

SUMMARY
Increased glucose metabolism in immune cells not only serves as a hallmark feature of acute inflammation, but also profoundly affects disease outcome following bacterial infection and tissue damage. However, the role of individual glucose metabolic pathways during viral infection remains largely unknown. Here we demonstrate an essential function of the hexosamine biosynthesis pathway (HBP)-associated O-linked β-N-acetylglucosamine (O-GlcNAc) signaling in promoting antiviral innate immunity. Challenge of macrophages with vesicular stomatitis viruses (VSV) enhances HBP activity and downstream protein O-GlcNAcylation. Human and murine cells deficient of O-GlcNAc transferase, a key enzyme for protein O-GlcNAcylation, show defective antiviral immune responses upon VSV challenge. Mechanistically, OGT-mediated O-GlcNAcylation of the signaling adaptor MAVS on serine 366 (S366) is required for K63-linked ubiquitination of MAVS and subsequent downstream RLR-antiviral signaling activation. Thus, our study identifies a molecular mechanism by which HBP-mediated O-GlcNAcylation regulates MAVS function and highlights the importance of glucose metabolism on antiviral innate immunity.
In Brief (eTOC Blurb)
Tiangliang et al. demonstrate that enhanced activation of the hexosamine biosynthesis pathway (HBP) occurs upon vesicular stomatitis virus (VSV) infection. HBP activation subsequently promotes RLR-antiviral immunity via O-linked β-N-acetylglucosamine (O-GlcNAc) signaling. Mechanistically, O-GlcNAcylation of the signaling adaptor MAVS on serine 366 is required for its ubiquitination and downstream signaling activation.
Graphical Abstract
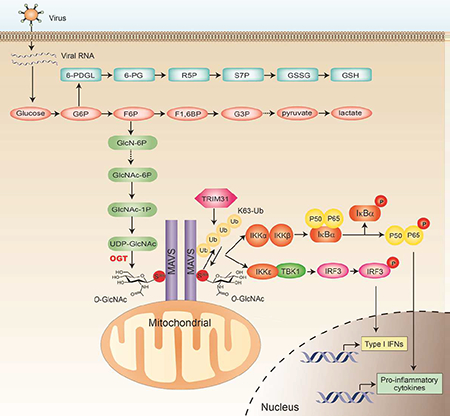
INTRODUCTION
Cell-autonomous energy metabolism and immune signaling are two co-evolutionized systems. Over the past several decades, many crucial cross-regulations between these two networks have been characterized either under naive condition or during immune activation in response to invading pathogens (Buck et al., 2017; O’Neill and Pearce, 2016). On the one hand, in response to pathogen infection or tissue damage, activation of immune signaling immediately alerts metabolic system to reprogram towards enhanced catabolic activity, which further conveys danger signals to other systems to initiate a series of restorative stress responses. On the other hand, elevated catabolic activity is required to meet the increased demand of biomolecules and energy for effective immune functions such as immune cell migration, phagocytosis, and cytokine production. Therefore, the possibility of modulating immune cell function through metabolic regulation represents a new approach for combatting pathogen- and tissue damage-induced inflammatory diseases. However, critical knowledge is required regarding the role of individual metabolism pathways in immune activation by various pathogen- or danger-associated molecular patterns (PAMPs or DAMPs).
Increased glucose uptake and utilization in immune cells serves as a hallmark feature of acute inflammation. After uptake through the effect of glucose transporter, glucose fluxes through three major pathways with distinct destinations and functions, including glycolysis, the pentose phosphate pathway (PPP), and the hexosamine biosynthesis pathway (HBP). HBP has been considered as a minor glucose metabolism pathway, since only 2–5% of the glucose that enters cells is directed to this pathway, eventually leading to the generation of its end product uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) (Hardiville and Hart, 2014). O-GlcNAc transferase (OGT) mediates the transfer of UDP-GlcNAc to serine or threonine residue of target proteins, known as protein O-GlcNAcylation (Levine and Walker, 2016; Yang and Qian, 2017). Previous studies have discovered essential roles of HBP and protein O-GlcNAcylation in many fundamental biological activities (Bond and Hanover, 2013; Hart et al., 2011). Due to a well established cross-regulation between O-GlcNAcylation and phosphorylation, it is expected that OGT-mediated O-GlcNAcylation plays a pivotal role in the immune signaling and inflammation. Previous studies have identified several signaling molecules involved in TLR pathway and NF-κB pathway as O-GlcNAcylation targets (Allison et al., 2012; Pathak et al., 2012; Ramakrishnan et al., 2013; Yang et al., 2008). We recently identified STAT3 O-GlcNAcylation as an important mechanism antagonizing its activation (Li et al., 2017). However, the overall effect of OGT in the innate immune function is still unknown.
During RNA virus infection, cytosolic RNA species are recognized by retinoic-acid inducible gene I (RIG-I)-like receptors (RLRs), including RIG-I and melanoma differentiation associated gene 5 (MDA5) (Brubaker et al., 2015; Moore and Ting, 2008; Roers et al., 2016; Wu and Chen, 2014). Activation of RIG-I and MDA5 leads to recruitment of the signaling adaptor protein MAVS (also referred to as IPS-1, VISA or Cardif) to activate downstream NF-κB and IRF3 signaling and induce type I interferon (IFN-I) production (Kawai et al., 2005; Meylan et al., 2005; Seth et al., 2005; Xu et al., 2005). Previous studies suggest that normal function of MAVS is markedly affected by its ubiquitination (You et al., 2009) and phosphorylation (Liu et al., 2015). We recently identified TRIM31 as a key E3 ubiquitin ligase catalyzing MAVS lysine-63 (K63)-linked ubiquitination, which is required for its antiviral immune function (Liu et al., 2017a). In this study, we characterized OGT-mediated MAVS O-GlcNAcylation on S366, which was induced by RNA virus infection, as a key mechanism for MAVS ubiquitination and activation of antiviral immune responses. Genetic deletion of OGT in myeloid cells markedly impaired antiviral innate immunity in vivo. Therefore, our findings demonstrate a mechanism whereby glucose metabolic reprogramming towards HBP pathway and boosting the O-GlcNAc signaling promotes antiviral immunity by targeting MAVS.
RESULTS
VSV infection enhances activities of glucose metabolic pathways
To achieve a comprehensive understanding of metabolic changes in response to viral challenge, we challenged mouse bone marrow-derived macrophages (BMMs) with vesicular stomatitis virus (VSV) and performed metabolomics assay (Shukla et al., 2017). Principal component analysis revealed a markedly altered metabolic profile induced by VSV (Figure 1A). Pathway-enrichment analysis identified several crucial metabolic pathways with significant changes such as nucleotide metabolism, amino and nucleotide sugar metabolism, glutathione metabolism, and glycerophospholipid metabolism (Figure 1B). Recent study indicates an essential role of glucose utilization in promoting antiviral defense response during influenza infection (Wang et al., 2016). We therefore specifically focused on VSV-induced glucose metabolism changes. Many intermediate metabolites involved in glycolysis and the PPP, two major glucose metabolic pathways, showed increased abundance upon VSV challenge (Figure 1D, 1E and 1F; Table S1). These findings were consistent with previous studies showing that viral infection promotes aerobic glycolysis and PPP (Bajwa et al., 2016; Jiang et al., 2016; Sanchez and Lagunoff, 2015). Moreover, we found that VSV infection also led to increased levels of metabolites in the HBP, which are included in the amino and nucleotide sugar metabolism pathway, indicating that VSV promotes HBP activity (Figure 1G). This result was further supported by analysis of 13C-glucose tracing by liquid chromatography-tandem-mass spectrometry (LC-MS/MS) assay, which revealed a significantly increased incorporation of glucose-derived carbon into the HBP end product UDP-GlcNAc in VSV-challenged macrophages compared with non-treated cells (Figure 1H). In addition, we detected a moderately increased transcript (Figure S1A) and protein (Figure S1B-D) levels of HBP rate-limiting enzyme glutamine fructose-6phosphate transaminase 1 (GFPT1) when BMMs were stimulated with VSV or poly(I:C), another widely used RLR agonist (Brubaker et al., 2015; Moore and Ting, 2008; Roers et al., 2016). Total protein O-GlcNAcylation in BMMs showed significant increase after the challenge with either VSV or poly(I:C) (Figure 1I). These findings demonstrate that viral infection promotes HBP activity and downstream OGT-mediated protein O-GlcNAcylation, as well as enhancing glycolysis and PPP.
