

Abstract
Peganum harmala L. is a perennial herbaceous plant and can be a future drug due to its wide medicinal purposes. Despite its economic importance, the molecular genetics of P. harmal have not yet been studied in detail. Genetic diversity of 12 P. harmala genotypes were investigated by using Inter-Simple Sequence Repeats (ISSR), PCR-RFLP of rDNA-ITS, PCR-SSCP of rDNA-ITS and Simple Sequence Repeat (SSR) markers. The level of polymorphism revealed by ITS-SSCP is the lowest, followed by ITS-RFLP then ISSR and the highest polymorphism level was reported for SSR marker. The AMOVA analysis implied that most of the variation occurred within the Populations. A value of inbreeding coefficient Fis estimated by the three co-dominant markers was nearly equal and offer an indication of the partial out-crossing reproductive system of P. harmala. Principal Coordinate Analysis (PCOA) plot revealed a clear pattern of clustering based on the locations of collected plants which coincide with the isolation by distance. The study revealed that ITS-SSCP and ISSR markers respectively were more informative than the other used markers in the assessment of genetic diversity of P. harmala. The results reflect the great diversity of P. harmala and data obtained from this study can be used for future collecting missions.
Keywords: Peganum harmala, Genetic diversity, ISSR, rDNA-ITS, SSR
1. Introduction
Peganum harmala L. (Peganaceae) is a perennial herbaceous plant and it has long been used for medicinal purposes as herbicide due to the presence of harmine [19]. It is one of the most frequently used medicinal plants for the relief of pain and as an antiseptic agent to treat hypertension, cardiac disease, some nervous system disorders such as Parkinson's disease, Lumbago asthma, colic, jaundice and as a stimulant emmenagogue [1], [5], [15], [33].
P. harmala is propagated by seeds, which have a very short span viability [28]. Because of increasing exploitation of P. harmala natural populations, there is a need to conserve the genetic stock and facing the problem of extinction [13], [28]. Despite, the medicinal uses of this species little is known about the breeding system, population genetic structure. Thus, the assessment and maintenance of its genetic diversity is prerequisite. Molecular techniques can play a role in uncovering the history, estimating the diversity, distinctiveness and population structure, and understanding of the distribution and extent of genetic variation within and between species [32]. So far there are no reports of using molecular markers for characterization of accessions of P. harmala.
In the present study, ISSR, ITS-RFLP, ITS-SSCP and SSR markers were used to assess the genetic diversity in P. harmala. Inter Simple Sequence Repeats (ISSR) markers have proved their effectiveness for population genetic studies and for studying taxonomic relationships at or below the species level [7], [14], [16].
The Internal Transcribed Spacer (ITS) of the nuclear ribosomal DNA region is a large segment consisting of 18S rDNA, 5.8S rDNA and 26S rDNA, congregated together with internal transcribed spacers, ITS1 and ITS2, between genes. ITS-rDNA region has been extensively used to determine genetic diversity and to classify several plants species because they are highly variable [2], [24], [27]. In this respect, two techniques are used to detect polymorphism in ribosomal DNA region; PCR-RFLP and PCR-SSCP. The PCR-RFLP technique detects polymorphisms in DNA regions, which have been amplified by specific oligonucleotide primers and restricted with different endonucleases. ITS-RFLP is a potent tool for the taxonomic study, population heterogeneity and the identification and monitoring of specific accession [18], [24]. Single strand conformation polymorphism (SSCP) detects single-base sequence changes by forming a different banding pattern dependent on its size and structure on a non-denaturing polyacrylamide gel [10].
Microsatellites (SSRs) are highly abundant, co-dominant inheritance, and have enormous extent of allelic diversity depending on the number of motif repetitions [34]. They have high potential for use in genetic diversity and evolutionary studies and determine self-pollination easily and accurately [23], [30], [39].
The ribosomal markers are small-sized molecules and have a low level of recombination compared with genomic markers. Using several marker techniques increase the sensitivity and resolution of detection genetic distinctiveness and revealing genetic variation through increased genome coverage. The present study aimed to compare the effectiveness of ISSR, ITS PCR-RFLP, ITS PCR-SSCP and SSRs markers, in assessing and analyzing the nature and the extent of genetic diversity of wild P. harmala populations collected from a narrow geographical region.
2. Materials and methods
2.1. Plant materials
A total of 12 accessions of A. halimus were collected from four populations. Three populations (1, 2 and 3) grow naturally in the Western Mediterranean desert of Egypt and population 4 grow inland at El-Katamya Plateau (Table 1). Samples were very scarce so three individuals were collected from each population. Genetic diversity of 12 P. harmala genotypes were investigated by using one dominant marker; Inter-Simple Sequence Repeats (ISSR), and three co-dominant markers; PCR-RFLP of rDNA-ITS, PCR-SSCP of rDNA-ITS and Simple Sequence Repeat (SSR) markers.
Table 1.
Location of the collection sites of P. harmala populations and their respective geographic coordinates in Egypt.
| Populations | Site | No | Longitude (N) Latitude (E) |
|---|---|---|---|
| Population 1 | Alexandria, Borg Al-arab, Egypt | 3 | 30°54′33.50″ 29°31′2.70″ |
| Population 2 | Alexandria, Borg Al-arab, Egypt | 3 | 30°54′7.50″ 29°32′42.20″ |
| Population3 | Alexandria, Omayed, Egypt | 3 | 30°47′3.31″ 29°12′9.30″ |
| Population4 | Cairo, Al-Kattamya mountain, Egypt | 3 | 29°58′22.90″ 31°48′36.20″ |
No = Sample size.
