

SUMMARY
The Hippo pathway controls the activity of YAP/TAZ transcriptional coactivators through a kinase cascade. Despite the critical role of this pathway in tissue growth and tumorigenesis, it remains unclear how YAP/TAZ–mediated transcription drives proliferation. By analyzing the effects of inactivating LATS1/2 kinases, the direct upstream inhibitors of YAP/TAZ, on mouse brain development and applying cell-number–normalized transcriptome analyses, we discovered that YAP/TAZ activation causes a global increase in transcription activity, known as hypertranscription, and upregulates many genes associated with cell growth and proliferation. In contrast, conventional read-depth–normalized RNA-sequencing analysis failed to detect the scope of the transcriptome shift and missed most relevant gene ontologies. Following a transient increase in proliferation, however, hypertranscription in neural progenitors triggers replication stress, DNA damage, and p53 activation, resulting in massive apoptosis. Our findings reveal a significant impact of YAP/TAZ activation on global transcription activity and have important implications for understanding YAP/TAZ function.
In Brief
Using cell-number–normalized transcriptome analysis, Lavado et al. show that inactivation of Hippo pathway LATS1/2 kinases during brain development causes YAP/TAZ–driven global hypertranscription, upregulating many genes involved in cell growth and proliferation. Hypertranscription in neural progenitors inhibits differentiation and triggers replication stress and DNA damage, resulting in massive apoptosis.
GRAPHIC ABSTRACT
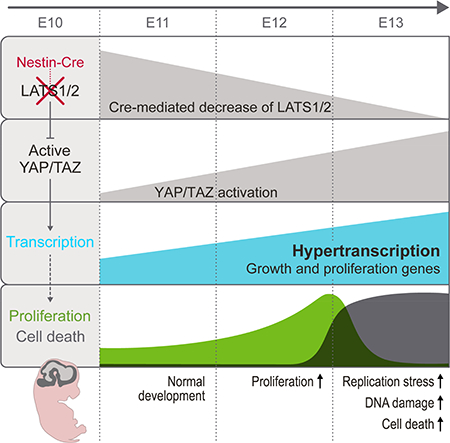
INTRODUCTION
The Hippo pathway regulates the development, homeostasis, regeneration, and tumorigenesis of various tissues across species (Pfleger, 2017; Yu et al., 2015). At its core are a kinase cascade and a transcription factor complex (Meng et al., 2016). The upstream kinases MST1 and MST2 activate the downstream kinases LATS1 and LATS2 (LATS1/2), which in turn phosphorylate the homologous transcriptional coactivators YAP and TAZ (YAP/TAZ)—the key effectors of the Hippo pathway—resulting in their cytoplasmic sequestration or degradation. When the Hippo kinase cascade is inactivated, unphosphorylated YAP/TAZ enter the nucleus, where they interact with the TEAD family of DNA-binding factors and activate gene expression. The most prominent function of YAP/TAZ is to promote cell proliferation and survival. Accordingly, animal models of Hippo pathway inactivation or YAP/TAZ activation almost always exhibit overgrowth or tumorigenic phenotypes, and YAP/TAZ activation has been observed in nearly all types of human solid tumor and is associated with tumor aggression and poor outcomes (Zanconato et al., 2016). Despite this, the genes that are consistently and strongly induced by YAP/TAZ in different contexts are often those related to the extracellular matrix (ECM), cell adhesion, and epithelial-to-mesenchymal transition (EMT) and are rarely those related to proliferation (Cai et al., 2015; Lavado et al., 2013; Lee et al., 2016; Ota and Sasaki, 2008; Su et al., 2015), raising the question of how YAP/TAZ activation drives proliferation in so many contexts.
As LATS1/2 directly phosphorylate YAP/TAZ, they are probably the most important gatekeepers of YAP/TAZ activation in many contexts. Indeed, mice lacking Lats1/2 in the developing gut (Cotton et al., 2017), kidney (Reginensi et al., 2016), and liver (Lee et al., 2016); in growing blood vessels (Kim et al., 2017); and in the adult liver (Chen et al., 2015; Lee et al., 2016) and heart (Heallen et al., 2013) all show YAP/TAZ activation. This in turn promotes the proliferation of gut mesenchymal progenitors, immature liver biliary epithelial cells, vascular endothelial cells, and adult cardiomyocytes in the corresponding tissues and organs. Surprisingly, in the adult mouse liver, YAP/TAZ activation induced by Lats1/2 deletion triggered hepatocyte senescence and death (Lee et al., 2016). Although polyploidy and markers of DNA damage and p53 activation were detected, the cause of these defects was unclear.
In the developing mammalian brain, apical neural progenitor cells (NPCs), including neuroepithelial cells and radial glial cells (RGCs), form an epithelial layer along the ventricles— a region known as the ventricular zone (VZ) (Kriegstein and Alvarez-Buylla, 2009). An RGC can undergo proliferative division to expand itself or neurogenic division to generate a new RGC and either a neuron or an intermediate progenitor cell (IPC). IPCs, residing in the subventricular zone (SVZ), produce more neurons through rounds of neurogenic division. Newborn neurons migrate outward through the intermediate zone (IZ) and settle at appropriate locations in the cortical plate (CP) to complete their differentiation. Precise orchestration of NPC proliferation, differentiation, and survival is critical for generating a brain with diverse types and the correct numbers of cells.
We investigated the role of LATS1/2 in mouse brain development. We found that the loss of Lats1/2 in apical NPCs results in their transient expansion, quickly followed by massive apoptosis, reduced differentiation, and tissue disintegration. These defects result from YAP/TAZ activation, as additional Yap;Taz deletion restores brain development. By applying cell-number– normalized (CNN) transcriptome analysis, we discovered that YAP/TAZ activation boosts global transcription activity and upregulates many genes associated with increased biosynthetic capacity and proliferation. In contrast, conventional RNA-sequencing (RNA-seq) analysis failed to detect the scope of the transcriptome shift and missed most relevant gene ontologies. Furthermore, this global transcription increase in NPCs causes DNA replication stress and DNA damage and triggers apoptosis. Our study reveals a mechanism by which YAP/TAZ–mediated transcription drives proliferation and highlights cell type–specific responses to YAP/TAZ activation.
RESULTS
Lats1/2 Deletion Activates YAP/TAZ, Expands RGCs, and Triggers Massive Apoptosis
To determine the role of LATS1/2 during mammalian brain development, we generated Lats1/2 conditional knockout (KO) mice by using Nestin-Cre, which is expressed in apical NPCs starting on E10.5 (Tronche et al., 1999). Brains of single KO mice or those lacking three Lats alleles developed normally (Figure S1D and data not shown), whereas double KO (dKO, Lats1/2F/F;Nestin-Cre) brains exhibited severe phenotypes and were the focus of this study. Compared to the levels in control (no Cre) brains, phospho-YAP levels gradually decreased and YAP/TAZ levels gradually increased in embryonic day (E) 12.5 and E13.5 dKO brains (Figure 1A), consistent with the established Hippo kinase cascade. In control E12.5 brains, YAP/TAZ were specifically expressed in Sox2+ RGCs (Bylund et al., 2003) and were largely cytoplasmic (Figure 1B). In Lats1/2 dKO brains, large portions of YAP/TAZ proteins shifted to the nucleus. Subcellular fractionation further confirmed a significant nuclear enrichment of YAP/TAZ proteins in dKO brains (Figure S1A). Thus, Lats1/2 deletion results in the upregulation of YAP/TAZ proteins and their nuclear translocation.
Figure 1. Lats1/2 Deletion Activates YAP/TAZ, Expands RGCs, and Triggers Massive Apoptosis.

(A) Quantitative Western blots comparing protein levels in control (no Cre) and Lats1/2 dKO (Lats1/2F/F;Nestin-Cre) brains.
(B) Immunostaining showing YAP/TAZ localization in E12.5 cortices.
(C) Quantification of E12.5 cortical ventricular zone (VZ) area based on Pax6 immunostaining. (D) Immunostaining and quantification of the S-phase length (Ts) and total cell-cycle length (Tc) of E12.5 Sox2+Tbr2− RGCs by cumulative EdU/BrdU labeling.
(E) H&E staining showing gross morphologic defects in dKO brains.
(F) Immunostaining for cleaved caspase-3 to detect apoptosis in E13.5 cortices. Dashed lines demarcate the ventricular surface; dotted lines mark the border between the ventricular/subventricular zone (VZ/SVZ) and the cortical plate (CP).
(G) Immunostaining and quantification of Sox2+Tbr2− RGCs, Tbr2+ IPCs, and P27+ neurons in E13.5 cortices. Dashed boxes indicate regions quantified.
Data are shown as mean ± SEM; *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant; unpaired two-tailed Student’s t-test. See also Figure S1.
Next, we examined the effect of Lats1/2 deletion on cortical RGC proliferation and differentiation. At E12.5, the numbers of Sox2+Tbr2− RGCs, Tbr2+ IPCs (Englund et al., 2005), and P27+ neurons (Nguyen et al., 2006) per 500-μm cortex length were unchanged in dKO brains (Figure S1B). However, the area of cortical VZ demarcated by Pax6 immunosignal (Englund et al., 2005) was increased in the rostral region of dKO brains (Figures 1C and S1C), suggesting lateral expansion of the RGC population. We then measured RGC cell-cycle length by a cumulative EdU/BrdU–labeling paradigm (Pilaz et al., 2016). Both the S-phase (Ts) and total cell-cycle (Tc) lengths of dKO RGCs were shortened (Figure 1D). Thus, Lats1/2 deletion accelerates the cell cycle of E12.5 cortical RGCs, leading to their expansion.
The morphology of Lats1/2 dKO brains deteriorated quickly. At E13.5, there was obvious disruption of the neuroepithelial structure and ventricular enlargement (Figure 1E). At E15.5, the rostral region had disintegrated into a disorganized mass of cells, and only a thin layer of cells was left in the rest of the brain (Figure 1E). Such severe dysplasia prompted us to examine whether cell death was increased in dKO brains. Cleaved caspase-3 immunostaining revealed massive apoptosis in E13.5 dKO brains, especially at the border between the VZ/SVZ and the Tuj1+ CP (Figure 1F). RGCs were increased and IPCs decreased in E13.5 dKO cortices (Figure 1G), suggesting that Lats1/2 deletion expands RGCs and impairs their differentiation into IPCs, although IPC reduction may be secondary to apoptosis.
Apoptosis in LATS-Deficient Brains Depends on p53 Activation
To uncover the cause of LATS-deficiency phenotypes, we used Affymetrix microarrays to compare the transcriptomes of E12.5 control and dKO cortices before the onset of apoptosis and tissue dysplasia (Figure S2A) and found upregulation of “p53 signaling pathway” by gene set enrichment analysis (Table S1 and Figure S2B). Immunostaining confirmed an increase in p53 proteins in E13.5, but not E12.5, dKO brains (Figures 2A and S2A), probably because microarrays are more sensitive than immunostaining. Because p53 is stabilized in response to various cellular stresses, including DNA damage (Mello and Attardi, 2018), we stained brain sections for the DNA damage markers γH2AX (Rogakou et al., 2000; Rogakou et al., 1998) and phospho-KAP1 (White et al., 2006; Ziv et al., 2006). Both markers revealed widespread DNA damage in E13.5 dKO brains (Figure 2A).