Figure 1.

RLR activation promotes glucose metabolism in macrophages. (A and B) Total metabolite profiling determined by LC-MS/MS metabolomics assay was assessed by principle component analysis (A) and pathway-enrichment analysis (B) in bone marrow-derived macrophages (BMMs) generated from C57BL/6 mice stimulated with vesicular stomatitis virus (VSV) (multiplicity of infection (MOI) = 1) for 4 h. (C) A summary of three glucose metabolic pathways, including the glycolysis (middle), PPP (left) and HBP (right). (D to G) Heatmap of metabolites (D) and fold changes in intermediate metabolites of the glycolysis (E), PPP (F), or HBP (G). (H) Fold change in 13C-labelled UDP-GlcNAc between non-treated and VSV-challenged BMMs in the presence of 13C6-glucose. (I) Immunoblotting (left) and densitometric analysis based on five independent experiments (right) to quantify ratio of total O-GlcNAc to actin in BMMs left untreated or treated with VSV or transfected with poly(I:C) (4 μg/ml) by lipofectamine 2000 for indicated periods. * P < 0.05, versus controls (two-tailed Student’s t-test (E to H)). Data are from one experiment representative of three experiments (A and B, D to G; mean ± s.d. of six biological replicates) or two experiments (H; mean ± s.d. of four biological replicates) or represent five independent experiments (I).
OGT promotes RLR-mediated antiviral immune responses
Increased HBP activity and protein O-GlcNAcylation upon VSV challenge prompted us to hypothesize that OGT-mediated O-GlcNAc signaling may play a role in antiviral immune responses. We generated mice with a myeloid-specific Ogt deletion (OgtΔmye) by crossing the Ogtfl/fl mice (Shafi et al., 2000) with lysosome M-Cre mice. Ogtfl/fl mice were used as wild type (WT) controls. OgtΔmye BMMs showed similar cell surface levels of macrophage differentiation markers CD11b and F4/80, as well as activation markers CD80, CD86 and MHCII, suggesting no significant effect of OGT deletion on BMM generation/differentiation (Figure S2A-B). In response to VSV challenge for 3 or 6 hours, OgtΔmye BMMs generated significantly fewer transcripts of genes encoding a series of antiviral effectors including Ifna4, Ifnb1, Il6, Tnfa, Isg15, Cxcl10 and Mx1 compared to WT BMMs (Figure 2A). OgtΔmye BMMs consistently produced lower amount of IFN-β, IL-6 and TNF-α proteins at 16-hour after VSV challenge (Figure 2B). No significant difference in cell viability was detected between Ogtfl/fl and OgtΔmye BMMs with or without VSV stimulation (Figure S2C). BMMs activated by transfected poly(I:C) showed similar phenotype (Figure 2C and 2D).
Figure 2.

OGT deficiency impairs antiviral immune responses in vitro. (A to D) BMMs generated from Ogtfl/fl and OgtΔmye mice were left untreated or stimulated with VSV (MOI = 1) (A and B) or transfected with 4 μg/ml poly(I:C) by lipofectamine 2000 (C and D) for indicated periods. Gene transcripts in the cells (A and C), IFN-β, IL-6 and TNF-α proteins in the supernatants (B and D) were measured with RT-PCR and ELISA, respectively. (E and F) Gene transcripts in the cells (E) and cytokines in the supernatants (F) from Ogtfl/fl or OgtΔmye peritoneal macrophages left untreated or stimulated with VSV for indicated periods. * P < 0.05, versus controls (two-tailed Student’s t-test (A to F)). Data are from one experiment representative of five experiments (A to F; mean ± s.d. of four biological replicates).
We isolated naïve peritoneal macrophages and observed a similar defect in the production of inflammatory mediators in VSV-challenged OgtΔmye peritoneal macrophages compared with similarly treated WT cells (Figure 2E and 2F). In contrast to similarly differentiated WT and OgtΔmye BMMs (Figure S2A-B), Ogt deficiency caused a 20% decrease in the percent and total number of peritoneal macrophages (Fig. S2DF). Cell surface level of CD11b, but not F4/80, was moderately decreased in OgtΔmye PMac compared to WT cells (Fig. S2G-H). These findings indicate that OGT deletion cause a moderate defect in peritoneal macrophage differentiation and the underlying mechanism warrants further investigation.
We next tested if defective IFN-I production in OgtΔmye macrophages is specific to RLR activation. When cells were stimulated with transfected DNA species poly(dA:dT) or interferon stimulatory DNA (ISD), or cGAMP, to activate cGAS-STING signaling pathway (Barber, 2015; Chen et al., 2016), cytokine production were comparable between WT and OgtΔmye BMMs (Figure S3A and B). Interestingly, in response to the stimulation with TLR agonists Pam3Cys (TLR2), LPS (TLR4) or CpG oligonucleotide (TLR9), OgtΔmye BMMs showed higher transcript levels of IFN-I and IFN-I-responsive genes compared to WT BMMs (Figure S3C). No difference in cytokine gene expression (Figure S3D) or STAT1 phosphorylation (Figure S3E) was observed between WT and OgtΔmye BMMs stimulated with recombinant mouse IFN-β. No defect in the transcripts of IFN-I and IFN-I-responsive genes was observed in OGT-KO THP-1 in response to the challenge with various TLR agonists (Figure S3F) or recombinant IFN-β (Figure S3G). These results suggest defective IFN-I production in OgtΔmye macrophages is specific to RLR activation.
Activation of antiviral immune signaling such as the TBK-1-IKKε, NF-κB and MAPK pathways has been well defined to promote the production of inflammatory mediators in response to viral infection (Brubaker et al., 2015; Moore and Ting, 2008; Roers et al., 2016; Wu and Chen, 2014). We found that OgtΔmye BMMs showed decreased phosphorylation of TBK1, IKKε, IRF3 and STAT1 in response to VSV challenge compared with similarly treated WT BMMs (Figure 3A). VSV-induced activation of the NF-κB pathway evidenced by phosphorylation of IKK, IκBα and p65 (Figure 3B), and phosphorylation of ERK (Figure 3C), were also attenuated in OgtΔmye BMMs than those in WT BMMs. No difference in p38 and JNK phosphorylation was detected (Figure 3C). OgtΔmye BMMs activated by transfected poly(I:C) showed similar defect in activation of the TBK-1-IKKε and NF-κB signaling (Figure S4). Furthermore, OgtΔmye peritoneal macrophages also exhibited defective activation of the TBK-1-IKKε (Figure 3D), NF-κB (Figure 3E), and ERK (Figure 3F) signaling upon VSV stimulation. Immunofluorescence staining revealed nuclear translocation of phosphorylated IRF3 (p-IRF3) (Figure 3G, upper panel) and p-p65 (Figure 3G, lower panel) following poly(I:C) or VSV stimulation in WT BMMs, but not in OgtΔmye BMMs. In sum, these findings demonstrate an essential role of OGT in activation of RLR-mediated antiviral immune signaling and production of inflammatory cytokines.