2.2. DNA extraction and ISSR-PCR amplifications
DNA was extracted from 100 mg of seeds using I-Genomic Plant DNA Extraction Mini Kit (INRTON), following the manufacturer’s instructions. The genomic DNA was used as a template in PCR amplification using standard ISSR-PCR protocol, with 25 µL sample volumes consisting of: 50 ng of genomic DNA, 5 µL of 5× amplification buffer; 0.5 µL of 25 mM MgCl; 2 µL of 10 mM of each dNTP; 6 µL 10 pmol of ISSR primer 0.1 µL Taq (Promega, USA), the volume was completed up to 25 ul with sterile distilled water. Three anchored inter-SSR primers were used (Table 2). The thermal cycling for the inter-SSR primers was as follows: denaturation at 95 °C for 2 min; 40 cycles at 95 °C for 15 s, 30 °C for 15 s, 72 °C for 1 min, a final elongation step of 5 min at 72 °C. The amplified products were separated by electrophoresis in a 2% agarose gel, stained with ethidium bromide and photographed under UV illumination.
Table 2.
Primer sequence, total number of bands (TB), number of polymorphic bands (PB), percentage of polymorphic bands (PB%), polymorphic information content (PIC) and marker index (MI), for ISSR, ITS-SSCP primers and ITS-RFLP restriction enzymes.
| Marker system | Primer | Primers sequence (5́–3́) | TB | PB | PB% | PIC | EMR | MI | |
|---|---|---|---|---|---|---|---|---|---|
| ISSR | ISSR3 | CACCACCACGC | 14 | 13 | 92.85 | 0.486 | 7.580 | 3.63 | |
| ISSR4 | GTGTGTGTGTGTCG | 10 | 9 | 90.00 | 0.339 | 6.260 | 2.123 | ||
| ISSR6 | GAGAGAGAGAGACC | 18 | 15 | 83.33 | 0.431 | 9.370 | 4.39 | ||
| Mean | 14 | 12.33 | 88.72 | 0.431 | 7.736 | 3.381 | |||
| ITS-RPLP | ITS 1 ITS4 |
EcoRV | 5́…GAT▾ATC..0.3́ 3́…CTA▲TAG..0.5́ |
4 | 4 | 100 | 0.395 | 2.166 | 0.472 |
| AluI | 5́…AG▾CT..0.3́ 3́…TC▲GA..0.5́ |
4 | 4 | 100 | 0.444 | 1.080 | 0.426 | ||
| HaeIII | 5́…GG▾CC..0.3́ 3́…CC▲GG..0.5́ |
1 | 1 | 0 | - | - | - | ||
| Mean | 4 | 4 | 100 | 0.419 | 1.623 | 0.449 | |||
| ITS-SSCP | ITS 1 ITS4 |
TCCGTA GGTGAACCTTGCGG TCCTCC GCTTATTGATATGC |
4 | 4 | 100 | 0.444 | 1.333 | 0.592 | |
| SSR | Pe17 | F: AAAATCATTTCAGGGTGC R: TACTTTGAGCCAGGTGCC |
5 | 4 | 80 | 0.480 | 2.400 | 1.152 | |
| Pe39 | F: TAGCAGAATCACAGAGTT R: CTAGAAATCCCACCAAAA |
4 | 2 | 50 | 0.186 | 3.550 | 0.666 | ||
| Pe145 | F: AATGGGGACGTGTTGTTA R: TGCAGATGGACGATGTTT |
5 | 1 | 20 | 0.095 | 2.163 | 0.090 | ||
| Pe217 | F: AAAAGCAGAACGCTCCCC R: CGGTGCCACGAAATAGTA |
4 | 2 | 50 | 0.218 | 0.950 | 0.382 | ||
| Mean | 4.50 | 2.25 | 0.244 | 2.163 | 0.572 | ||||
2.3. Amplification ITS1-5.8S-ITS2 rDNA gene
The internal transcribed spacer ITS1 and ITS2 regions and the 5.8S ribosomal DNA (rDNA) regions were amplified by using universal primers ITS1 (5′-TCCGTA GGTGAACCTTGCGG-3′) and ITS4 (5′-TCCTCC GCTTATTGATATGC-3′) [36]. Amplifications were performed in 25 μL volumes containing 50 ng of template DNA, 12.5 μl of PCR master mix buffer (2×) (Bioline, Germeny), and 10 pmol for each primer. PCR cycles were as follows: initial denaturation for 3 min at 95 °C followed by 30 cycles of 1 min at 95 °C, annealing at 55 °C for 1 min and extension at 72 °C for 1 min, and 5 min at 72 °C. PCR products were then separated electrophoretically on agarose gel using 2% (w/v) agarose in 0.5 × TBE buffer [29]. The gel was stained with ethidium bromide.
2.4. ITS PCR-RFLP amplification
The PCR product (approx. ≥850 bp) was digested using three restriction enzymes EcoRV, HaeIII and AluI (Table 2). Reactions contained 5 μl of amplified PCR product and 1 μl of restriction enzyme, 2.5 μl enzyme buffer, the volume was completed up to 20 ul with sterile distilled water. Digestions were incubated 45–60 min at 37 °C and stored at −20 °C until use.
2.5. ITS PCR-SSCP amplification
A 3 μl of the PCR product (approx. ≥850 bp) was added to 10 μl of denaturation solution (94% formamide, 0.05% xylene cyanol) and heated at 95 °C for 5 min then suddenly placed on ice. Samples were loaded on a nondenaturing polyacrylamide gel (GeneGel Excel 12, 5/24; Amersham Pharmacia Biotech). Electrophoresis was performed in a temperature-controlled electrophoresis system (GenePhor; Amersham Pharmacia Biotech) at 6 °C with a first run at 600 V, 25 mA, and 15 W for 10 min and then at 600 V, 37 mA, and 21 W for 3 h. Gel was stained with ethidium bromide and photographed under UV illumination.
