

Abstract
The chromatography of deoxyribonuclease (DNase) from small intestine of camel Camelus dromedarius by DEAE-Sepharose separated three isoforms DNase 1, DNase 2 and DNase 3. The DNase 3 was purified to homogeneity by chromatography on Sephacryl S-200. The molecular weight of DNase 3 was 30 kDa using gel filtration and SDS-PAGE. The pH optimum of DNase 3 was reported at 7.0 using Tris-HCl buffer. The temperature optimum of DNase 3 was found to be 50 °C. The enzyme was stable up to 50 °C for one h incubation. The Km value was 28.5 µg DNA, where this low value indicated the high affinity of enzyme toward DNA as substrate. No activity of DNase 3 was determined in the absence of metal cations. Mg2+ and Ca2+ caused significant enhancement in the enzyme activity by 90 and 75%, respectively. The mixture of Mg2+ and Ca2+ caused 100% of enzyme activity. Ni2+, Co2+, Ba2+, Zn2+ and Cd2+ showed very strong inhibitory effect on enzyme activity. In conclusion, the characterization of DNase 3 indicated that the enzyme is considered as a member of DNase I family. The low Km value of the DNA suggested that the high digestion of DNA of camel forage by small intestine DNase 3.
Keywords: Deoxyribonuclease, Small intestine, Camel
1. Introduction
Deoxyribonucleases (DNases) have been classified into DNase I and DNase II families. DNase I enzymes (EC 3.1.21.1) had neutral pH and required Mg2+ and Ca2+ cations. DNases I family included DNase1L1, DNase1L2 and DNase1L3 [5], [25], [28]. DNase II family (EC 3.1.22.1) hydrolyzed DNA at acidic pH and in the absence of Mg2+ and C2+ cations [5], [30], [32]. DNase II family included DNase 2a [17], DNase 2b [19] and L-DNase II [27].
DNase I hydrolyzed DNA to oligonucleotides with 5′-phospho and 3′-hydroxy termini [11], [29]. DNase I is existed principally in tissues of the alimentary canal included intestine, pancreas and salivary glands, where it hydrolyzed DNA [9]. DNase I was also detected in human small intestine [24]. Although the enzyme has been found in all other studied tissues its role in non-digestive tissues remains uncertain [26]. DNase I may be participated in apoptotic cell death [1], [13], [20]. DNase II is found in a wide variety of animal tissues. They are active and present in lysosome and believed to act as a barrier to transfection for DNA or vectors entering the cell by endocytosis [7], [23] and may degrade foreign DNAs and play a role in the replication of DNA [2]. DNase II digested DNA of apoptotic cells after phagocytosis [12].
The feeding of camels depended on low quality natural forages, which existed in deserts and dry lands. Camels can survive in very dry environmental conditions with the minimal amount of water [3]. Most studies of digestion of camel focused on microbial digestion in the rumen [6], [21], [22]. We previously studied some of digestive enzymes in camel such as carbohydrases in pancreas and disaccharidase in small intestine [15], [14]. In the present study, deoxyribonuclease, as digestive enzyme, from small intestine of camel Camelus dromedarius has been purified and characterized.
2. Materials and methods
2.1. Camel small intestine
Camel small intestine was obtained from Cairo slaughter house. The small intestine was saved directly into an ice box for transportation to the laboratory and transferred to frozen storage at −80 °C.
2.2. Deoxyribonuclease assay
Deoxyribonuclease (DNase) activity measurements were carried out according to Yasuda et al. [29]. The one ml reaction mixture consisted of 20 µg calf thymus DNA, 10 mM MgCl2, 10 CaCl2, 50 mM Tris-HCl buffer, pH 7.0 and 2–10 µg protein. The change in absorbance at 260 nm was followed at 30 s intervals. One unit of DNase activity was defined as the amount of enzyme which increases the O.D. 1.0 per min under standard assay conditions.
2.3. Purification of DNase from camel small intestine
2.3.1. Preparation of crude extract
The DNase crude extract was prepared by homogenization of 5 g camel small intestine in 15 ml of 20 mM Tris–HCl buffer, pH 7.0 using a homogenizer. The homogenate was centrifuged at 10,000 g and the supernatant was designated as crude extract. The crude extract was subjected to dialysis against the same buffer.
