress Flsq formation while promoting concurrent transfer of two electrons.2 The expected shift in the active site Thr was not evident in the crystal structure of HhIYD but still likely mediates its redox chemistry since substitution of the Thr with Ala destabilized its Flsq.14 This result was apparent from the lack of Flsq observed during reduction of the HhIYD variant in the presence of F‐Tyr and by its gain of nitroreductase activity that seems diagnostic of hydride transfer in this superfamily. The structural and chemical consequences of substituting this key Thr have now been examined in detail using HsIYD as described in this article. A co‐crystal with F‐Tyr confirms the relevance of this ligand as a substrate mimic in redox studies and finally, the significance of repositioning the active site Thr is demonstrated with WT HsIYD by a diagnostic change in catalytic specificity.
Results and discussion
Co‐crystal structure of F‐Tyr and HsIYD containing an Ala for Thr substitution (T239A)
IYD like most all other representatives of the nitroreductase superfamily exists as an α2‐homodimer in which two identical active sites are formed along the subunit interface. The core architecture is highly conserved throughout the superfamily and its diversity is primarily established by the presence or absence of inserts at three distinct locations that establish loop structures surrounding the active site.4 One such loop is present in IYD and used for binding and covering the active site [E1 in Fig. 3(A)]. The co‐crystal structure of F‐Tyr and the T329A variant of HsIYD was determined for the dual purpose of identifying possible perturbations caused by loss of the key Thr and binding of the inert substrate analog F‐Tyr. Characterization of the redox properties has required the use of F‐Tyr rather than I‐Tyr to prevent turnover of the Flhq‐containing enzyme. The structure of T239A HsIYD with F‐Tyr was determined at a resolution of 2.3 Å (PDB code, 5YAK, Table S1) and is illustrated in Figure 3(A). The aromatic region of F‐Tyr stacks over the Flox and its zwitterion interacts with both the E1 loop and Flox in analogy to I‐Tyr in the native enzyme (PDB code, 4TTC).11 The general features of these two structures are nearly equivalent and superpose with an RMSD of 0.23 Å for a 2918 atom comparison (Fig. S1).
Figure 3.
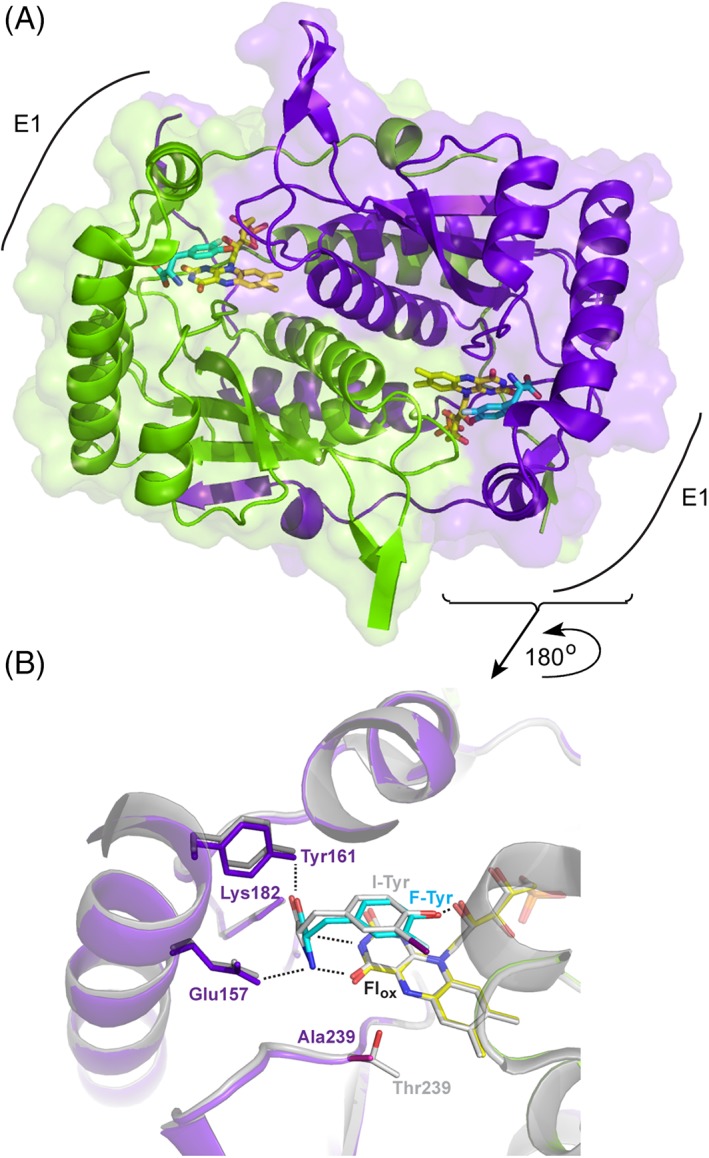
Structure of T239A HsIYD. The two polypeptides of the α2‐homodimer are illustrated in purple and green. Atom type is distinguished by color in the active site bound FMN and the carbon atoms of F‐Tyr are depicted in cyan. (A) The homodimer of the T239A variant of HsIYD containing F‐Tyr (pdb 5YAK). (B) The active site region surrounding F‐Tyr bound to T239A HsIYD is overlaid with the co‐crystal structure of WT HsIYD and I‐Tyr (grey, pdb 4TTC).11
F‐Tyr establishes all of the same polar contacts with the T239A variant identified earlier in the co‐crystal of I‐Tyr and WT HsIYD.11 Similar to I‐Tyr, F‐Tyr likely binds in its phenolate form and is stabilized by co‐ordination to the 2′‐hydroxyl of the Fl's ribityl group.11 Additionally, the zwitterion of F‐Tyr interacts with the same Glu157, Tyr161, and Lys182 as did I‐Tyr [Fig. 3(B)]. Closure of the E1 lid and sequestration of the active site ligand from the solvent is also nearly identical for F‐Tyr and I‐Tyr as evident from their superimposition [Fig. 3(B)]. Residues that typically surround the iodo substituent of I‐Tyr do not significantly condense around the fluoro substituent of F‐Tyr despite its relatively small size and short C–F bond distance. For example, the closest approach of Y212 to the halogen is 4 Å in the structures containing I‐Tyr11 and F‐Tyr. Similarly, distances between the halogen and the proximal α‐carbon of G129 (3.8 Å and 3.5 Å) and the backbone nitrogen of A130 (3.6 Å and 3.4 Å) for I‐Tyr and F‐Tyr, respectively, differ by less 10%. However, the fluoro group of F‐Tyr resides closer to the N5 of Flox by ~0.5 Å relative to the iodo group (3.7 Å vs. 4.3 Å) and conversely, the iodo group resides closer to the β‐carbon of Y211 and indole nitrogen of W169 by ~ 0.5 Å (3.7 Å vs. 4.3 Å and 4.1 Å vs. 4.6 Å, respectively). Overall, F‐Tyr is a reasonable analog of I‐Tyr as characterized by its active site coordination and stabilization of the closed form of loop E1. This validated the further use of F‐Tyr in characterizing the redox chemistry of IYD.11, 14
Replacement of Thr by Ala in the T239A variant has minimal impact on the local structure of IYD relative to that of WT HsIYD with I‐Tyr (Fig. 4). The backbone orientation in the active site remains constant and the amide nitrogen of residue 239 maintains equivalent distances to N5 (3.5 Å) and O4 (2.9 Å) of Flox. All remaining residues that interact with Flox including Ser residues at positions 102 and 128 and Arg residues at positions 100, 101, 104, and 279 superpose with their counterparts in the structure of WT HsIYD. No groups apparently shift to minimize the void created by the loss of a methyl and hydroxyl groups created by the Thr to Ala substitution. Also, no electron density surrounding the Fl N5 was observed during refinement of the crystal structure that might have indicated the presence of water in place of the Thr side chain for the T239A variant (Fig. 5). Such water might not be apparent if it were highly dynamic or disordered but then it would also not likely contribute significantly to bonding with Fl N5. The effects of the Thr to Ala substitution described here for the human HsIYD also likely explain the inability of a bacterial HhIYD to stabilize its Flsq intermediate after equivalent substitution of its corresponding Thr residue with Ala.14 Further consequences of the Ala for Thr substitution in HsIYD are described below.
