

Abstract
The reaction of an aryne with an alkyne to generate a benzocyclobutadiene (BCB) intermediate is rare. We report here examples of this reaction, revealed by Diels-Alder trapping of the BCB by either pendant or external, electron-deficient alkynes. Mechanistic delineation of the reaction course is supported by DFT calculations. A three-component process joining a benzyne, first, with an electron-rich and, then, an electron-poor alkyne was uncovered. Reactions in which the BCB functions in a rarely observed role as a 4π diene component in Diels-Alder reactions are reported. The results also shed new light on aspects of the hexadehydro-Diels-Alder reaction used to generate the benzynes.
Keywords: benzocyclobutadiene, cyclization, benzyne, Diels–Alder, alkyne cascades
Graphical Abstract
Benzocyclobutadiene (BCB) has been accessed via a [2+2] cycloaddition of a benzyne and an alkyne. The BCB generated by this method exhibited rare reactivity—namely, as a 4π component in a Diels–Alder reaction to furnish highly substituted polyaromatic compounds as the product. DFT calculations revealed many mechanistic details of this cascade process.
o-Benzyne (1) and its analogs have been widely studied and used in organic synthesis. The major classic reaction modes of the distorted triple bond involve nucleophilic addition and [4+2] or 1,3-dipolar cycloadditions.[1] Another highly reactive intermediate benzocyclobutadiene (2, BCB) is not nearly as explored due to its relative inaccessibility.[2] Moreover, reactions of BCBs, once formed, are often not highly selective and give rise to an array of products. In principle, BCB can be generated by reaction of a benzyne with an alkyne via a net [2+2] cycloaddition. However, reports of such transformations are extremely rare.[3,4] This is surprising because the reaction of a benzyne with an alkyne is computed to be significantly exergonic (cf. 58.5 kcal•mol−1 for 9 to 10, Figure S1 in the SI) and to proceed with a low activation energy.[5] Therefore, the paucity of reports of alkyne + benzyne [2+2] reactions may reflect inefficiency in the subsequent manifold of reactions into which an initially formed BCB intermediate enters rather than inherent difficulty in its generation. In the initial discovery, Stiles et al. (Figure 1a) rationalized the formation of dibenzocyclooctatetraenes 3 by dimerization of BCB intermediates 2.[3] In the only other example of this type of process to our knowledge, Shindo and Alabugin recently reported the reaction of various benzynes with electron-rich ynolates 4 to give triptycenes 6, which they proposed to arise via initial formation of formal [2+2] adducts 5 (Figure 1b).[4]
Figure 1.

Only previous examples of benzyne + alkyne net [2+2] cyclization (a, b) and our first encounter: dimerization of 7 (c).
The hexadehydro-Diels–Alder (HDDA) reaction provides a reagent-free and fully atom-economical method for producing a benzyne intermediate from a poly-yne substrate,[6] which can be immediately trapped by an in situ arynophile.[7] The formation of benzyne is the rate-limiting step in an HDDA generation/trapping cascade,[8] which means that the benzyne is present in only a low steady-state concentration along with a larger amount of the poly-yne precursor, depending upon the stage of conversion. Therefore, in the absence of a trapping reagent, we presume that the HDDA-benzyne engages one of the several alkynes in the substrate, generally in a non-selective fashion to generate a host of BCB intermediates, resulting in intractable product mixtures. Thus, we were surprised to observe that heating the triyne 7 in the absence of any trapping partner gave one major product (1H NMR spectrum of the crude product mixture)—the dimer 8, which was isolated in 45% yield (Figure 1c). Five alkyne units in two molecules of 7 had been transformed into a naphthalene core bearing one tBu-ethynyl substituent. This purely thermal process raises a number of intriguing mechanistic questions.
Our proposed mechanism for this unexpected transformation is shown in Figure 2. The consumption of 7 proceeded with a comparable half-life to that of a similar triyne substrate in a typical HDDA cycloisomerization.[9] Thus, formation of the HDDA benzyne 9 derived from triyne 7 presumably began the process. To form the observed regioisomer of naphthalene 8, benzyne 9 would need to selectively undergo a [2+2] cycloaddition with one of the three triple bonds in 7 and with a preference for one of two possible orientations. This would produce the BCB intermediate 10, which could then be trapped regioselectively in a [4+2] cycloaddition as the 4π component by the intramolecularly tethered propiolate dienophile to afford the hemi-Dewar naphthalene 11. Intermediate 11 would be expected to rapidly open to 8.[10]
Figure 2.

