

Abstract
Although tumour suppressor gene hypermethylation is a universal feature of cancer cells, little is known about the necessary molecular triggers. Here, we show that Wilms' tumour 1 (WT1), a developmental master regulator that can also act as a tumour suppressor or oncoprotein, transcriptionally regulates the de novo DNA methyltransferase 3A (DNMT3A) and that cellular WT1 levels can influence DNA methylation of gene promoters genome-wide. Specifically, we demonstrate that depletion of WT1 by short-interfering RNAs leads to reduced DNMT3A in Wilms' tumour cells and human embryonal kidney-derived cell lines. Chromatin immunoprecipitation assays demonstrate WT1 recruitment to the DNMT3A promoter region and reporter assays confirm that WT1 directly transactivates DNMT3A expression. Consistent with this regulatory role, immunohistochemical analysis shows co-expression of WT1 and DNMT3A proteins in nuclei of blastemal cells in human fetal kidney and Wilms' tumours. Using genome-wide promoter methylation arrays, we show that human embryonal kidney cells over-expressing WT1 acquire DNA methylation changes at specific gene promoters where DNMT3A recruitment is increased, with hypermethylation being associated with silencing of gene expression. Elevated DNMT3A is also demonstrated at hypermethylated genes in Wilms’ tumour cells, including a region of long-range epigenetic silencing. Finally, we show that depletion of WT1 in Wilms' tumour cells can lead to reactivation of gene expression from methylated promoters, such as TGFB2, a key modulator of epithelial–mesenchymal transitions. Collectively, our work defines a new regulatory modality for WT1 involving elicitation of epigenetic alterations which is most likely crucial to its functions in development and disease.
Introduction
Normal development requires stringently regulated epigenetic programming that controls stage and cell-specific gene expression. In cancer, erroneous epigenetic programming can deregulate expression of genes encoding key growth regulators and give rise to cells ‘locked-in’ to a stem-cell like state resistant to normal growth and apoptotic signals (1). Two fundamental questions for epigenetic regulation are: (i) how is the epigenetic machinery (de)regulated, and (ii) how might individual genes selected for modulation? There is emerging evidence that developmental transcription factors modulate epigenetic marks such as DNA methylation by directing DNA methyltransferases (DNMTs) to specific gene promoters and also by transcriptionally regulating expression of DNMTs. Examples include gene repression resulting from MYC through its protein–protein interaction with DNA methyltransferase 3A (DNMT3A) (2), STAT3 via DNMT1 protein interaction and DNMT1 transactivation (3,4) and DAXX via DNMT1 interaction and DNMT1 transactivation (5). With regard to control of de novoDNMTs, HOXB3 in humans (6), and Vezf1 in mice (7), have both been shown to transactivate DNMT3B (6,7); notably, however, little is known about the regulation of DNMT3A.
Studies in Wt1-targetted mice have shown that Wt1 is essential for the normal development of the kidney and urogenital system, mesothelium, heart and lungs, with homozygous wt1 deletion being embryonically lethal as a result of developmental failure (8). WT1 is also important in the development of other tissues, such as the liver, retina and in sex determination (9–11) and in regulating epithelial–mesenchymal transitions (EMT) (12). The involvement of WT1 in carcinogenesis is underlined by the mutation of the gene in ∼10% of Wilms’ tumours (WTs) (13) and acute myeloid leukaemias (14), suggesting tumour suppressor gene (TSG) function, but also by other tumours where WT1 is over-expressed, such as ovarian, lung and colorectal cancers (14), gliomas (15), melanomas (16) and hepatocellular carcinomas (17). WT1 proteins are expressed as four main isoforms arising from two alternate splicing events, inclusion or exclusion of exon 5, encoding 17 amino acids and the insertion or exclusion of 3 amino acids (lysine–threonine–serine, KTS) between zinc fingers 3 and 4 (+KTS/−KTS isoforms). WT1 without either splice is commonly referred to as WT1-A, whereas WT1-B includes exon 5 amino-acids. Similarly WT1-C and WT1-D include +KTS, with WT1-C excluding exon 5 amino-acids and WT1-D including them. The –KTS isoforms have greater affinity for DNA, and their influence on differentiation and development is mediated via transcriptional activation and repression of downstream target genes. The +KTS isoforms exert a less well-characterized post-transcriptional regulatory influence through involvement in RNA metabolism (18), but can also mediate some transcriptional control (19). We also identified a genomically imprinted alternative WT1, AWT1, which retains WT1 exonic structure and splicing patterns from exons 2–10. AWT1 proteins are truncated by 147 amino acids at the amino-terminus relative to WT1. In most WTs, AWT1 displays relaxation of imprinting (20).
Given the critical involvement of WT1 in development and disease, we hypothesized that WT1 may be involved in modulating epigenetic regulation. Recently, evidence for the developmental importance of Wt1-mediated epigenetic regulation has begun to emerge, with epicardial and renal cell-type-specific Wnt4 expression and chromatin states having been shown to be contingent on Wt1 (21). Furthermore, the tumour suppressor menin was shown to epigenetically silence PAX2 expression through repressive protein complexes comprising WT1, DNMT1 and EZH2 (22). In this study, we focus on examining whether epigenetic states are regulated by WT1 in a tumour cell context, and present data that demonstrate that WT1 directly regulates the de novo methytranferase DNMT3A (23), and that cellular WT1 status influences genome-wide promoter methylation.
RESULTS
WT1 depletion alters the levels of de novo DNA methyltransferases
In order to investigate whether transcription of genes encoding epigenetic modifiers depends on WT1 levels, we transfected the WiT49 Wilms' tumour cell line with WT1-targeting short-interfering RNA (siRNA) and analysed mRNA expression of genes encoding DNA and histone modifying proteins. A striking 6-fold reduction in DNMT3A mRNA was observed together with 4-fold reduction in DNMT3L which has been shown to stimulate DNMT3A de novo DNA methyltransferase activity (24), although basal expression of DNMT3L, was much lower. In contrast to DNMT3A transcripts, DNMT1, DNMT3B and histone methyltransferases genes such as EZH2, EHMT1 and EHMT2 were not markedly affected by WT1 knockdown (Fig. 1A).
Figure 1.