Figure 4.
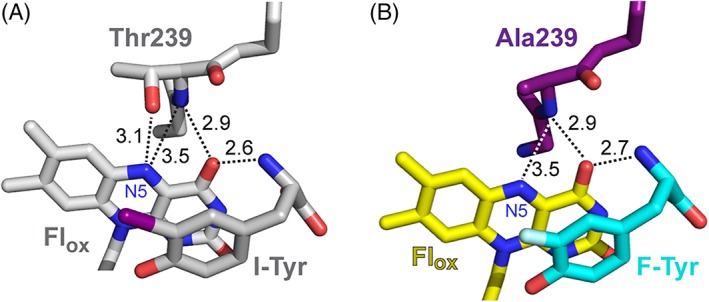
The environment of the N5 position of Flox in the co‐crystals of (A) WT HsIYD with I‐Tyr11 and (B) its T239A variant with F‐Tyr.
Figure 5.
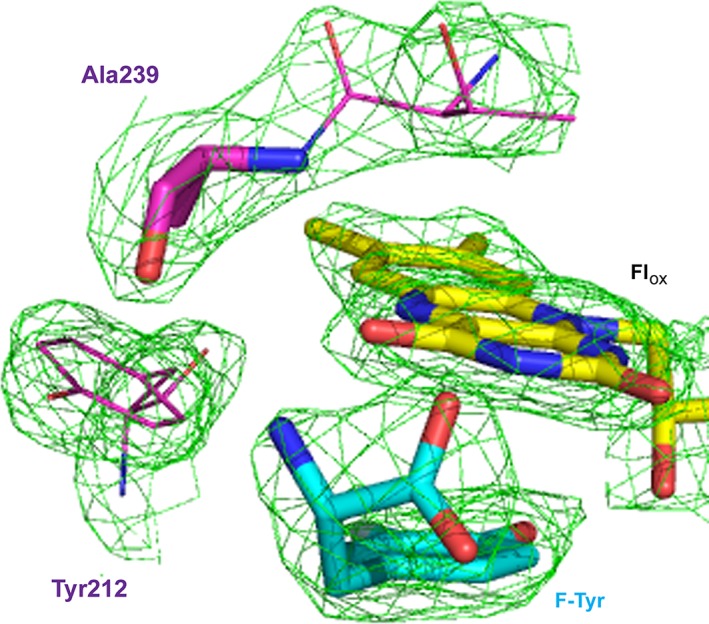
Omit map (2Fo–Fc contoured at 1 σ) of the region between Ala239 and Flox in the co‐crystal of T239A HsIYD and F‐Tyr illustrated in the colors defined in Figure 3.
Altering the redox properties of IYD by substitution of Thr239
From the very first structural studies of IYD, the substrate was expected to modulate the redox chemistry of Fl by significantly altering its surrounding environment.12 However, a change between two and one electron processes was not initially anticipated. In the absence of substrate, the redox active isoalloxazine ring of Fl is highly exposed to solvent and shares only a single polar interaction with the protein created between Arg104 and N1,O2 of Fl.11 In the presence of substrate, exposure becomes extremely limited. The zwitterion of substrate establishes polar interactions with the pyrimidine portion of the isoalloxazine ring and, as described above, a Thr shifts into hydrogen bonding distance of Fl N5. Collectively, these additional interactions stabilize the Flsq intermediate of WT HsIYD (Table 1).11 Hydrogen bonding to the Fl N5 was expected to be particularly important for this stabilization based on precedence from many other flavoproteins that contain hydrogen bond donors or acceptors at this position.15, 16, 17 The significance of this interaction in IYD was first tested by substituting the active site Thr for an Ala in the bacterial HhIYD but its efficiency for dehalogenation (k cat/K m) decreased only 15‐fold relative to WT.14 Another group of enzymes within the nitroreductase superfamily named BluB contains an analogous side chain hydrogen bond from a Ser to Fl N5 and consequently also has the possibility of stabilizing a Flsq intermediate as it reacts with molecular oxygen to form dimethylbenzimidazole.18 Replacement of its active site Ser with Gly suppresses turnover by ~30‐fold.19 However, the effect of these substitutions on the redox potential of Fl had not been reported previously and has now been examined for HsIYD.
Table 1.
Reduction potentials of HsIYD and its T239A and T239S variantsa
| −F‐Tyr | + F‐Tyr | |||
|---|---|---|---|---|
| HsIYD | Flox/hq (mV) | Flox/sq (mV) | Flsq/hq (mV) | |
| WTb | −200 ± 4 | −156 ± 2 | −310 ± 4 | |
| T239S | −206 ± 4 | ~ −234 | ndc | |
| Flox/hq (mV) | ||||
| T239A | −215 ± 4 | −272 ± 4 | ||
Data are relative to the standard hydrogen electrode and represent average E m values based on three independent determinations.
From Hu et al.11
Not determined due to the concurrent presence of Flox, Flsq, and Flhq.
The standard xanthine/xanthine oxidase protocol was used to reduce human HsIYD slowly under anaerobic conditions.20, 21 The T239A variant in the absence of F‐Tyr generated its fully reduced Flhq form without accumulation of the one electron reduced Flsq (Fig. S2) as expected from the prior behavior of WT HsIYD.11 Repetition of this reduction in the presence of a standard dye indicated that the redox potential for this process (Flox/hq) is only slightly lower than that for WT enzyme (Table 1) and these together frame the midpoint potential of free Fl in solution (Flox/hq − 205 mV).22
The T239A variant does not accumulate the Flsq intermediate after addition of F‐Tyr in contrast to the response by WT HsIYD [Fig. 6(A)].11 Thus, the loss of hydrogen bonding to Fl N5 resulting from the T239A substitution significantly destabilizes the one electron reduced intermediate similar to that observed for the bacterial HhIYD.14 As the co‐crystal structure of T239A HsIYD and F‐Tyr illustrates, no other significant perturbations are observed from this amino acid substitution (Fig. 4). While the change in the Fl environment induced by F‐Tyr did not facilitate single electron transfer, the change still had a substantial effect on the redox potential of Fl. The Flox/hq couple decreased by 57 mV in the presence of F‐Tyr (Table 1 and Fig. 7). This is best explained by the π‐stacking that forms between Fl and the electron rich aromatic substrate. Such an effect has been observed with other flavoproteins and ascribed to the unfavorable electrostatics associated with the system's gain of additional electrons during Fl reduction.23, 24 The zwitterion of F‐Tyr could also affect the Flox/hq couple since both the cationic ammonium and anionic carboxylate groups directly coordinate to the pyrimidine portion of the isoalloxazine. However, their influence may be minimized by their opposing abilities to stabilize an increase in electron density resulting from the reduction of Flox.