Figure 3.

OGT is critical for the activation of antiviral immune signaling and antiviral innate immune responses in vivo. (A to C) Phosphorylation of IFN-I signaling molecules including TBK1, IKKε, IRF3 and STAT1 (A), NF-κB (B), and MAPK (C) signaling molecules in Ogtfl/fl and OgtΔmye BMMs left untreated or stimulated with VSV (MOI = 1) for indicated periods. (D to F) Phosphorylated TBK1, IKKε, IRF3 and STAT1 (D), NF-κB (E), and MAPK (F) signaling molecules in Ogtfl/fl and OgtΔmye peritoneal macrophages treated with VSV for indicated periods. (G) Immunofluorescence staining of phosphorylated IRF3 (upper panel) or p65 (lower panel) in Ogtfl/fl and OgtΔmye BMMs left untreated or challenged with VSV for 4 h. Scale bar, 20 μm. (H) Survival of Ogtfl/fl (n = 18) and OgtΔmye (n = 20) mice after intraperitoneal injection with VSV (1 × 108 PFU per mouse). (I to L) Transcripts of Ifna4 and Ifnb1 in the spleen (I), liver (J), or lungs (K) and IFN-β protein in serum (L) from Ogtfl/fl and OgtΔmye mice challenged with either PBS or VSV (2 × 107 PFU) for 24 h (n = 6 per group). (M and N) VSV RNA in the spleen, liver and lungs (M) and histological analysis of the lung tissue (N) in Ogtfl/fl and OgtΔmye mice challenged with VSV (n = 5 per group). Scale bar: 20 μm. * P < 0.05, versus controls (Kaplan-Meier (H) or two-tailed Student’s t-test (A to F, I to M)). Data are from one experiment representative of three independent experiments and are expressed as mean ± s.d.
To examine the role of myeloid-derived OGT in antiviral immune responses in vivo, we intraperitoneally injected either VSV or PBS into WT and OgtΔmye mice. OgtΔmye mice showed significantly increased mortality rate compared to WT mice (Figure 3H). RT-PCR analysis revealed robust induction of Ifna4 and Ifnb1 gene transcription in the spleen (Figure 3I), liver (Figure 3J), and lungs (Figure 3K) from WT mice 24 hours after challenge, indicating efficient induction of antiviral immunity. In contrast, cytokine expression, as well as serum IFN-β level (Figure 3L), was significantly blunted in OgtΔmye mice. Meanwhile, OgtΔmye mice contained significantly higher level of VSV transcript in the spleen, liver and lungs, indicating insufficient control of viral replication (Figure 3M). Histological analysis of the lung tissue showed greater infiltration of immune cells and injury in OgtΔmye mice compared with WT mice (Figure 3N). These findings indicate that OGT in myeloid cells is crucial for the induction of antiviral innate immune response in vivo. It is worth noting that in addition to defective antiviral immune signaling, defective macrophage in vivo differentiation may also contribute to the increased susceptibility in viral-infected OgtΔmye mice.
We further assessed the requirement of OGT in antiviral innate immune responses in human cells. RIG-I-MAVS signaling-mediated antiviral immune responses function in both hematopoietic and non-hematopoietic stromal cells (Brubaker et al., 2015; Moore and Ting, 2008; Roers et al., 2016; Wu and Chen, 2014). We employed CRISPR/Cas9-based gene targeting strategy with the use of single guide RNA (gRNA) specific to human OGT gene to generate OGT-knockout (KO) THP-1 and HT29 cells. Cells containing non-targeting gRNA were used as WT controls. Consistent with an important antiviral immune function of OGT in mouse cells, OGT deficiency led to defective activation of the TBK-1-IKKε (Figure 4A and 4D) and NF-κB (Figure 4B and 4E) signaling in response to VSV challenge in both THP-1 and HT29 Cells. Consequently, VSV-induced cytokine production including IFNB1, IL6 and TNFA was significantly attenuated in OGT-KO THP-1 (Figure 4C) and HT29 (Figure 4F) cells compared to WT cells. To test whether decreased cytokine production in OGT-KO human cells was caused by any off-target effect, we employed a lentivector-based transduction strategy to reconstitute OGT-KO THP-1 cells with human OGT with synonymous mutation that was not recognized by OGT gRNA. We confirmed the successful OGT reconstitution and consequently rescue of protein O-GlcNAcylation in OGT-KO THP-1 cells (Figure 4G). Compared to empty vector control, OGT reconstitution restored VSV-induced cytokine production in OGT-KO THP-1 cells, suggesting that decreased cytokine production in OGT-KO human cells was indeed caused by the loss of OGT (Figure 4H). These results suggest that OGT is important for the induction of antiviral immune response in human cells.
Figure 4.

OGT promotes antiviral immune responses in human cells. (A to F) Immunoblotting of phosphorylated TBK1, IKKε, IRF3 and STAT1 (A and D), and NF-κB (B and E) signaling molecules in OGT-KO and WT control THP-1 (A to C) or HT29 (D to F) cells challenged with VSV (MOI = 1) for indicated periods. Transcripts of IFNB1, IL6 and TNFA in the cells were measured with RT-PCR (C and F). (G and H) Protein O-GlcNAcylation (G) and cytokine transcripts (H) in OGT-KO THP-1 cells virally transduced with either empty vector or expression vector for human OGT with synonymous mutation that was not recognized by OGT gRNA, followed by VSV challenge for 6 h. * P < 0.05, versus controls (two-tailed Student’s t-test (C, F and H)). Data are from one experiment representative of three experiments (A, B, D, E and G) or represent four independent experiments (C, F and H; mean ± s.d. of four biological replicates).
OGT promotes RLR signaling via its enzymatic activity
OGT comprises an N-terminal region containing multiple tetratricopeptide repeat (TPR) units and a C-terminal region consisting of two catalytic domains (Lazarus et al., 2011). Previous study has suggested that, in addition to its classical enzymatic effect, OGT can function as a scaffold protein via N-terminal TPR domain independent of its enzymatic activity (Yang et al., 2002). Thus, we sought to determine whether the promoting effect of OGT on antiviral signaling depended on its enzymatic activity by employing both genetic and pharmacological approaches. We first generated a mutation with lysine 908 replaced by alanine (K908A) on human OGT (Clarke et al., 2008; Martinez-Fleites et al., 2008). As previously described, K908A mutation totally abolished the enzymatic function of OGT without damaging its protein abundance in 293T cells (Figure S5A). Through a lentivector-based transduction strategy, reconstitution of OgtΔmye BMMs with WT OGT fully rescued cytokine production on the transcript (Figure 5A) and protein (Figure 5B) level, and restored activation of the TBK-1-IKKε and NF-κB signaling upon VSV challenge (Figure 5C), but those reconstituted with OGT K908A mutant failed to do so.
Figure 5.