Figure 2. Apoptosis in LATS-Deficient Brains Depends on p53 Activation.

(A) Images of TUNEL assay and immunostaining for DNA damage markers in E13.5 control and Lats1/2;Nestin-Cre dKO brains. Dashed lines demarcate the ventricular surface; solid lines demarcate the outer edge of the cortical plate.
(B) TUNEL assay, immunostaining, and quantification showing the effects of deleting p53 in the Lats1/2;Emx1-Cre dKO background (mean ± SEM). ZO-1 and β-catenin label apical junctions.
*P < 0.05; **P < 0.01; ***P < 0.001.
(C) Immunostaining for the RGC marker Pax6, the pan-neuronal marker Tuj1, and the layerspecific neuronal markers Satb2 and Tbr1, showing defects in E15.5 Lats1/2;p53 KO brains. The blue color in (B) and (C) is DAPI signal. See also Figure S2 and Table S1.
To determine whether the apoptosis in Lats1/2 dKO brains depended on p53, we generated Lats1/2;p53 triple KO (tKO) mice with Emx1-Cre, which drives cortex-specific deletion starting on E9.5 (Gorski et al., 2002). Mice carrying Emx1-Cre can be maintained as homozygous, increasing the chance of producing tKO mice. Deleting Lats1/2 with Emx1-cre also led to massive apoptosis (Figure 2B); this was similar to the result with Nestin-Cre but occurred earlier (at E12.5) because Emx1-Cre is turned on before Nestin-Cre. The additional removal of p53 greatly diminished apoptosis (Figure 2B). γH2AX was even more elevated in tKO cortex than in Lats1/2 dKO cortex (Figure 2B), indicative of aberrant survival of DNA-damaged cells in the absence of p53. These data suggest that apoptosis in Lats1/2 dKO brains is largely p53 dependent and that DNA damage is upstream or independent of p53 activation.
Despite apoptosis being prevented, Lats1/2;p53 tKO cortex was far from normal. At E12.5, both dKO and tKO cortices showed structural disorganization, apical junction disruption, and depletion of Tbr2+ IPCs (Figures 2B and S2C). At E15.5, both dKO and tKO cortices completely lacked layering organization and contained markedly fewer Satb2+ and Tbr1+ neurons than control cortex (Figure 2C). In summary, the loss of Lats1/2 in NPCs triggers widespread DNA damage and p53-dependent apoptosis. It also causes severe disorganization of brain structures and impairs differentiation, independent of p53-induced apoptosis.
Dysplasia of LATS-Deficient Brains Results from YAP/TAZ Activation
The apoptosis and dysplasia phenotypes of Lats1/2 dKO brains were unexpected because Hippo kinase cascade inactivation and subsequent YAP/TAZ activation typically lead to tissue overgrowth. To test whether these phenotypes were due to YAP/TAZ activation, we deleted Yap and Taz in the Lats1/2;Emx1-Cre dKO background. At postnatal day 0 (P0), Lats1/2 dKO cortex was markedly smaller than control cortex and was completely disorganized, with Pax6+ RGCs being misplaced outside the VZ, cortical neurons lacking layering organization, and their axons being severely reduced and unable to form normal projections (Figures 3A and S3A). Deleting Yap largely restored the size of the cortex, but its organization was notably defective, with numerous patches of RGCs intermingled with neurons throughout the CP (Figures 3A and S3A).
Figure 3. Dysplasia of LATS-Deficient Brains Results from YAP/TAZ Activation.

(A) Pax6 and Tuj1 immunostaining images of one hemisphere and neurofilament (NF) immunostaining images of the midline region of P0 cortices, showing the effects of deleting Yap and Taz in the Lats1/2 dKO background. Roman numerals indicate the number of Yap;Taz alleles deleted; Ctrl: control. The same naming scheme is used in (B) and (C). Dashed lines outline the cortex. CC: corpus callosum.
(B) Immunostaining of E13.5 brains showing the effects of deleting Yap and Taz in the Lats1/2 dKO background. Dashed lines demarcate the ventricular surface. The blue color is DAPI signal. (C) Quantification of E13.5 cleaved caspase-3 signals and E13.5 and P0 Pax6 signals (mean ± SEM). Dashed lines indicate the mean values of control (Ctrl) samples. Comparisons are against the results with controls unless otherwise indicated. *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant.
See also Figure S3.
Removing a further Taz allele largely restored the layering of cortical neurons, although some RGCs were still dispersed outside the VZ and the corpus callosum (the axon tract connecting the cortical hemispheres) was still missing (Figures 3A and S3A). Completely removing Yap and Taz essentially restored cortex morphology (Figures 3A and S3A). These results indicate that YAP/TAZ activation is required for the dysplasia of LATS-deficient brains. The near-complete restoration of brain morphology by Yap;Taz deletion is in stark contrast to the severe disorganization and impaired differentiation of Lats1/2;p53 tKO cortex, suggesting that YAP/TAZ activation mediates most, if not all, of the effects subsequent to Lats1/2 loss, whereas p53 activation accounts for apoptosis induction only.
We further quantified apoptosis and RGC numbers upon additional Yap;Taz deletion (Figures 3B and3C). Apoptosis was strongly elevated in E13.5 Lats1/2 dKO cortex as compared to control cortex. Although apoptosis remained highly elevated after the additional deletion of one allele of Yap, it was reduced when both Yap alleles were deleted and returned to basal levels when three or four Yap;Taz alleles were deleted. The changes in RGC numbers exhibited developmental dynamics. At E13.5, RGCs were severely depleted in dKO cortex. They were restored to the numbers seen in controls when one Yap allele was deleted, increased further when both Yap alleles were deleted, and returned to the control numbers when three or four Yap;Taz alleles were deleted. At P0, dKO and control cortices contained similar numbers of RGCs, which were strongly expanded when one or two Yap alleles were deleted, expanded to a lesser degree when three Yap;Taz alleles were deleted, and decreased when all four alleles were deleted. These data revealed a clear dosage effect of Yap;Taz allele numbers, suggesting that the consequences of LATS inactivation depend on the levels of active YAP/TAZ: very high levels of active YAP/TAZ trigger massive apoptosis and severely deplete RGCs, whereas high-to-moderate levels of active YAP/TAZ expand RGCs and moderately elevate apoptosis.
To test whether YAP/TAZ activation was sufficient to induce apoptosis and block differentiation, we overexpressed YAP and its variants in the developing cortex by in utero electroporation. Although wild-type (WT) YAP did not induce cell death, YAP5SA, which harbors mutations in 5 LATS phosphorylation sites and is thus resistant to LATS-mediated inhibition (Zhao et al., 2007), triggered significant cell death (Figure S3B). The YAP5SA/S94A mutant, which cannot bind TEAD (Zhao et al., 2007), failed to induce cell death, suggesting that the ability of YAP5SA to induce cell death depends on TEAD binding and probably on TEADmediated transcription. Consistent with our previous report (Cao et al., 2008), YAP overexpression maintained the RGC state and inhibited differentiation, as indicated by a larger fraction of transfected cells residing in the VZ/SVZ, a smaller fraction in the CP, and the presence of fewer Tbr2+ IPCs when compared to the results of control GFP transfections (Figures S3B and S3C). YAP5SA had even stronger effects: essentially all transfected cells remained as RGCs in the VZ and few differentiated into IPCs (Figures S3B and S3C), which resembled the phenotypes of Lats1/2 dKO brains (Figure 1G). Compared to control transfections, the YAP5SA/S94A mutant did not alter the fraction of cells in the VZ/SVZ or the number of IPCs, but it did cause retention of transfected cells in the IZ (Figures S3B and S3C); whether this reflects a normal function of YAP is unclear. These data indicate that ectopic, high levels of YAP/TAZ activation are sufficient to induce apoptosis and block differentiation, and that TEADmediated transcription is probably required for this process.
LATS-Deficient Neural Stem Cells in Culture Recapitulate In Vivo Phenotypes
Next, we employed neural stem cell (NSC) cultures in more mechanistic studies. We derived NSCs from E13.5 control and Lats1/2;Nestin-Cre dKO cortices. In adherent cultures, YAP/TAZ predominantly resided in the cytoplasm of control NSCs but showed strong nuclear localization in dKO NSCs (Figure 4A). p53 was detected at the background level in control cells but showed intense nuclear signals in dKO cells (Figure 4A). When grown in differentiation medium, dKO cells failed to undergo neuronal differentiation (Figure 4B), which resembled the differentiation defect observed in vivo. When grown as neurospheres, dKO spheres were smaller than control spheres and showed higher levels of cleaved caspase-3 immunosignal (Figures 4C and S4A). If all visible cell clusters were included, the numbers of neurospheres formed by control and dKO NSCs were similar; however, the viability and number of live cells were significantly lower in dKO cultures (Figures 4C), recapitulating the in vivo phenotype of increased apoptosis. NSCs derived from E12.5 dKO cortices exhibited similar defects (Figure S4B), indicating that phenotypically they progressed in culture as they did in vivo.
Figure 4. LATS-Deficient Neural Stem Cells in Culture Recapitulate In Vivo Phenotypes.

(A) Immunostaining of NSCs in adherent cultures comparing YAP/TAZ localization and p53 levels in control and Lats1/2;Nestin-Cre dKO cells.
(B) Immunostaining of NSCs grown in differentiation medium for the neuronal marker Tuj1 and the progenitor marker nestin.
(C) Representative images and quantification of neurospheres 3 days after plating (mean ± SEM).
(D) Procedure for NSC transfection, sorting, and assay.
(E) Immunostaining showing p53 levels in cells of the indicated genotypes transfected with mCherry or Cre-mCherry plasmid.
(F) Quantification of NSCs with the indicated floxed alleles transfected with mCherry or CremCherry plasmid 3 days after plating in neurosphere culture (mean ± SEM). Dashed boxes in (A) and (E) indicate enlarged regions. *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant. See also Figure S4
We then tested whether the phenotypes exhibited by Lats1/2 dKO NSCs in culture were mediated by p53 and YAP/TAZ. We transfected NSCs derived from Lats1/2F/F, Lats1/2F/F;p53F/F, and Lats1/2F/F;YapF/F;TazF/F cortices with plasmids encoding mCherry (control) or Cre-mCherry (Cre), then sorted mCherry+ cells and grew them in neurosphere cultures (Figure 4D). Like NSCs derived from Lats1/2 dKO cortices, Lats1/2F/F cells transfected with Cre showed p53 upregulation and reduced viability and cell numbers when compared to control-transfected cells (Figures 4E and4F). Lats1/2F/F;p53F/F and Lats1/2F/F;YapF/F;TazF/F cells transfected with Cre no longer exhibited p53 activation or less viability than the corresponding controls. Interestingly, whereas the cell number was restored in Cre-transfected Lats1/2F/F;YapF/F;TazF/F cells, it was still significantly reduced in Cre-transfected Lats1/2F/F;p53F/F cells (Figure 4F), suggesting that the latter still had a proliferation defect. These results demonstrate that LATS-deficient NSCs in culture recapitulate many aspects of Lats1/2 dKO phenotypes in vivo.