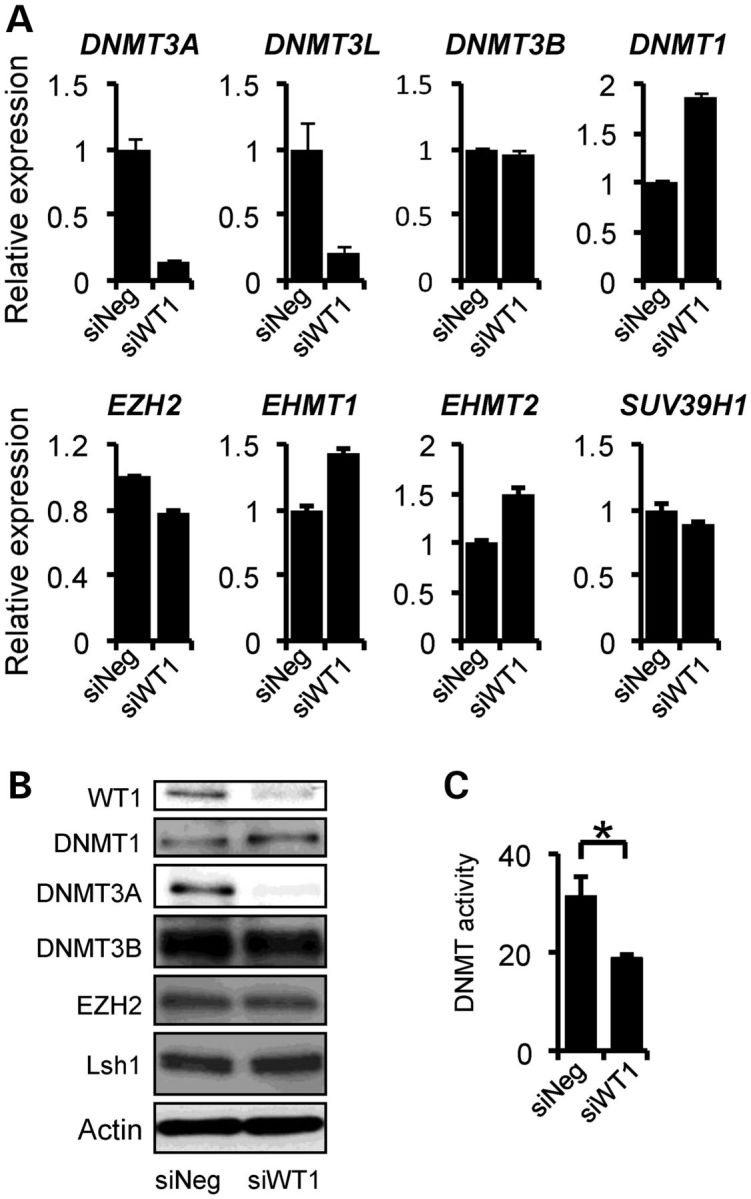
WT1-dependent changes in epigenetic modifiers in Wilms' tumour cell line WiT49. (A) Quantitative PCR showing levels of epigenetic modifier mRNAs in WiT49 cells following WT1 siRNA-induced knockdown (siWT1) compared with negative control (siNeg). (B) Immunoblotting showing decreased DNMT3A in WiT49 cells after WT1 knockdown. (C) DNA methyltransferase enzymatic assay demonstrating decrease in activity accompanying WT1 knockdown (P < 0.05).
Immunoblotting analysis of WiT49 cells with WT1 knockdown proteins showed robust and specific reduction in DNMT3A protein levels after WT1 knockdown, with little or no change of other epigenetic modifiers tested (Fig. 1B). To exclude the possibility of off-target effects, a second WT1-targeting siRNA was also tested and also showed WT1-dependent reduction in DNMT3A (Supplementary Material, Fig. S1A). We then evaluated whether cellular DNA methyltransferase enzymatic activity was affected by WT1 knockdown and found that WT1 depletion did indeed significantly reduce DNA methyltransferase activity (Fig. 1C).
In order to further examine WT1-mediated regulation of DNMT3A in isogenic cell lines with low and high expression of WT1, we constructed human embryonal kidney (HEK293)-derived cell lines with inducible WT1-B (W203 and W210). We also cloned a vector control (IP100) and cell lines with inducible transgenes for the WT1-related transcription factor, EGR1 (E6) and a cell line with dual EGR1 and WT1 expression (EW41). In the absence of induction with doxycycline, WT1-B cell lines exhibited ‘leaky’ expression of the transgenes comparable with cells known to express physiologically high levels of WT1, such as the mouse metanephric mesenchyme line (M15) and WiT49 cells (Supplementary Material, Fig. S1B). Immunoblotting of these independent clonal lines revealed that elevated basal WT1 in cell lines W203, W210 and EW41 was correlated with higher expression of DNMT3A protein, in contrast to the vector control IP100 cells and the EGR1 over-expressing E6 cells (Fig. 2A). As observed with WiT49 cells, when WT1 was knocked down in W210 cells, DNMT3A protein levels were greatly reduced whereas other epigenetic modifiers did not change significantly (Fig. 2B). Cellular DNA methytransferase enzymatic activity was also concomitantly reduced (Fig. 2C). Intron 1 of DNMT3A also contains a micro-RNA, miR-1301, whose expression was also decreased in both W210 and WiT49 cells following WT1 depletion (Supplementary Material, Fig. S1C), suggesting that miR-1301 is regulated by the DNMT3A 5′-promoter (see below). Together, these data demonstrate that DNMT3A RNA and protein levels are positively regulated by WT1 in heterologous cell systems.
Figure 2.

WT1-dependent DNMT3A expression in human embryonic kidney (HEK293) isogenic cell lines. (A) Immunoblotting demonstrating elevated DNMT3A protein in WT1 over-expressing cell lines (W210, W203) and EW41 (EGR1 plus WT1 over-expression), relative to IP100 and E6 (EGR1 over-expression). (B) Immunoblotting showing decreased DNMT3A in W210 cells after WT1 knockdown. (C) Decreased DNA methyltransferase enzymatic activity accompanying WT1 knockdown (P < 0.05).
WT1 directly activates DNMT3A transcription
We assessed whether DNMT3A may be a direct transcriptional target of WT1. The DNMT3A gene produces two main isoforms, DNMT3A1 and DNMT3A2 (25), regulated by three different promoters (26). Promoters 1 and 2 regulate DNMT3A1 transcripts with variant 5′ exons and produce proteins of 130 kDa. Promoter 3 regulates DNMT3A2 encoding an ES-cell-specific 100 kDa protein (Fig. 3A). We conducted quantitative scanning chromatin immunoprecipitation (ChIP) assays to examine whether WT1 bound to the upstream and downstream DNMT3A promoter regions in WiT49 cells. As shown in Figure 3B, WT1 was recruited at the 5′-promoters, but not at the 3′-promoter. Examination of the DNA sequence in this region revealed several high-affinity WT1 consensus sites, upstream and downstream of the transcriptional start sites (Supplementary Material, Fig. S2A), similar to WT1-target gene requirements reported previously (27). Consistent with the increased expression of DNMT3A in W210 cells relative to IP100 cells, WT1 recruitment in the 5′-region was greatly elevated in W210 cells relative to IP100 cells (Fig. 3C). We next co-transfected WiT49 cells with a DNMT3A 5′-promoter reporter plasmid together with WT1-targeting siRNA. Knockdown led to decreased promoter activity, confirming positive regulation of 5′-promoters by physiological WT1 levels. A similar result was shown in W210 cells (Fig. 3D). In order to demonstrate that transactivation was dependent on DNA binding, we co-transfected the DNMT3A reporter together with expression constructs for WT1-B, a Denys-Drash mutant WT1 (R394W) and AWT1-B. Transactivation of the reporter was observed with WT1-B, but not with the DNA-binding impaired mutant WT1 or with AWT1-B (Fig. 3E). Immunohistochemistry on Wilms' tumour sections demonstrated that WT1 and DNMT3A are co-expressed in the same cell type in vivo. Nuclear co-expression of WT1 and DNMT3A was observed in the blastemal cells of triphasic tumours, whereas the epithelial and stromal components showed little or no staining or weak cytoplasmic DNMT3A staining (Fig. 4). Transcript levels for WT1 and DNMT3A in WTs with wild-type WT1 were also assessed and found to correlate significantly (R = 0.74, P = 0.002; Supplementary Material, Fig. S2B). Taken together, these experiments demonstrate that DNMT3A is a target for direct transcriptional regulation by WT1, and suggest that genomic DNA methylation may, at least in part, depend on WT1.