OGT promotes antiviral immune signaling via its enzymatic activity. (A to C) Transcripts of Ifna4, Ifnb1, Il6 and Tnfa in the cells (A), IFN-β, IL-6 and TNF-α proteins in the supernatants (B), and phosphorylated TBK1, IRF3, IκBα, IKKα/β and p65 (C) in OgtΔmye BMMs virally transduced with either empty vector or expression vector for OGT WT or K908A mutant, followed by the challenge with VSV (MOI = 1). (D to I) Transcripts of Ifna4, Ifnb1, Il6 and Tnfa in the cells (D and G), IFN-β, IL-6 and TNF-α proteins in the supernatants (E to H), and phosphorylated TBK1, IRF3, IκBα, IKKα/β and p65 (F and I) in Ogtfl/fl and OgtΔmye BMMs pretreated with or without either PUGNAc (50 μM) (D to F) or OSMI-1 (20 μM) (G to I) for 2 h, followed by VSV challenge. * P < 0.05, versus controls (two-tailed Student’s t-test (A, B, D, E, G and H). Data are from one experiment representative of three independent experiments (A to G). Data are from one experiment representative of three experiments (A, B, D, E, G and H; mean ± s.d. of three biological replicates) or represent two independent experiments (C, F and I).
PUGNAc and thiamet-G (TMG) are two widely used inhibitors of the O-GlcNAcylase (OGA) that potently enhances protein O-GlcNAcylation (Hart et al., 2011; Li et al., 2017), whereas OSMI-1 is a recently discovered OGT inhibitor and decreases protein O-GlcNAcylation (Angelova et al., 2015; Ortiz-Meoz et al., 2015). In line with the importance of OGT enzymatic activity in promoting VSV-induced IFN-I signaling, pretreatment of BMMs with PUGNAc, which markedly promoted protein O-GlcNAcylation, resulted in elevated cytokine production on the transcript (Figure 5D) and protein (Figure 5E) levels and enhanced activation of the TBK-1-IRF3 and NF-κB signaling (Figure 5F) in response to VSV challenge for 3 or 6 hours. TMG pretreatment caused similar increase in cytokine production and signaling activation (Figure S5 (BC)). In contrast, VSV-induced cytokine production (Figure 5G and 5H), as well as TBK1-IKKε and NF-κB activation (Figure 5I), were blunted in BMMs pretreated with OSMI-1 compared to vehicle control. In sum, the findings with the use of both genetic and pharmacological approaches indicate that OGT promotes RLR signaling through its enzymatic activity.
O-GlcNAcylation of MAVS on S366 is critical for RLR signaling
We sought to dissect at which level OGT exerts its promoting effect on the RLR-mediated IFN signaling. A human IFNB1 promoter reporter assay revealed that OGT caused enhanced luciferase activity induced by DDX58 (encoding RIG-I), IFIH1 (encoding MDA5), or MAVS (Figure 6A). In contrast, OGT showed no effect on luciferase activity driven by TBK1, IKBKE (encoding IKKε), or IRF3, suggesting that OGT is functioning on the MAVS level. Consistently, O-GlcNAcylation of exogenously expressed MAVS could be detected in 293T cells, and this signal was much enhanced when OGT was co-expressed (Figure 6B). The O-GlcNAc signal on MAVS was abolished when anti-O-GlcNAc antibody was pre-incubated with 500 mM N-acetylglucosamine (GlcNAc), indicating the specificity of O-GlcNAc signal. OGT-mediated MAVS O-GlcNAcylation required its enzymatic activity, since OGT K908A mutant was not able to catalyze MAVS O-GlcNAcylation (Figure 6C). In contrast, neither RIG-I (Figure S6A) nor MDA5 (Figure S6B) was O-GlcNAcylated, even when OGT was co-expressed. We further generated N-terminal (1–270 aa) and C-terminal (271–540 aa) truncated fragments of MAVS, and detected O-GlcNAcylation on its C-terminus (Figure 6D). Finally, VSV challenge induced a marked elevation of endogenous MAVS O-GlcNAcylation in WT BMMs, but not OgtΔmye BMMs, indicating an OGT-dependent MAVS O-GlcNAcylation promoted by VSV (Figure 6E). These results demonstrate that MAVS is O-GlcNAcylated by OGT depending on its enzymatic function, and VSV challenge induces MAVS O-GlcNAcylation.
Figure 6.

O-GlcNAcylation of MAVS on S366 is critical for RLR signaling. (A) Luciferase activities in 293T cells transfected for 30 h with an IFNB1-luciferase reporter plasmid together with RIG-I N-terminus (RIG-I (N)), MDA5, MAVS, TBK1, IKKε, or IRF3, in the presence or absence of OGT. (B to D) 293T cells were transfected for 30 h with the indicated combination of Flag-tagged MAVS full-length (B and C) or N- and C-terminus (D), and Myc-tagged OGT WT (B and D) or K908A mutant (C). Immunoprecipitated MAVS was assessed for O-GlcNAcylation with specific anti-O-GlcNAc antibody. (E) Total MAVS was immunoprecipitated from Ogtfl/fl and OgtΔmye BMMs challenged with or without VSV (MOI = 1) for 4 h, followed by immunoblotting with anti-O-GlcNAc antibody. (F) LC-MS/MS analysis of Flag-tagged MAVS co-transfected with Myc-tagged OGT identified either T365 or S366 as MAVS O-GlcNAcylation site. MS/MS spectrum of the 2+ ion at m/z 924.99542 corresponding to O-GlcNAcylated MAVS peptide AGMVPSKVPTSMVLTK. (G and H) A series of point mutations of MAVS assayed for O-GlcNAcylation. (I to L) Total and phosphorylated IRF3 and NF-κB p65 (I and K) and IFN-β production (J and L) in Mavs−/− mouse embryonic fibroblasts (MEFs) transduced with either empty vector or GFP-tagged MAVS WT or S366A mutant (I and J), pretreated with or without thiamet G (10 μM) for 2 h (K and L), then followed by VSV challenge. (M) Cross-species sequence alignment of MAVS revealed conserved O-GlcNAcylation site S366. * P < 0.05, versus controls (two-tailed Student’s t-test (A) and ANOVA with Bonferroni post-tests (J)). Data are from one experiment representative of four experiments (A and J; mean ± s.d. of four biological replicates) or three experiments (B to L, G to I) or represent two independent experiments (F).
We sought to identify the O-GlcNAcylation site(s) on MAVS through mass spectrometry (MS) analysis. Several detectable O-GlcNAcylation sites were shared between the full-length and C-terminal fragment of MAVS revealed by the liquid chromatography coupled to tandem MS (LC-MS/MS) (Figure 6F). A series of mutant constructs of MAVS were then generated with those individual residues as potential O-GlcNAcylation sites mutated to alanine (Figure 6G). An initial screen revealed that a double mutation (T365A/S366A) caused the loss of O-GlcNAc signal without affecting MAVS protein abundance, indicating that MAVS was O-GlcNAcylated on either T365 or S366, or both (Figure 6G). Further analysis with the use of single mutations identified S366 as an O-GlcNAcylation site on MAVS (Figure 6H). To test if loss of MAVS O-GlcNAcylation affects downstream antiviral signaling, we reconstituted MAVS-knockout (Mavs−/−) mouse embryonic fibroblasts (MEFs) with either MAVS WT or S366A mutant through a lentivector-based transduction strategy. VSV challenge failed to induce the phosphorylation of IRF3 and NF-κB p65 (Figure 6I) and IFN-β production (Figure 6J) in Mavs−/− MEFs, confirming MAVS as a key mediator of antiviral innate immunity against RNA viruses (Kawai et al., 2005; Meylan et al., 2005; Seth et al., 2005; Xu et al., 2005). Mavs−/− MEFs reconstituted with WT MAVS restored the ability to phosphorylate IRF3 and p65 and produce IFN-β upon VSV challenge, whereas those reconstituted with MAVS S366A mutant failed to do so (Figure 6I and 6J). Furthermore, we reason that if additional OGT target other than MAVS exists which contributes to RLR-induced IFN-I generation, activation the O-GlcNAc signaling should enhance VSV-induced antiviral response in the presence of loss-of-O-GlcNAcylation mutant of MAVS. However, activation the O-GlcNAc signaling by thiamet G pretreatment failed to cause any increase in IRF3 and p65 phosphorylation (Figure 6K) or IFN-β production (Figure 6L) in Mavs−/− MEFs reconstituted with MAVS S366A mutant. These results indicate that MAVS is at least the major, if not only one, functional OGT target that promotes RLR-mediated antiviral response. Interestingly, MAVS S366 is well conserved among metazoan species (Figure 6M), suggesting the importance of O-GlcNAcylation on S366 as an evolutionarily conserved mechanism promoting MAVS biological activity. It should be noted that other O-GlcNAcylation site(s) may exist on MAVS, which can be indirectly affected by MAVS S366 O-GlcNAcylation. In sum, these results demonstrate that MAVS O-GlcNAcylation on S366 is critical for the induction of RLR-mediated antiviral immune signaling.