Figure 6.
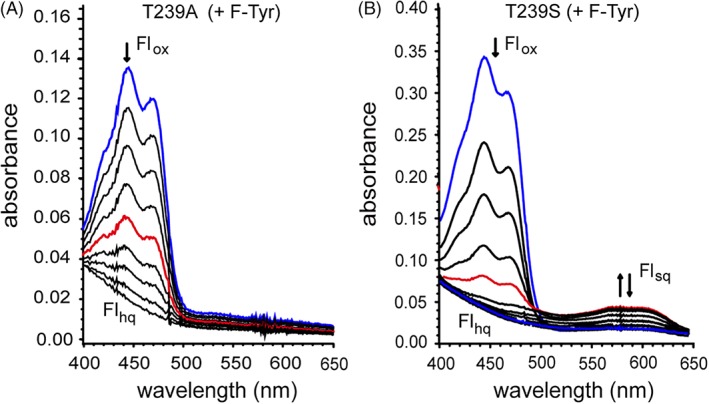
Reduction of (A) the T239A and (B) T239S variants of HsIYD in the presence of F‐Tyr (0.6 μM). IYD samples containing 900 μM xanthine, 2 μM methyl viologen, 200 mM potassium chloride and 100 mM potassium phosphate pH 7.4 were degassed with argon and reduction was initiated by addition of xanthine oxidase (40 μg/ml).20,21 The initial spectra representing Flox are highlighted in blue and the spectra with a maximum A 590 representing Flsq is highlighted in red.
Figure 7.
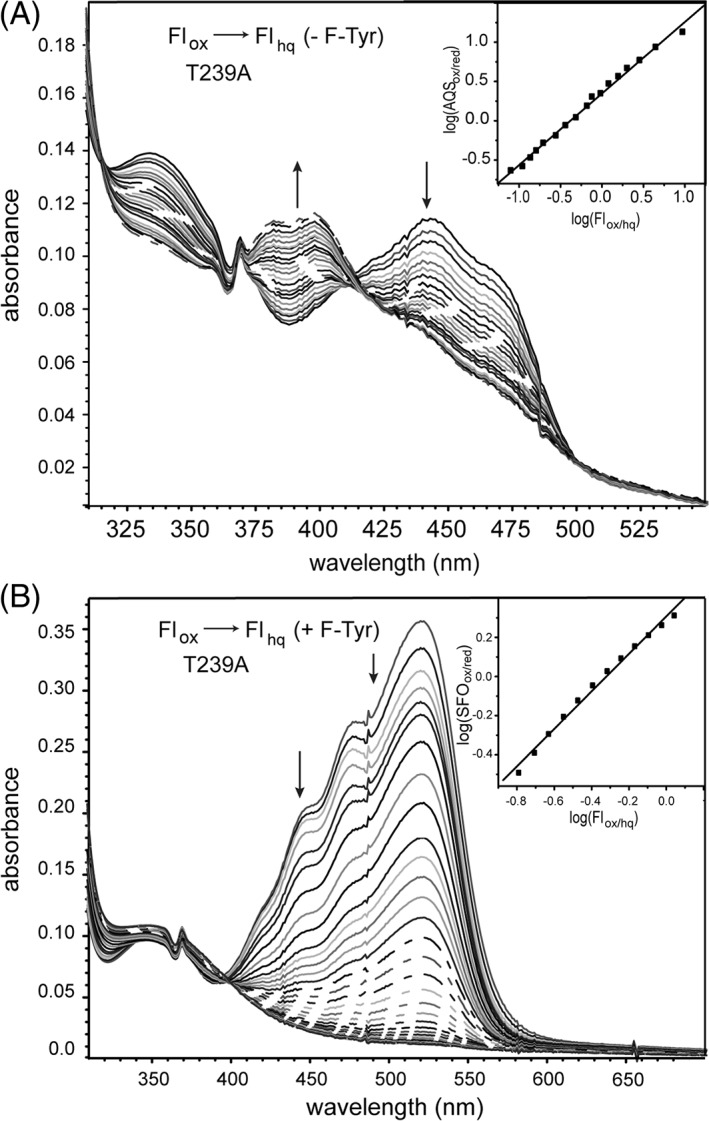
Redox titration of the T239A variant of HsIYD. (A) Enzyme was reduced by xanthine/xanthine oxidase in the presence of the reference dye AQS. Reduction of AQS was monitored at 328 nm, an isosbestic point for Flox/hq in HsIYD T239A, and conversely reduction of Flox was measured at 355 nm, the isosbestic point of AQSox/red. Inset: the linear best fit of log(Flox/Flhq) versus log(AQSox/AQSred) was used to calculate the midpoint potential.20,21 (B) Equivalent analysis was repeated in the presence of F‐Tyr (0.6 mM). Reduction of the standard dye SFO was monitored at 520 nm and compared to the reduction of Flox that was monitored at 445 nm after correcting for the absorbance of SFO. Inset: linear best fit of log(Flox/Flhq) versus log(SFOox/SFOred) was used to calculate the midpoint potential.20,21
A Thr equivalent to Thr239 in HsIYD is highly conserved throughout the IYD subgroup of the nitroreductase superfamily,4 even though Ser has the potential to satisfy the required hydrogen bonding to Fl N5 and is the most common residue for this function in the subgroup BluB.19 A T239S variant of HsIYD was consequently generated to test the stringency for Thr at this position. In the absence of F‐Tyr, the Ser‐containing HsIYD behaved equivalently to all other IYDs and did not accumulate detectable levels of Flsq (Fig. S2). Similarly, T239S HsIYD did not affect the Flox/hq couple and its midpoint potential remained indistinguishable from that of WT HsIYD [Table 1 and Fig. 8(A)]. In the presence of F‐Tyr, Flsq was evident during reduction but did not accumulate to the extent observed for WT HsIYD [Fig. 6(B)].11 This result is consistent with a destabilization of the Flsq intermediate and the ~78 mV decrease in the Flox/sq couple relative to the WT system [Table 1 and Fig. 8(B)]. Further reduction of the T239S variant generated a mixture of Flox, Flsq, and Flhq that complicated quantitative determination of the Flsq/hq potential. Despite the common hydrogen bonding properties of Ser and Thr, these residues are not fully interchangeable in the active site of IYD. Perhaps the additional methyl group of Thr helps to maintain the hydroxyl group in an optimal orientation for the Fl. However, not even the constraints established by the Thr methyl group are always adequate since the crystal structure of bacterial HhIYD revealed distinct rotamer populations for the Thr methyl and hydroxy groups.7 Not all flavoproteins are equivalently sensitive to a similar Ser for Thr substitution. For example, neither the redox properties nor side chain orientation is affected when a Thr responsible for hydrogen bonding to Fl N5 is replaced by Ser in pyranose 2‐oxidase.25
Figure 8.