2.3.2. Column chromatography
The dialyzate was applied directly to a diethylaminoethanol (DEAE)-Sepharose column (10 × 1.6 cm i.d.) pre-equilibrated with the same buffer. The adsorbed material was eluted with a stepwise gradient of NaCl ranging from 0.0 to 0.2 M prepared in the same buffer at a flow rate of 30 ml/h and 3-ml fractions were collected. The pooled fractions (0.2 M NaCl) with the highest specific activity of DNase were concentrated through dialysis against solid sucrose and applied on a Sephacryl S-200 column (90 × 1.6 cm i.d.) previously equilibrated with the same buffer and developed at a flow rate of 20 ml/h, and 3.0 ml fractions were collected.
2.4. Protein determination
Protein was quantified by the method of Bradford [4]. Bovine serum albumin was used as the protein standard.
2.5. Molecular weight determination
Molecular weight was determined by gel filtration technique using Sephacryl S-200. The column (90 × 1.6 cm i.d.) was calibrated with cytochrome C (12,400), carbonic anhydrase (29,000), bovine serum albumin (67,000), alcohol dehydrogenase (150,000) and β-amylase (200,000). Dextran blue (2,000,000) was used to determine the void volume (Vo). Subunit molecular weight was estimated by SDS-polyacrylamide gel electrophoresis [10]. SDS-denatured phosphorylase b (94,000), bovine serum albumin (67,000), ovalbumin (43,000), carbonic anhydrase (30,000), soybean trypsin inhibitor (20,000) and α-lactalbumin (14,200) were used for the calibration curve.
2.6. Characterization of DNase 3
The effect of pH on DNase 3 activity was determined in the pH range from 4.5 to 9.0 using 50 mM sodium acetate buffer (pH 4.5–6.0), sodium phosphate buffer (pH 6.5–7.5) and Tris-HCl buffer (pH 7.0–9.0). The optimal temperature for DNase activity was determined by incubating the enzyme-substrate mixtures at various temperatures (10–80 °C) in 50 mM Tris-HCl buffer, pH 7.0. Thermal stability of DNase was measured in terms of residual activity after incubation of DNase at different temperatures (10–80 °C) for 1 h prior to substrate addition. The Km value was determined from Lineweaver-Burk plot using DNA concentrations from 20 to 100 µg. The effect of metal cations on DNase activity was investigated by preincubating the enzyme with 10 mM Mg2+, Ca2+, Zn2+, Co2+, Hg2+, Ba+, Cd2+ and Ni2+ for one h prior to substrate addition.
3. Results and discussion
Table 1 summarizes the purification steps of DNase from camel small intestine. Three isoforms DNase1, DNase 2 and DNase 3 were appeared from the chromatography on DEAE-Sepharose column (Fig. 1). Further purification was restricted to DNase 3 with highest specific activity compared with DNase 1 and DNase 2. DNase 3 with the highest specific activity of 8750 units/mg protein and 8.16 purification fold was detected from chromatography on Sephacryl S-200 column (Fig. 2).
Table 1.
Purification scheme of DNase from small intestine of camel.
| Purification step | Total protein (mg) | Total unitsa | Specific activity (unit/mg protein) | Fold purification | % Recovery |
|---|---|---|---|---|---|
| Crude extract | 3.15 | 3375 | 1071 | 1.0 | 100 |
| Chromatography on DEAE-Sepharose | |||||
| 0.0 M NaCl (DNase 1) | 0.78 | 1300 | 1666 | 1.55 | 38.5 |
| 0.1 M NaCl (DNase 2) | 0.336 | 833 | 2479 | 2.31 | 24.6 |
| 0.2 M NaCl (DNase 3) | 0.15 | 670 | 4466 | 4.16 | 19.8 |
| Gel filtration on Sephacryl S-200 | |||||
| (DNase 3) | 0.06 | 525 | 8750 | 8.16 | 15.5 |
One unit of DNase activity is defined as the amount of enzyme that increases the optical density 1.0 per min under standard assay conditions.
Fig. 1.

A typical elution profile for the chromatography of camel small intestine DNase on DEAE-Sepharose column (10 × 1.6 cm i.d.) previously equilibrated with 20 mM Tris-HCl buffer, pH 7.0. Fractions of 3 ml were collected at flow rate 30 ml/h. ∘----∘ O.D at 280 nm, •---• Units/fraction.
Fig. 2.

A typical elution profile for the chromatography of camel small intestine DNase of DEAE-Sepharose fraction 0.2 M NaCl on Sephacryl S-200 column (90 × 1.6 cm i.d.) previously equilibrated with 20 mM Tris-HCl buffer, pH 7. Fractions of 3.0 ml were collected at flow rate 20 ml/h. ∘----∘ O.D at 280 nm, •---• Units/fraction. The void volume was detected in fraction number 25.