O-GlcNAcylation of MAVS on S366 promotes its K63-linked ubiquitination
We next sought to determine the molecular mechanism by which MAVS O-GlcNAcylation promotes downstream antiviral immune signaling. Our recent study identified TRIM31-mediated K63-lined ubiquitination of MAVS as a critical protein modification mechanism promoting antiviral immune responses (Liu et al., 2017a). Interestingly, during the overexpression experiments in 293T cells, we readily observed slower-migrating smear of MAVS induced by OGT when overexposed, indicating a possibility that OGT affects MAVS ubiquitination (data not shown). We therefore tested the hypothesis that O-GlcNAcylation of MAVS is vital for its ubiquitination using both overexpression and endogenous system. When 293T cells were transfected with plasmids expressing Flag-tagged MAVS and hemagglutinin (HA)-tagged ubiquitin, MAVS ubiquitination was readily detected. Co-expression of OGT induced further enhancement in total and K63-linked ubiquitination of MAVS but showed no effect on K48-linked ubiquitination (Figure 7A). We further accessed the effect of OGT on MAVS ubiquitination using either K63-only or K48-only ubiquitin mutants, which contain replacement of all lysine residues by alanine except lysine at position 63 and 48, respectively. OGT increased MAVS ubiquitination in the presence of K63-only ubiquitin (Figure 7B), but not K48-only ubiquitin (Figure 7C), suggesting that OGT promotes K63-linked ubiquitination of MAVS.
Figure 7.

O-GlcNAcylation of MAVS on S366 promotes its K63-linked ubiquitination. (A to C) MAVS ubiquitination in 293T cells were transfected with Flag-tagged MAVS and Myc-tagged OGT, in the presence of HA-tagged ubiquitin WT (A), K63-only (B) or K48only (C) mutants. (D) Total MAVS was immunoprecipitated from Ogtfl/fl and OgtΔmye BMMs challenged with or without VSV (MOI = 1) for 4 h, followed by immunoblotting with specific anti-ubiquitin antibodies, as indicated. (E) Total or K63-only or K48-only ubiquitinated MAVS was assessed in 293T cells transfected with Flag-tagged MAVS together with either OGT WT or K908A mutant. (F) MAVS K63-linked ubiquitination assayed in Mavs−/− MEFs transduced with either empty vector or GFP-tagged MAVS WT or S366A mutant, followed by VSV challenge for 4 h. (G to I) Ubiquitination of MAVS in 293T cells co-transfected with Flag-tagged MAVS WT (G and H) or S366A mutant (I), GFP-tagged TRIM31 and HA-tagged ubiquitin, together with either OGT WT (G) or K908A mutant (H). (J and K) MAVS K63 ubiquitin or O-GlcNAcylation (J) and IFNB1 transcript in single TRIM31-KO or OGT/TRIM31-DKO THP-1 cells challenged with VSV. * P < 0.05, versus controls (two-tailed Student’s t-test (K)). Data are from one experiment representative of four experiments (A to K).
To investigate whether OGT is required for MAVS ubiquitination during VSV challenge, we immunoprecipitated endogenous MAVS from WT or OgtΔmye BMMs challenged with or without VSV for 4 hours, followed by immunoblotting with a series of anti-ubiquitin antibodies. VSV infection caused robust increase in total and K63-linked ubiquitination of MAVS in WT macrophages, as previously described (Liu et al., 2017a; Paz et al., 2009). However, those increases did not occur in OgtΔmye BMMs, suggesting that OGT was required for MAVS ubiquitination during VSV challenge (Figure 7D). Further experiment with the use of OGT enzymatic-dead K908A mutant revealed that OGT-mediated enhancement in MAVS total and K63 ubiquitination requires the enzymatic function of OGT (Figure 7E). When Mavs−/− MEFs reconstituted with either WT MAVS or MAVS with loss-of-O-GlcNAcylation S366A mutation were challenged with VSV, MAVS S366A showed an attenuated K63-linked ubiquitination compared with WT MAVS (Figure 7F). This result suggested a requirement of MAVS O-GlcNAcylation on VSV-induced MAVS K63 ubiquitination and was consistent with the key role of MAVS O-GlcNAcylation on antiviral immune signaling (Figure 7I and 7J). When 293T cells were transfected with plasmids expressing GFP-tagged TRIM31 and MAVS, co-expression of OGT induced further enhancement in MAVS K63 ubiquitination (Figure 7G), which was dependent on OGT enzymatic activity (Figure 7H). Interestingly, coimmunoprecipitation assay showed that OGT promoted the association between MAVS and TRIM31 (Figure 7G) through OGT enzymatic activity (Figure 7H). Compared to WT MAVS, S366A mutant lost its association with TRIM31 (Figure 7I). These findings indicate that OGT-mediated MAVS O-GlcNAcylation facilitates MAVS targeting by TRIM31 to initiate K63-linked ubiquitination and downstream antiviral signaling. Finally, to dissect the functional relationship between OGT-mediated MAVS O-GlcNAcylation and TRIM31-mediated MAVS ubiquitination, we utilized CRISPR/Cas9-based gene targeting strategy to generate single TRIM31-KO or OGT/TRIM31-double KO (DKO) THP-1 cells. While MAVS ubiquitination was markedly attenuated by OGT deletion, MAVS O-GlcNAcylation remained largely intact in TRIM31-KO cells (Figure 7J). Importantly, comparing TRIM31-KO or OGT/TRIM31-DKO, OGT deletion failed to decrease MAVS ubiquitination or IFN-β production in the absence of TRIM31 (Figure 7J and 7K). Together these findings suggest that induced MAVS O-GlcNAcylation on S366 mediated by OGT is an upstream step for MAVS K63 ubiquitination and activation of antiviral immune signaling during RNA viral infection.
We next asked whether the promoting effect of O-GlcNAcyaltion on MAVS anti-viral signaling occurred in a physiological condition by lowering glucose level in cell culture medium. BMMs in medium with low glucose level or without glucose showed a markedly decreased total O-GlcNAc signal (Figure S7A), indicating that glucose availability could affect OGT-mediated protein O-GlcNAcylation. Importantly, VSV infection-induced MAVS O-GlcNAcylation, as well as MAVS ubiquitination, attenuated when glucose level decreased in medium (Figure S7B). Moreover, decreasing glucose level in cell culture medium also led to decreased cytokine generation upon VSV challenge, including Ifna4, Ifnb1, Il6, Tnfa, Isg15, Cxcl10 and Mx1 (Figure S7C). The correlation between MAVS O-GlcNAcylation, ubiquitination and cytokine production suggests that promoting effect of MAVS O-GlcNAcylation on RLR-mediated antiviral immune responses exists in physiological conditions.