Although LATS1/2 have been implicated in mitosis (Hergovich and Hemmings, 2012), Lats1/2 dKO NSCs showed very low levels of mitosis defects (Figures S4C−S4E) that are unlikely to account for the severe viability and proliferation defects of these cells.
LATS-Deficient NSCs Exhibit Prolonged S Phase and DNA Replication Stress
To uncover the cellular defects of Lats1/2 dKO NSCs, we first observed their behavior directly by live imaging. We transfected NSCs with a modified Fucci plasmid that labels the plasma membrane of all transfected cells green and the nuclei of those in the S/G2/M phases orange (Sakaue-Sawano et al., 2008) and focused on cells displaying orange fluorescence at the beginning of the 14-h imaging period (Figures 5A–5C, Movies S1–S4). Of the 191 control NSCs analyzed, most divided within the imaging period and only a small fraction did not divide or died (Figures 5D and S5A). In contrast, of the 186 dKO NSCs analyzed, fewer than half divided. Moreover, among the cells that divided, it took significantly longer for dKO cells than for control cells to enter mitosis (indicated by cell rounding; arrows in Figures 5A and5B) (Figure 5E), suggesting that the S/G2 phases were extended in dKO cells. We further assessed the cellcycle phases by measuring the DNA content by propidium iodide (PI) staining and flow cytometry. A larger fraction of dKO cells than of control cells was in S phase, whereas the fraction of cells in G2/M phases was similar for control and dKO cells (Figure 5F), consistent with an extended S phase. We then quantified the in vivo cell-cycle length of E13.5 RGCs by the cumulative EdU/BrdU-labeling method described above (Figure 1D). In contrast to the effects at E12.5, both Ts and Tc were extended in dKO RGCs at E13.5 (Figure 5G), corroborating the results obtained with cell cultures.
Figure 5. LATS-Deficient NSCs Exhibit Prolonged S Phase and DNA Replication Stress.

(A–C) Live imaging snapshots of Fucci-labeled NSCs showing division (A and B) and death (C). Arrows indicate cell rounding at the start of mitosis; arrowheads show the daughter cells.
(D) Quantification of cell behaviors observed by live imaging. The number above each bar in the upper panel is the cell number. Fisher’s exact test was used in the lower panel.
(E) Quantification of the time interval between the beginning of imaging and the start of mitosis (mean ± SD).
(F) PI analysis of cell-cycle profiles (mean ± SEM).
(G) Quantification of Ts and Tc of E13.5 RGCs in vivo by cumulative EdU/BrdU labeling (mean ± SEM).
(H) Top: Diagram of DNA fiber assay. Bottom: Representative images of DNA fibers.
(I) Left panel: Replication fork speeds of individual fibers (mean ± SD). Right panel: Average fork speeds for each culture (mean ± SEM).
(J) Box plot of sister-fork ratios (the longer sister fork divided by the shorter one). Bars: median values; boxes: 25th–75th percentiles; whiskers: 10th–90th percentiles. Arrowheads: sister forks.
(K) Two-segment ratios (the longer segment of each DNA fiber divided by the shorter one) arranged in increasing order.
(L) Viability dose-response curve for sorted live NSCs treated with the CDK4/6 inhibitor PD 0332991 for 24 h (mean ± SEM). Dashed lines indicate NSC viabilities at 0 h.
(M) Cell number dose-response curve for sorted live NSCs treated with the CDK4/6 inhibitor PD 0332991 for 24 h (mean ± SEM). The dashed line shows the number of live cells plated at 0 h. In (J) and (K), the permutation-based Wilcox rank-sum test and Ansari-Bradley test were used to compare the median and dispersion, respectively. Comparisons in (L) and (M) are against the results of vehicle-treated dKO NSCs. *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant. See also Figure S5 and Movies S1–S4.
To assess the relevance of the S-phase extension, we examined its response to p53 and Yap;Taz deletion. Cre-transfected Lats1/2F/F cells, when compared to control mCherry transfection, showed cell-cycle profile changes similar to those seen in Lats1/2 dKO cells: an increase in the S-phase fraction, no change in the G2/M fraction, and a corresponding decrease in the G0/G1 fraction (Figure S5C). Deleting p53 did not affect these changes, whereas deleting Yap;Taz erased them (Figure S5C). These results indicate that the S-phase extension is not secondary to p53 activation; thus, it may be a relevant cellular phenotype linking YAP/TAZ activation to p53 activation.
The prolonged S phase of LATS-deficient NPCs prompted the hypothesis that they had defects in DNA replication. This was reinforced by the notion that genes driving cell proliferation often induce DNA replication stress (Macheret and Halazonetis, 2015). To test this, we measured replication fork speeds with a DNA fiber assay that involves labeling progressing replication forks via sequential incorporation of two nucleotide analogs (Figure 5H) (Nieminuszczy et al., 2016). The average fork speed of dKO NSCs was less than that of control cells (Figure 5I), which is indicative of replication stress. Another indication of replication stress is increased asymmetry between sister forks emanating in opposite directions from the same origin (Figure 5J, arrowheads). Normally these sister forks tend to travel at similar speeds. Thus, their length ratio (the length of the longer fork divided by that of the shorter one) is close to 1, as was the case in control cells (Figure 5J). Under replication stress, fork stalling and collapse affect the sister forks independently, causing asymmetry in the sister-fork length (ratio > 1). Indeed, sister-fork asymmetry was greatly increased in dKO cells, with both the median and dispersion of sister-fork ratios being significantly larger (Figure 5J). We obtained similar results by calculating the length ratios of the two labeled segments in one fiber (image in Figure 5H): whereas the vast majority of fibers in control cells had a ratio close to 1, the median and dispersion of the ratios in dKO cells were significantly increased (Figures 5K). These results indicate that Lats1/2 dKO NSCs experience replication stress. Moreover, preventing replication stress by blocking S-phase entry with a CDK4/6 inhibitor (Toogood et al., 2005) significantly increased the viability and cell number of dKO NSCs (Figures 5L and5M), whereas replicationstress inducers reduced the viability and cell number and induced DNA damage in WT NSCs (Figures S5D and S5E), suggesting that replication stress contributes to the defects of Lats1/2 dKO NSCs.
Hypertranscription Contributes to Defects of LATS-Deficient NSCs
We next investigated the potential mechanistic link between YAP/TAZ activation and replication stress. Given that YAP/TAZ are potent transcription activators, we were intrigued by reports that increased global transcription activity, also known as hypertranscription (Percharde et al., 2017a), could induce replication stress (Kotsantis et al., 2016; Stork et al., 2016). Therefore, we monitored the global transcription activity in control and Lats1/2 dKO NSCs. We quantified nascent RNA synthesis by using nuclear incorporation of 5-ethynyluridine (EU) for 1 h (Jao and Salic, 2008) and found that RNA synthesis was significantly elevated in dKO cells (Figures 6A and 6B). This increase was not affected by the additional removal of p53 but was eliminated by the additional removal of Yap;Taz (Figure 6C), indicating that hypertranscription was induced by YAP/TAZ activation and was upstream or independent of p53 activation. Fast-proliferating cells often have elevated RNA synthesis (Percharde et al., 2017a); however, this cannot be why RNA synthesis was higher in Lats1/2 dKO cells because they proliferated more slowly than control cells.
Figure 6. Hypertranscription Contributes to Defects of LATS-Deficient NSCs.

(A) Representative images of EU staining after 1 h of labeling.
(B) Quantification of nuclear EU intensity. Left panel: EU intensity of individual cells (mean ± SD). Right panel: Average intensity of each culture.
(C) Average EU intensity of NSCs with the indicated floxed alleles transfected with mCherry or Cre-mCherry plasmid.
(D) Dose-response curves for NSCs treated with the transcription inhibitor DRB for 24 h. Comparisons are against the results of vehicle-treated dKO NSCs. The dashed line shows the number of live cells plated at 0 h.
(E) Effects of treating dKO NSCs with DRB on EU incorporation, γH2AX, and p53 activation.
(F) Effects of treating dKO NSCs with DRB on the replication fork speeds, sister-fork ratios, and two-segment ratios. For the fork ratios, Wilcox rank-sum test and Ansari-Bradley test were used to compare the median and dispersion, respectively.
(G) Immunostaining and quantification of NSCs of the indicated genotypes grown in differentiation medium with transcription inhibitors for 3 days.
Data are shown as mean ± SEM unless otherwise noted; *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant. See also Figure S6.
To determine whether hypertranscription was causally involved in the defects of Lats1/2 dKO NSCs, we inhibited transcription with an elongation inhibitor, DRB (Bensaude, 2011). After 24 h of DRB treatment, the viability and cell number of control NSCs were reduced as expected, but—remarkably—those of dKO cells increased significantly (Figure 6D). DRB also reduced DNA damage, as detected by γH2AX staining, and p53 activation in dKO cells (Figure 6E). Replication stress in dKO cells was partially alleviated; although the fork speed was not improved, fork asymmetry was significantly reduced by DRB (Figure 6F). Flavopiridol, another elongation inhibitor, and triptolide, an initiation inhibitor (Bensaude, 2011), also improved the viability and cell number and alleviated replication stress in dKO cells (Figures S6A–S6C). We then tested whether hypertranscription was also involved in the differentiation defect of dKO cells. For this experiment, we used Cre-transfected Lats1/2F/F;p53F/F NSCs to avoid the complication of cell death. These cells failed to differentiate when grown in differentiation medium (Figure 6G), similar to Lats1/2 dKO NSCs (Figure 4B). Both DRB and flavopiridol greatly enhanced neuronal differentiation of these cells, although they had only mild effects on control (p53F/F;Cre and mCherry-transfected Lats1/2F/F;p53F/F) cells (Figure 6G), indicating that they are not strong differentiation inducers per se. Together, these data suggest that YAP/TAZ– induced hypertranscription is an important cause of LATS-deficiency phenotypes.
Despite the increase in nascent transcription, protein synthesis was not elevated in Lats1/2 dKO NSCs (Figure S6D), possibly because of the slower proliferation rate of dKO cells. Interestingly, EU pulse labeling in WT E14.5 embryos revealed markedly higher levels of RNA synthesis in RGCs than in differentiating neurons (Figure S6E), raising the possibility that YAP/TAZ contribute to the high levels of RGC transcription during normal development.