Proposed mechanism for dimerization of triyne 7 to the alkynylnaphthalene derivative 8.
To explore some of the generality of the reaction, we prepared a series of analogs of ester 7, differing in the substituents at the termini of the diyne and diynophile (12, Figure 3). Notably, a cyclopropyl group remains intact (13d), presumably because the spin of the diradical intermediates is substantially delocalized, thereby slowing the rate of the potential ring-opening reaction.[11] Aryl substituents (13i–13o) are compatible. The presence of a TMS group in the aryl-substituted triynes led to improved yields and facilitated the solubility and handling of some of the products. Some of the products exhibited blue fluorescence upon exposure to a 365 nm light source (see SI for two examples).[12]
Figure 3.

Examples of dimerizations of ester-tethered triynes 12. [a] dr = 1:1. [b] a small amount of a regioisomer (see SI for 13i’) was observed (1H NMR analysis of the crude product mixture). [c] 140 °C, 48 h. [d] 150 °C, 24 h. TBS = tert-butyldimethylsilyl, TMS = trimethylsilyl, TIPS = triisopropylsilyl.
DFT calculations were performed to inform the mechanistic thinking (Figure S1). Conversion of 7 to 9 is seen to be exergonic by 47.7 kcal•mol−1.[9,13] The reaction of 9 with a second molecule of 7 to form the BCB intermediate 10 was seen to be a stepwise net [2+2] cyclization proceeding through a diradical (S17).[5],[14] Because an alkyne is a strong radical-stabilizing group,[15] the lowest energy transition structure (TS) was computed to be that implied by arrows “a” (Figure 2). The subsequent conversion of 10 to the hemi-Dewar naphthalene 11, a formal Diels-Alder cycloaddition, was also computed to be stepwise (cf. arrows “b”).[16] A TS for the concerted process was located but found to have a 24.8 kcal•mol−1 higher barrier compared to the diradical pathway. Finally, the fragmentation of 11 was computed to afford naphthalene 8 via a low barrier TS (9.2 kcal•mol−1). Notably, the overall transformation that converts the five C≡C bonds in two molecules of 7 to the naphthalene 8 is computed to be 199 kcal•mol−1 (!) exergonic.
To bring additional light to bear on our hypothesized mechanism, hetero-dimerization experiments were carried out. The HDDA precursors 14a–d and triynes 7a–d (Figure 4a) have the indicated mismatched half-lives for their rates of cycloisomerization. We presumed that, when heated together at 80 °C, triyne 7 would remain essentially intact, while tetrayne 14 would cyclize to the benzyne 15. In the event, 15 was trapped by 7 to give 16a–d in a process paralleling the benzocyclobutadiene trapping stage in the homodimerization of 7. Similarly, two faster-reacting poly-ynes were also tested with the propiolate derivative 7d (Figure 4b); these gave rise to the polyaromatic compounds (18, 20). The product fluorenone 18 is noteworthy because it arises from a naphthyne intermediate, formed by way of a domino-HDDA, double cycloisomerization[17] of a pentayne precursor (see SI). Additionally (Figure 4c), the propiolate diyne 7e captured the benzyne derived from 14a to give 21, a product suggesting that the bulky mesityl substituent had steered the intramolecularly linked propiolate to approach the BCB in an unusual orientation (see the dashed line) to afford a net [2+2+2] product along with a minor isomer that derives from the normal pathway (ca. 5:1, see SI).
Figure 4.

Hetero-dimer formation between a fast-reacting HDDA substrate (e.g., 14) and propiolate 7 and its analogs. Ms = methanesulfonyl.
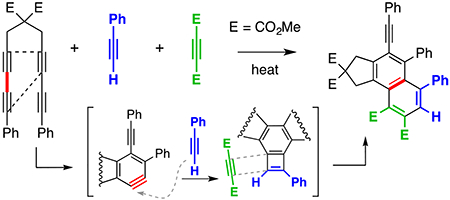
We next hypothesized that an initial stepwise [2+2] reaction between a benzyne and a simple alkyne (but one bearing a radical stabilizing group) would also form a BCB intermediate, which might then be trapped preferentially by a second, electron-deficient alkyne. This was first demonstrated (Figure 5a) in a three-component reaction involving the HDDA-precursor 22, the (relatively) electron-rich 2,4-hexadiyne (23a), and electron-poor dimethyl acetylenedicarboxylate (24). The naphthalene derivative 25a (38%) was produced; its structure was assigned on the basis of nOe and HMBC analyses (see SI). This reaction most likely proceeds through three reactive intermediates: the benzyne 26, the BCB 27, and the hemi-Dewar naphthalene 28. Similarly, we surmised that simple arylethynes 23b–d might also serve as effective initial alkyne traps for the benzyne (Figure 5b). Indeed, heating 22 with one of 23b–d and 24 gave 25b–d. Each of these reactions could have produced 8 constitutionally isomeric, 3-component adducts, but the only such product observed (direct LC-MS analysis of the reaction mixture) was that having the skeleton common to 25b–d.
Figure 5.