DISCUSSION
We found here that activation of the RLR-MAVS signaling upon RNA virus challenge led to a quick increase in activities of glucose metabolic pathways, indicating that glucose metabolism may function in antiviral immune responses. This finding is consistent with two recent studies showing that elevated aerobic glycolysis upon viral infection is important for antiviral immune responses in mouse and human cells (Bajwa et al., 2016; Jiang et al., 2016). In addition, metabolic reprogramming towards increased glucose uptake, glycolysis and PPP has been well defined in innate immune cells that are classically activated by TLR agonists or bacterial infection (Everts et al., 2014; Lachmandas et al., 2016; O’Neill and Pearce, 2016). In contrast, alternative activation of innate immune cells, known as M2 polarization and usually associated with wound healing and tissue repair, generates a metabolic signature that mainly relies on mitochondrial metabolism consuming fatty acid and glutamine (Liu et al., 2017a). Therefore, increased glucose metabolism seems to be a common feature of innate immune responses against PAMPs or DAMPs. We sought to examine the role of individual metabolism pathways in innate immune system and found an essential role of HBP-associated O-GlcNAc signaling on antiviral immunity by targeting MAVS. Our study demonstrates that pharmacological activation of O-GlcNAc signaling promotes antiviral immune responses in macrophage culture system, thus supporting a promising therapeutic potential of OGT in viral infection-associated diseases.
Despite a similar increase pattern of glucose metabolic activity in response to either viral or bacterial challenge, one recent study observed an opposite effect of glucose metabolism on host survival between viral and bacterial in vivo challenge (Wang et al., 2016). Overnight fasting or pharmacological inhibition of glucose utilization promoted animal survival in LPS or bacterial-induced sepsis, but exacerbated animal survival upon influenza virus challenge. This observation indicates a possibility that different regulation of glucose metabolic activity between bacterial and viral challenge may exist. Indeed, in metabolomics experiment and flux assay parallel to the ones with VSV challenge (Figure 1), we observed a decreased HBP activity and protein O-GlcNAcylation in LPS-stimulated macrophages, although both glycolysis and PPP metabolic activities are expectedly increased. The underlying mechanism of the above observation is still unclear, but may potentially involve the alteration in enzymatic activity of GFPT, the first and rate-limiting enzyme in HBP. It is also expected that OGT may exert distinct functionality by targeting different groups of signaling molecules upon bacterial versus viral challenge. Our current study revealed MAVS as a target of OGT-mediated O-GlcNAcylation in viral-challenged macrophages. MAVS is a mitochondria-localized adaptor protein that forms a prion-like functional complex to activate downstream NF-κB and IRF3 signaling (Wu and Chen, 2014). We identified S366 as the key O-GlcNAcylation site on MAVS by unbiased LC-MS/MS analysis, followed by confirmatory studies of testing individual T→A or S→A mutants. Meanwhile, it is possible that other O-GlcNAcylation site(s) may exist on MAVS, which can be indirectly affected by MAVS S366 O-GlcNAcylation. Importantly, loss of O-GlcNAcylation by S366A mutation leads to enhanced activation of antiviral immune signaling, which suggests that O-GlcNAcylation exerts an intrinsic promoting effect on MAVS antiviral function. These findings thus expand our current understanding of metabolic regulation of immune signaling pathways and highlight an essential role of immunometabolism in promoting antiviral immunity.
Despite a neglectable glucose utilization change in M2 macrophages (Rodriguez-Prados et al., 2010), a recent study revealed that enhanced HBP activity, leading to increased UDP-GlcNAc generation, is a hallmark feature of M2 macrophages (Jha et al., 2015). It is reasonable to suspect an increased enzymatic activity of GFPT as a potential mechanism to promote HBP flux activity without global glucose uptake increase in M2 macrophages. Further assays identified that protein N-glycosylation pathway, which shares UDP-GlcNAc as a common substrate but exhibits totally distinct functions from O-GlcNAcylation pathway (Wellen and Thompson, 2010), was required for M2 polarization (Jha et al., 2015). Our study detected a similar elevation in HBP activity and UDP-GlcNAc availability in viral-infected macrophages. Further studies may focus on the individual effects of N-glycosylation pathway versus OGT-mediated GlcNAcylation in innate immune functions and their disease relevance.
Previous biochemical studies have identified several key molecules in the TLR and NF-κB signaling pathway as targets for O-GlcNAcylation, including IKKβ, p65, c-Rel and TAB1 (Allison et al., 2012; Pathak et al., 2012; Ramakrishnan et al., 2013; Yang et al., 2008). The overall effect of protein O-GlcNAcylation on inflammation has been proposed to enhance activation of inflammatory immune signaling (Yang et al., 2008). However, it remains unclear whether or not additional mechanism other than regulating protein phosphorylation exists by which O-GlcNAc signaling modulates protein functionality. This is of particular importance to innate immune signaling pathways due to the essential roles of several additional forms of posttranslational modification other than phosphorylation, including ubiquitination, acetylation, et al. We found here that viral infection caused a robust increase in MAVS O-GlcNAcylation in macrophages, and O-GlcNAcylation of MAVS on S366 is an essential step for its ubiquitination, subsequently, activation of downstream antiviral innate immune signaling. Mechanistically, we revealed that O-GlcNAcylation of MAVS on S366 promoted the association between MAVS and TRIM31, a recently discovered E3 ubiquitin ligase for MAVS by our group (Liu et al., 2017a). Therefore, our study demonstrates a working model in which OGT-mediated O-GlcNAcylation modulates the functionality of proteins through promoting their K63-linked ubiquitination. In summary, our findings provide a mechanistic link between OGT-mediated glucose metabolic pathway and antiviral innate immune signaling by targeting MAVS, and expand our current understanding of importance of glucose metabolic regulation in viral infection-associated diseases.
STAR★METHODS
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to will be fulfilled by the Lead Contact, Haitao Wen (haiwen75@gmail.com).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell Culture
HEK293T (ATCC CRL-11268), L929 (ATCC CCL-1) and Vero (ATCC CCL-81) cells were purchased from ATCC and maintained under a humidified atmosphere of 5% CO2 at 37°C in Dulbecco’s modified Eagle’s medium (DME M) supplemented with 10% fetal bovine serum (Sigma-Aldrich). THP-1 cells were purchased from ATCC and maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (Sigma-Aldrich). BMMs were generated from Ogtfl/fl and OgtΔmye mice in the presence of L-929 conditional medium, as previously described (Li et al., 2017). Peritoneal macrophages were isolated by peritoneal lavage with 10-ml sterile DPBS containing 2% FBS. Mavs−/− MEFs have been previously described (Sun et al., 2006).
Mice
OgtΔmye mice were generated by crossing the Ogtfl/fl mice (Shafi et al., 2000) with lysosome M-Cre mice. C57BL/6 mice and lysosome M-Cre mice were purchased from Jackson Laboratories. All animals were housed under specific pathogen-free conditions at 21°C and 31% humidity. Mice were weaned at 28 da ys after birth, separated into same sex groups and housed in plastic cages in a ventilated rack. Food (Chow TD. 7912; Harlan Teklad) and water were provided ad libitum, and health checks were performed daily. Cages were changed every two weeks. Mice were treated in accordance with the guidelines established by the University of Nebraska Medical Center and National Institute of Health Guide for the Care and Use of Laboratory Animals and the Institutional Animal Care and Use Committee (IAUCU).