Global Transcriptome Shift in LATS-Deficient Brains
To further verify the presence of hypertranscription in LATS-deficient NPCs in vivo and to determine the scope of the transcriptional changes, we performed cell-number–normalized (CNN) RNA-seq (Percharde et al., 2017b) of E12.5 brains. At this stage, the morphologic differences between control and Lats1/2;Nestin-Cre dKO brains were subtle, and cell death, DNA damage, and p53 activation were undetectable in dKO brains by immunostaining (Figure S2A). We collected equal numbers of cells from control and dKO telencephalons and added equal amounts of synthetic External RNA Controls Consortium (ERCC) spike-in poly(A) RNAs before RNA extraction. The total RNA yield of dKO cells was significantly higher than that of control cells (Figure 7A). Consistent with this, RNA-seq read counts showed that the fraction of ERCC reads among the total reads was larger in control than in dKO samples (Figure S7A). However, control and dKO brains expressed similar numbers of genes, regardless of whether the expressed genes were defined as those having at least 1 count per million (cpm) RNA-seq reads (Figure 7B) or as those having the promoter chromatin mark H3K4me3 within 5 kb of the transcription start site (TSS) (Figure S7B).
Figure 7. CNN RNA-seq Reveals Global Transcriptome Shift in LATS-Deficient Brains.

(A) Quantification of total RNA content per cell for E12.5 control and Lats1/2;Nestin-Cre dKO telencephalic cells (mean ± SEM).
(B) Venn diagram showing overlap of expressed genes defined by having at least 1 cpm RNAseq reads in E12.5 control and dKO brains.
(C) Gene expression heatmaps for the top 3000 genes that were most varied in read-depth–normalized and ERCC–normalized data.
(D) MA plot of differentially expressed genes (FDR < 0.05). Colored dots highlight genes with FDR < 0.01 and with the following fold changes (FC): maroon, log2FC > 1; red, log2FC > 0.5; blue, log2FC < −0.5; black, log2FC < −1.
(E) Histograms showing gene expression changes in E12.5 dKO telencephalic cells relative to equal numbers of control cells as measured by NanoString without normalization (left panel) or after normalization to housekeeping genes (right panel).
(F) Comparison of NanoString and CNN RNA-seq data. Pearson correlation analysis.
(G) Top GO Biological Processes terms enriched in the ERCC-4268 and RD-522 gene sets.
(H) Top transcription factors whose targets (predicted based on ENCODE ChIP-seq data) were enriched in the ERCC-4268 and RD-522 gene sets.
(I) qRT-PCR analysis of some of the upregulated genes identified by CNN RNA-seq (mean ± SEM).
*P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant. See also Figure S7, and Tables S2 and S3.
To compare the effects of ERCC normalization and conventional read-depth normalization, we generated heatmaps of data obtained with both methods (Figure 7C). Whereas read-depth normalization detected similar numbers of up- and down-regulated genes in dKO vs. control brains, ERCC normalization revealed that many genes were upregulated in dKO brains and only a few were downregulated. An MA plot of significantly changed genes [false discovery rate (FDR) < 0.05] called by ERCC normalization also shows a clear global increase in transcript levels (Figure 7D and Table S2).
Next, we employed NanoString technology to directly quantify gene expression differences in another set of equal numbers of control and dKO telencephalic cells without an enzymatic reaction, such as amplification (Geiss et al., 2008). The raw NanoString counts showed that most of the 770 genes in the NanoString panel were upregulated, whereas normalizing the raw counts to housekeeping genes resulted in similar numbers of up- and down-regulated genes (Figure 7E). The trends of changes detected by CNN RNA-seq and NanoString correlated well (Figure 7F). These results validated the global upregulation of transcript levels in Lats1/2 dKO brains.
To exclude the possibility that the increased RNA levels were simply due to increased RNA stability in dKO cells, we analyzed intron reads, because regulation of RNA stability affects mature mRNAs but should not affect intron levels (Wu and Brewer, 2012). Intron levels were globally elevated in dKO cells (Figure S7D and S7E). RNA splicing in dKO cells was normal (Figure S7F), indicating that the intron level increase was not due to abnormal intron retention. These results further confirmed the presence of hypertranscription in Lats1/2 dKO brains.
Many Genes Associated with Increased Biosynthetic Capacity and Proliferation are Upregulated in LATS-Deficient Brains
To determine what types of genes were upregulated in dKO brains, we performed gene ontology (GO) analysis of those genes with FDR < 0.01 by using Enrichr (Chen et al., 2013; Kuleshov et al., 2016). The 4268 upregulated genes [log2 fold change (log2FC) > 0.5] based on ERCC normalization (ERCC-4268) showed significant enrichment of terms for rRNA processing, translation, DNA replication, cell cycle, etc. (Figure 7G and Table S2). In contrast, none of these terms were enriched in the 522 upregulated genes (log2FC > 0.5) based on read-depth normalization (RD-522), and the most significantly enriched terms were ECM organization, Hippo signaling, and EMT, which is consistent with published reports (Lee et al., 2016; Su et al., 2015) (Figure 7G and Table S2). GO analysis of the 550 most strongly upregulated genes (log2FC > 1) by ERCC normalization, ERCC-550, found terms similar to those found by RD-522 (Table S2), which is not surprising because these two sets largely overlap (Figure S7C). These analyses indicate that YAP/TAZ activation upregulates a large set of genes associated with cell growth and proliferation. This effect is not detected by traditional read-depth normalization or by focusing on the most strongly upregulated genes, underscoring the importance of using appropriate normalization methods and analyzing the whole set of changed genes when global transcriptome shifts are present.
ERCC normalization revealed that more than 100 cytoplasmic and mitochondrial ribosomal protein genes were upregulated in dKO brains (Table S2). We performed CNN qRT-PCR for some of these genes and confirmed their upregulation (Figure 7I). These changes were undetectable when Gapdh was used for qRT-PCR normalization. In fact, both CNN RNA-seq and CNN qRT-PCR detected upregulation of Gapdh in dKO (Table S2 and Figure 7I). CNN qRT-PCR also detected upregulation of 18S rRNA and several primary (unspliced) Pol I and Pol II transcripts in dKO. Changes in rRNA levels could not be detected by our RNA-seq approach because rRNAs were depleted during library preparation.
YAP/TAZ and TEAD predominantly bind distal enhancers rather than promoters (Galli et al., 2015; Stein et al., 2015). They cooperate with other transcription factors such as AP-1 (a dimer of JUN and FOS) and Myc (Croci et al., 2017; Stein et al., 2015; Zanconato et al., 2015). To gain insights into the transcriptional mechanism underlying hypertranscription in dKO brains, we examined the results of a transcription-factor (TF)/target-gene enrichment analysis performed by Enrichr based on ENCODE TF ChIP-seq data (Chen et al., 2013). Surprisingly, the ERCC4268 gene set showed extraordinary overlap with E2F and Myc-Max targets (Figure 7H and Table S2). Ingenuity pathway analysis of ERCC-4268 also identified Myc as the top upstream regulator (P = 2.93E−81). These results agree with a report that Myc and YAP–TEAD coordinately regulate genes required for cell proliferation (Croci et al., 2017). Indeed, ERCC4268 overlaps significantly with the 2196 genes identified by Croci et al. as being upregulated by Myc and YAP co-overexpression in 3T9 fibroblasts (3T9–2196) (843 genes overlap, P = 1.5E−29 by Fisher’s exact test). Given that Myc promotes transcriptional amplification or hypertranscription in cancer cells, lymphocytes, and mouse primordial germ cells (Lin et al., 2012; Nie et al., 2012; Percharde et al., 2017b), we investigated whether it was involved in the hypertranscription of Lats1/2 dKO NSCs. We treated NSCs with a potent Myc inhibitor, 10058F4 (Yin et al., 2003). Over a wide dosage range, the Myc inhibitor had no effect on NSC viability, cell number, or EU incorporation (Figure S7G), although it did impair the colony morphology, proliferation, and pluripotency marker expression of mouse embryonic stem cells (mESCs), consistent with the reported functions of Myc in mESCs (Smith et al., 2010) (Figures S7H and S7I). These data suggest that Myc is not required for YAP/TAZ–induced hypertranscription. Corroborating this finding, Myc and Max genes were upregulated less than 2fold in dKO brains (Table S2 and Figure S7J), whereas in the studies cited above, Myc protein or mRNA levels were elevated more than 10-fold over baseline. Together, these results suggest that many genes upregulated in Lats1/2 dKO brains are Myc targets; however, their upregulation does not depend on Myc when YAP/TAZ are highly activated.
Applying the same TF/target-gene analysis to the RD-522 and ERCC-550 gene sets revealed significant overlap with TEAD4 and JUN targets, but not with E2F or Myc targets (Figure 7H and Table S2), and there is no significant overlap between them and 3T9–2196 (P > 0.1). ERCC4268 also significantly overlaps with TEAD4 and JUN targets, but to a much lesser degree than with E2F and Myc targets (Figure 7H and Table S2). Because Enrichr predicts and ranks target genes based on the distance of the TF ChIP-seq peaks from the TSS (Chen et al., 2013), it preferentially identifies TF/target-gene pairs in which the TF binds close to the TSS of the target gene. Therefore, these results suggest that the genes that were strongly upregulated in Lats1/2 dKO brains (ERCC-550 and RD-522) tend to have TEAD and JUN binding sites near their TSSs. Verteporfin, which disrupts YAP–TEAD interaction (Liu-Chittenden et al., 2012), increased the viability and cell number and lowered nascent transcription of dKO NSCs (Figure S7K), suggesting that TEAD is involved in the hypertranscription of LATS-deficient cells, although this experiment does not indicate whether the upregulated genes were direct TEAD targets.
DISCUSSION
By studying the effects of Lats1/2 deletion on mouse brain development, we made two important discoveries. First, YAP/TAZ activation resulting from Lats1/2 deletion leads to massive apoptosis of NPCs instead of the overgrowth typically associated with YAP/TAZ activation. Second, YAP/TAZ activation leads to hypertranscription and upregulates many genes associated with increased biosynthetic capacity and proliferation. These findings raise several important questions.
First, is hypertranscription a common effect of YAP/TAZ activation? Although microarray and RNA-seq have been routinely used to study transcriptional changes caused by YAP/TAZ activation, these approaches cannot detect global shifts unless appropriate normalization methods are applied (Lovén et al., 2012; Percharde et al., 2017a). By comparing normalization using ERCC spike-in RNAs added based on the cell number with traditional read-depth normalization, we demonstrated that many transcripts are elevated in LATS-deficient brains and that this change cannot be detected by read-depth normalization. Moreover, ERCC normalization revealed upregulation of many genes associated with cell growth and the cell cycle, consistent with the known function of YAP/TAZ in promoting proliferation, whereas read-depth normalization detected mainly changes in genes involved in ECM organization and EMT. Thus, our approach yielded insights into YAP/TAZ function. We propose that similar approaches be used to reassess the transcriptional effects of YAP/TAZ activation in other contexts. For example, is YAP/TAZ–driven hypertranscription present in human tumors showing YAP/TAZ activation? Experiments to address this question must be designed carefully to ensure that any observed global transcriptome shift is not due to differences in the cell state, such as the proliferation rate (Kress et al., 2015).