2.6. SSR-PCR amplification
Amplification of DNA used standard Polymerase Chain Reaction (PCR) protocols, with 25 µL sample volumes consisting of: approximately 50 ng of genomic DNA, 5 µL of 5× amplification buffer; 1.5 µL of 25 mM MgCl; 2 µL of 10 mM of each dNTP; 0.5 U Taq (Promega, USA); and 1 µL 10 pmol of primer. Four SSR primers were selected based on microsatellite polymorphism according to HAN & AN [11] (Table 2). The thermal cycling SSR primers was as follows: 30 cycles at 95 °C for 2 min, 53 °C for 1 min, 72 °C for 1 min, a final elongation step of 10 min at 72 °C. The amplified products were separated on 2% agarose gel, stained with ethidium bromide and photographed under UV illumination.
2.7. Statistical analysis
Alleles were sized using TotalLab version 1.11 software (Nonlinear Dynamics Ltd., Durham, USA) in the presence of a 1.5 kb DNA ladder. Bands were binary scored presence (1) or absence (0) characters to assemble the matrix of the ISSR phenotypes and the number of total bands (TB), the number of polymorphic bands (PB) and the percentage of polymorphic bands (PB%) were calculated. Two parameters: polymorphic information content (PIC) and marker index (MI) were calculated to measure the performance of the used markers according to Roldan-Ruiz et al. [25] and Varshney et al. [35].
The parameters of genetic diversity were calculated using POPGENE 3.2 software [38]. The observed heterozygosity (Ho) is available for co-dominant markers; ITS-RFLP, ITS-SSCP and SSR. Analysis of molecular variance (AMOVA) within and among populations was done using ARLEQUIN V. 1.1 [8]. Principals coordinate analysis (PCoA) [9] was conducted using PAST program [3] on single and combined data of the used markers. The Mantel test was applied using XLSTATARTVIS Software to test the significance of the correlation between the geographic distance and the genetic distance matrices. The genetic dissimilarities were calculated according to Nei & Li [20] using NTSYS software [26]. The similarity between matrices based on different marker system (ISSR, ITS-RFLP, ITS-SSCP and SSR) was calculated using the standardized Mantel coefficient [17].
3. Results
3.1. ISSR profiling
Three ISSR primers (Table 2 and Fig. 1) generated a total of 42 products with an average of 14 products per primer, of which 37 (88.72%) products, with an average of 12.33 products per primer, were polymorphic, and 5 (11.90%) products were monomorphic. The number of products generated by these arbitrary primers was found to range from 10 to 18 of different sizes ranging from 100 to 1500 bp (Fig. 1). The primer ISSR6 yielded the maximum (18) and primer ISSR4, giving the minimum number of amplicon (10). The percentage of polymorphism ranged from 83.33% for primers ISSR6 to 92.85% for primers ISSR3 with an average of 88.72% polymorphism per primer. In this study, high PIC value of 0.49 for primer ISSR3 and low PIC value of 0.33 for primer ISSR4, with an average value of PIC per primer 0.43 was obtained. The highest effective multiplex ratio (EMR) 9.37 was observed with the primer ISSR6 and the lowest 6.26 was observed with the primer ISSR4 with an average EMR of 7.74 per primer. The highest MI was observed with the primer ISSR6 (4.39) and lowest in the primer ISSR4 (2.12) with an average MI of 3.38 per primer was obtained (Table 2).
Fig. 1.

ISSR profiles generated from genomic DNA of P. harmala populations with primers; M: DNA marker.
The observed number of alleles (Na) and effective number of alleles (Ne) ranged between 1.214–1.452 and 1.171–1.361, respectively (Table 3). Similarly, Nei’s gene diversity (h) and Shannon’s Information index (I) ranged between 0.105–0.201 with overall diversity of 0.333 and 0.136–0.287 with an average value of 0.491, respectively. It was found that the genetic variation in populations 4 (I = 0.136) growing in Al-Kattamya mountain lower than the other populations. The percentage of polymorphic loci (PPL) was estimated in the range of 21.43%–45.24% (Table 3). The gene flow value (Nm) and the diversity (Gst) among populations were found to be 0.737 and 0.406, respectively (Table 4).
Table 3.
Genetic diversity and differentiation parameters for four wild populations of P. harmala.