The homogeneity of the purified DNase 3 was demonstrated by the presence of one single protein band on SDS-polyacrylamide gel (Fig. 3). The molecular weight of DNase 3 was estimated to be 30 kDa by gel filtration. This molecular weight was confirmed by SDS-PAGE (Fig. 3) and estimated to be 30 kDa as single subunit. The similar molecular weights of DNases I were reported for rabbit (35 kDa) [31], porcine pancreas (34.5) [16] and hen pancreas (33 kDa) [18]. On the contrary, DNase II from Euglena gracilis and human small intestine had molecular weights of 45 kD and 28–32 kDa, respectively [8], [23].
Fig. 3.
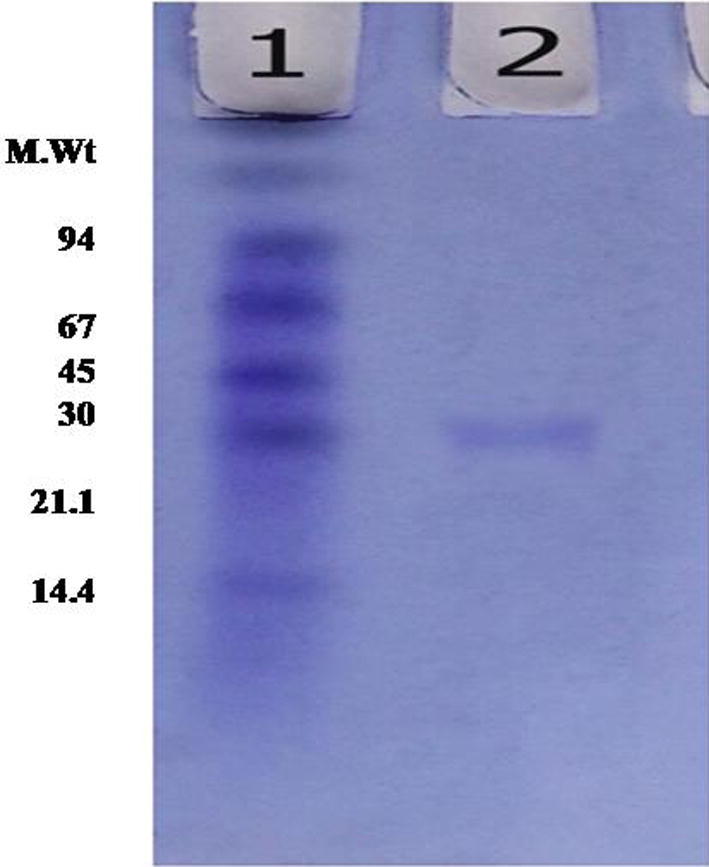
SDS-PAGE for purification and molecular weight determination of camel small intestine DNase. (1) Standard proteins, (2) Sephacryl S-200 DNase 3.
The pH optimum of DNase 3 was reported at 7.0 using Tris-HCl buffer (Fig. 4). At the same pH using sodium phosphate buffer, DNase 3 had no maximum activity. The results suggested that the type of buffer may be affected the pH optimum of the enzyme. Similar and neutral pH optima were reported for DNase I from porcine and hen pancreas [16], [18]. Takeshita et al. [26] reported that the pH optima of DNase I from human, pig, bovine, rabbit, rat and mouse were ranged from 6.5 to 7.0. The acidic pH optima at 5.3 and 4.6 were detected for DNase II from Euglena gracilis and porcine pancreas [8]. Human small intestine DNase II had pH optimum at 6.0 [23].
Fig. 4.

pH optimum of camel small intestine DNase 3. The enzyme activity was measured at various pHs using the standard assay method as previously described. Each point represents the average of two experiments.
The temperature optimum of DNase 3 was reported at 50 °C (Fig. 5). The thermal stability of DNase 3 was investigated (Fig. 6). The enzyme was stable up to 50 °C. Ikeda and Takata [8] reported that the temperature optima at 50 and 40 °C were detected for DNase II from Euglena gracilis and porcine pancreas. The Euglena DNase II is stable up to 60 °C for 15 min incubation and its activity was completely lost after 15 min incubation at 80 °C. The low temperature optimum at 30 °C was detected for human small intestine DNase II [23]. The kinetic of the camel small intestine DNase 3 for DNA was determined by Lineweaver-Burk plot (Fig. 7). The Km value was 28.5 µg DNA, where this low value indicated the high affinity of enzyme toward DNA as substrate.