How does YAP/TAZ activation lead to hypertranscription? YAP/TAZ may directly bind and upregulate many genes. Alternatively, they may bind and upregulate just a few genes that in turn activate many others. Distinguishing between these possibilities is not easy, because YAP/TAZ and TEAD mostly bind distal enhancers and accurately assigning distal enhancers to their target genes on a genomic scale is challenging. Nevertheless, our bioinformatic analyses provided some important insights. The genes upregulated in LATS-deficient brains were highly enriched for Myc targets, which is interesting because Myc has been shown to promote transcriptional amplification or hypertranscription (Lin et al., 2012; Nie et al., 2012; Percharde et al., 2017b). YAP/TAZ and Myc both promote transcription elongation by recruiting P-TEFb to its target genes (Galli et al., 2015; Rahl et al., 2010). However, Myc at physiologic levels binds mainly active promoters and starts binding active distal enhancers when overexpressed (Kress et al., 2015), whereas YAP/TAZ and TEAD predominantly bind distal enhancers (Galli et al., 2015; Stein et al., 2015). We propose a model in which many genes involved in cell growth and proliferation are co-regulated by Myc and YAP/TAZ–TEAD, with Myc binding at their promoters and YAP/TAZ–TEAD binding at their distal enhancers. Myc and YAP/TAZ may act synergistically (Croci et al., 2017) or independently, e.g., by responding to different signals or having different dynamics (early vs. late or transient vs. long-lasting responses). The mode of interplay between Myc and YAP/TAZ may depend on their respective levels and the cell type. For example, although our data suggest that Myc is not required for YAP/TAZ-induced hypertranscription in LATS-deficient NSCs, it might be required when YAP/TAZ levels are lower or in other cell types.
What are the cellular consequences of YAP/TAZ–induced hypertranscription? Based on our finding that the upregulated genes are highly enriched for those associated with increased cell growth and the cell cycle, we propose that hypertranscription is a mechanism by which YAP/TAZ activation drives proliferation. Indeed, LATS-deficient NPCs exhibit an accelerated cell cycle, albeit transiently. The upregulated genes are also enriched for those associated with negative regulation of the apoptotic process (Figure 7G and Table S2), consistent with the established role of YAP/TAZ in promoting cell survival. In NPCs, however, hypertranscription eventually leads to DNA replication stress, DNA damage, and apoptosis. These detrimental effects might result from the induction of specific genes. Alternatively, elevated transcription activity itself could exacerbate the conflicts between replication and transcription as the two large machineries move along the same DNA template in proliferating cells, causing replication stress and DNA damage (Hamperl and Cimprich, 2016). The response of a given cell type to YAP/TAZ–induced hypertranscription most likely depends on how well that cell type handles and tolerates transcription-replication conflicts, replication stress, and DNA damage. NPCs are particularly sensitive to DNA damage, and their threshold for activating apoptosis is very low (McKinnon, 2017), which is probably why NPCs undergo cell death despite the upregulation of anti-apoptotic genes. Hypertranscription may trigger replication stress and DNA damage in other cell types, e.g., the Lats1/2-deficient adult hepatocytes that show increased senescence and death (Lee et al., 2016). On the other hand, in tumors with YAP/TAZ activation, hypertranscription may promote tumor progression by driving proliferation and survival and by inducing replication stress, the latter of which in turn generates genomic instability and selects for escape from apoptosis (Macheret and Halazonetis, 2015).
Lastly, how are LATS and YAP/TAZ regulated under physiologic and pathologic conditions? Our data suggest that, in early stages of brain development, LATS are kept on and YAP/TAZ are largely shut off. However, this dynamic may change at different developmental stages or in different regions of the nervous system. Indeed, P0 Lats1/2;Yap;Taz KO cortices are thinner than control cortices and contain fewer RGCs (Figures 3A–3C and S3A), which may reflect a requirement for YAP/TAZ in later stages of brain development. The Hippo pathway core components are regulated by a wide range of signals and cellular processes (Meng et al., 2016). Although this suggests that the Hippo pathway can sense and integrate diverse types of signals, the relative impact of these signals on YAP/TAZ activity almost certainly depends on the cell type and physiologic conditions. Much work is needed if we are to obtain a clear and comprehensive understanding of the operational logic behind this complex signaling network centered around YAP/TAZ regulation.
STAR METHODS
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by, the Lead Author, Dr. Xinwei Cao (Xinwei.cao@stjude.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The following mouse strains were used: Lats1flox and Lats2flox (Heallen et al., 2011), Nestin-Cre (JAX stock #003771), Emx1-Cre (JAX stock #005628), p53flox (JAX stock #008462), and Yapflox and Tazflox (Xin et al., 2011). All mice were maintained in a mixed genetic background. All animal procedures were approved by the Institutional Animal Care and Use Committee of St. Jude Children’s Research Hospital. Mouse housing and husbandry conditional followed the standards set by the Animal Resource Center at St. Jude Children’s Research Hospital. Embryonic (E) day 12–15 and postnatal (P) day 0 mice were used for this study. Their sex was not determined.
Neural Stem Cell (NSC) Cultures
Cortices of E12.5 or E13.5 mouse embryos were dissected in cold DMEM/F12. Each culture was derived from an independent embryo. Cells were dissociated with StemPro Accutase (Thermo Fisher Scientific A1110501) and plated at 50,000 cells/ml in petri dishes and grown in NSC maintenance medium composed of DMEM/F12 (Thermo Fisher Scientific 11320033), N2 (Thermo Fisher Scientific 17502048), B27 (Thermo Fisher Scientific 17504044), 20 ng/ml FGF2 (Miltenyi Biotec 130–093-841) and 20 ng/ml EGF (Miltenyi Biotec 130–093-825) at 37°C, 5% CO2. After 3 days, neurospheres were dissociated with StemPro Accutase into single cell suspensions and plated at 50,000 cells/ml in NSC maintenance medium either in petri dishes for neurosphere cultures or on poly-L-Lysine (30 μg/ml) and laminin (3 μg/ml) coated coverslips for adherent cultures. These cells are considered passage-1 NSCs.
Mouse Embryonic Stem Cell (ESC) Cultures
JM8A3.N1 mouse ES cells (Pettitt et al., 2009) were grown in DMEM high glucose (Thermo Scientific 11960044) supplemented with 15% FBS (Thermo Scientific 10439024), non-essential amino acids (NEAA) (Thermo Scientific 11140050), L-glutamine (Thermo Scientific 25030081), 0.1 mM 2-mercaptoethanol (Sigma M6145) and 2000 U/ml LIF (Millipore ESG1107) on irradiated mouse embryonic fibroblasts in 12-well plates.
METHOD DETAILS
Plasmids
pCAG3-IRES-EGFP (pCIG3) was generated by inserting an MCS-IRES-EGFP cassette into pCAGGS. YAP (Addgene #19045), YAP5SA (Addgene #27371), and YAP5SA/S94A (Addgene #33103) were gifts from Kun-Liang Guan. The coding sequences were PCR-amplified and inserted into pCIG3. pCAGGS-mCherry (Addgene #41583) was a gift from Phil Sharp. pCAGCre-T2A-mCherry and a modified Fucci plasmid (myr-hmAG1-T2A-hmKO2-Geminin) were gifts from David Solecki.
Quantitative Western Blot and Subcellular Fractionation
E12.5 and E13.5 mouse brains were dissected and total cell lysates were prepared in 20 mM HEPES (pH 7.4), 150 mM NaCl, 2% SDS and 5% glycerol supplemented with AEBSF and Halt protease and phosphatase inhibitors (Thermo Fisher Scientific). Protein concentrations were measured by BCA assay (Thermo Scientific). 20 μg of protein per lane were subjected to SDSPAGE, probed with primary antibodies and infrared conjugated secondary antibodies (LI-COR, IRDye 680LT and 800CW), and detected using the ODYSSEY imaging system (LI-COR). Quantification was performed with Image Studio software ver 5.2 (LI-COR). Cytoplasmic and nuclear fractionation was performed by using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific 78833) according to the manual. For each E13 brain, 200 μl of CER I, 11 μl of CER II, and 100 μl of NER were used. 5% of each fraction was loaded per lane for quantitative Western blots. Each lane in Western blot images and each data point in quantification graphs correspond to an individual embryo.
In Utero Electroporation
Pregnant mice carrying E12.5 embryos were deeply anesthetized with 2.5–3% of isoflurane and the uterine horns exposed. Approximately 2 μl of DNA (2 μg/μl) mixed with 0.1% fast green (Sigma) in PBS was injected into a lateral ventricle of the embryo brain using a pulled glass micropipette. Embryo head was placed within a 3-mm tweezer-type electrodes, and square-wave electric pulses (30 V, 50 msec) were delivered 5 times at 950 msec intervals using an electroporator (BTX ECM 830, Harvard Apparatus). Embryos were returned to dam and allowed to resume normal development before being harvested for subsequent analyses.
Histology, Immunostaining, TUNEL Assay, and Image Acquisition
Mouse brains were dissected in PBS, fixed overnight in 4% paraformaldehyde (PFA) at 4°C, and washed in PBS. For paraffin sections, fixed brains were dehydrated in 70% ethanol overnight, embedded in paraffin, and sectioned at 7-μm thickness. For histological analysis, sections were rehydrated and stained with hematoxylin and eosin (H&E). For immunostaining, after rehydration, sections were incubated for 30 min in 10 mM Sodium Citrate (pH 6.0) at 95°C for antigen retrieval. After cooling down at room temperature (RT) for 20 min, sections were washed twice in PBS, blocked and permeabilized in PBS with 3% normal donkey serum and 0.2% Triton X-100 for 1 h, and incubated with primary antibodies at 4 °C overnight. Sections were then washed in PBS 3 times with 15-min intervals and incubated with appropriate fluorescence–conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) at 1:1000 dilution and DAPI (1:1000 dilution) for 2–3 h at RT. After three washes in PBS, sections were coverslipped in ProLong Gold (Thermo Scientific Invitrogen). For DAB (3,3′diaminobenzidine tetrahydrochloride) staining, a horseradish peroxidase (HRP)–conjugated secondary antibody (Jackson ImmunoResearch Laboratories) was used at 1:250 dilution. After washes in PBS, sections were incubated in the ImmPACT DAB Peroxidase (HRP) Substrate (Vector Laboratories SK-4105) to desired color development and immediately washed in tap water. For frozen sections, fixed brains were cryoprotected in 30% sucrose overnight, embedded in Tissue Freezing Medium (Thermo Scientific), and sectioned at 10–14-μm thickness.