| Marker system | Populations | No | Polymorphicloci | Percentagepopulation level (PPL%) | Observednumber of alleles (Na) | Number of effective alleles (Ne) | Shannon’s index of diversity (I) |
Nei’s genediversity (h) |
|---|---|---|---|---|---|---|---|---|
| ISSR | Population 1 | 3 | 19 | 45.24 | 1.452 | 1.361 | 0.287 | 0.201 |
| Population 2 | 3 | 16 | 38.10 | 1.381 | 1.304 | 0.242 | 0.169 | |
| Population 3 | 3 | 18 | 42.86 | 1.428 | 1.342 | 0.272 | 0.190. | |
| Population 4 | 3 | 9 | 21.43 | 1.214 | 1.171 | 0.136 | 0.0952 | |
| Mean | 3 | 15.5 | 36.91 | 1.369 | 1.295 | 0.234 | 0.155 | |
| Over all loci | 12 | 37 | 88.10 | 1.881 | 1.584 | 0.491 | 0.333 | |
| ITS-RFLP | Population 1 | 3 | 2 | 66.67 | 2.000 | 1.461 | 0.439 | 0.259 |
| Population 2 | 3 | 1 | 33.33 | 1.667 | 1.667 | 0.366 | 0.222 | |
| Population 3 | 3 | 1 | 33.33 | 2.000 | 1.785 | 0.505 | 0.305 | |
| Population 4 | 3 | 1 | 33.33 | 1.500 | 1.500 | 0.346 | 0.250 | |
| Mean | 3 | 1.25 | 41.66 | 1.792 | 1.603 | 0.414 | 0.259 | |
| Over all loci | 12 | 2 | 66.67 | 3.000 | 2.481 | 0.831 | 0.456 | |
| ITS-SSCP | Population 1 | 3 | 1 | 33.33 | 3.000 | 2.000 | 0.867 | 0.500 |
| Population 2 | 3 | 1 | 3.33 | 3.000 | 2.571 | 1.011 | 0.611 | |
| Population 3 | 3 | 1 | 33.33 | 2.000 | 1.381 | 0.450 | 0.277 | |
| Population 4 | 3 | 0 | 0.00 | 1.000 | 1.000 | 0.000 | 0.000 | |
| Mean | 12 | 0.75 | 17.49 | 2.25 | 1.738 | 0.582 | 0.347 | |
| Overall loci | 3 | 1 | 33.33 | 4.000 | 2.526 | 1.083 | 0.604 | |
| SSRs | Population 1 | 3 | 4 | 100 | 2.500 | 2.130 | 0.791 | 0.500 |
| Population 2 | 3 | 4 | 100 | 2.000 | 2.000 | 0.693 | 0.500 | |
| Population 3 | 3 | 4 | 100 | 2.000 | 2.000 | 0.693 | 0.500 | |
| Population 4 | 3 | 3 | 75 | 1.75 | 1.750 | 0.519 | 0.375 | |
| Mean | 3 | 3.75 | 93.75 | 2.06 | 1.97 | 0.674 | 0.468 | |
| Over all loci | 12 | 4 | 100 | 2.750 | 2.344 | 0.905 | 0.559 |
No = Sample size.
Table 4.
Estimates of observed heterozygosity (Ho) and expected heterozygosity (HE), in breeding coefficient (Fis) and gene flow (Nm).
| Marker system | Populations | Ho | HE | Fis |
|---|---|---|---|---|
| ISSR | Nm = 0.737 | |||
| ITS-RFLP | Population 1 | 0.222 | 0.311 | 0.286 |
| Population 2 | 0.222 | 0.266 | 0.165 | |
| Population 3 | 0.500 | 0.366 | -0.366 | |
| Population 4 | 0.500 | 0.300 | -0.666 | |
| Mean | 0.361 | 0.31 | 0.371 | |
| Over all loci | 0.277 | 0.475 | 0.416 | |
| Nm = 0.737 | ||||
| ITS-SSCP | Population 1 | 0.667 | 0.667 | 0.331 |
| Population 2 | 0.333 | 0.733 | 0.549 | |
| Population 3 | 0.333 | 0.333 | -0.667 | |
| Population 4 | 0.00 | 0.000 | 0.000 | |
| Mean | 0.333 | 0.433 | 0.386 | |
| Over all loci | 0.333 | 0.630 | 0.471 | |
| Nm = 0.105 | ||||
| SSRs | Population 1 | 0.833 | 0.600 | 0.388 |
| Population 2 | 1.000 | 0.600 | 0.667 | |
| Population 3 | 1.000 | 0.600 | 0.667 | |
| Population 4 | 0.750 | 0.450 | -0.652 | |
| Mean | 0.896 | 0.563 | 0.471 | |
| Over all loci | 0.895 | 0.584 | -0.532 | |
| Nm = 1.285 |
The estimate of genetic structure (Fst = 0.368) of populations was significantly different from zero (P < .0001) (Table 5). AMOVA analysis showed that most of genetic variation (63.19%) was observed within the populations, whereas the variance among populations was 36.80% (Table 5). The estimate of gene flow Nm based on Gst was 0.406 calculated by Popgene program (Table 4). This result is equivalent to Fixation Indices (Fst) 0.368 P < .0001 calculated with the Arlequin program, which suggests a very low amount of differentiation among populations. The Mantel Test showed a significant correlation between the genetic distance and the geographic distance (r = 0.382, P = .007).
Table 5.
Hierarchical analysis of molecular variance (AMOVA) within and among wild populations of P. harmala. The P values are the probabilities of having a greater variance component than the observed values by chance alone and are based on 1023 random permutations of the data matrix.
| Source of variation | ISSR |
ITS-RFLP |
ITS-SSCP |
SSRs |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variancecomponents | Percentagevariation | P | Variancecomponents | Percentagevariation | P | Variancecomponents | Percentagevariation | P | Variancecomponents | Percentagevariation | P | |
| Among populations | 3.009 | 36.8 | *** | 0.385 | 48.68 | ** | 0.126 | 36.58 | NS | 0.222 | 18.82 | *** |
| Among individuals within populations | 5.166 | 63.19 | *** | 0.052 | −6.58 | NS | 0.052 | 15.00 | NS | −0.833 | −70.59 | NS |
| Within individuals | 0.458 | 57.89 | ** | 0.166 | 48.32 | ** | −1.179 | 151.76 | NS | |||
| Fst | 0.368 | *** | 0.486 | ** | 0.365 | NS | 0.188 | *** | ||||
NS = not significant.
P < .0001.
P < .005.
3.2. ITS region PCR-RFLP
Using ITS1 and ITS4 primers, a single amplification product of rDNA with fragment size 850 bp was observed. Restriction analysis of P. harmala ITS using three different endonucleases (EcoRV, HaeIII and AluI) resulted in distinctly different band patterns for each enzyme tested (Fig. 2). Three genotypes were observed for EcoRV enzyme and four for AluI enzyme in populations 1, 2 and 3. Only one genotype was detected in population 4 in the digestion with the two enzymes EcoRV and AluI. The restriction fragments corresponding to the restriction sites by EcoRV were 800 bp, 738 bp, 455 bp and 300 bp; by AluI were 750 bp, 700 bp, approximately and by HaeIII was undigested. Different alleles were found among individuals of all populations except population 4 in which all individuals share the same alleles in the two enzymes used (Fig. 3). The PIC value and MI were 0.395, 0.44 and 0.472, 0.426 for EcoRV and AluI respectively. The effective multiplex ratio (EMR) (2.166) of EcoRV enzyme was higher than AluI enzyme.