Figure 3.

DNMT3A is directly transcriptionally regulated by WT1. (A) Schematic of the human DNMT3A gene. The 5′-promoter for DNMT3A1 at exons 1a and 1b and the 3′-promoter for DNMT3A2 are shown in expansions together with ChIP amplicons tested (numbered 1–7). The microRNA miR-1301 residing in the large 5′-intron is also shown. (B) Scanning quantitative ChIP analysis demonstrating WT1 recruitment at the DNMT3A 5′-promoters in WiT49 cells. (C) High WT1 recruitment at the DNMT3A 5′-promoters in W210 cells (white bars) but not in IP100 cells (black diamonds). (D) Decreased DNMT3A promoter activity accompanying WT1 knockdown in WiT49 cells (left panel) and W210 cells (right panel). (E) The DNMT3A promoter is transactivated by over-expression of WT1-B, and not by AWT1-B. Transactivation is impaired by a common WT1 pathological mutant, Denys-Drash R394W in HEK293 cells.
Figure 4.

DNMT3A and WT1 are co-expressed in the developing kidney and in Wilms' tumour. Immunohistochemical analysis of fetal kidney (FK) and Wilms' tumour (WT) demonstrating nuclear co-expression of WT1 and DNMT3A in blastemal cells (bl). In fetal kidney, overlapping staining is also seen the glomerular primitive Bowman capsule (ca) and the primitive podocytes of the glomerulus (po). Stromal (st) and epithelial (ep) compartments show little or no nuclear staining for WT1 and DNMT3A.
WT1 status affects genome-wide promoter methylation
In order to evaluate whether DNA methylation is affected by the altered DNMT3A levels in WT1 over-expressing cells, we conducted genome-wide promoter methylation analysis of IP100 and W210 cell lines using methyl-cytosine immunoprecipitation-microarray analysis (MCIP-chip). We identified 68 genes that acquired methylation in W210 cells relative to IP100 cells, whereas 28 genes showed decreased methylation (Supplementary Material, Fig. S3A and Table S1). Using combined bisulphite and restriction analysis (COBRA), we validated gain of methylation in eight genes in W210 cells, including CDH9, ESR1, MLH1, SIM1 and TGFB2. Cells over-expressing the related transcription factors EGR1 and AWT1 did not consistently exhibit DNA methylation changes at the levels observed in WT1 over-expressing cells (Supplementary Material, Fig. S3B).
Pyrosequencing of bisulphite-converted DNAs was then used for high-resolution quantitative analysis of DNA methylation changes at the MLH1, SIM1 and TGFB2 gene promoter regions. All three genes showed increased methylation in the WT1-expressing lines (W210, W203 and EW41) relative to the low expression lines, IP100 and E6 (Fig. 5A). DNA hypermethylation was also confirmed by bisulphite sequencing of the ESR1 promoter (Supplementary Material, Fig. S3C), where gain of methylation in the WT1 over-expressing cells coincided with a previously reported WT1-binding site (28). Expression analysis of MLH1, SIM1 and TGFB2 demonstrated reduced transcription of all genes in WT1 over-expressing cells relative to vector-only IP100, EGR1-only E6 and AWT1-only TWI-3 cell lines (Fig. 5B). We next analysed whether DNMT3A was increased at promoters exhibiting hypermethylation in W210 cells relative to IP100 cells. As shown in Figure 5C, MLH1 and TGFB2 promoters showed higher DNMT3A recruitment in W210 cells; SIM1, which is partially methylated in IP100 cells, maintained high levels of DNMT3A in both IP100 and W210 cells. The TAF7 gene, which encodes a basal transcription factor [TATA box-binding protein (TBP)-associated factor], showed low and unchanged DNMT3A at its promoter in both IP100 and W210 cells. WT1 recruitment at the methylated genes in W210 cells was not increased significantly compared with IP100 cells, in contrast to the elevated recruitment at the DNMT3A promoter (Fig. 3C). Thus, promoters of specific genes in WT1-over-expressing HEK293 cells exhibit enhanced DNMT3A recruitment and acquire DNA methylation, leading to silencing of gene expression.
Figure 5.

Increased DNA methylation and gene silencing associated with WT1 expression. (A) High-resolution bisulphite pyrosequencing demonstrating increased DNA methylation at SIM1, MLH1 and TGFB2. Each data point represents an individual CpG dinucleotide. Open circles, IP100; open triangles, E6; black squares, W210; black triangles, W203; black diamonds, EW41. (B) Quantitative gene expression analysis showing silencing of genes hypermethylated in association with high WT1 levels, i.e. W210, W203 and EW41 cells. (C) ChIP analysis showing increased recruitment of DNMT3A at SIM1, MLH1 and TGFB2, which are methylated gene promoters in W210 cells (black bars) compared with IP100 (white bars). The unmethylated TAF7 gene is also shown. Note that WT1 recruitment levels are not altered between IP100 and W210 cells.
DNMT3A is associated with gene silencing in Wilms' tumour
DNMT3A has been shown to be over-expressed in many cancers, including bladder and kidney (29), melanoma (30), ovarian cancer (31) and retinoblastoma (32). Despite this, evidence for its recruitment to hypermethylated TSGs is restricted to a few examples, such as CDKN1A (2) and RASSF1 (33). As it has also been shown that Dnmt3a can also facilitate gene transcription (34), we sought to establish the association of DNMT3A with gene silencing in Wilms' tumour cells. We therefore examined recruitment of DNMT3A across the protocadherin (PCDH) locus of long-range epigenetic silencing (LRES) in WiT49 cells, which we previously showed to encode tumour suppressor proteins in Wilms' tumour and colorectal cancer (35,36). DNMT3A binding was high within the LRES, but low proximal and distal to the LRES region, displaying a reciprocal pattern relative to histone 3–lysine 4 dimethylation (H3K4me2), a marker for actively transcribed genes (Fig. 6A). DNMT3A enrichment was also high at hypermethylated genes at non-LRES genes at other genomic loci, including SIM1 and RASSF1, and low at MLH1, the latter being unmethylated in WiT49 cells (Fig. 6B and Supplementary Material, Fig. S4A). Knockdown of WT1 in WiT49 cells led to decreased DNMT3A binding at the methylated promoters, further re-enforcing the regulation of DNMT3A by WT1 (Fig. 6C). DNMT3A is therefore implicated in TSG silencing, most likely in conjunction with other chromatin modifiers.