Three-component reactions: benzyne + electron-rich alkyne + BCB “dienophile”. Five equivalents of each (relative to the HDDA substrate) were used in each experiment. DMAD = Dimethyl acetylenedicarboxylate.
We then showed it is not essential to use an alkyne as the electron-deficient BCB trapping agent for this transformation. For example, reactions of 22 with 23a/23b in the presence, now, of maleimide 29 as the third component led to dihydronaphthalene products 25e/25f (Figure 5c). To demonstrate that these reactions are not unique to only the benzyne 26, the ester-linked triyne HDDA substrate 7d was also examined. When heated in the presence of DMAD (24) and 2,4-hexadiyne (23a), this gave 25g (Figure 5d), the direct analog of product 25a. Interestingly, when this reaction was performed in the absence of 24, a three component product was still observed—namely, 25h. This 2:1 adduct has incorporated two equivalents of 2,4-hexadiyne (23a) and the benzyne from 7d. The regioselectivity of this process supports the view that net [4+2] trapping of the BCB is a highly asynchronous event with considerable diradical character—the non-participating, second alkyne in 23a is a powerful radical stabilizing group.[15]
Finally, we have observed that the high, strain relief-driven reactivity of a benzyne is not a prerequisite for cyclobutadiene formation.[18] In particular, when heated in the presence of DMAD (24), the tetrayne 30 (Figure 6) gave the adducts 33a and 33b as the only tractable products. These most likely arise from a [4+2] reaction between the fused cyclobutadiene (CB) 32 and 24. It is surprising that the tetrayne 30, which contains a four-atom tether between its internal alkynes, gives a CB to the exclusion of a benzyne.[19] This is the first time we have gained insight to why there is a nearly absolute requirement that HDDA substrates contain a three-atom tether linking the diyne and diynophile moities—a four-atom linker is more capable of accommodating formation of a fused cyclobutadiene (cf. 32) and the diradical (cf. 31) does so (black arrow) in preference to cyclizing at the distal terminus of the propargylic radical (gray arrow), even though the resulting benzyne is computed to be (see SI) 30.5 kcal•mol−1 more stable than the isomeric CB 32. Additionally, trapping of 32 with N-phenylmaleimide (34) was explored. Initially, a mixture of multiple stereoisomeric products, composing what we surmised to be 2:1 adducts from DA reaction of the maleimide with 35, was observed. This complication was avoided when the reaction was performed in the presence of manganese dioxide, an oxidant that effectively intercepted diene 35 by its conversion to the phthalimide 36.[20]
Figure 6.