Fig. 2.

RFLP pattern of ITS-rDNA gene with ALuI and EcoRV restriction endonucleases of four P. harmala populations; M: DNA marker, * Amplified product of ITS-rDNA gene; AB, BB, CC, DD, AD: name of genotype.
Fig. 3.

Allele frequencies recorded for four P. harmala populations at ALuI and EcoRV restriction enzymes.
The observed number of alleles (Na) and effective number of alleles (Ne) ranged between 1.5–2.0 and 1.461–1.785, respectively. Nei’s gene diversity (h) and Shannon’s Information index (I) ranged between 0.222 and 0.305 with overall diversity of 0.346–0.505 with an average value of 0.456 and 0.831 respectively. The percentage of polymorphic loci (PPL) was 33.33% for all populations except population 1 which was 66.76% (Table 3).
The estimate of genetic structure (Fst = 0.486) of populations is significantly different from zero (P = .002) (Table 5). Analysis of AMOVA showed that 51.31% genetic variation occurred within the populations, whereas the variance among populations was 48.70% (Table 5), which was in accordance with the Fst (0.486%). The estimate of gene flow Nm based on Fst was 0.219 (Table 4).
The observed heterozygosity (Ho) over all loci was higher than those expected by Hardy–Weinberg model in populations 3 and 4; however, it was lower in populations 1 and 2 (Table 4). Estimates of inbreeding coefficient (Fis) for all populations were non- significantly negative for all loci. ITS-RFLP using AulI and EcoRV enzymes distinctly characterizes P. harmala populations 1 and 2. The Mantel test showed a non-significant correlation between genetic distance and geographic distance (r = −0.162, P = .15).
3.3. ITS region PCR- SSCP profiling
Five ITS-SSCP patterns with allele size ranged from 1000 bp–1800 bp were observed in P. harmala individuals (Fig. 4). The patterns were denominated as genotypes AA, BB, AC, AD and BC. Three genotypes (namely AA, AC and AD) were seen in populations 1; three in population 2 (namely AA, AC and BB), two genotypes (namely BB and BC) were identified in population 3, while only one genotype (namely BB) was found in population 4 (Fig. 4). Fig. 5 shows that the allele and genotype frequencies varied between the populations studied here; except for allele D, which was found only in heterozygous individual of population 1, suggesting a presence of a rare allele or a mutation specific to this population (Table 2). In this marker system, the PIC value of 0.44 was recorded. The effective multiplex ratio (EMR) and the MI were 1.33 and 0.592 respectively (Table 2).
Fig. 4.

Polyacrylamide gel showing the PCR-SSCP patterns of alleles of the internal transcribed spacer (ITS) of the nuclear ribosomal DNA gene amplified from genomic DNA of four P. harmala populations. The SSCP-derived genotypes are indicated at the top of each lane. Bands A, B, C and D in lanes indicate the alleles identified in this work.
Fig. 5.
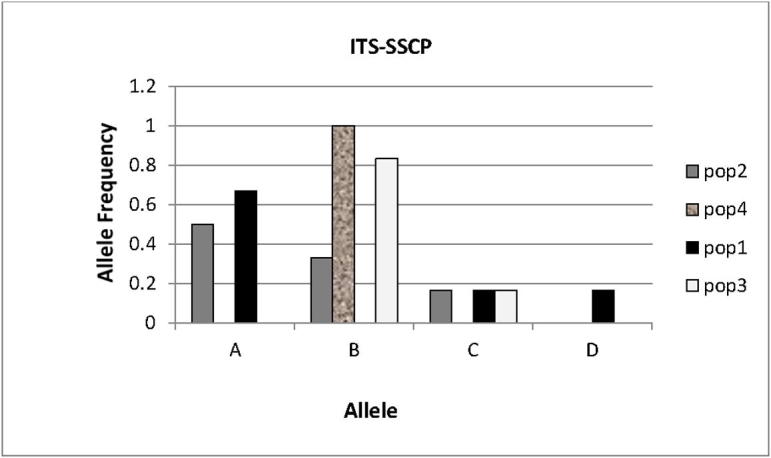
Allele frequencies recorded for four P. harmala populations at the PCR-SSCP of the internal transcribed spacer (ITS) of the nuclear ribosomal DNA gene.
The observed number of alleles (Na) and effective number of alleles (Ne) ranged between 1.0–3.0 and 1.0–2.571, respectively. Nei’s gene diversity (h) and Shannon’s Information index (I) ranged between 0.00 and 0.611 with overall diversity of 0.604 and 0.00–0.867 with an average value of 1.083, respectively. The percentage of polymorphic loci (PPL) (33.33) was constant for populations 1, 2 and 3, while there was no polymorphism detected for population 4 (Table 3).
The estimate of genetic structure (Fst = 0.365) of populations is non- significantly different from zero (P = .07) (Table 5). Analysis of AMOVA showed that 63.32% of genetic variation was observed within the populations, whereas the variance among populations was 36.58% (Table 5), which was in accordance with the Fst (36.2%) calculated by Popgene program. The estimate of gene flow Nm based on Fst was 0.105 (Table 4).
The observed heterozygosity (Ho) over all loci was equal to those expected by Hardy–Weinberg model in all studied populations (Table 4). Estimates of inbreeding coefficient (Fis) for populations 2 and 4 indicated an excess of heterozygosity as they were significantly negative for all loci. However, estimates of inbreeding coefficient (Fis) for populations 1 and 3 were non-significantly negative for all loci (Table 4). The Mantel test showed a significant correlation between genetic distance and geographic distance (r = 0.296, P = .008).