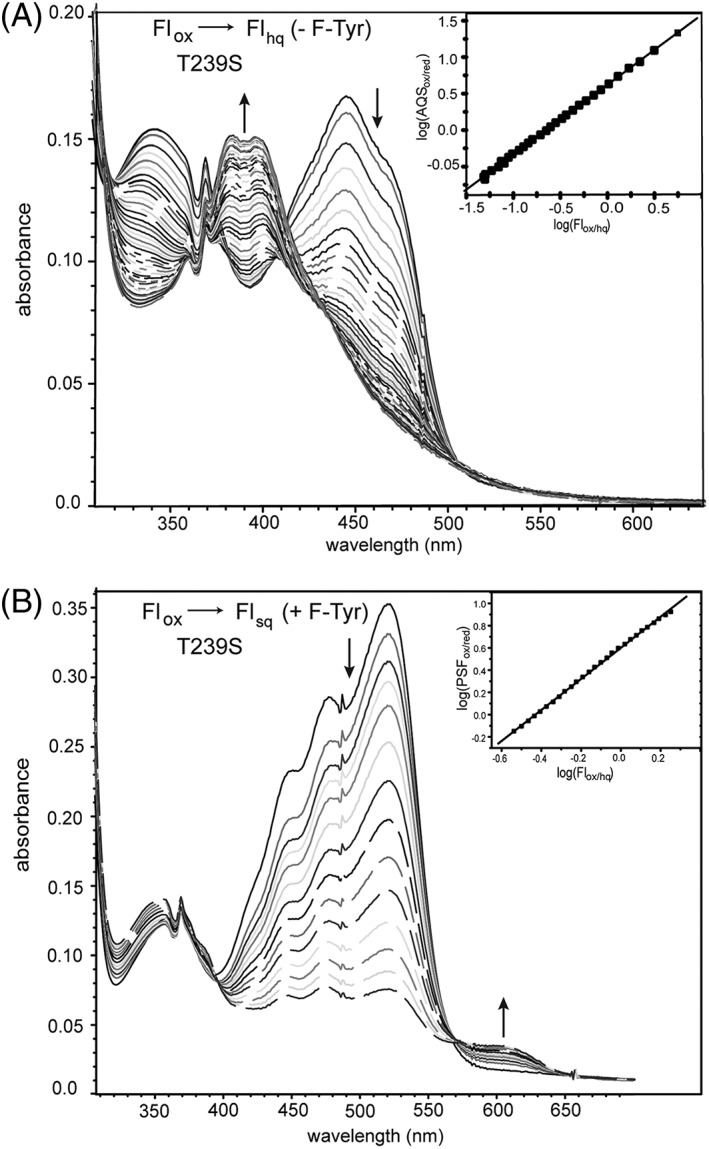
Redox titration of the T239S variant of HsIYD. (A) Enzyme was reduced by xanthine/xanthine oxidase in the presence of the reference dye AQS. Reduction of AQS was monitored at 330 nm, an isosbestic point for Flox/hq in HsIYD T239S, and conversely reduction of Flox was measured at 355 nm, the isosbestic point of AQSox/red. Inset: the linear best fit of log(Flox/Flhq) versus log(AQSox/AQSred) was used to calculate the midpoint potential.20,21 (B) Equivalent analysis was repeated in the presence of F‐Tyr (0.6 mM). Reduction of PSF was monitored at 494 nm, an isosbestic point for Flox/sq and reduction of Flox to Flsq was monitored at 455 nm. Inset: the linear best fit of log(Flox/Flsq) versus log(PSFox/PSFred) was used to calculate the midpoint potential.20,21
Modulating active site binding and catalysis by hydrogen bonding at Fl N5
Substrates of IYD, unlike those of most other flavoproteins, bind directly to Fl and the protein and thus, any perturbations to the Fl environment have the potential to affect halotyrosine binding as well as turnover. However, no change in binding was observed for the T239A variant. Both I‐Tyr and the inert substrate mimic F‐Tyr bound to WT HsIYD and its T239A variant with equal affinity (Fig. S3 and Table 2). Surprisingly, the more conservative substitution of Thr with Ser affected binding more than the change from Thr to Ala. The dissociation constants of F‐Tyr and I‐Tyr increased by a similar two‐fold for the T239S variant relative to those for WT HsIYD (Fig. S4 and Table 2). In all examples to date, F‐Tyr typically binds IYD at least 10‐fold more weakly than chloro‐, bromo‐, and iodotyrosine.5, 7, 8, 11 This decrease in F‐Tyr affinity is consistent with the loss of van der Waals interactions between the halogen and the surrounding protein. Side chains do not appear to pack around the small fluoro substituent and instead remain at a distance that would accommodate the large iodo substituent. The difference in acidity of I‐Tyr and F‐Tyr may also contribute to their relative binding affinities since only the phenolate form is expected to bind IYD.11 However, this could only account for a two‐fold effect on K d since the concentration of the phenolate form of I‐Tyr should be only twice that of F‐Tyr at pH 7.4 based on the greater acidity of 2‐iodophenol relative to 2‐fluorophenol (0.33 pK a units).26 The minimal ability of fluoro groups to participate in halogen bonding is unlikely to affect the affinity for IYD since even the iodo group of I‐Tyr does not participate in this type of interaction from the structures characterized to date.7, 11, 12, 13 A Trp (W169) is proximal to the iodo substituent in the T239A structure (closest approach of 4.6 Å) but neither its π‐system nor its side chain nitrogen is oriented appropriately for halogen bonding.27
Table 2.
Binding and catalytic dehalogenation by HsIYD and its T239A and T239S variants
| K d | Catalytic constantsa | ||||
|---|---|---|---|---|---|
| HsIYD | F‐Tyr (μM) | I‐Tyr (μM) | k cat (min−1) | K m (μM) | k cat/K m (min−1 μM−1) |
| WTb | 1.3 ± 0.4 | 0.15 ± 0.09 | 13 ± 1 | 31 ± 6 | 0.4 ± 0.1 |
| T239S | 3.3 ± 0.5 | 0.37 ± 0.07 | 12 ± 2 | 17 ± 4 | 0.7 ± 0.2 |
| T239A | 1.5 + 0.2 | 0.20 ± 0.05 | 3.4 ± 0.5 | 55 ± 10 | 0.06 ± 0.01 |
Determined by [125I]‐iodide release from I2‐Tyr.