Figure 6.

Association of DNMT3A with tumour-specific methylation changes in Wilms' tumour WiT49 cells. (A) ChIP across the PCDH LRES on chromosome 5q31. High DNMT3A binding is apparent at hypermethylated genes (black bars) and lower binding at unmethylated genes (open bars). Histone 3 lysine 4 dimethylation (35) across the locus is shown by the grey points. (B) DNMT3A recruitment at non-LRES hypermethylated (SIM1 and RASSF1) and unmethylated (MLH1) genes. (C) WT1 knockdown leads to decreased DNMT3A recruitment at methylated genes. High DNMT3A is present at the hypermethylated genes (SIM1, RASSF1, PCDHA13 and PCDHGA2, black bars) and not at the unmethylated MLH1 (white bar). Note the intermediate level DNMT3A recruitment at TGFB2 (grey bar). DNMT3A levels after WT1 knockdown are indicated by hatched bars.
DNA methylation of genes with dense CpG island promoters and high DNA methylation such as SIM1 and RASSF1 was not altered by WT1 knockdown, and although changes in their expression levels were not detectable by QRT–PCR, endpoint PCR revealed derepression of SIM1 and RASSF1expression (Supplementary Material, Fig. S4B and C). Other genes such as CDH9 and PCDHGA12 could not be reactivated (data not shown). In the case of TGFB2 however, decreased methylation of the promoter was observed with bisulphite pyrosequencing in WT1 knockdown samples, and TGFB2 transcript levels were greatly elevated concomitant with methylation changes (Fig. 7A). Demethylation of the TGFB2 promoter and its re-expression was evident with 5-aza-2-deoxycytidine treatment (Supplementary Material, Fig. S4D). WiT49 cells treated with WT1 and DNMT3A siRNAs expressed >50-fold more FN1/fibronectin, a marker for mesenchymal cells arising from TGF-β induced epithelial-to-mesenchymal transition (Fig. 7B) (37). Treated cells also underwent profound morphological changes, acquiring a mesenchymal appearance (Fig. 7C) Together, our data confirm that DNMT3A is associated with TSG silencing in cancer cells, and that WT1 depletion can trigger gene reactivation at a subset of genes, such as TGFB2, and thereby lead to profound alterations in cellular phenotypes.
Figure 7.

Activation of TGFB2 and EMT. (A) Quantitative analysis of TGFB2 DNA methylation and expression changes associated with WT1 depletion demonstrating decreased methylation (black diamonds) and increased expression of TGFB2 (white bars). Two independent WT1 siRNAs are indicated by numbering. (B) Increased fibronectin/FN1 mRNA expression accompanying knockdowns demonstrated by quantitative RT–PCR, shown as fold change relative to negative control siRNA (siNeg). (C) Acquisition of mesenchymal phenotype observed with phase contrast microscopy following WT1/DNMT3A depletion.
DISCUSSION
While it is recognized that WT1 is crucially involved in the development of diverse tissue systems, and critical cellular processes requiring coordinated growth and differentiation control, such as EMT, angiogenesis, tumour suppression and oncogenesis, the mechanisms and intermediaries by which it exerts influence over the prerequisite complex gene expression programmes remain relatively obscure (18,38). Similarly, although DNA methyltransferases are frequently over-expressed in cancer (39), thereby leading to tumour–suppressor gene hypermethylation (40), little is known about the mechanisms by which DNMT deregulation occurs. In this study, we have shown that the de novo DNA methyltransferase DNMT3A is a potentially critical target gene for transcriptional regulation by WT1, and present strong evidence for WT1 over-expression triggering de novo methylation events.
Regulation of the de novo methyltransferases DNMT3A and DNMT3B by the general transcription factors Sp1 and Sp3 has previously been reported (41), but in terms of cell-type specific or oncogenic transcription factors, evidence is restricted to HOXB3 (in humans) and Vezf1 (in mice) regulation of DNMT3B (6,7). Our finding that WT1 regulates DNMT3A is therefore of particular relevance to cancers where WT1 is over-expressed, and also to developmental lineages requiring WT1. Our studies of HEK293 model cell lines demonstrated very selective methylation of promoters and increased DNMT3A recruitment contingent on WT1. Of the 68 genes undergoing de novo DNA methylation in W210 cells, 32 were also methylated in WiT49 cells and 6 genes (ANKAR, BHLHB9, CDH9, HRH2, LIMCH1 and SIM1) exhibited hypermethylation when comparing WiT49 cells with fetal kidney MCIP-chip (data not shown); further genes with lower methylation levels in WiT49 cells such as TGFB2 may have been below the array detection threshold. The partial overlap of de novo methylation targets in W210 cells and WiT49 cells may be attributable to HEK293 cells representing cells of a neural lineage rather than embryonic kidney (42). In contrast to increased DNMT3A promoter binding, WT1 recruitment at W210 methylated promoters was not higher in W210 cells compared with IP100 cells, suggesting that WT1 is not solely required for targeting of DNMT3A, although we cannot formally exclude this possibility as WT1 might vacate promoters which become methylated. Interestingly, CDH9 has been shown to be expressed in early kidney development and interstitial cells outlining the Bowman's capsule in normal adult kidney (43), and SIM1 is also expressed in fetal kidney (44); thus epigenetic regulation through the WT1/DNMT3A axis may, by targeting genes such as CDH9 and SIM1, modulate cell differentiation during renal development.