Immunostaining was carried out as described above without rehydration and antigen retrieval. TUNEL assay was performed using the ApoTag Plus In Situ Apoptosis Fluorescein Detection Kit (Millipore S7111). Images of H&E–stained sections were acquired using a Zeiss Stereo Discovery V8 microscope equipped with an AxiocamMRc camera and processed with Axiovision. Low magnification fluorescent images were acquired using an Olympus BX63 microscope equipped with an Orca Flash4.0 camera. High magnification fluorescent images were acquired using a Zeiss LSM 780 confocal microscope.
Image Quantifications
All images used for cell counting were acquired using a Zeiss LSM 780 confocal microscope and a 40x objective lens. Cell numbers were counted manually using ImageJ. For each genotype, 3–4 embryos with one section of comparable positions per embryo were quantified. For cortical ventricular zone area measurements at E12.5, images of Pax6 DAB staining were acquired using an Olympus BX63 microscope. Areas of interest were outlined and measured using the Olympus CellSens Dimension analysis program (Olympus). For each genotype, 3–4 embryos with one section of each rostrocaudal position per embryo were quantified. For quantification of TUNEL, cleaved caspase-3, and Pax6 fluorescent signals, image acquisition was performed below saturation with the same settings for all the samples. The areas containing TUNEL and cleaved caspase-3 signals were measured using the Imaris 9.1 software (Bitplane) and shown as percentages of DAPI+ areas. The areas containing Pax6 signals in the cortex were measured using Imaris and shown as total areas. For each genotype, 3 animals with 8 (E13.5) or 6 (P0) images per animal were analyzed. For quantification of the in utero electroporation experiments, processed image stacks were segmented and GFP+ cells were automatically counted and visually confirmed using Imaris. For each transfection, 3 embryos with one section per embryo were quantified. Data points in all quantification graphs correspond to individual embryos.
Calculation of in Vivo Cell Cycle Parameters
Pregnant dams were injected intraperitoneally with EdU (10 μg/g body weight) and followed by BrdU (50 μg/g body weight) 1.5 h later. Dams were killed at 2 h following EdU administration, and embryos were fixed. EdU was detected by the Click-it Plus EdU Alexa Fluor 488 Imaging Kit (Thermo Scientific Invitrogen), followed by immunostaining with Sox2, Tbr2 and BrdU antibodies. To calculate the cell cycle parameters of RGCs (Sox2+Tbr2−), the total number of Sox2+Tbr2− cells (Cyclingfraction), the number of Sox2+Tbr2− cells in the S phase (Sfraction, Sox2+Tbr2−EdU+BrdU+) and the number of Sox2+Tbr2− cells in the “leaving fraction” (Lfraction, Sox2+Tbr2−EdU+BrdU−) were counted manually using ImageJ. S-phase length (TS) and total cell-cycle length (TC) were then calculated using the following equations:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
In vivo EU labeling and detection
A pregnant dam carrying E14.5 embryos was injected intraperitoneally with EU (150 μg/g body weight) and embryos were collected 3 h later (Jao and Salic, 2008). Embryos and the dam were fixed and processed as described above. EU was detected by the Click-it RNA Alexa Fluor 488 Imaging Kit (Thermo Scientific Invitrogen C10329), followed by immunostaining with Pax6, Tbr2 and Ki67 antibodies.
NSC Culture Assays
All assays were performed on passage-1 NSC cultures.
Immunostaining
Free floating neurospheres or NSCs grown on poly-L-Lysine (30 μg/ml) and laminin (3 μg/ml) coated coverslips were fixed in 4% PFA for 15 min at RT, permeabilized in PBS with 0.001% Triton X-100 for 10 min, blocked in PBS with 10% fetal calf serum for 30 min, and incubated with primary and secondary antibodies for 1–3 h at RT.
Differentiation assay
Passage-1 NSCs were plated in differentiation medium composed of DMEM/F12, N2, B27, and 5% horse serum (Thermo Fisher Scientific 26050088) at 50,000 cells/ml on poly-L-Lysine and laminin coated coverslips for 1, 3, or 5 days. For differentiation assays in the presence of transcription inhibitors, cells were grown for 3 days and medium was changed every day with fresh inhibitors.
Quantification of neurosphere numbers and viability
Neurosphere number was manually counted 3 days after plating using gridded scoring dishes (Stem Cell Technologies 27500). For viability and cell number quantifications, neurospheres were dissociated with StemPro Accutase into single cell suspensions and an aliquot was mixed with trypan blue before being measured by the Countess automated cell counter (Thermo Fisher Scientific).
Electroporation
NSCs grown as neurospheres were dissociated with StemPro Accutase into single cell suspensions. 5 million cells were electroporated with 2 μg of mCherry or Cre-T2A-mCherry plasmid using the Mouse Neural Stem Cell Nucleofector kit according to the manufacturer’s protocol (Lonza VPG-1004) and plated in NSC maintenance medium. For each plasmid, 2 electroporations per culture were performed to produce enough mCherry+ cells for subsequent assays. The following day, mCherry+ cells were purified by FACS, re-plated at 50,000 cells/ml, cultured for 3 days and then used for analyses.
Cell cycle profile analysis by propidium iodide (PI) staining and flow cytometry
105 dissociated NSCs were stained with PI and analyzed by St. Jude Children’s Research Hospital Flow and Cell Sorting Shared Resource. Results were plotted using the Modfit software.
EU incorporation assay
Passage-1 NSCs were plated in NSC maintenance medium on poly-L-Lysine and laminin coated coverslips and, on the following day, labeled with 1 mM of EU for 1 h. Cells were then fixed and EU signal detected using the Click-It RNA imaging kit (Thermo Fisher Scientific C10329). Unlabeled cells (negative control) were also subjected to EU detection and showed minimal background signal. Image acquisition was performed below saturation with the same laser settings for all the samples using a Zeiss LSM 780 confocal microscope and a 40x objective lens. DAPI signal was used to delimit the nucleus and mean nuclear EU signal intensities per pixel per nucleus were quantified using Imaris. For each condition (genotype and treatment), 3 cultures with 100 cells per culture were analyzed.
Inhibitor treatments
Passage-1 NSCs were plated at 50,000 cells/ml in 6-well plates in NSC maintenance medium containing inhibitors at indicated dosages for 24 h. For CDK4/6 inhibitor PD 0332991 treatment, dead cells were removed using the Dead Cell Removal Kit (Miltenyi Biotec 130–090-101) before plating. 3 control and 3 Lats1/2 dKO cultures were treated for each condition.
Quantifications of mitotic defects, DNA damage, and p53 activation
Passage-1 NSCs were plated in NSC maintenance medium at 50,000 cells/ml on poly-L-Lysine and laminin coated coverslips. The following day, immunostaining was performed as described above. To examine mitotic defects, cells were stained with phospho-Histone H3 Ser10 (pHH3) and γ-Tubulin antibodies and imaged with a Zeiss LSM 780 microscope at 63x with a 1.5 optical zoom. Chromosome distribution, morphology, and microtubule organization was used to determine mitotic phases. 3 cultures per genotype with 100 mitotic cells per culture were analyzed. To examine DNA damage and p53 activation, cells were stained with γH2AX and p53 antibodies and imaged with a Zeiss LSM 780 microscope at 40x. DAPI signal was used to delimit the nucleus area and mean nuclear γH2AX signal intensities per pixel per nucleus were quantified with Imaris. The presence of nuclear p53 signal was quantified manually. 3 cultures per condition (genotype and treatment) with 50 cells per culture were analyzed.
Protein synthesis assay
Nascent protein synthesis was assessed using the surface sensing of translation (SUnSET) assay as described (Schmidt et al., 2009). Passage-1 NSCs were grown on poly-L-Lysine and laminin coated coverslips and incubated with 10 μg/ml of puromycin for 10 min or with 25 μM of cyclohexamide for 5 min and then with 10 μg/ml of puromycin for 10 min. Cells were fixed and permeabilized as described above, and stained with a mouse anti-Puromycin antibody using a MOM kit (Vector Laboratories). Image acquisition was performed below saturation with the same camera settings for all the samples using an Olympus BX63 microscope and a 40x objective. Total Puromycin immunofluorescence signals per cell were measured using Imaris. For each condition (genotype and treatment), 3 cultures with 50 cells per culture were analyzed.
Live Imaging of NSCs
E13.5 cortices were dissociated with StemPro Accutase and cells electroporated with a modified Fucci plasmid using the Mouse Neural Stem Cell Nucleofector kit (Lonza VPG-1004). The plasmid encodes a mAG (the monomeric version of Azami Green) fused to the Src myristoylation signal and, connected via a T2A peptide, a mKO2 [a fast-folding variant of mKO (monomeric version of Kusabira Orange)] fused to human Geminin1−110 that accumulates in S/G2/M phases of a cell cycle. After electroporation, cells were resuspended in NSC maintenance medium and plated at 20,000 cells/ml in a μ-Slide 2 Well (Ibidi 80286) coated with 50 μg/ml of fibronectin (Thermo Fisher Scientific PHE0023) and covered with a DIC lid for μSlides (Ibidi 80055). After an overnight incubation, mKO2+ (orange) cells were imaged for 14 h by time-lapse microscopy using a Nikon C2 confocal microscope with an environmental chamber at 37°C, 5% CO2 using a 20x objective lens with 15-min imaging intervals. For each imaging session, cells derived from one control and one Lats1/2 dKO embryos were imaged. 4 sessions, using cells derived from 4 different control and dKO embryos, were performed and a total of 191 control and 186 dKO cells recorded. Data were analyzed using the NIS-Elements software.
DNA Fiber Assay and Data Analysis
This assay was performed as previously described (Nieminuszczy et al., 2016; Schwab and Niedzwiedz, 2011). Passage-1 NSCs were incubated with 25 μM of IdU for 20 min followed with 250 μM of CldU for 20 min in NSC maintenance medium at 37°C. Cells were then harvested by centrifugation and resuspended to 5×105 cells/ml. 2 μl of the cell suspension was air-dried on a glass slide for 3 min and lysed in 200 mM Tris-HCl (pH 7.5), 50 mM EDTA and 0.5% SDS solution for 2 min. Slides were tilted at a 15° angle to let the fibers spread, air-dried for 5 h, and treated with methanol/acetic acid (3:1) for 10 min and 2.5 M HCl for 80 min. IdU and CldU were detected using specific antibodies. DNA fibers were imaged using a Zeiss LSM 780 confocal microscope at 63x with a 2x optical zoom. For each genotype, 3 cultures were analyzed. From each culture, 100 to 125 fibers were imaged and measured. Fiber length was measured with ImageJ. For the statistical analyses of sister-fork ratios and 2-segment ratios, since the data deviated from normal distribution (determined by Shapiro test for normality, both control and dKO groups were associated with P < 0.0001), the tests for comparing the median and dispersion were computed by using nonparametric tests. The permutation-based Wilcox rank-sum test was used for median test and the permutation-based Ansari-Bradley test was used for dispersion test. These analyses were performed using R 3.4.2. Codes are available upon request.