From Hu et al.11
The T239S variant of HsIYD was fully competent at promoting reductive dehalogenation of diiodotyrosine (I2‐Tyr) despite its two‐fold lower affinity for halotyrosines (Table 2). Moreover, the catalytic efficiency of this variant was greater than WT by almost two‐fold resulting from a decrease of its K m value (Fig. S5). Values of k cat for WT and T239S HsIYD remained equivalent despite the lower potential of the Flox/sq couple for the Ser variant. In contrast, catalytic activity was compromised by the loss of the side chain hydroxyl group in the T239A variant. Both k cat and K m decreased (3.8‐ and 1.8‐folds, respectively) for a net loss in efficiency of less than seven‐fold. This effect seems modest for a variant that cannot sustain an observable concentration of Flsq but the ability to accumulate this intermediate and the ability to promote single electron transfer processes are not necessarily synonymous. An analogous substitution of a Ser with Gly in the BluB subgroup of the nitroreductase superfamily also did not abolish its catalytic activity but rather diminished the efficiency by ~30‐fold as noted above.19
A mechanism of reductive dehalogenation involving the sequential transfer of single electrons remains most consistent with all data gathered on IYD to date (Fig. 9).2 This also best explains the inability of the 5‐deaza analog of Fl to support catalysis since this species is restricted to hydride transfer reactions.28, 29 The modest effects of T239A on catalysis suggest that the loss of hydrogen bonding to Fl N5, while important for stabilizing the Flsq intermediate, may not substantially contribute to the rate determining step of reductive dehalogenation. The slow step in turnover has not yet been identified and could involve the closure of the active site lid, substrate protonation or electron transfer. Cleavage of the aryl‐halide bond is not likely rate determining since I‐Tyr and bromotyrosine are processed at similar rates despite differences in their C–X bond strengths.6, 8 Similarly, product release is not likely rate determining since the second‐order rate constant of I‐Tyr turnover measured by rapid kinetics is approximately equivalent to the corresponding k cat/K m measured under steady‐state conditions.6
Figure 9.
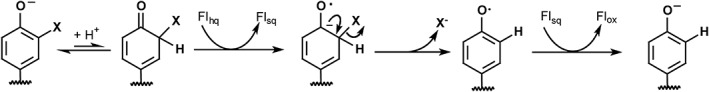
Proposed mechanism of reductive dehalogenation.2
While accumulation of Flsq may not be a major determinant in the catalytic efficiency of IYD, it is likely related to catalytic specificity. Hydrogen bonding between Fl N5 and a side chain of Ser or Thr correlates in the nitroreductase superfamily with the promotion of one electron processes to catalyze reductive dehalogenation (IYD subgroup) and oxygen activation for dimethylbenzimidazole formation (BluB subgroup). Alternative hydrogen bonding between a backbone amide NH and Fl N5 allows for hydride transfer and nitroreduction.10 Accordingly, destabilization of Flsq as described in this work likely explains why a similar Thr to Ala substitution in bacterial HhIYD allowed hydride transfer to become competitive based on a decrease of dehalogenase activity and gain of nitroreductase activity.14 Thus, changes in hydrogen bonding allow for a switch between disparate activities.
Switching catalytic specificity of IYD under control of substrate
For WT HsIYD, the switch between one‐ and two‐electron transfer relies on the hydrogen bonding of Thr239 but this, in turn, is controlled by a mechanism dependent on the substrate. Halotyrosine binding is necessary to close the active site lid and shift Thr239 into hydrogen bonding distance to Fl N5.11 The inability for 2‐iodophenol to induce similar changes in conformation was used to rationalize its very slow rate of dehalogenation.7 The co‐crystal structure of 2‐iodophenol and bacterial HhIYD confirmed this expectation. The aromatic group of 2‐iodophenol stacks above Fl analogously to I‐Tyr but the lid sequence remains dynamic and undetected by X‐ray crystallography.7 Consistent with this observation, 2‐iodophenol was also not able to stabilize detectable levels of Flsq during reduction of human HhIYD.7 2‐Iodophenol binds with even less affinity to human HsIYD than to its bacterial homolog.7 Similar to the results with bacterial HhIYD, human HsIYD is now shown to process 2‐iodophenol with very low efficiency as illustrated under single turnover conditions by oxidation of Flhq [Fig. 10(A)]. Less than 50% of the reduced Flhq of HsIYD could be oxidized after 8 h in the presence of 10 equivalents of 2‐iodophenol. In contrast, oxidation of the remaining Flhq was accomplished within 1 h and one equivalent of I‐Tyr [Fig. 10(A)]. These data are consistent with the inability of 2‐iodophenol to establish sufficient interactions with IYD to activate the switch for single electron transfer.
Figure 10.
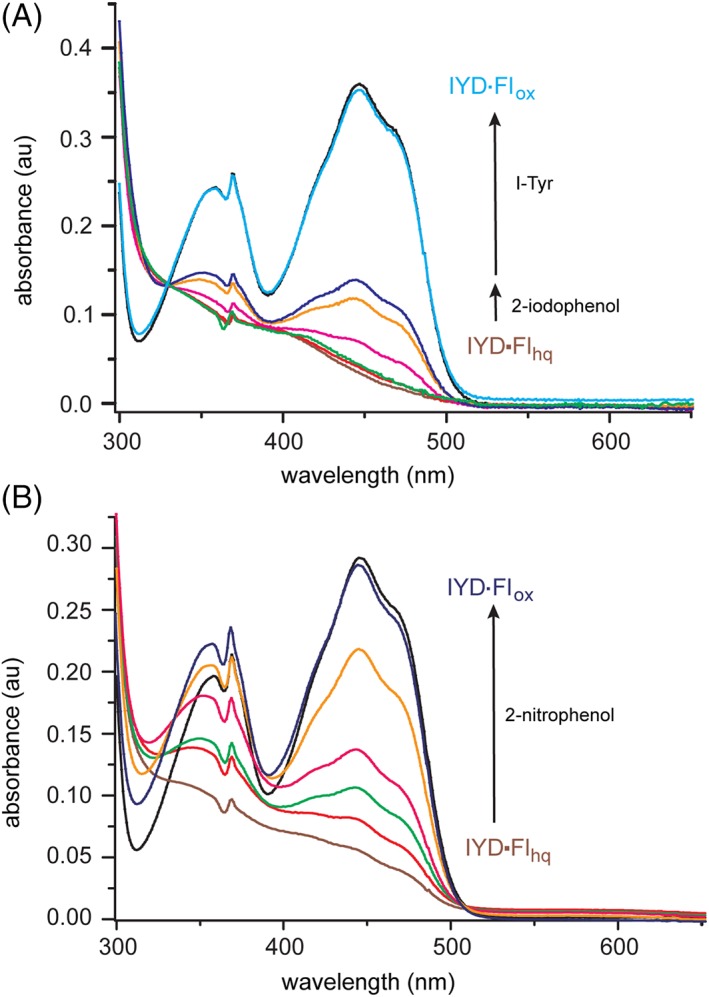
Oxidation of HsIYD•Flhq by 2‐iodophenol and 2‐nitrophenol. (A) HsIYD (30 μM, black) in 100 mM MES pH 6.0, 500 mM NaCl and 10% glycerol was reduced with a minimal concentration of dithionite under anaerobic conditions (600 μl, brown) and then checked for excess dithionite by adding a small trace of air‐saturated buffer (6 μl, red). Next, a spectrum was recorded 2 h after addition of 2‐iodophenol (30 μM, green), again 2 h after a further addition of 2‐iodophenol (20 μM, magenta) and 2 h after each of two more additions of 2‐iodophenol (100 μM tan, 150 μM, dark blue). A spectrum typical of Flox was regenerated 1 h after addition of I‐Tyr (30 μM, light blue). (B) HsIYD (24 μM, black) was reduced under the same conditions (brown) and then a spectrum was recorded immediately after addition of 2‐nitrophenol (12 μM, red) and again after 2 h (green). Spectra were further recorded 2 h (magenta), 4 h (tan) and 8 h (dark blue) after a subsequent addition of a final aliquot of 2‐nitrophenol (12 μM).