Although the de novo DNMTs are presumptive oncogenes based on their ability to mediate TSG silencing, descriptive or functional data for DNMT3A oncogenicity are limited. Unlike Dnmt3a over-expression, Dnmt3b-mediated hypermethylation in an ApcMin/+ model of intestinal neoplasia was shown to increase tumour burden (45). Furthermore, DNMT3A mutations found in acute myeloid leukaemia appear to decrease DNA methylation (46), and Dnmt3a deletion in a mouse model of lung cancer led to increased tumour progression without affecting initiation (47). This might support a TSG role for DNMT3A in certain cancers, although it is worth noting that DNMT3A expression in intestinal cancers appears to be relatively low (29) and that mir-1301, which we have shown is co-ordinately regulated by the 5′-promoter, appears not to be conserved in mice. While the targets of this miRNA remain to be discovered, it is distinctly possible that they also will indirectly modify epigenetic states. Our data show a strong association between DNMT3A recruitment and TSG hypermethylation; DNMT3A was enriched on promoters of hypermethylated genes within the PCDH LRES and was absent from neighbouring unmethylated genes. Interestingly, the unmethylated genes appear to be protected from DNA hypermethylation by high H3K4me2 marking, which has recently been shown to possess limited ability to catalytically activate DNMT3A relative to unmethylated H3K4 (48). The interplay of epigenetic modifiers and marks necessary for gene silencing is also alluded to by the resistance to re-expression following DNMT3A depletion exhibited by hypermethylated genes, similar to that demonstrated for EZH2 (49). Notably, however, re-expression of SIM1 and RASSF1 could be triggered by WT1 knockdown in the absence of DNA methylation changes. We speculate that this may be attributable to the inhibition of reading DNA methylation by proteins such as the methyl-binding domain inhibitor protein MBDin (50), or through the methylation-independent, histone deacetylase-associated repressive ability of DNMT3A reported previously (51). While DNA methylation is generally considered to ‘lock’ genes into a silenced state, it is clear from recent studies with histone deacetylase inhibitors that re-expression of silenced genes can occur without promoter hypermethylation being erased (52). In contrast to SIM1 and RASSF1, a decrease in promoter methylation accompanying WT1/DNMT3A reduction was evident at TGFB2, this promoter exhibiting low-intermediate methylation in our and other analyses (53). TGF-β signalling can profoundly enhance or repress cancer cell growth and is also known to drive EMT (54), as we observed in our experiments. With respect to Wilms' tumour, which is considered to arise from a differentiation block of blastemal cells, deregulated WT1/DNMT3A might disable the differentiation of blastemal cells into other components such as stromal cells by epigenetically curtailing TGFB2 expression and TGF-β-induced changes. Interestingly, stromal cells have been shown to express high levels of TGFB2 and its transcriptional regulator LBX1 (55,56), which binds the TGFB2 promoter in the region we have shown to undergo methylation. Although our studies have focussed on Wilms' tumour, they emphasize the need for further studies to examine the role of WT1/DNMT3A-mediated epigenetic regulation in broader cancer and developmental cell contexts where WT1 has been implicated in EMT such as mammary epithelial cells (57) and epicardial cells (12).
Recent studies have implicated Wt1 in an ‘epigenetic flip-flop’ at the Wnt4 locus during the control of cardiac and renal development (21), and also in the complex containing Menin, EZH2 and DNMT1 which silences PAX2 expression (22). Our study mechanistically defines DNMT3A as a critical epigenetic modifier downstream of WT1, and considerably expands the potential ‘regulome’ of WT1 given the diverse and complex roles recently ascribed to DNMT3A, including facilitating transcription of genes by non-promoter CpG island methylation (34) and also increasing gene transcription from certain promoters by actively demethylating DNA in complexes with thymine-DNA glycosylase (58,59). The multiple regulatory roles of DNMT3A are likely determined by diverse protein complexes and by chromatin modifications, and these may also influence the ability of WT1 to cascade signals to a variety of gene targets necessary for its pleiotropic regulatory effects. While the combined transcriptional, post-transcriptional and epigenetic regulatory roles of WT1 are complex, our studies underline the importance of further integrated analyses to extend the paradigm of epigenetic control by developmental transcription factors reported herein.
MATERIALS AND METHODS
Patient samples
All tissues were obtained as snap frozen samples from the Bristol Children's Hospital. Human fetal tissue samples were from 19 to 31 weeks of gestation. All human tissues were acquired with appropriate local research ethics committee approval.
Plasmids and short-interfering RNAs
WT1, AWT1 and EGR1 full-length cDNAs were amplified from human fetal kidney mRNA, verified by DNA sequencing and cloned into the pBIG-3i tetracycline inducible expression vector (60). The R394W mutant WT1-B was made using site-directed mutagenesis (Stratagene). The DNMT3A promoter-luciferase plasmid was previously described (26). siRNAs were transfected at a final concentration of 25 nm using DharmaFECT 1 (Thermo Scientific). Sequences of siRNAs used are given in Supplementary Material, Table S2.
Cell culture, plasmids, transfections and reporter assays
HEK293 cells (adenovirus transformed human embryonic kidney cells) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 2 mmol/l l-glutamine, 0.1 mg/ml penicillin/streptomycin, at 37°C under 5% CO2. WiT49 cells required 15% fetal calf serum, 6 ul/l β-mercaptoethanol and insulin–transferrin–selenium.
To generate stable cell lines, recombinants were transfected into HEK293 cell lines using Fugene 6 (Promega), and independent clones were selected using 100 ug/ml of Hygromycin B (Boehringer Mannheim). After verification, cell lines were maintained using 50 ug/ml of Hygromycin B. For transient transfections reporter activity assays, 105 cells/well were seeded in 24-well plates and 100 ng of reporter plasmids were co-transfected with 100 ng of expression plasmids with Fugene 6, or with 50 nm of siRNAs using DharmaFECT DUO (Thermo Scientific). Luciferase samples were assayed after 48 h using Dual-luciferase reporter kit (Promega) and a Modulus Luminometer (Turner Biosystems). Experiments were performed at least thrice in triplicate.
Methyl-CpG immunoprecipitation (MCIP) and ChIP
For enrichment of methylated DNA from IP100 and W210 cells, genomic DNAs were extracted and fragmented to a size range of 200–500 bp using a Diagenode Bioruptor. Four micrograms of sonicated genomic DNA and purified recombinant methyl-CpG binding domain (MBD) protein (61) were incubated at 4°C overnight in immunoprecipitation buffer, and then for a further 2 h with goat anti-mouse IgG magnetic beads (N.E. Biolabs). After purification, MCIP DNA was then sent to Nimblegen for labelling and hybridization to Nimblegen HG18 Refseq promoter arrays. Data were analysed using ChipMonk v1.2.1 tiling array analysis software (http://www.bioinformatics.bbsrc.ac.uk/projects/chipmonk). To identify hypermethylated CGIs, a log2ratio cut-off of 1.5 and a window of 500 bp were used to carry out the replicate t-test (P < 0.05) on probes within 200 bp of predicted CpG islands (http://genome.ucsc.edu). Additional statistical analysis was carried out using R (http://www.r-project.org/). The array data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE39713.
Chromatin for WT1 and DNMT3A recruitment analysis was prepared using the Magna ChIP Kit (Millipore). Antibodies used were anti-WT1 (S. Roberts) and anti-DNMT3A (Sigma). Quantification using real-time PCR was carried out as described previously (35). Primer sequences are available in Supplementary Material, Table S2.