3.4. SSR profiling
Four SSR primers used and were found to be polymorphic in 12 P. harmala genotypes. All of the loci were successfully amplified in P. harmala. The four primer pairs generated a total of 13 alleles in all individuals (Fig. 6). Distribution of alleles in all loci for all populations was not normal distribution, suggesting that mutation is not stepwise in the P. harmala populations (Fig. 7). The genotypes for the four SSR loci (SSR17, SSR39, SSR145 and SSR217) were detected as follows; two genotypes were found in population 1, one in populations 3, 4. One genotype was seen for all SSR loci in population 2; except at SSr217 where two genotypes were seen. The number of alleles produced by different primers was three; except in Primer Pe17 which produced four alleles with an average of 2.3 alleles per primer (Fig. 7). PIC value was the highest for the primer SSR17 (0.444) while, the lowest PIC value recorded for the primer ISSR145 (0.095). The mean PIC value for ten SSR primers was 0.244 which was quite low (Table 2). The highest effective multiplex ratio (EMR) 3.55 was observed with the primer SSR 39 while the lowest 0.95 was observed with the primer SSR 145 with an average EMR of 2.163 per primer. The highest MI was observed with the primer SSR17 (1.152) and lowest in the primer SSR217 (0.383) with an average MI of 0.573 per primer was obtained (Fig. 3; Table 3). These four polymorphic microsatellite loci presented here are the first study set of microsatellite markers after isolation by HAN & AN [11] for P. harmala, and they are useful for investigating population genetics.
Fig. 6.

SSR markers profile of four loci for four P. harmala populations; M: DNA marker.
Fig. 7.

Allele frequencies recorded for four P. harmala populations at four microsatellite loci.
The observed number of alleles (Na) and effective number of alleles (Ne) ranged between 1.57–2.50 and 1.75–2.13, respectively (Table 3). Nei’s gene diversity (h) and Shannon’s Information index (I) ranged between 0.375 and 0.500 with overall diversity of 0.559 and 0.519–0.791 with an average value of 0.906, respectively. The percentage of polymorphic loci (PPL) was 100% for populations 1, 2 and 3, while it was 75% for population 4 (Table 3).
The estimate of genetic structure (Fst = 0.188) of populations is significantly different from zero (P < .0001). AMOVA analysis showed that most of genetic variation (95.29%) occurred within the populations, whereas the variance among populations was 18.82% (Table 5). The estimate of gene flow Nm based on Fst was 1.285 (Table 4). The observed heterozygosity (Ho) over all loci was higher than those expected by Hardy–Weinberg model in all studied populations (Table 4). The value of inbreeding coefficient (Fis) for all populations indicated a general excess of heterozygosity as they were non- significantly negative for all loci. The Mantel Test showed a significant correlation between genetic distance and geographic distance (r = 0.330, P = .014).
Data for co-dominant markers (ITS-RFLP, ITS-SSCP and SSR) were treated as dominant. The combined data analysis derived on the basis of ISSR, ITS-RFLP, ITS-SSCP and SSR data illustrated that the first and second PCoA axis explained 42.16% and 23.98% of the total variation, respectively (Fig. 8). The second PCoA axis obviously separated population 4 from the other populations. Individuals of all populations clustered each in its own cluster.
Fig. 8.

Principle coordinates (PCoA) of combined ISSR, ITS-RFLP, ITS-SSCP and SSRs markers data for four P. harmala populations collected from Egypt.
The relationship between the genetic distances based on ISSR, ITS-RFLP, ITS-SSCP and SSR markers matrices was calculated using the Mantel Test [17] (Table 6). In fact we found a low matrix correlation coefficient ranged from r = 0.043 between ITS-SSCP and SSR markers to r = −0.275 between ISSR and ITS-SSCP markers. However, a moderate matrix correlation was found between ISSR and SSR markers (r = 0.458).
Table 6.
Matrix correlations between sets of genetic dissimilarity data between ISSR, ITS-RFLP, ITS-SSCP and SSR markers.
| Marker systems | ISSR | ITS-RFLP | ITS-SSCP | SSR |
|---|---|---|---|---|
| ISSR | 0 | |||
| ITS-RFLP | −0.270* | 0 | ||
| ITS-SSCP | −0.275* | 0.059 NS | 0 | |
| SSR | 0.458** | -0.126 NS | 0.043 NS | 0 |
NS = not significant.
P < .005.
P < .05.
4. Discussion
Peganum harmala is being depleted from the wild population due to the sustainable utilization of this medicinal plant, so that information on genetic structure and the diversity of populations are essential for their conservational program. Although P. harmala can be a future drug, only one study was carried out using RAPD marker on wild plants from the same geographical region [6]. Among the four marker systems (ISSR, ITS-RFLP, ITS-SSCP and SSR) used in the current study to evaluate the P. harmal genetic structure and diversity, the level of polymorphism revealed by ITS-SSCP (33.33%) is the lowest followed by ITS-RFLP (66.67%), then ISSR (88.10) and finally the highest polymorphism was recorded for SSR marker (100%). The Percentage of polymorphism (87.30) using eight RAPD primers in a previous study by [6] in populations of P. harmala collected from the same geographical range coincide with the percentage of ISSR marker polymorphism (88.10) recorded in the present study. The mean value of Nei’s gene diversity index (h) for co-dominant markers (ITS-RFLP, ITS-SSCP and SSR) was nearly equal (0.46, 0.60 and 0.56 respectively) and higher than dominant marker ISSR (0.33). The same value of Nei’s gene diversity index (h) for ISSR (0.33) was detected by using RAPD marker (0.31) [6]. The estimated value is near to the h value (0.174–0.328) estimated for out-crossing plants [31], which is consistent with the general characteristics of P. harmala as the species pollinated by animals. The observed heterozygosity (Ho) estimates by SSR was fourfold larger than ITS-RFLP and threefold larger than ITS-SSCP. Similarly the comparison among different markers by NYBOM [21] showed that dominant markers (AFLP, ISSR and RAPD) heterozygosity were quite close whereas SSR-based estimates were threefold larger.