Affymetrix Microarray and Data Analysis
Total RNA was extracted from 3 control and 3 Lats1/2 dKO E12.5 cortices using Trizol. 100 ng of total RNA from each sample was converted to biotin-labeled cDNA using the Affymetrix WT protocol, then hybridized to an Affymetrix Mouse Gene 2.0 ST GeneChip (Life Technologies). Signals from scanned arrays were normalized and summarized by the RMA algorithm using the Affymetrix Expression Console software v1.1. Differentially expressed transcripts were identified by ANOVA (Partek Genomics Suite v6.6) and the false discovery rate (FDR) estimated using the Benjamini-Hochberg method. Because none of the transcripts achieved FDR < 0.05, those with P ≤ 0.05 and log2 fold change (log2FC) ≥ 0.5 or ≤ −0.5 were listed in Table S1. Gene set enrichment analysis was performed using curated pathways obtained from MSigDB (Broad Institute) as previously described (Subramanian et al., 2005). Ranking of genes was calculated using the signal-to-noise algorithm, and a P-value for each gene set was estimated by comparing the observed enrichment score to that obtained from a null distribution computed from 1000 permutations of genes within gene sets. FDR was estimated as previously described (Subramanian et al., 2005).
Cell Number Normalized (CNN) RNA-seq and Data Analysis
Telencephalons from E12.5 embryos were dissected, dissociated with StemPro Accutase, and passed through a 40-μm cell strainer to obtain single-cell suspensions. Cells were counted and 1.0 × 106 cells were pelleted by centrifugation. Diluted ERCC spike-in mix (1:20, Thermo Fisher Scientific 4456740) was added to cell pellets and Trizol prior to RNA extraction at a ratio of 1 μl of diluted ERCC spike-in mix per million cells. RNA was purified using a Direct-zol RNA kit (Zymo Research R2052) and eluted in 25 μl of elution buffer.
Library preparation and sequencing were performed by the Genome Sequencing Facility at St. Jude Children’s Research Hospital. RNA quality was checked by a 2100 Bioanalyzer RNA 6000 Nano assay (Agilent) before library generation. Libraries were prepared from 350 ng of total RNA using the TruSeq Stranded Total RNA Library Prep Kit (Illumina 20020597), then quantified using the Quant-iT PicoGreen dsDNA assay (Life Technologies). Samples were sequenced on Illumina HiSeq4000, obtaining 100 million 100-bp paired-end reads. RNAs from 5 control and 4 dKO embryos were sequenced.
Sequences were mapped to the mm9 genome with additional ERCC sequences using the STAR aligner (Dobin et al., 2013). Transcript level data was counted using HTSEQ (Anders et al., 2015). Raw counts were divided into transcript counts and ERCC counts. For quality control purposes, the linearity of the ERCC spike-in controls were considered and two spike-ins that consistently across all samples did not conform to the expected order (which is likely to be a manufacturer’s error) were excluded from the ERCC raw counts. Transcript read counts and ERCC read counts were then used to determine normalization factors. Data were voom fitted and contrasted to determine differentially expressed genes using the empirical Bayes method in limma in R 3.4.0 (Law et al., 2014). Data visualizations were performed in STATA/MP 14.2 (StataCorp). Intron read counts were analyzed in the same manner. The splicing score of each transcript was calculated using the following equation:
Bioinformatics analyses of differentially expressed genes were performed using Enrichr http://amp.pharm.mssm.edu/Enrichr/ (Chen et al., 2013; Kuleshov et al., 2016). Enrichment scores were displayed as combined scores, which is a combination of the P-value computed using the Fisher’s exact test and the z-score computed by assessing the deviation from the expected rank, in the main text. Full lists with a FDR (adjusted P value) cutoff of 0.01 are provided in Table S2.
NanoString Analysis
Total RNA from 3 control and 3 Lats1/2;Nestin-Cre dKO E12.5 telencephalons, each sample containing 1.0 × 106 dissociated cells, was extracted as described above. 7.5 μl of total RNA was subjected to NanoString nCounter analysis. The mouse PanCancer Pathways Panel was chosen because it contained ~200 genes that were found to be upregulated (log2FC > 0.5) by CNN RNAseq analysis. Raw counts were converted to log2 values and used to calculate log2FC of dKO vs. control. Data were plotted either without further normalization (raw) or normalized to designated housekeeping genes in the panel.
CNN qRT-PCR
Total RNA from 4 control and 4 Lats1/2 dKO E12.5 telencephalons, each sample containing 1.0 × 106 dissociated cells, was extracted as described above. 2 μl of total RNA was used to generate 200 μl of cDNA by using SuperScript III reverse transcriptase (Life Technologies). qPCR reactions were performed using 4.5 μl of cDNA and iTaq Universal SYBR Green Supermix (Bio-Rad) on a CFX96 Real-Time System (Bio-Rad). Each reaction was performed in technical triplicates and averaged. Average values were then grouped based on genotype and the fold change between control and dKO calculated. PCR primers were designed using IDT’s Realtime PCR tool and tested using a serial dilution (1 to 1:1024) of cDNA generated from 1 μg of total RNA. Primers were assessed based on linear amplification on a standard curve plot, amplification efficiency between 90−110%, and peak uniformity in a dissociation curve. Primers sequences are available upon request.
Chromatin Immunoprecipitation and Sequencing (ChIP-seq) and Data Analysis
Each E12.5 mouse brain was dissociated in 1 ml of cold PBS by pipetting. Protein−DNA complexes were crosslinked by incubating in 1% formaldehyde/PBS (Sigma F1635) for 10 min at RT with gentle rotation. Crosslinking was stopped with 125 mM glycine/PBS for 5 min and washed 3 times with cold PBS. Cells were lysed in 1 ml lysis buffer (50 mM HEPES-KOH [pH 7.5], 140 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.5% NP-40, 0.25% Triton X-100) supplemented with protease and RNAse inhibitors for 10 min at 4ºC with gentle rotation. Nuclei were pelleted at 1800 rcf for 2 min, washed with 1 ml wash buffer (10 mM Tris-HCl [pH 8.0], 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA) supplemented with protease and RNAse inhibitors for 10 min, and lysed in 900 μl nuclear lysis buffer (10 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na-Deoxycholate, 0.5% N-Lauroylsarcosine) for 10 min.
Lysed nuclei (300 μl/1.5 ml sonication tube) were sonicated for 5 cycles of 15-sec on and 45-sec off in a Qsonica Q800R2 sonicator. Chromatin was quantitated and diluted with nuclear lysis buffer to ~500 ng/μl and further sonicated for 10 cycles of 15-sec on and 45-sec off to obtain an average DNA fragment size of ~300 bp. Sonicated chromatin was centrifuged at 14,000 rpm for 10 min and the supernatant chromatin was either used for ChIP or flash frozen and stored at −80ºC.
10% of chromatin from each ChIP reaction was used as the input control. For each ChIP reaction, 30 μg of chromatin was incubated with 1.5 μg of H3K4me3 antibody (Millipore 07473) or normal rabbit IgG (Cell signaling #2729S) overnight at 4ºC and with 25 μl of Protein-A Dynabeads (Invitrogen) for 1 h. Beads were washed at RT with RIPA buffer (50 mM HEPESKOH [pH 7.6], 500 mM LiCl, 1 mM EDTA, 1% NP-40, 0.7% Na-deoxycholate) 5 times and with TE buffer (10 mM Tris-HCl [pH 8.0], 0.1 mM EDTA) once. Chromatin was eluted by incubating in 300 μl of elution buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA, 1% SDS) for 5 min at RT with vortexing. After reverse-crosslinking at 65°C overnight with the addition of 18 μl NaCl (5 M) and 2 μl proteinase K (10 mg/ml), DNA was extracted using a PCR purification kit (QIAGEN #28106) and quantified using the Quant-iT PicoGreen dsDNA assay.
Sequencing libraries were prepared from 10 ng of DNA using the NEBNext ChIP-Seq Library Prep Reagent Set for Illumina with NEBNext Q5 Hot Start HiFi PCR Master Mix according to the manufacturer’s instructions (New England Biolabs) with the following modifications: a second 1:1 Ampure cleanup was added after adaptor ligation and the Ampure size selection step prior to PCR eliminated. Completed libraries were analyzed for insert size distribution on a 2100 BioAnalyzer High Sensitivity kit (Agilent Technologies) or Caliper LabChip GX DNA High Sensitivity Reagent Kit (PerkinElmer). Libraries were quantified using the Quant-iT PicoGreen ds DNA assay (Life Technologies) and Kapa Library Quantification kit (Kapa Biosystems). Fifty-cycle single-end sequencing was performed on an Illumina HiSeq 2500 or HiSeq 4000.
ChIP-Seq reads were aligned to mm9 (MGSCv37) using the BWA software (version 0.7.12r1039, default parameter). Duplicated reads were marked using the Picard software (version 2.6.0-SNAPSHOT) and only non-duplicated reads used for analysis using SAMtools (parameter “-q 1 -F 1024” version 1.2). Data quality control (QC) follows ENCODE criteria. Relative strand correlation (RSC) values were calculated and the fragment size estimated under the support of R (version 2.14.0) by SPP (version 1.1) with packages caTools (version 1.17) and bitops (version 1.0–6). At least 10 million unique reads with RSC > 1 was required to pass QC. 2 control and 2 Lats1/2 dKO samples passed QC. Bigwig files were generated by extending each read to the best fragment size (the smallest fragment size estimated by SPP) and manually inspected using IGV genome browser for clear peaks and low background noise. MACS2 (version 2.1.1.20160309) was used to call peaks using the best fragment size with a cutoff of FDR < 0.05. Peak calling results for H3K4me3 ChIP were merged into two bed files, one for control and one for Lats1/2 dKO (bedtools 2.23.0). Peak annotation was performed on the merged results with Homer version 4.5. Peaks within +/− 5 kb of the transcription start site (TSS) were retained and deduplicated by gene name and the membership compared between dKO peaks and control peaks (STATA/MP 14.2).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using the GraphPad Prism 7 software unless otherwise noted in above sections. Statistical significance (P values) were calculated by unpaired two-tailed Student t-test unless otherwise noted. Statistical significance was denoted as follows: *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant. Area-proportional Venn diagrams were generated by using the eulerAPE software http://www.eulerdiagrams.org/eulerAPE/ .
Supplementary Material
A control cell that divided within the imaging period. Cells were transfected with a modified Fucci plasmid. The green color is from Azami Green fused to the Src myristoylation signal. The orange-red color is from Kusabira Orange fused to human Geminin1−110 that accumulates in S/G2/M phases of a cell cycle.
A Lats1/2;Nestin-Cre dKO cell that divided within the imaging period.
A dKO cell that died.
A dKO cell that did not divide within the imaging period.
Affymetrix microarray analysis comparing E12.5 control and Lats1/2;Nestin-Cre dKO cortices. Differentially expressed genes (P < 0.05) and associated Gene Set Enrichment Analysis (GSEA) using Hallmark gene sets and Canonical Pathways are listed.
CNN RNA-seq comparing E12.5 control and Lats1/2;Nestin-Cre dKO telencephalons. Differentially expressed genes called by ERCC normalization and read-depth normalization are listed for comparison.