Methylation and expression analysis
DNA was sodium bisulphite converted using the EZ DNA Methylation-Gold Kit (Zymo Research, CA, USA). Amplicons for COBRA and bisulphite pyrosequencing were made using the Hot Start Red Taq PCR system (Sigma). Bisulphite pyrosequencing and quantitative real-time RT–PCR was performed as previously described (36). The pyrosequencing assay for TGFB2 was purchased from Qiagen. MicroRNA was assayed using ABI TaqMan® MicroRNA Assays for has-miR-1301 (ID #2827) and has-RNU6B (ID #1093) as a control. The TaqMan® MicroRNA RT kit was used to perform the reverse transcription according to the manufacturer's instructions with 20 ng RNA for each reverse transcription reaction. Primers and restriction assays are available in Supplementary Material, Table S2.
DNA methyltransferase enzymatic activity was assayed using the EpiQuik DNA Methyltransferase (DNMT) Activity Kit (Epigentek).
Immunoblotting and immunohistochemistry
Protein extraction and immunoblotting were carried out essentially as previously described (35). Primary antibodies used were DNMT3A (Cell Signalling), WT1 (Abcam) and β-actin (Sigma).
For immunohistochemistry, 2 µm sections of tissue were fixed for 24 h in 10% neutral buffered formaldehyde. Immunohistochemistry was performed with a Leica Microsystem Bond III automated machine using the Bond Polymer Refine Detection Kit (Ref DS9800) followed by Bond Dab Enhancer (Ref AR9432). The slides were dewaxed with Bond Dewax Solution (Ref AR9222). Heat-mediated antigen retrieval was performed using Bond Epitope Retrieval Solution (2) for 30 min (WT1) and 20 min (DNMT3A). Primary antibody dilutions were 1:100 for WT1 (6FH2, Dako), and 1:250 for DNMT3A (H-295, Santa Cruz). Three fetal kidneys and five triphasic Wilms' tumours were assessed and gave similar staining patterns. Negative control sections omitted the primary antibody (Supplementary Material, Fig. S2C).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by Kidney Research UK grant RP42/2009, Cancer Research UK grant C20791/A12743, and a Dorothy Hodgkin postgraduate fellowship. A.-S.M., K.S. and J.H.P. were internship undergraduates from the Universities of Jena, Heidelberg and Bristol, respectively.
Supplementary Material
Acknowledgements
We thank Herman Yeger for WiT49 cells, Dr Yanagisawa and Dr Lobie for DNMT3A luciferase reporter plasmids and Dr Rehli for cells expressing recombinant MBD. We also thank Andrew Ward for helpful discussions and comments.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Lotem J., Sachs L. Epigenetics and the plasticity of differentiation in normal and cancer stem cells. Oncogene. 2006;25:7663–7672. doi: 10.1038/sj.onc.1209816. [DOI] [PubMed] [Google Scholar]
- 2.Brenner C., Deplus R., Didelot C., Loriot A., Vire E., De Smet C., Gutierrez A., Danovi D., Bernard D., Boon T., et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 2005;24:336–346. doi: 10.1038/sj.emboj.7600509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Q., Wang H.Y., Marzec M., Raghunath P.N., Nagasawa T., Wasik M.A. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc. Natl Acad. Sci. USA. 2005;102:6948–6953. doi: 10.1073/pnas.0501959102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Q., Wang H.Y., Woetmann A., Raghunath P.N., Odum N., Wasik M.A. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood. 2006;108:1058–1064. doi: 10.1182/blood-2005-08-007377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puto L.A., Reed J.C. Daxx represses RelB target promoters via DNA methyltransferase recruitment and DNA hypermethylation. Genes Dev. 2008;22:998–1010. doi: 10.1101/gad.1632208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palakurthy R.K., Wajapeyee N., Santra M.K., Gazin C., Lin L., Gobeil S., Green M.R. Epigenetic silencing of the RASSF1A tumor suppressor gene through HOXB3-mediated induction of DNMT3B expression. Mol. Cell. 2009;36:219–230. doi: 10.1016/j.molcel.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gowher H., Stuhlmann H., Felsenfeld G. Vezf1 regulates genomic DNA methylation through its effects on expression of DNA methyltransferase Dnmt3b. Genes Dev. 2008;22:2075–2084. doi: 10.1101/gad.1658408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kreidberg J.A., Sariola H., Loring J.M., Maeda M., Pelletier J., Housman D., Jaenisch R. WT-1 is required for early kidney development. Cell. 1993;74:679–691. doi: 10.1016/0092-8674(93)90515-R. [DOI] [PubMed] [Google Scholar]
- 9.Ijpenberg A., Perez-Pomares J.M., Guadix J.A., Carmona R., Portillo-Sanchez V., Macias D., Hohenstein P., Miles C.M., Hastie N.D., Munoz-Chapuli R. Wt1 and retinoic acid signaling are essential for stellate cell development and liver morphogenesis. Dev. Biol. 2007;312:157–170. doi: 10.1016/j.ydbio.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 10.Wagner K.D., Wagner N., Vidal V.P., Schley G., Wilhelm D., Schedl A., Englert C., Scholz H. The Wilms’ tumor gene Wt1 is required for normal development of the retina. EMBO J. 2002;21:1398–1405. doi: 10.1093/emboj/21.6.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hammes A., Guo J.K., Lutsch G., Leheste J.R., Landrock D., Ziegler U., Gubler M.C., Schedl A. Two splice variants of the Wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell. 2001;106:319–329. doi: 10.1016/S0092-8674(01)00453-6. [DOI] [PubMed] [Google Scholar]
- 12.Martinez-Estrada O.M., Lettice L.A., Essafi A., Guadix J.A., Slight J., Velecela V., Hall E., Reichmann J., Devenney P.S., Hohenstein P., et al. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat. Genet. 2010;42:89–93. doi: 10.1038/ng.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rivera M.N., Haber D.A. Wilms’ tumour: connecting tumorigenesis and organ development in the kidney. Nat. Rev. Cancer. 2005;5:699–712. doi: 10.1038/nrc1696. [DOI] [PubMed] [Google Scholar]
- 14.Yang L., Han Y., Suarez Saiz F., Minden M.D. A tumor suppressor and oncogene: the WT1 story. Leukemia. 2007;21:868–876. doi: 10.1038/sj.leu.2404624. [DOI] [PubMed] [Google Scholar]
- 15.Clark A.J., Ware J.L., Chen M.Y., Graf M.R., Van Meter T.E., Dos Santos W.G., Fillmore H.L., Broaddus W.C. Effect of WT1 gene silencing on the tumorigenicity of human glioblastoma multiforme cells. J. Neurosurg. 2009;112:18–25. doi: 10.3171/2008.11.JNS08368. [DOI] [PubMed] [Google Scholar]