Fig. 5.

Temperature optimum of camel small intestine DNase 3. The enzyme activity was measured at various temperatures using the standard assay method as previously described. Each point represents the average of two experiments.
Fig. 6.

Effect of temperature on the thermal stability of camel small intestine DNase 3. Each point represents the average of two experiments.
Fig. 7.

A typical Lineweaver–Burk plot of camel small intestine DNase 3 activity to substrate concentrations. Each point represents the average of two experiments.
The influence of metal cations on camel small intestine DNase 3 activity is shown in Table 2. No activity of DNase 3 was determined in the absence of metal cations. Mg2+ and Ca2+ caused significant enhancement in the enzyme activity by 90 and 75%, respectively. The mixture of Mg2+ and Ca2+ caused 100% of enzyme activity. Ni2+, Co2+, Ba2+, Zn2+ and Cd2+ showed very strong inhibitory effect on enzyme activity. Hg2+ was completely inhibited the enzyme activity. Several studies reported that the activity of DNase I required Mg2+ and Ca2+ cations [16], [18]. Takeshita et al. [26] reported that the DNase I activity of human, pig, bovine, rabbit, rat and mouse required Mg2+ and Ca2+ cations. The inhibition of human small intestine DNase II activity by 20% and 40% in the presence of 10 mM Mg2+ and Ca2+ cations was detected [23].
Table 2.
Effect of 10 mM metal cations on DNase 3 from small intestine of camel.
| Metal cation | % Relative activity |
|---|---|
| Non | 0.0 |
| Mg+2 | 90 |
| Ca+2 | 75 |
| Mg+2 + Ca+2 | 100 |
| Ni+2 | 12 |
| Co+2 | 5 |
| Ba+2 | 8 |
| Zn+2 | 7 |
| Cd+2 | 10 |
| Hg+2 | 0.0 |
4. Conclusion
The characterization of the purified camel small intestine DNase 3 especially the neutral pH optimum at 7.0 and its activity which required Mg2+ and Ca2+ indicated the enzyme a member of DNase I family. The low Km value of the DNA appeared the high affinity of enzyme toward DNA, which suggested the high digestion of DNA of camel forage by small intestine DNase 3.
Footnotes
Peer review under responsibility of National Research Center, Egypt.
References
- 1.Aslan E., Arslanyolu M. Eur. J. Protistol. 2015;51:173–185. doi: 10.1016/j.ejop.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 2.Bernardi G., Appella E., Zito R. Biochemistry. 1965;4:1725–1729. [Google Scholar]
- 3.Bhatt V.D., Dande S.S., Patil N.V., Joshi C.G. Mol. Biol. Rep. 2013;40:3363–3371. doi: 10.1007/s11033-012-2411-4. [DOI] [PubMed] [Google Scholar]
- 4.Bradford M.M. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 5.Fischer H., Scherz J., Szabo S., Mildner M., Benarafa C., Torriglia A., Tschachler E., Eckhart L. PLoS ONE. 2011;6:1–9. doi: 10.1371/journal.pone.0017581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gharechahi J., Zahiri H.S., Noghabi K.A., Salekdeh G.H. System. Appl. Microbiol. 2015;38:67–76. doi: 10.1016/j.syapm.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 7.Howell D.P-G., Krieser R.J., Eastman A., Barry M.A. Mol. Therapy. 2003;8:957–963. doi: 10.1016/j.ymthe.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Ikeda S., Takata N. Comp. Biochem. Physiol. B. 2002;131:519–525. doi: 10.1016/s1096-4959(02)00026-x. [DOI] [PubMed] [Google Scholar]
- 9.Lacks S.A. J. Biol. Chem. 1981;256:2644–2648. [PubMed] [Google Scholar]
- 10.Laemmli U.K. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 11.M. Laskowski, Enzymes 3rd ed. 4 (1971) 289–311.