An alternative to 2‐iodophenol was then used to identify the intrinsic properties of IYD in the absence of the substrate‐induced activation. A complementary strategy involving substitution of the key Thr with Ala in bacterial HhIYD already revealed an alternative nitroreductase activity common to many members of its superfamily.14 The potential for WT human HsIYD to reveal an analogous activity was tested with 2‐nitrophenol. This ligand binds weakly to HsIYD (K d of 2.7 mM, Fig. S6) but with a similar magnitude as 2‐iodophenol (K d of 1.4 mM).7 However, unlike 2‐iodophenol, 2‐nitrophenol is fully competent at oxidizing the Flhq of WT HsIYD [Fig. 10(B)]. This oxidation was readily observed with a single equivalent of 2‐nitrophenol. However, even this amount can be considered a potential excess since nitroreductases generally yield the two‐electron reduced nitroso intermediate transiently before promoting a more rapid second reduction to the hydroxylamine product.30, 31, 32 This ability of 2‐nitrophenol to undergo reduction by a WT IYD is not duplicated with 2‐nitrotyrosine since this tyrosine derivative is expected to activate the one electron mechanism of Flhq oxidation by the closure of the active site lid of WT IYD.14
The general strategy of substrate control has previously been identified in a variety of flavoproteins. For example, efficient reduction of p‐hydroxybenzoate hydroxylase by NADPH requires the presence of p‐hydroxybenzoate and avoids non‐productive activation of molecular oxygen.33 Active site lid and loop sequences are also known to facilitate substrate capture and sequestration for efficient Fl‐dependent catalysis.34, 35 Closure of the active site lid of IYD is distinct from most examples since the conformational change does not accelerate an intrinsic reaction of Fl but rather switches the redox processes available to Fl. The dehalogenation activity of IYD depends on the presence of the full halotyrosine substrate. Only such compounds are capable of inducing the necessary conformational change and hydrogen bonding to Fl N5 for promoting single electron transfer associated with dehalogenation (Fig. 9).
IYD illustrates a level of substrate control that serves an obvious purpose in vertebrates. These organisms all require the recovery of iodide from I‐Tyr and I2‐Tyr but consumption of other iodinated compounds such as thyroid hormone and its biosynthetic intermediates would disrupt thyroid function. This deleterious activity is prevented by the inability of the common iodophenol group to close the active site lid and stabilize Flsq.7 However, in the absence of the physiological substrate or the appropriate residue to hydrogen bond with Fl N5, a promiscuous nitroreductase activity can be detected.36
Material and methods
Materials
Oligonucleotides were obtained from Intergrated DNA Technologies. Rosetta™ 2(DE3) competent Escherichia coli cells were purchased from Novagen (San Diego, CA). All enzymes were obtained from New England Biolabs except for xanthine oxidase that was obtained from Sigma‐Aldrich Chemicals (St. Louis, MO). All other biochemical reagents were purchased at the highest grade and used without further purification unless specified. F‐Tyr and Nile Blue were obtained from Astatech, Inc. and Santa Cruz Biotechology, Inc. (Santa Cruz, CA), respectively. 2‐Iodophenol and 2‐nitrophenol were obtained from Acros Organics and Sigma‐Aldrich Chemicals, respectively.
General methods
Catalytic deiodination of I2‐Tyr was determined by a standard protocol that quantifies the release of [125I]‐iodide from [125I]‐I2‐Tyr as reported previously.37, 38 Assays are maintained at pH 7.4 with 100 mM potassium phosphate and initiated by addition of the reductant, dithionite, as described in Figure S5. Ligand binding to HsIYD was monitored by quenching the fluorescence of the active site Flox (λ ex = 450 nm and λ em = 527 nm) as described previously.5, 39 Briefly, HsIYD was titrated independently with I‐Tyr, F‐Tyr, and 2‐nitrophenol over a range of concentrations centered at that necessary for 50% quenching of the emission when possible. Fluorescence intensities (F) were normalized by dividing by the initial fluorescence (F0) (obtained in the absence of ligand) and plotted against the ligand concentration ([L]) using Origin 7. Dissociation constants (K d) were obtained from nonlinear fitting to the following equation:39
| (1) |
Mutagenesis of HsIYD
The parent HsIYD identified as WT represents the native HsIYD lacking Residues 1–31 that constitute a transmembrane anchor. This is expressed as a fusion with a C‐terminal His6 and an N‐terminal SUMO using the plasmid pETSUMO‐hIYD described previously.11 Enzyme variants were generated by site‐directed mutagenesis of this plasmid. The T239A variant was produced by using a forward primer of 5′‐CTGGTGACTGTCACTACGGCGCCTCTCAACTGTGGCC‐3′ and its complement 5′‐GGCCACAGTTGAGAGGCGCCGTAGTGACAGTCACCAG‐3′. The T239S variant was created by using a primer of 5′‐GGTGACTGTC‐ACCACGAGTCCTCTCAACTGTGGCC‐3′ and its complement 5′‐GGCCACAGTTGAGAGGACTCGTGGTGACAGTCACC‐3′. Mutations were confirmed by DNA sequencing and the desired plasmids were used to transform electro‐competent E. coli Rosetta 2(DE3) for protein expression.
Protein expression and purification
Transformed cells were grown in LB media with kanamycin and chloramphenicol at 37°C to an OD600 of 0.6–0.8. Protein expression was then induced by adding isopropyl ß‐d‐1‐thiogalactopyranoside (0.2 mM) and further incubation at 18°C for 4 h. Cells were harvested by centrifugation, resuspended in 50 mM sodium phosphate pH 8.0, 500 mM NaCl and 10% glycerol and flash frozen for storage at −80°C. Purification has been described previously and relies on nickel ion affinity chromatography, cleavage of the SUMO fusion with Ulp1 protease and a final gel filtration column.11 Fractions containing IYD were pooled and the concentration of IYD was estimated from the A 280 value after correcting for the contribution of bound FMN (A 280/A 450 = 1.57)11 and an extinction coefficient (ε 280 = 37,930 M−1 cm−1) estimated by ExPASY ProtParam tool for HsIYD and used for the T239A and T239S variants as well.40 The concentration of FMN derived from its A 450 (ε 450 = 12,500 M−1 cm−1).41 Purified IYD was concentrated to ~10 mg/ml by using Amicon® Ultra centrifugal filters prior to storage at 4°C.