Virus
Vesicular stomatitis virus (VSV) was amplified using Vero cells (Diallo et al., 2012), which were maintained under a humidified atmosphere of 5% CO2 at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Sigma-Aldrich).
METHOD DETAILS
qRT-PCR
Total RNA was extracted from in vitro cultured BMMs, peritoneal macrophages or MEFs using Trisure (Bioline). cDNA synthesis was performed with Moloney murine leukemia virus reverse transcriptase (Invitrogen) at 38°C fo r 60 min. RT-PCR was performed using SYBR Green PCR Master Mix in an Applied Biosystems StepOnePlus detection system. The fold difference in mRNA expression between treatment groups was determined by a standard delta-delta Ct method. β-actin was analyzed as an internal control. All samples were run in triplicates and data represent at least three independent experiments. Data analyses were blinded as to the provenance and treatment of the samples.
The primer sequences of individual genes are listed in the Table S2.
Immunoblotting
Electrophoresis of proteins was performed by using the NuPAGE system (Invitrogen) according to the manufacturer’s protocol. Briefly, cultured BMMs were collected and lysed with RIPA buffer. Proteins were separated on a NuPAGE gel and were transferred onto nitrocellulose membranes (Bio-Rad). Appropriate primary antibodies and HRPconjugated secondary antibodies were used and proteins were detected using the Enhanced Chemiluminescent (ECL) reagent (Thermo Scientific). The images were acquired with ChemiDoc MP System (Bio-Rad).
ELISA
Cytokines generated by in vitro cultured BMMs, peritoneal macrophages and MEFs were quantified using the ELISA Set for mouse IL-6 and TNF-α (BD Biosciences) according to the manufacturer’s protocol. ELISA for mouse IFN-β in cell culture supernatants and serum were performed using rat monoclonal anti-mouse IFN-β (Cosmo Bio USA #7891) as capture antibody and rabbit polyclonal anti-mouse IFN-β (R&D Systems #32400–1) as detection antibody, as previously described (Rothenfusser et al., 2005).
Plasmids and Molecular Cloning
The expression plasmids for RIG-I (full-length and N-terminus), MDA5, MAVS, IRF3, TBK1 and IFNB1 luciferase reporter were kindly provided by Z. Chen (University of Texas Southwestern Medical Center). pCMV vector expressing human OGT has been described (Chen et al., 2013). pcDNA3 vector containing FLAG-tagged IKKε (#26201), HA-tagged ubiquitin WT (#18712) was purchased from Addgene.
To generate the N- and C-terminal fragments of MAVS, pcDNA3 vector containing FLAG-tagged full-length MAVS was used as a template for PCR. To generate the lentivector expressing MAVS or OGT, using pcDNA3-MAVS or pCMV-OGT as templates, full-length MAVS or OGT was subcloned into the pWPXLd lentivector (Addgene #12258) in proper reading frame adjacent to GFP. All primers used for cloning are listed in the Table S3. To generate a series of MAVS mutant constructs and OGT K908A mutant, Phusion Site-Directed mutagenesis Kit was used according to the manufacturer’s instructions (Thermo Scientific). Primers used during mutagenesis PCR are listed in the Table S4. The complete nucleotide sequences of MAVS mutants and OGT K908A mutant were double-checked by sequencing.
Cell Transfection
As indicated in the Figure Legends, 293T cells were transfected for 30 h with a combination of expression plasmids for OGT WT or K908A mutant, MAVS WT or mutants, ubiquitin WT or mutants, and TRIM31 with X-tremeGENE HP DNA Transfection Reagent (Roche). For lentivector-based transduction, 293T cells were employed to package the pWPXLd lentivirus expressing OGT WT or K908A mutant, MAVS WT or S366A mutant, which were further used to transduce BMMs or MEFs.
Luciferase Assay
293T cells were transfected with IFNB1 luciferase reporter construct together with expression plasmids for RIG-I (N-terminus), MDA5, MAVS, TBK1, IKKε or IRF3, in the presence or absence of OGT using FuGENE6 (Roche). Empty pcDNA3 was used to maintain equal DNA amounts for transfection. Cells were harvested at 30 h post transfection. Luciferase activity was measured according to the manufacturer’s instructions (Promega). Data were normalized for transfection efficiency by calculating the ratio between firefly luciferase activity and Renilla luciferase activity, as previously described (Liu et al., 2017a).
CRISPR/Cas9 Knockdown
The human OGT-targeted single gRNA sequence, 5’- AATCACGCAGTGGTGCACGG, or TRIM31-targeted single gRNA sequence, 5’-TCAGGTTCAACTCGCTGTTG, was cloned into the lentiCRISPR v2 backbone (Addgene #52961). Then, the empty vector control, OGT-targeted lentiviruses or TRIM31-targeted lentiviruses were packaged in 293T cells using the envelop-vector pMDL and VSV-G packaging vector. Transduction of THP-1 and HT29 human cells was performed in the presence of polybrene. Single cell colonies were selected by unlimited dilution. Cells with effective OGT or TRIM31 deletion were used for further assays. To reconstitute OGT KO THP-1 cells with OGT, cells were transduced with a lentiviral plasmid encoding human OGT sequence with synonymous mutation to ensure that OGT gRNA could not introduce Cas9-mediated gene editing in the OGT transgene.
VSV Challenge in vivo
Both male and female Ogtfl/fl and OgtΔmye mice between the ages of 8–10 weeks were infected with VSV (1 × 108 PFU/mouse for survival study and 2 × 107 PFU/mouse for immunological studies) by a single intraperitoneal injection, as previously described (Liu et al., 2017a). Animal survival was monitored for 12 days post VSV injection. Serum and tissues including spleen, liver and lungs were collected at 24 h post injection for immunological and histological analyses. No sex-based differences after VSV challenge were observed in both Ogtfl/fl and OgtΔmye mice.
Metabolomics Analysis
3 × 106 BMMs from male C57BL/6 mice (8 weeks of age) left untreated or challenged with VSV for 4 h were harvested. The metabolite extraction was performed as previously described (Gunda et al., 2016; Shukla et al., 2017). The media was aspirated, and the cells were washed twice with LC-MS grade water to remove media remnants before lysing the cells. The polar metabolites were extracted from cells using cryogenically cold 80% methanol/water mixture. The metabolite extracts were resuspended in 50% methanol/water mixture and analyzed using LC-MS/MS. A single reaction monitoring (SRM) LC-MS/MS method with AB SCIEX 5500 QTRAP® (Framingham, MA) was used for analysis, as described previously (Gunda et al., 2016).
The data acquisition was carried out using Analyst™1.6 software (AB SCIEX, Framingham, MA), and peaks were integrated with Multiquant™ (AB SCIEX, Framingham, MA). The peak areas were normalized with the respective protein concentrations, and the resultant peak areas were subjected to relative quantification analyses with MetaboAnalyst 3.0 (Xia et al., 2012). Further, principal component analysis, heatmap and pathway impact analysis was performed using the features provided in MetaboAnalyst 3.0 software. It should be noted that this metabolomics method is not able to distinguish between epimers such as UDP-GlcNAc versus UDP-GalNAc.