- 12.MacLea K.S., Krieser R.J., Eastman A. Biochem. Biophys. Res. Commun. 2002;292:415–421. doi: 10.1006/bbrc.2002.6687. [DOI] [PubMed] [Google Scholar]
- 13.Mannherz H.G., Peitsch M.C., Zanotti S., Paddenberg R., Polzar B. Curr. Top. Microbiol. Immunol. 1995;198:161–174. doi: 10.1007/978-3-642-79414-8_10. [DOI] [PubMed] [Google Scholar]
- 14.Mohamed S.A., Fahmy A.S., Salah H.A. Comp. Biochem. Physiol. 2007;146B:124–130. doi: 10.1016/j.cbpb.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Mohamed S.A., Fahmy A.S., Salah H.A. Comp. Biochem. Physiol. B. 2005;140:73–83. doi: 10.1016/j.cbpc.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 16.Mori S., Yasuda T., Takeshita H., Nakajima T., Nakazato E., Mogi K., Kaneko Y., Kishi K. Biochim. et Biophys. Acta. 2001;1547:275–287. doi: 10.1016/s0167-4838(01)00196-0. [DOI] [PubMed] [Google Scholar]
- 17.Nagata S. Ann. Rev. Immunol. 2005;23:853–875. doi: 10.1146/annurev.immunol.23.021704.115811. [DOI] [PubMed] [Google Scholar]
- 18.Nakashima Y., Yasuda T., Takeshita H., Nakajima T., Hosomi O., Mori S., Kishi K. Int. J. Biochem. Cell Biol. 1999;31:1315–1326. doi: 10.1016/s1357-2725(99)00051-5. [DOI] [PubMed] [Google Scholar]
- 19.Nishimoto S., Kawane K., Watanabe-Fukunaga R., Fukuyama H., Ohsawa Y., Uchiyama Y., Hashida N., Ohguro N., Tano Y., Morimoto T., Fukuda Y., Nagata S. Nature. 2003;424:1071–1074. doi: 10.1038/nature01895. [DOI] [PubMed] [Google Scholar]
- 20.Polzar B., Peitsch M.C., Loos R., Tschopp J., Mannherz H.G. Eur. J. Cell Biol. 1993;62:397–405. [PubMed] [Google Scholar]
- 21.Samsudin A.A., Wright A.D., Al Jassim R. Appl. Environ. Microbiol. 2012;78:8836–8839. doi: 10.1128/AEM.02420-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samsudin A.A., Evans P.N., Wright A.D., Al Jassim R. Environ. Microbiol. 2011;13:3024–3035. doi: 10.1111/j.1462-2920.2011.02579.x. [DOI] [PubMed] [Google Scholar]
- 23.Shikara M., Al-Jaff K.H.M. Turk. J. Biochem. 2012;37:188–195. [Google Scholar]
- 24.Shimada O., Ishikawa H., Tosaka-Shimada H., Yasuda T., Kishi K., Suzuki S. J. Histochem. Cytochem. 1998;46:833–840. doi: 10.1177/002215549804600706. [DOI] [PubMed] [Google Scholar]
- 25.Shiokawa D., Tanuma S. Biochemistry. 2001;40:143–152. doi: 10.1021/bi001041a. [DOI] [PubMed] [Google Scholar]
- 26.Takeshita H., Mogi K., Yasuda T., Nakajima T., Nakashima Y., Mori S., Hoshino T., Kishi K. Biochem. Biophys. Res. Commun. 2000;269:481–484. doi: 10.1006/bbrc.2000.2300. [DOI] [PubMed] [Google Scholar]
- 27.Torriglia A., Lepretre C. Front. Biosci. 2009;14:4836–4847. doi: 10.2741/3572. [DOI] [PubMed] [Google Scholar]
- 28.Yang W. Q. Rev. Biophys. 2011;44:1–93. doi: 10.1017/S0033583510000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yasuda T., Awazu S., Sato W., Iida R., Tanaka Y., Kishi K. J. Biochem. 1990;108:393–398. doi: 10.1093/oxfordjournals.jbchem.a123212. [DOI] [PubMed] [Google Scholar]
- 30.Yasuda T., Nadano D., Awazu S., Kishi K. Biochim. Biophys. Acta. 1992;1119:185–193. doi: 10.1016/0167-4838(92)90390-y. [DOI] [PubMed] [Google Scholar]
- 31.Yasuda T., Takeshita H., Nakajima T., Hosomi O., Nakashima Y., Kishi K. Biochem. J. 1997;325:465–473. doi: 10.1042/bj3250465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yasuda T., Takeshita H., Lida R., Nakajima T., Hosomi O., Nakashima Y., Kishi K. J. Biol. Chem. 1998;273:2610–2616. doi: 10.1074/jbc.273.5.2610. [DOI] [PubMed] [Google Scholar]