Crystallography of HsIYD T239A with F‐Tyr
To obtain a co‐crystal of F‐Tyr and the T239A variant of HsIYD, the protein was pre‐incubated with 3 mM F‐Tyr for at least 20 min (4°C). Co‐crystals formed in ~ 2 days at 20°C by the hanging drop diffusion method with a ratio of 0.8 μl T239A HsIYD (11.7 mg/ml, 50 mM sodium phosphate pH 7.4, 100 mM NaCl, 1 mM DTT, 10% glycerol) to 1 μl precipitant containing 0.17 M sodium acetate, 85 mM Tris–HCl pH 8.5, 22.5% w/v polyethylene glycol 4000 and 15% glycerol. Crystals were suspended in cyroloops mounted on copper magnetic bases (Hampton Research, Alisa Viejo, CA) and flash cooled in liquid nitrogen. X‐ray diffraction data were collected at the National Synchrotron Light Source beamline X‐25. All data were processed using the HKL2000 package.42 Molecular replacement was performed by PHASER within the CCP4 program suite using a dimer of mouse IYD containing I‐Tyr (PDB code 3GFD) with all heteroatoms removed and B‐factors set to 20.0 (Å2).12, 43, 44 Three iterations of PHASER were performed to yield a model with six monomers per asymmetric unit, as predicted by Matthews coefficient calculations. Iterative manual model building and the addition of water molecules, FMN, and the F‐Tyr ligand were performed in COOT and refined by Refmac5 in CCP4.45, 46 The final refinement statistics are summarized in Table S1. All structural figures were prepared with PyMOL.
Reduction and redox titration of IYD variants under anaerobic conditions
All titrations were performed at 25°C using a standard xanthine/xanthine oxidase reducing system.20, 21, 47 Individual samples within sealable quartz cuvettes contained 10–15 μM IYD, 900 μM xanthine, 2 μM methyl viologen, 200 mM potassium chloride and 100 mM potassium phosphate pH 7.4. Molecular oxygen was removed from these solutions by continuously flushing with argon for at least 20 min prior to addition of IYD. The reaction was initiated by addition of 40 μg/ml xanthine oxidase. Spectra changes were monitored every 2 min over 2 h. Midpoint potentials were measured as described by Massey20, 21 using reference dyes as indicators, anthraquinone‐2‐sulfonate (AQS, −225 mV), phenosafranine (PSF, − 259 mV), and safranin O (SFO, −280 mV).45 The concentration of each species was determined at the wavelengths described in the relevant figure. All midpoint potentials were calculated based on the Nernst equation and reported versus the standard hydrogen electrode.21 Values represent the average of three independent determinations and the error corresponds to the standard deviation.
Conclusions
The inert substrate analog F‐Tyr was expected to mimic the binding of the physiological substrate I‐Tyr to IYD and thus when F‐Tyr was discovered to stabilize the Flsq intermediate of IYD during redox titration, I‐Tyr was expected to act similarly. The assumption that F‐Tyr induced the same conformational changes in IYD that were discovered earlier for I‐Tyr11, 12 has now been confirmed by characterizing the co‐crystal structure of F‐Tyr and a T239A variant of HsIYD. F‐Tyr adopts a conformation nearly identical to that of I‐Tyr in WT HsIYD (Fig. 3).11 Characterization of the co‐crystal emphasizes the power of hydrogen bonding at Fl N5 to control the pathways available for catalysis. Loss of hydrogen bonding by a Thr side chain previously correlated to destabilization of the Flsq intermediate and the structure of T239A HsIYD in complex with F‐Tyr now demonstrates that no other changes are evident to influence the reactivity of Fl (Figs. 4 and 5). Even a minor perturbation of the hydrogen bonding to Fl N5 by a Thr to Ser substitution significantly destabilized the Flsq intermediate. Surprisingly, this substitution did not adversely affect catalytic dehalogenation and loss of this hydrogen bond by a Thr to Ala substitution decreased k cat/K m by less than seven‐fold. Alignment of the appropriate hydrogen bonding in WT IYD is dependent on substrate identity and provides a mechanism for specificity in thyroid tissue where other iodinated species abound. In the absence of halotyrosine, IYD exhibits a weak ability to promote an alternative nitroreduction that is expected to require hydride rather than single electron transfer.10 Thus, reductive dehalogenation catalyzed by IYD can be controlled by the substrate as well as by its protein and cofactor, as demonstrated previously.14, 29
Supporting information
Appendix S1: Supporting Information.
Acknowledgments
We thank Annie Heroux who led data collection on beamline X‐25 of the National Synchrotron Light Source, a U.S. Department of Energy Office of Science User Facility operated by Brookhaven National Laboratory under Contract no. DE‐AC02‐98CH10886. The authors have no conflict of interest to report.
Short statement for a broader audience: The environment surrounding a flavin cofactor typically determines its ability to promote one and two‐electron transfer processes. Crystallography, redox titration, site‐directed mutagenesis and reaction kinetics are used to describe a related method of control based on ligand binding to iodotyrosine deiodinase. This enzyme promotes single electron transfer for catalytic dehalogenation but also supports two‐electron transfer in the presence of nitrophenol to promote nitroreduction.
Grant sponsor: National Institutes of Health; Grant number: AI119540.
Present address: Jimin Hu, National Pesticide Engineering Research Center, College of Chemistry, Nankai University, Tianjin, China
References
- 1. Rokita SE. Flavoprotein Dehalogenases In: Hille R, Miller SM, Palfey B, Eds, 2013. Handbook of Flavoproteins. Berlin: DeGruyter; p. 337–350. [Google Scholar]
- 2. Sun Z, Su Q, Rokita SE (2017) The distribution and mechanism of iodotyrosine deiodinase defied expectations. Arch Biochem Biophys 632:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Phatarphekar A, Buss JM, Rokita SE (2014) Iodotyrosine deiodinase: a unique flavoprotein present in organisms of diverse phyla. Mol BioSyst 10:86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Akiva E, Copp JN, Tokuriki N, Babbit PC (2017) Evolutionary and molecular foundations of multiple contemporary functions of the nitroreductase superfamily. Proc Natl Acad Sci USA 114:E9549–E9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McTamney PM, Rokita SE (2009) A mammalian reductive deiodinase has broad power to dehalogenate chlorinated and brominated substrates. J Am Chem Soc 131:14212–14213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bobyk KD, Ballou DP, Rokita SE (2015) Rapid kinetics of dehalogenation promoted by iodotyrosine deiodinase from human thyroid. Biochemistry 54:4487–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ingavat N, Kavran JM, Sun Z, Rokita SE (2017) Active site binding is not sufficient for reductive deiodination by iodotyrosine deiodinase. Biochemistry 56:1130–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Phatarphekar A, Rokita SE (2016) Functional analysis of iodotyrosine deiodinase from Drosophila melanogaster . Protein Sci 25:2187–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Phatarphekar A, Su Q, Eun SH, Chen X, Rokita SE (2018) The importance of a halotyrosine dehalogenase for Drosophila fertility. J Biol Chem 293:10314–10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koder RL, Haynes CA, Rodgers ME, Rodgers DW, Miller A‐F (2002) Flavin thermodynamics explain the oxygen insensitivity of enteric nitroreductase. Biochemistry 41:14197–14205. [DOI] [PubMed] [Google Scholar]