A four-atom tether thwarts the HDDA reaction by allowing for faster formation of the CB 32.
In conclusion, we have described a series of polyalkyne cascade processes via benzocyclobutadiene (BCB) intermediates. These demonstrate the feasibility of generating BCBs from certain (thermally generated) benzynes and appropriate alkyne trapping partners. A rare mode of BCB trapping—namely, as a 4π component in DA reactions—was also uncovered. This results in the production of alkynyl naphthalene derivatives under purely thermal conditions. DFT calculations support a stepwise formation of the BCB and guided us in the design of several multicomponent reactions. These results provide new mechanistic insights about thermal alkyne chemistry.
Supplementary Material
Acknowledgements
This work was supported by the U.S. Dept. of Health and Human Services [National Institute of General Medical Sciences (R01 GM65597 then R35 GM127097)] and the National Science Foundation (CHE-1665389). Computational work was made possible by the University of Minnesota Supercomputing Institute (MSI). Some of the NMR data were obtained with an instrument funded by the NIH Shared Instrumentation Grant program (S10OD011952). We thank Victor G. Young, Jr. (University of Minnesota) for the X-ray diffraction analysis.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Hoffmann RW, Dehydrobenzene and Cycloalkynes; Organic Chemistry; a Series of Monographs 11; Academic Press, New York, 1967;Garcia-López J-A, Greaney MF, Chem. Soc. Rev 2016, 45, 6766,and refs 3–16 therein to previous reviews.
- [2].a) Cava MP, Mitchell MJ, Cyclobutadiene and Related Compounds; Academic, New York, 1967; [Google Scholar]; b) Toda F, Garratt P, Chem. Rev 1992, 92, 1685. [Google Scholar]
- [3].a) Stiles M, Burckhardt U, Haag A, J. Org. Chem, 1962, 27, 4715; [Google Scholar]; b) Stiles M, Burckhardt U, J. Am. Chem. Soc 1964, 86, 3396. [Google Scholar]
- [4].Umezu S, Gomes GP, Yoshinaga T, Sakae M, Matsumoto K, Iwata T, Alabugin IV, Shindo M, Angew. Chem. Int. Ed 2017, 56, 1298. [DOI] [PubMed] [Google Scholar]
- [5].Cahill KJ, Ajaz A, Johnson RP, Aust. J. Chem 2010, 63, 1007. [Google Scholar]
- [6].Bradley AZ, Johnson RP, J. Am. Chem. Soc 1997, 119, 9917;Miyawaki K, Suzuki R, Kawano T, Ueda I, Tetrahedron Lett. 1997, 38, 3943;Torikai K, Otsuka Y, Nishimura M, Sumida M, Kawai T, Sekiguchi K, Ueda I, Bioorg. Med. Chem 2008, 16, 5441,and references therein;Tsui JA, Sterenberg BT, Organometallics 2009, 28, 4906.
- [7].a) Hoye TR, Baire B, Niu DW, Willoughby PH, Woods BP, Nature 2012, 490, 208; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Baire B, Niu D, Willoughby PH, Woods BP, Hoye TR, Nat. Protoc 2013, 8, 501; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yun SY, Wang K, Lee N, Mamidipalli P, Lee D, J. Am. Chem. Soc 2013, 135, 4668; [DOI] [PubMed] [Google Scholar]; For reviews: [Google Scholar]; d) Holden C, Greaney MF, Angew. Chem., Int. Ed 2014, 53, 5746; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Diamond OJ, Marder TB, Org. Chem. Front 2017, 4, 891. [Google Scholar]
- [8].a) Woods BP, Baire B, Hoye TR, Org. Lett 2014, 16, 4578; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Willoughby PH, Niu D, Wang T, Haj MK, Cramer CJ, Hoye TR, J. Am. Chem. Soc 2014, 136, 13657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Marell DJ, Furan LR, Woods BP, Lei XY, Bendelsmith AJ, Cramer CJ, Hoye TR, Kuwata KT, J. Org. Chem 2015, 80, 11744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].The parent hemi-Dewar naphthalene is reported to open to naphthalene with a t1/2 of ca. 5 h at 38 °C:Grimme W, Heinze U, Chem. Ber 1978, 111, 2563.
- [11].Halgren TA, Roberts JD, Horner JH, Martinez FN, Tronche C, Newcomb M, J. Am. Chem. Soc 2000, 122, 2988. [Google Scholar]
- [12].Xu F, Hershey KW, Holmes RJ, Hoye TR, J. Am. Chem. Soc 2016, 138, 12739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Ajaz A, Bradley AZ, Burrell RC, H. Li WH, Daoust KJ, Bovee LB, DiRico KJ, Johnson RP, J. Org. Chem 2011, 76, 9320; [DOI] [PubMed] [Google Scholar]; b) Liang Y, Hong X, Yu PY, Houk KN, Org. Lett 2014, 16, 5702; [DOI] [PubMed] [Google Scholar]; c) Skraba-Joiner SL, Johnson RP, J. Org. Chem 2015, 80, 11779. [DOI] [PubMed] [Google Scholar]
- [14].Yao ZK, Yu ZX, J. Am. Chem. Soc 2011, 133, 10864. [DOI] [PubMed] [Google Scholar]
- [15].a) Bernstein HJ, Spectrochim. Acta 1962, 18, 161; [Google Scholar]; b) Pasto DJ, Krasnansky R, Zercher C, J. Org. Chem 1987, 52, 3062; [Google Scholar]; c) Henry DJ, Parkinson CJ, Mayer PM, Radom L, J. Phys. Chem. A 2001, 105, 6750; [Google Scholar]; d) Zipse H, Top. Curr. Chem 2006, 263, 163. [Google Scholar]
- [16].Limanto J, Khuong KS, Houk KN, Snapper ML, J. Am. Chem. Soc 2003, 125, 16310. [DOI] [PubMed] [Google Scholar]
- [17].Xiao X, Hoye TR, Nature Chem. 2018, DOI: 10.1038/s41557-018-0075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Related substrates that might have produced a fused CB have been shown to proceed, instead, through a propargylic ene reaction, a process not feasible for 30:Saaby S, Baxendale IR, Ley SV, Org. Biomol. Chem 2005, 3, 3365;Robinson JM, Sakai T, Okano K, Kitawaki T, Danheiser RL, J. Am. Chem. Soc 2010, 132, 11039.A four-atom tethered diyne without additional alkynyl substituents required 265 °C (4 h) to form a CB intermediate:Lee C, Leung M, Lee G, Liu Y, Peng S, J. Org. Chem 2006, 71, 8417.
- [19].As a control, 30 was heated in the presence of excess furan, an excellent benzyne trap, and no evidence for benzyne formation was seen.
- [20].Corey EJ, Lazerwith SE, J. Am. Chem. Soc 1998, 120, 12777. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.