- 16.Wagner N., Panelos J., Massi D., Wagner K.D. The Wilms’ tumor suppressor WT1 is associated with melanoma proliferation. Pflugers Arch. 2008;455:839–847. doi: 10.1007/s00424-007-0340-1. [DOI] [PubMed] [Google Scholar]
- 17.Perugorria M.J., Castillo J., Latasa M.U., Goni S., Segura V., Sangro B., Prieto J., Avila M.A., Berasain C. Wilms’ tumor 1 gene expression in hepatocellular carcinoma promotes cell dedifferentiation and resistance to chemotherapy. Cancer Res. 2009;69:1358–1367. doi: 10.1158/0008-5472.CAN-08-2545. [DOI] [PubMed] [Google Scholar]
- 18.Roberts S.G. Transcriptional regulation by WT1 in development. Curr. Opin. Genet. Dev. 2005;15:542–547. doi: 10.1016/j.gde.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 19.Wagner N., Wagner K.D., Xing Y., Scholz H., Schedl A. The major podocyte protein nephrin is transcriptionally activated by the Wilms’ tumor suppressor WT1. J. Am. Soc. Nephrol. 2004;15:3044–3051. doi: 10.1097/01.ASN.0000146687.99058.25. [DOI] [PubMed] [Google Scholar]
- 20.Dallosso A.R., Hancock A.L., Brown K.W., Williams A.C., Jackson S., Malik K. Genomic imprinting at the WT1 gene involves a novel coding transcript (AWT1) that shows deregulation in Wilms’ tumours. Hum. Mol. Genet. 2004;13:405–415. doi: 10.1093/hmg/ddh038. [DOI] [PubMed] [Google Scholar]
- 21.Essafi A., Webb A., Berry R.L., Slight J., Burn S.F., Spraggon L., Velecela V., Martinez-Estrada O.M., Wiltshire J.H., Roberts S.G., et al. A Wt1-controlled chromatin switching mechanism underpins tissue-specific wnt4 activation and repression. Dev. Cell. 2011;21:559–574. doi: 10.1016/j.devcel.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu B., Zeng D.Q., Wu Y.A., Zheng R., Gu L., Lin X.A., Hua X.X., Jin G.H. Tumor Suppressor Menin Represses Paired Box Gene 2 expression via Wilms tumor suppressor protein-polycomb group complex. J. Biol. Chem. 2011;286:13937–13944. doi: 10.1074/jbc.M110.197830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okano M., Bell D.W., Haber D.A., Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/S0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 24.Chedin F., Lieber M.R., Hsieh C.L. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc. Natl Acad. Sci. USA. 2002;99:16916–16921. doi: 10.1073/pnas.262443999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen T.P., Ueda Y., Xie S.P., Li E. A novel Dnmt3a isoform produced from an alternative promoter localizes to euchromatin and its expression correlates with active de novo methylation. J. Biol. Chem. 2002;277:38746–38754. doi: 10.1074/jbc.M205312200. [DOI] [PubMed] [Google Scholar]
- 26.Yanagisawa Y., Ito E., Yuasa Y., Maruyama K. The human DNA methyltransferases DNMT3A and DNMT3B have two types of promoters with different CpG contents. Biochim. Biophys. Acta Gene Struct. Exp. 2002;1577:457–465. doi: 10.1016/S0167-4781(02)00482-7. [DOI] [PubMed] [Google Scholar]
- 27.Hartwig S., Ho J., Pandey P., Macisaac K., Taglienti M., Xiang M., Alterovitz G., Ramoni M., Fraenkel E., Kreidberg J.A. Genomic characterization of Wilms’ tumor suppressor 1 targets in nephron progenitor cells during kidney development. Development. 2010;137:1189–1203. doi: 10.1242/dev.045732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han Y., Yang L., Suarez-Saiz F., San-Marina S., Cui J., Minden M.D. Wilms’ tumor 1 suppressor gene mediates antiestrogen resistance via down-regulation of estrogen receptor-alpha expression in breast cancer cells. Mol. Cancer Res. 2008;6:1347–1355. doi: 10.1158/1541-7786.MCR-07-2179. [DOI] [PubMed] [Google Scholar]
- 29.Robertson K.D., Uzvolgyi E., Liang G., Talmadge C., Sumegi J., Gonzales F.A., Jones P.A. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 1999;27:2291–2298. doi: 10.1093/nar/27.11.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng T., Kuang Y., Wang L., Li J., Wang Z., Fei J. An essential role for DNA methyltransferase 3a in melanoma tumorigenesis. Biochem. Biophys. Res. Commun. 2009;387:611–616. doi: 10.1016/j.bbrc.2009.07.093. [DOI] [PubMed] [Google Scholar]
- 31.Bai X., Song Z., Fu Y., Yu Z., Zhao L., Zhao H., Yao W., Huang D., Mi X., Wang E., et al. Clinicopathological significance and prognostic value of DNA methyltransferase 1, 3a, and 3b expressions in sporadic epithelial ovarian cancer. PLoS ONE. 2012;7:e40024. doi: 10.1371/journal.pone.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qu Y., Mu G.Y., Wu Y.W., Dai X., Zhou F., Xu X.Y., Wang Y., Wei F.C. Overexpression of DNA methyltransferases 1, 3a, and 3b significantly correlates with retinoblastoma tumorigenesis. Am. J. Clin. Path. 2012;134:826–834. doi: 10.1309/AJCPHGQ69FXDFWII. [DOI] [PubMed] [Google Scholar]
- 33.Li H.W., Rauch T., Chen Z.X., Szabo P.E., Riggs A.D., Pfeifer G.P. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J. Biol. Chem. 2006;281:19489–19500. doi: 10.1074/jbc.M513249200. [DOI] [PubMed] [Google Scholar]
- 34.Wu H., Coskun V., Tao J., Xie W., Ge W., Yoshikawa K., Li E., Zhang Y., Sun Y.E. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science. 2010;329:444–448. doi: 10.1126/science.1190485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dallosso A.R., Hancock A.L., Szemes M., Moorwood K., Chilukamarri L., Tsai H.H., Sarkar A., Barasch J., Vuononvirta R., Jones C., et al. Frequent long-range epigenetic silencing of protocadherin gene clusters on chromosome 5q31 in Wilms’ tumor. PLoS Genet. 2009;5:e1000745. doi: 10.1371/journal.pgen.1000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dallosso A.R., Oster B., Greenhough A., Thorsen K., Curry T.J., Owen C., Hancock A.L., Szemes M., Paraskeva C., Frank M., et al. Long-range epigenetic silencing of chromosome 5q31 protocadherins is involved in early and late stages of colorectal tumorigenesis through modulation of oncogenic pathways. Oncogene. 2012 doi: 10.1038/onc.2011.609. [advance online publication] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hocevar B.A., Brown T.L., Howe P.H. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999;18:1345–1356. doi: 10.1093/emboj/18.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hohenstein P., Hastie N.D. The many facets of the Wilms’ tumour gene, WT1. Hum. Mol. Genet. 2006;15(Spec No. 2):R196–R201. doi: 10.1093/hmg/ddl196. [DOI] [PubMed] [Google Scholar]