NanoString nCounter analysis comparing RNAs extracted from equal numbers of E12.5 control and Lats1/2;Nestin-Cre dKO telencephalic cells. Raw counts and normalized (to housekeeping genes) counts are shown.
Highlights.
Loss of Hippo pathway LATS kinase leads to YAP/TAZ–driven global hypertranscription
YAP/TAZ–driven hypertranscription upregulates many growth and proliferation genes
Hypertranscription impairs neural progenitor differentiation and causes apoptosis
Cell-number–normalized methods are required to detect global hypertranscription
ACKNOWLEDGMENTS
We thank Dr. Richard Ashmun, Dr. Jennifer Peters, Jim Houston, Maria Kinsey, the Flow Cytometry & Cell Sorting facility, the Hartwell Center, and the Cell & Tissue Imaging Center at St. Jude Children’s Research Hospital for assistance with data acquisition and analysis. We thank Dr. Young-Goo Han for suggestions regarding the manuscript, Dr. Keith A. Laycock for scientific editing of the manuscript, and Joshua Stokes and Jessica Anderson for assistance with graphic design. A.L., J.Y.P., J.P., R.K., and X.C. are supported by R01 NS086938 and ALSAC.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
Microarray, RNA-seq, and ChIP-seq data associated with this manuscript can be found at the GEO repository under accession number GSE120016.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with highthroughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaude O (2011). Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity? Transcription 2, 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bylund M, Andersson E, Novitch BG, and Muhr J (2003). Vertebrate neurogenesis is counteracted by Sox1–3 activity. Nat. Neurosci 6, 1162–1168. [DOI] [PubMed] [Google Scholar]
- Cai J, Maitra A, Anders RA, Taketo MM, and Pan D (2015). β-Catenin destruction complex–independent regulation of Hippo-YAP signaling by APC in intestinal tumorigenesis. Genes Dev 29, 1493–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Pfaff SL, and Gage FH (2008). YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 22, 3320–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhang N, Xie R, Wang W, Cai J, Choi KS, David KK, Huang B, Yabuta N, Nojima H, et al. (2015). Homeostatic control of Hippo signaling activity revealed by an endogenous activating mutation in YAP. Genes Dev. 29, 1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton JL, Li Q, Ma L, Park JS, Wang J, Ou J, Zhu LJ, Ip YT, Johnson RL, and Mao J (2017). YAP/TAZ and Hedgehog coordinate growth and patterning in gastrointestinal mesenchyme. Dev. Cell 43, 35–47.e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croci O, De Fazio S, Biagioni F, Donato E, Caganova M, Curti L, Doni M, Sberna S, Aldeghi D, Biancotto C, et al. (2017). Transcriptional integration of mitogenic and mechanical signals by Myc and YAP. Genes Dev. 31, 2017–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, and Hevner RF (2005). Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J. Neurosci 25, 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli GG, Carrara M, Yuan WC, Valdes-Quezada C, Gurung B, Pepe-Mooney B, Zhang T, Geeven G, Gray NS, de Laat W, et al. (2015). YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol. Cell 60, 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, et al. (2008). Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol 26, 317–325. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, and Jones KR (2002). Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci 22, 6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtan AM, Lu V, Bhutkar A, and Sharp PA (2012). In vivo structure–function analysis of human Dicer reveals directional processing of precursor miRNAs. RNA 18, 1116–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, and Cimprich KA (2016). Conflict resolution in the genome: How transcription and replication make it work. Cell 167, 1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heallen T, Morikawa Y, Leach J, Tao G, Willerson JT, Johnson RL, and Martin JF (2013). Hippo signaling impedes adult heart regeneration. Development 140, 4683–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, and Martin JF (2011). Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332, 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergovich A, and Hemmings BA (2012). Hippo signalling in the G2/M cell cycle phase: Lessons learned from the yeast men and sin pathways. Semin. Cell Dev. Biol 23, 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao CY, and Salic A (2008). Exploring RNA transcription and turnover in vivo by using click chemistry. Proc. Natl. Acad. Sci. USA 105, 15779–15784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim YH, Kim J, Park DY, Bae H, Lee DH, Kim KH, Hong SP, Jang SP, Kubota Y, et al. (2017). YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J. Clin. Invest 127, 3441–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsantis P, Silva LM, Irmscher S, Jones RM, Folkes L, Gromak N, and Petermann E (2016). Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun 7, 13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress TR, Sabò A, and Amati B (2015). MYC: connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 15, 593–607. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, and Alvarez-Buylla A (2009). The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci 32, 149–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavado A, He Y, Paré J, Neale G, Olson EN, Giovannini M, and Cao X (2013). Tumor suppressor Nf2 limits expansion of the neural progenitor pool by inhibiting Yap/Taz transcriptional coactivators. Development 140, 3323–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Park JO, Kim TS, Kim SK, Kim TH, Kim MC, Park GS, Kim JH, Kuninaka S, Olson EN, et al. (2016). LATS-YAP/TAZ controls lineage specification by regulating TGFβ signaling and Hnf4α expression during liver development. Nat. Commun 7, 11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, and Young RA (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, and Pan D (2012). Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 26, 1300–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, Levens DL, Lee TI, and Young RA (2012). Revisiting global gene expression analysis. Cell 151, 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M, and Halazonetis TD (2015). DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol 10, 425–448. [DOI] [PubMed] [Google Scholar]
- McKinnon PJ (2017). Genome integrity and disease prevention in the nervous system. Genes Dev. 31, 1180–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello SS, and Attardi LD (2018). Deciphering p53 signaling in tumor suppression. Curr. Opin. Cell Biol 51, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Moroishi T, and Guan KL (2016). Mechanisms of Hippo pathway regulation. Genes Dev. 30, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L, Besson A, Heng JI, Schuurmans C, Teboul L, Parras C, Philpott A, Roberts JM, and Guillemot F (2006). p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev. 20, 1511–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al. (2012). c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieminuszczy J, Schwab RA, and Niedzwiedz W (2016). The DNA fibre technique— tracking helicases at work. Methods 108, 92–98. [DOI] [PubMed] [Google Scholar]
- Ota M, and Sasaki H (2008). Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development 135, 4059–4069. [DOI] [PubMed] [Google Scholar]
- Percharde M, Bulut-Karslioglu A, and Ramalho-Santos M (2017a). Hypertranscription in development, stem cells, and regeneration. Dev. Cell 40, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percharde M, Wong P, and Ramalho-Santos M (2017. b). Global hypertranscription in the mouse embryonic germline. Cell Rep. 19, 1987–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt SJ, Liang Q, Rairdan XY, Moran JL, Prosser HM, Beier DR, Lloyd KC, Bradley A, and Skarnes WC (2009). Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nat. Methods 6, 493–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger CM (2017). The Hippo pathway: a master regulatory network important in development and dysregulated in disease. Curr. Top. Dev. Biol 123, 181–228. [DOI] [PubMed] [Google Scholar]
- Pilaz LJ, McMahon JJ, Miller EE, Lennox AL, Suzuki A, Salmon E, and Silver DL (2016). Prolonged mitosis of neural progenitors alters cell fate in the developing brain. Neuron 89, 83–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, and Young RA (2010). c-Myc regulates transcriptional pause release. Cell 141, 432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginensi A, Enderle L, Gregorieff A, Johnson RL, Wrana JL, and McNeill H (2016). A critical role for NF2 and the Hippo pathway in branching morphogenesis. Nat. Commun 7, 12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, and Bonner WM (2000). Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem 275, 9390–9395. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, and Bonner WM (1998). DNA doublestranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868. [DOI] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. [DOI] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, and Pierre P (2009). SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275–277. [DOI] [PubMed] [Google Scholar]
- Schwab RA, and Niedzwiedz W (2011). Visualization of DNA replication in the vertebrate model system DT40 using the DNA fiber technique. JoVE e3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KN, Singh AM, and Dalton S (2010). Myc represses primitive endoderm differentiation in pluripotent stem cells. Cell Stem Cell 7, 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, Agarinis C, Schmelzle T, Bouwmeester T, Schübeler D, and Bauer A (2015). YAP1 exerts its transcriptional control via TEAD-mediated activation of enhancers. PLoS Genet. 11, e1005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork CT, Bocek M, Crossley MP, Sollier J, Sanz LA, Chédin F, Swigut T, and Cimprich KA (2016). Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. eLife 5, e17548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su T, Bondar T, Zhou X, Zhang C, He H, and Medzhitov R (2015). Two-signal requirement for growth-promoting function of Yap in hepatocytes. eLife 4, e02948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toogood PL, Harvey PJ, Repine JT, Sheehan DJ, VanderWel SN, Zhou H, Keller PR, McNamara DJ, Sherry D, Zhu T, et al. (2005). Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem. 48, 2388–2406. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, and Schütz G (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet 23, 99–103. [DOI] [PubMed] [Google Scholar]
- White DE, Negorev D, Peng H, Ivanov AV, Maul GG, and Rauscher FJ III (2006). KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions. Cancer Res. 66, 11594–11599. [DOI] [PubMed] [Google Scholar]
- Wu X, and Brewer G (2012). The regulation of mRNA stability in mammalian cells: 2.0. Gene 500, 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ, Richardson JA, BasselDuby R, and Olson EN (2011). Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal 4, ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Giap C, Lazo JS, and Prochownik EV (2003). Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 22, 6151–6159. [DOI] [PubMed] [Google Scholar]
- Yu FX, Zhao B, and Guan KL (2015). Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163, 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Cordenonsi M, and Piccolo S (2016). YAP/TAZ at the roots of cancer. Cancer Cell 29, 783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, and Piccolo S (2015). Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol 17, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. (2007). Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, BekkerJensen S, Bartek J, and Shiloh Y (2006). Chromatin relaxation in response to DNA doublestrand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol 8, 870–876. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A control cell that divided within the imaging period. Cells were transfected with a modified Fucci plasmid. The green color is from Azami Green fused to the Src myristoylation signal. The orange-red color is from Kusabira Orange fused to human Geminin1−110 that accumulates in S/G2/M phases of a cell cycle.
A Lats1/2;Nestin-Cre dKO cell that divided within the imaging period.
A dKO cell that died.
A dKO cell that did not divide within the imaging period.
Affymetrix microarray analysis comparing E12.5 control and Lats1/2;Nestin-Cre dKO cortices. Differentially expressed genes (P < 0.05) and associated Gene Set Enrichment Analysis (GSEA) using Hallmark gene sets and Canonical Pathways are listed.
CNN RNA-seq comparing E12.5 control and Lats1/2;Nestin-Cre dKO telencephalons. Differentially expressed genes called by ERCC normalization and read-depth normalization are listed for comparison.
NanoString nCounter analysis comparing RNAs extracted from equal numbers of E12.5 control and Lats1/2;Nestin-Cre dKO telencephalic cells. Raw counts and normalized (to housekeeping genes) counts are shown.