The AMOVA analysis, based on the used four markers, implied that most of the variation occurred within the examined populations. The presence of such percentage of variation was found in an earlier study of P. harmala populations using RAPD marker [6]. The AMOVA showed that the ISSR and ITS-SSCP markers has the same amount of variation (ISSR = 36.80%; ITS-SSCP = 36.58%) occurred among populations (one to half), while in SSR marker nearly one fourth of the variation occurred among populations (SSR = 18.82%). The result concurs with the higher crossing rate of P. harmala among populations. A value of estimate gene flow Nm < 1, Nm = 1 and Nm > 4 classified as low, moderate and extensive gene flow, respectively. The value obtained for Nm (estimate gene flow) based on the mean Fst (Nm = 0.737 for ISSR; Nm = 0.219 for ITS-RFLP; Nm = 0.105 for ITS-SSCP and Nm = 1.285 for SSR) indicates low to moderate gene flow among these wild populations of P. harmala. This amount of gene flow leads to the high percentage of variability within populations. The value of Nm for SSR was higher than the values estimated by other system markers due to the high variability and high mutation rate of SSRs. The abnormal distribution of SSR alleles in all studied loci for all populations was shown here in this study suggests that mutation is not stepwise in P. harmala. The stepwise mutation model in SSR used to describe the ability of alleles to mutate up or down by one or a small number of repeat units. Therefore, the more complex the repeat structure is, the lower the probability that stepwise mutational actions will occur [4], [30].
Self-pollination has an impact on plant genetic diversity at the population level instead of the species level [37]. The mean population value of inbreeding coefficient (Fis) obtained for the three co-dominant markers (Fis = 0.416 for ITS-RFLP; Fis = 0.471 for ITS-SSCP; Fis = 0.532 for SSR) was nearly equal and offer an indication of the partial outcrossing (self-incompatible) reproductive system of P. harmala.
Principal coordinates analysis (PCoA) remained more or less the same in separate ISSR, ITS-RFLP, ITS-SSCP and SSR data and combined all data. A clear pattern of clustering was detected based on the locations of collected plants which coincide with the isolation by distance (Mantel test for ISSR, r = 0.382, P = .007; for ITS-SSCP, r = −0.296, P = .008; for SSR, r = 0.330, P = .014). This is clearly shown in Principal Coordinate Analysis (PCoA2), which separated population 4 from the other populations probably due to the lowest genetic diversity of the population and the isolated distance due to the high elevation of its location. It also worth noting that by ITS-RFLP and ITS-SSCP markers, the individuals of this population shared the same alleles. The percentage of inbreeding coefficient and moderate amount of gene flow may also explain the aggregate of individuals in its own population. The correlation between the genetic distance and geographical distance for SSR marker coincide with abnormal distribution of alleles (see Fig. 7) and could be explained by high allele frequencies due to partial selfing or geneotypes are unequal in viability and fertility (show some selection) and small population size may be another cause.
Comparison of PIC values for the four markers indicated that the average of PIC values for ITS-SSCP primer (0.444) was the highest followed by ISSR primers (0.431) and ITS-RFLP primers (0.353), while the lowest average of PIC value was recorded for and SSR primers (0.244). There was a similar degree of polymorphism information content provided by the ISSR, ITS -RFLP and SSR markers; however the lowest mean PIC value of ITS-SSCP markers produced might have been due to the low number of polymorphic loci evaluated for this marker. Unexpected, the marker index values of dominant marker ISSR (3.38) was higher than co-dominant markers; ITS-RFLP, ITS-SSCP and SSR (0.347, 0.590 and 0.572 respectively). On the basis of higher PIC values (0.444, 0.431), Shannon diversity index (I) (1.08, 0.905) and marker index (MI) (0.592, 3.381); ITS-SSCP and ISSR markers respectively were more informative than the other used marker systems in the assessment of genetic diversity of P. harmala. Although the ITS region is highly conserved within the species [12]; using PCR-SSCP for the molecular characterization identified P. hamala accessions. The mean polymorphism information content (PIC) is nearly equal in ITS PCR-SSCP and ITS-RFLP (0.444 and 0.419 respectively) due to the fact that two marker techniques targeted the same fragments of the genome. The mean PIC value provided by these two co-dominant markers is similar to the degree of polymorphism information content of dominant marker ISSR. The coincidence between these values agrees with the results published by Pejic et al. [22].
A low matrix correlation was found between sets of genetic similarity data; except between ISSR and SSR markers suggesting that the used sets of markers revealed not linked estimates of genetic relationships. The ISSR and SSRs profiles are generated from microsatellite rich regions of the genome, while ITS regions are a large segment of ribosomal DNA region and are highly variable. Thus, these methods involve regions having different genome coverage and evolutionary histories. The results obtained in this study revealed high agreement between co-dominat markers (ITS-RFLP, ITS-SSCP and SSRs) estimates as Nei’s genetic diversity (h) and inbreeding coefficient (Fis). However, inconsistency was detected in the estimates of gene flow (Nm) and the correlation between genetic distance and geographical distance, since it is significant for all markers, except for ITS-RFLP marker. Based on our data ITS-SSCP and ISSR markers are more informative than the other markers used. The great diversity of P. harmala and the data obtained from this study can be used for future collecting missions. However, the results point to the need to adopt different strategies for selecting markers and choosing an upper number for each marker for a reliable estimation of genetic diversity.