- 39.Miremadi A., Oestergaard M.Z., Pharoah P.D.P., Caldas C. Cancer genetics of epigenetic genes. Hum. Mol. Genet. 2007;16:R28–R49. doi: 10.1093/hmg/ddm021. [DOI] [PubMed] [Google Scholar]
- 40.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev. Genet. 2007;8:286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 41.Jinawath A., Miyake S., Yanagisawa Y., Akiyama Y., Yuasa Y. Transcriptional regulation of the human DNA methyltransferase 3A and 3B genes by Sp3 and Sp1 zinc finger proteins. Biochem. J. 2005;385:557–564. doi: 10.1042/BJ20040684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shaw G., Morse S., Ararat M., Graham F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002;16:869–871. doi: 10.1096/fj.01-0995fje. [DOI] [PubMed] [Google Scholar]
- 43.Thedieck C., Kalbacher H., Kuczyk M., Muller G.A., Muller C.A., Klein G. Cadherin-9 is a novel cell surface marker for the heterogeneous pool of renal fibroblasts. PLoS ONE. 2007;2:e657. doi: 10.1371/journal.pone.0000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chrast R., Scott H.S., Chen H., Kudoh J., Rossier C., Minoshima S., Wang Y., Shimizu N., Antonarakis S.E. Cloning of two human homologs of the Drosophila single-minded gene SIM1 on chromosome 6q and SIM2 on 21q within the Down syndrome chromosomal region. Genome Res. 1997;7:615–624. doi: 10.1101/gr.7.6.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Linhart H.G., Lin H., Yamada Y., Moran E., Steine E.J., Gokhale S., Lo G., Cantu E., Ehrich M., He T., et al. Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev. 2007;21:3110–3122. doi: 10.1101/gad.1594007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hajkova H., Markova J., Haskovec C., Sarova I., Fuchs O., Kostecka A., Cetkovsky P., Michalova K., Schwarz J. Decreased DNA methylation in acute myeloid leukemia patients with DNMT3A mutations and prognostic implications of DNA methylation. Leuk. Res. 2012 doi: 10.1016/j.leukres.2012.05.012. [advance online publication] [DOI] [PubMed] [Google Scholar]
- 47.Gao Q., Steine E.J., Barrasa M.I., Hockemeyer D., Pawlak M., Fu D.D., Reddy S., Bell G.W., Jaenisch R. Deletion of the de novo DNA methyltransferase Dnmt3a promotes lung tumor progression. Proc. Natl Acad. Sci. USA. 2011;108:18061–18066. doi: 10.1073/pnas.1114946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li B.Z., Huang Z., Cui Q.Y., Song X.H., Du L., Jeltsch A., Chen P., Li G., Li E., Xu G.L. Histone tails regulate DNA methylation by allosterically activating de novo methyltransferase. Cell Res. 2011;21:1172–1181. doi: 10.1038/cr.2011.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGarvey K.M., Greene E., Fahrner J.A., Jenuwein T., Baylin S.B. DNA methylation and complete transcriptional silencing of cancer genes persist after depletion of EZH2. Cancer Res. 2007;67:5097–5102. doi: 10.1158/0008-5472.CAN-06-2029. [DOI] [PubMed] [Google Scholar]
- 50.Lembo F., Pero R., Angrisano T., Vitiello C., Iuliano R., Bruni C.B., Chiariotti L. MBDin, a novel MBD2-interacting protein, relieves MBD2 repression potential and reactivates transcription from methylated promoters. Mol. Cell Biol. 2003;23:1656–1665. doi: 10.1128/MCB.23.5.1656-1665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fuks F., Burgers W.A., Godin N., Kasai M., Kouzarides T. Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J. 2001;20:2536–2544. doi: 10.1093/emboj/20.10.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raynal N.J., Si J., Taby R.F., Gharibyan V., Ahmed S., Jelinek J., Estecio M.R., Issa J.P. DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory. Cancer Res. 2012;72:1170–1181. doi: 10.1158/0008-5472.CAN-11-3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu L., Kron K.J., Pethe V.V., Demetrashvili N., Nesbitt M.E., Trachtenberg J., Ozcelik H., Fleshner N.E., Briollais L., van der Kwast T.H., et al. Association of tissue promoter methylation levels of APC, TGFbeta2, HOXD3 and RASSF1A with prostate cancer progression. Int. J. Cancer. 2011;129:2454–2462. doi: 10.1002/ijc.25908. [DOI] [PubMed] [Google Scholar]
- 54.Elliott R.L., Blobe G.C. Role of transforming growth factor Beta in human cancer. J. Clin. Oncol. 2005;23:2078–2093. doi: 10.1200/JCO.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 55.Li C.M., Kim C.E., Margolin A.A., Guo M., Zhu J., Mason J.M., Hensle T.W., Murty V.V., Grundy P.E., Fearon E.R., et al. CTNNB1 mutations and overexpression of Wnt/beta-catenin target genes in WT1-mutant Wilms’ tumors. Am. J. Pathol. 2004;165:1943–1953. doi: 10.1016/S0002-9440(10)63246-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu M., Smolen G.A., Zhang J., Wittner B., Schott B.J., Brachtel E., Ramaswamy S., Maheswaran S., Haber D.A. A developmentally regulated inducer of EMT, LBX1, contributes to breast cancer progression. Genes Dev. 2009;23:1737–1742. doi: 10.1101/gad.1809309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Burwell E.A., McCarty G.P., Simpson L.A., Thompson K.A., Loeb D.M. Isoforms of Wilms’ tumor suppressor gene (WT1) have distinct effects on mammary epithelial cells. Oncogene. 2007;26:3423–3430. doi: 10.1038/sj.onc.1210127. [DOI] [PubMed] [Google Scholar]
- 58.Metivier R., Gallais R., Tiffoche C., Le Peron C., Jurkowska R.Z., Carmouche R.P., Ibberson D., Barath P., Demay F., Reid G., et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 59.Kangaspeska S., Stride B., Metivier R., Polycarpou-Schwarz M., Ibberson D., Carmouche R.P., Benes V., Gannon F., Reid G. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–115. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- 60.Strathdee C.A., McLeod M.R., Hall J.R. Efficient control of tetracycline-responsive gene expression from an autoregulated bi-directional expression vector. Gene. 1999;229:21–29. doi: 10.1016/S0378-1119(99)00045-1. [DOI] [PubMed] [Google Scholar]
- 61.Gebhard C., Schwarzfischer L., Pham T.H., Schilling E., Klug M., Andreesen R., Rehli M. Genome-wide profiling of CpG methylation identifies novel targets of aberrant hypermethylation in myeloid leukemia. Cancer Res. 2006;66:6118–6128. doi: 10.1158/0008-5472.CAN-06-0376. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.