- 11. Hu J, Chuenchor W, Rokita SE (2015) A switch between one‐ and two‐electron chemistry of the human flavoprotein iodotyrosine deiodinase is controlled by substrate. J Biol Chem 290:590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thomas SR, McTamney PM, Adler JM, LaRonde‐LeBlanc N, Rokita SE (2009) Crystal structure of iodotyrosine deiodinase, a novel flavoprotein responsible for iodide salvage in thyroid glands. J Biol Chem 284:19659–19667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Buss JM, McTamney PM, Rokita SE (2012) Expression of a soluble form of iodotyrosine deiodinase for active site characterization by engineering the native membrane protein from Mus musculus . Protein Sci 21:351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mukherjee A, Rokita SE (2015) Single amino acid switch between a flavin‐dependent dehalogenase and nitroreductase. J Am Chem Soc 137:15342–15345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fraaije MW, Mattevi A (2000) Flavoenzymes: diverse catalysts with recurrent features. Trends Biochem Sci 25:126–132. [DOI] [PubMed] [Google Scholar]
- 16. van den Heuvel RH, Fraaije MW, Mattevi A, van Berkel WJ (2000) Asp‐170 is crucial for the redox properties of vanillyl‐alcohol oxidase. J Biol Chem 275:14799–14808. [DOI] [PubMed] [Google Scholar]
- 17. Wijaya IMM, Domratcheva T, Iwata T, Getzoof ED, Kandori H (2016) Single hydrogen bond donation from flavin N5 to proximal asparagine ensures FAD reduction in DNA photolyase. J Am Chem Soc 138:4368–4376. [DOI] [PubMed] [Google Scholar]
- 18. Collins HF, Biedendieck R, Leech HK, Gray M, Escalante‐Semerena JC, McLean KJ, Munro AW, Rigby SEJ, Warren MJ, Lawrence AD (2013) Bacillus megaterium has both a functional BluB protein required for DMNB synthesis and a related flavoprotein that forms a stable radical species. PLoS One 8:e55708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taga ME, Larsen NA, Howard‐Jones AR, Walsh CT, Walker GC (2007) BluB cannibalizes flavin to form the lower ligand of vitamin B12 . Nature 446:449–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Massey V. A simple method for the determination of redox potentials In: Curti B, Ronchi S, Zanetti G, Eds, 1991. Flavins Flavoproteins Proceedings of the International Symposium. Berlin: Gruyter & Co.; p. 59–66. [Google Scholar]
- 21. van den Heuvel RHH, Fraaije MW, Van Berkel WJH (2002) Redox properties of vanillyl‐alcohol oxidase. Methods Enzymol 353:177–186. [DOI] [PubMed] [Google Scholar]
- 22. Draper RD, Ingraham LL (1968) A potentiometic study of flavin semiquinone equilibrium. Arch Biochem Biophys 125:802–8808. [DOI] [PubMed] [Google Scholar]
- 23. Zhou Z, Swenson RP (1996) The cumulative electrostatic effect of aromatic stacking interactions and the negative electrostatic environment of the flavin mononucleotide binding site is a major determinant of the reduction potential for the flavodoxin from Desulfovibrio vulgaris [Hildenborough]. Biochemistry 35:15980–15988. [DOI] [PubMed] [Google Scholar]
- 24. Lostao A, Gómez‐Moreno C, Mayhew SG, Sancho J (1997) Differential stabilization of the three FMN redox forms by tyrosine 94 and tryptophan 57 in flavodoxin from Anabeaena and its influence on the redox potentials. Biochemistry 36:14334–14344. [DOI] [PubMed] [Google Scholar]
- 25. Pitsawong W, Sucharitakul J, Prongjit M, Tan TC, Spadiut O, Haltrich D, Divne C, Chaiyen P (2010) A conserved active‐site threonine is important for both sugar and flavin oxidations of pyranose 2‐oxidase. J Biol Chem 285:9697–9705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stradins J, Hansanli B (1993) Anodic voltammetry of phenol and benzenthiol derivatives. 1. Influence of pH on electrooxidation potentials of substitutes phenols and evaluation of pKa from anodic voltammetry data. J Electroanal Chem 353:57–69. [Google Scholar]
- 27. Auffinger P, Hays FA, Westhof E, Ho SP (2004) Halogen bonds in biological molecules. Proc Natl Acad Sci USA 101:16789–16794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hersh LB, Walsh C (1980) Preparation, characterization and coenzymic properties of 5‐carba‐5‐deaza and 1‐carba‐1‐deaza analogs of riboflavin, FMN and FAD. Methods Enzymol 66:277–287. [DOI] [PubMed] [Google Scholar]
- 29. Su Q, Boucher PA, Rokita SE (2017) Conversion of a dehalogenase to a nitroreductase by swapping its flavin cofactor with a 5‐deazaflavin analog. Angew Chem Int Ed 56:10862–10866. [DOI] [PubMed] [Google Scholar]
- 30. Koder RL, Miller A‐F (1998) Steady‐state kinetic mechanism, stereospecificity, substrate and inhibitor specificity of Enterobacter cloacae nitroreductase. Biochim Biophys Acta 1387:395–405. [DOI] [PubMed] [Google Scholar]
- 31. Pitsawong W, Hoben JP, Miller A‐F (2014) Understanding the broad substrate repertoire of nitroreductase based on its kinetic mechanism. J Biol Chem 289:15203–15214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Park JT, Gómez Ramos LM, Bommarius AS (2015) Engineering towards nitroreductase functionality in ene‐reductase scaffolds. ChemBioChem 16:811–818. [DOI] [PubMed] [Google Scholar]
- 33. Husain M, Massey V (1979) Kinetic studies on the reaction of p‐hydroxybenzoate hydroxylase. J Biol Chem 254:6657–6666. [PubMed] [Google Scholar]
- 34. Ouedraogo D, Souffrant M, Vasquez S, Hamelberg D, Gadda G (2017) Importance of loop L1 dynamics for substrate capture and catalysis in Pseudomonas aeruginosa d‐arginine dehydrogenase. Biochemistry 56:2477–2487. [DOI] [PubMed] [Google Scholar]
- 35. Yu T‐Y, Mok KC, Kennedy KJ, Valton J, Anderson KS, Walker GC, Taga ME (2012) Active site residues critical for flavin binding and 5,6‐dimethylbenzimidazole biosynthesis in the flavin destructase enzyme BluB. Protein Sci 21:839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Copley SD (2017) Shining a light on enzyme promiscuity. Curr Opin Struct Biol 47:167–175. [DOI] [PubMed] [Google Scholar]
- 37. Rosenberg IN, Goswami A (1984) Iodotyrosine deiodinase from bovine thyroid. Methods Enzymol 107:488–500. [DOI] [PubMed] [Google Scholar]
- 38. Friedman JE, Watson JA Jr, Lam DW‐H, Rokita SE (2006) Iodotyrosine deiodinase is the first mammalian member of the NADH oxidase/flavin reductase superfamily. J Biol Chem 281:2812–2819. [DOI] [PubMed] [Google Scholar]
- 39. Warner JR, Copley SD (2007) Pre‐steady‐state kinetic studies of the reductive dehalogenation catalyzed by tetrachlorohydroquinone dehalogenase. Biochemistry 46:13211–13222. [DOI] [PubMed] [Google Scholar]
- 40. Pace CN, Vajdos F, Fee L, Grimseley G, Gray T (1995) How to measure and predict the molar absorption coefficient of a protein. Protein Sci 4:2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koziol J (1971) Fluorometric analyses of riboflavin and its coenzymes. Methods Enzymol 18:235–285. [Google Scholar]
- 42. Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- 43. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Cryst 40:658–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Cryst D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Cryst D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 46. Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Nicholls RA, Winn MD, Long F, Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst D67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Clark WM. Oxidation‐Reduction Potentials of Organic Systems. Baltimore: Williams & Wilkins, 1960;p. 184–203. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information.