Footnotes
Peer review under responsibility of National Research Center, Egypt.
References
- 1.Bukhari N., Choi J.H., Jeon C.W. Appl. Chem. 2008;12:101–104. [Google Scholar]
- 2.Chou C.H., Tsai C.C. Bot. Bull. Acad. Sin. 1999;40:319–327. [Google Scholar]
- 3.Davis J.C. John Wiley and Sons; New York: 1986. Statistics and data analysis in geology. [Google Scholar]
- 4.Di Rienzo A. Proc Natl Acad Sci USA. 1994;91:3166–3170. doi: 10.1073/pnas.91.8.3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eddouks M., Maghrani M., Lemhadri A. J Ethnopharmacol. 2002;82:97–103. doi: 10.1016/s0378-8741(02)00164-2. [DOI] [PubMed] [Google Scholar]
- 6.EL-Bakatoushi R., Hegazy A.K., Fawzi M. Afr J Biotechnol. 2011;10:15883–15890. [Google Scholar]
- 7.EL-Bakatoushi R., Alframawy A.M. Flora. 2013;208:464–477. [Google Scholar]
- 8.Excoffier L., Smouse P., Quattro J. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gower J.C. Biometrika. 1966;53:326–338. [Google Scholar]
- 10.Gruszczynska J., Brokowska K., Charon K.M. J Appl Genet. 2005;46:311–314. [PubMed] [Google Scholar]
- 11.Han J., An L.Z. Conserv Genet. 2009;10:1899–1901. [Google Scholar]
- 12.Hillis D.M., Dixon M.T., Rev Q. Biology. 1991;66:411–453. doi: 10.1086/417338. [DOI] [PubMed] [Google Scholar]
- 13.Khawar K.M., Ozel C.A., Balci S. Inter J Agri Biol. 2005;7:790–793. [Google Scholar]
- 14.Kozyrenko M.M., Gontcharova S.B., Gontcharov A.A. Flora. 2011;206:691–696. [Google Scholar]
- 15.Leporatti M.L., Ghedira K. J Ethnobiol Ethnomed. 2009;5:31. doi: 10.1186/1746-4269-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li A., Ge S. Ann Bot. 2001;87:585–590. [Google Scholar]
- 17.Mantel M. Cancer Res. 1967;27:209–220. [PubMed] [Google Scholar]
- 18.Mir B.A., Koul S., Kumar A. Ind J Biotechnol. 2010;9:325–328. [Google Scholar]
- 19.Moloudizargari M., Mikaili P., Aghajanshakeri S. Pharmaco Rev. 2013;7:199–212. doi: 10.4103/0973-7847.120524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nei M., Li W. Proc Natl Acad Sci USA. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nybom H. Mol Ecol. 2004;13:1143–1155. doi: 10.1111/j.1365-294X.2004.02141.x. [DOI] [PubMed] [Google Scholar]
- 22.Pejic I., Ajmone-Marsan P., Morgante M. Theor Appl Genet. 1998;97:1248–1255. [Google Scholar]
- 23.Perera M.F., García M.B., Romero Carolina Díaz. Rev Ind y Agríc de Tucumán Tomo. 2012;89(2):1–7. [Google Scholar]
- 24.Peyachoknagula S., Mongkolsiriwatanab C., Wannapinpongd S. Sci Asia. 2014;40:113–120. [Google Scholar]
- 25.Roldan-Ruiz I., Dendauw J., Vanbockstaele E. Mol Breed. 2000;6:125–134. [Google Scholar]
- 26.Rohlf FJ. NTSYSpc. Numerical taxonomy and multivariate analysis system exeter software, setauket, New York, vol. 2; 2000. p. 1.
- 27.Saar D.E., Polans N.O. Pisum Genet. 2000;32:42–45. [Google Scholar]
- 28.Saini R., Jaiwal P.K. J Exper Biol. 2000;38:499–503. [PubMed] [Google Scholar]
- 29.Sambrook J., Fritsch E.F., Maniatis T. Cold Spring Harbor Laboratory Press; New York: 1989. Cold spring harbor. [Google Scholar]
- 30.Schlötterer C., Tautz D. Nucleic Acids Res. 1992;20:211–215. doi: 10.1093/nar/20.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schoen D.J., Brown A.H.D. Proc Natl Acad Sci USA. 1991;88:4494–4497. doi: 10.1073/pnas.88.10.4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Somasundaram S., Kalaiselvam M. Annamalai University; India: 2011. International training course on mangroves and biodiversity; pp. 82–91. [Google Scholar]
- 33.Tahraoui A., El-Hilaly J., Israili Z.H. J Ethnopharmacol. 2007;110:105–117. doi: 10.1016/j.jep.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 34.Tautz D. Nucleic Acids Res. 1989;17:6463–6471. doi: 10.1093/nar/17.16.6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Varshney R.K., Chabane K., Hendre P.S. Plant Sci. 2007;173:638–649. [Google Scholar]
- 36.White T.J., Bruns T., Lee S. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis M.A., Gelfand D.H., Sninsky J.J., White T.J., editors. PCR protocols. A guide to methods and applications. Academic Press; San Diego: 1990. pp. 315–322. [Google Scholar]
- 37.Wright S.I., Lauga B., Charlesworth D. Mol Ecol. 2003;12:1247–1263. doi: 10.1046/j.1365-294x.2003.01743.x. [DOI] [PubMed] [Google Scholar]
- 38.Yeh FC, Yang RC, Boyle T. POPGENE. Microsoft windows based freeware for population genetic analysis. Release 1.31. University of Alberta, Edmonton; 1999.
- 39.Zhang M.Q., Zheng X.F., Yu A.L. Sugar Techn. 2004;6(4):251–259. [Google Scholar]