LC-MS/MS Analysis for 13C-UDP-GlcNAc
13C-labeling of UDP-GlcNAc from U-13C6-Glucose was detected from cell lysates using a LC-MS/MS-based scheduled reaction monitoring methodology. Briefly, the liquid chromatography constituted an Acquity UPLC BEH amide column (Waters Inc.) and a gradient buffer method consisting of buffer A (Acetonitrile) and buffer B (10 mM, Ammonium bicarbonate pH, 9.0) with the initial concentration of buffer A set to 85%, which was decreased in a gradient manner to 50% by 15 min. This was followed by an increase in the percent of buffer A to 85% by 23 min. The 30-minute LC method included an additional 7 min wash period. The mass spectrum for 12C-UDP-GlcNAc was acquired by electrospray ionization (ESI) in the negative ion mode and the 13C-labeled UDP-GlcNAc was analyzed based on the 13C-labeling fragmentation pattern of the respective parent and daughter ions.
Cell stimulation under different glucose levels
BMMs generated from male C57BL/6 mice (8 weeks of age) were placed in culture medium with 0, 5 or 25 mM glucose for 16 h, then left untreated or followed by VSV stimulation for 4 h. Total protein O-GlcNAcylation was detected by immunoblotting. Endogenous MAVS was immunoprecipitated, followed by immunoblotting with indicated antibodies. Gene transcripts in the cells were measured by RT-PCR.
MAVS O-GlcNAcylation Site Mapping
MS strategy was employed to identify MAVS O-GlcNAcylation sites, as described in our recent study (Li et al., 2017). Briefly, immunoprecipitated MAVS from 293T cells was subjected to SDS-PAGE. The band corresponding to MAVS was excised and the protein was reduced with DTT, alkylated with iodoacetamide, and digested with trypsin overnight, then subjected to LC-MS/MS analysis using a nanoAcquity (Waters Corp) coupled to an LTQ Orbitrap Velos (Thermo Scientific). The LTQ Orbitrap Velos was operated in data-dependent mode where the 10 most intense precursors were selected for subsequent collision-induced dissociation (CID) fragmentation. Raw data files processed using Proteome Discoverer (PD) version 2.0 (Thermo Scientific). Peak lists were searched against a Homo sapiens Uniprot database using Sequest. The following parameters were used to identify tryptic peptides for protein identification: 10 ppm precursor ion mass tolerance; 0.6 Da product ion mass tolerance; up to two missed trypsin cleavage sites; carbamidomethylation of Cys was set as a fixed modification; hexNAc (+203.0794 Da) of N/S/T, oxidation of M and phosphorylation of S/T/Y were set as variable modifications. The Percolator node was used to determine false discovery rates (FDR) and a peptide FDR of 5% were used to filter all results.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
All experiments were performed a minimum of three independent replications. Data were analyzed as mean ± s.d.. All data meet the assumptions of the tests. Student’s unpaired t-test was used to compare the means of two groups. One-way Analysis of Variance (ANOVA) with Bonferroni post-tests was used to compute statistical significance between multiple groups. GraphPad Prism software (Version 5.0) was used to determine statistical significance among multiple studies. P values of less than 0.05 were considered significant. The exact value of n, representing the number of experimental replication or mice in the experiments depicted, was indicated in the figure legends.
Table S1. Differentially expressed metabolites between Non-treated and VSV-infected groups. Related to Figure 1.
Supplementary Material
Highlights.
Glucose metabolic pathways display enhanced activation upon VSV infection
O-GlcNAcylation transferase (OGT) enzymatically promotes RLR-mediated antiviral immunity
OGT-mediated O-GlcNAcylation of MAVS on S366 promotes its K63-linked ubiquitination
MAVS K63-linked ubiquitination is critical for its role in RLR-antiviral signaling
ACKNOWLEDGMENTS
We are grateful to Z.J. Chen for the human IFNB1 promoter reporter, the expression plasmids for RIG-I, MDA5, MAVS, IRF3 and TBK1, and Mavs−/− MEFs; X. Yang for Ogtfl/fl mice and helpful discussions; X. Yu for expression plasmid for OGT; L. Kong for the technical support on in vivo experiments; M. Yuan for the metabolomics assay; Wen lab members for discussions; and N.E. Sarvetnick for advice and support. The NIH grant R01GM120496 supports the work performed in the lab of H.W. J.M.A. is supported by the NIH grants 5P01CA120964 and 5P30CA006516. The NIH grant P30CA016086 supports mass spectrometry assay performed in the UNC Lineberger Comprehensive Cancer Center Proteomics Center. P.K.S. is supported by the NIH grants R01CA163649, R01CA210439, and R01CA216853. P.K.S. is also a recipient of the American Association for Cancer Research (AACR)-Pancreatic Cancer Action Network (PanCAN) Career Development Award (30–20-25-SING), the Specialized Programs for Research Excellence (SPORE, 2P50 CA127297), and the Pancreatic Tumor Microenvironment Research Network (U54, CA163120). Y.L. is supported by the NIH/NIDCR DE024173 (K99/R00 Pathway to Independence Award). C.G. is supported by the Natural Science Foundation of China (31730026, 81525012, 81471538).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allison DF, Wamsley JJ, Kumar M, Li D, Gray LG, Hart GW, Jones DR, and Mayo MW (2012). Modification of RelA by O-linked N-acetylglucosamine links glucose metabolism to NF-kappaB acetylation and transcription. Proc Natl Acad Sci U S A 109, 16888–16893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelova M, Ortiz-Meoz RF, Walker S, and Knipe DM (2015). Inhibition of O-Linked N-Acetylglucosamine Transferase Reduces Replication of Herpes Simplex Virus and Human Cytomegalovirus. J Virol 89, 8474–8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajwa G, DeBerardinis RJ, Shao B, Hall B, Farrar JD, and Gill MA (2016). Cutting Edge: Critical Role of Glycolysis in Human Plasmacytoid Dendritic Cell Antiviral Responses. J Immunol 196, 2004–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN (2015). STING: infection, inflammation and cancer. Nat Rev Immunol 15, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond MR, and Hanover JA (2013). O-GlcNAc cycling: a link between metabolism and chronic disease. Annu Rev Nutr 33, 205–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker SW, Bonham KS, Zanoni I, and Kagan JC (2015). Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol 33, 257–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, Sowell RT, Kaech SM, and Pearce EL (2017). Metabolic Instruction of Immunity. Cell 169, 570–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Chen Y, Bian C, Fujiki R, and Yu X (2013). TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493, 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Sun L, and Chen ZJ (2016). Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 17, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Clarke AJ, Hurtado-Guerrero R, Pathak S, Schuttelkopf AW, Borodkin V, Shepherd SM, Ibrahim AF, and van Aalten DM (2008). Structural insights into mechanism and specificity of O-GlcNAc transferase. EMBO J 27, 2780–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diallo JS, Vaha-Koskela M, Le Boeuf F, and Bell J (2012). Propagation, purification, and in vivo testing of oncolytic vesicular stomatitis virus strains. Methods Mol Biol 797, 127–140. [DOI] [PubMed] [Google Scholar]
- Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJ, et al. (2014). TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol 15, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, and Maniatis T (2003). IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4, 491–496. [DOI] [PubMed] [Google Scholar]
- Gunda V, Yu F, and Singh PK (2016). Validation of Metabolic Alterations in Microscale Cell Culture Lysates Using Hydrophilic Interaction Liquid Chromatography (HILIC)-Tandem Mass Spectrometry-Based Metabolomics. PLoS One 11, e0154416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardiville S, and Hart GW (2014). Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab 20, 208–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GW, Slawson C, Ramirez-Correa G, and Lagerlof O (2011). Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem 80, 825–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, et al. (2015). Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430. [DOI] [PubMed] [Google Scholar]
- Jiang H, Shi H, Sun M, Wang Y, Meng Q, Guo P, Cao Y, Chen J, Gao X, Li E, and Liu J (2016). PFKFB3-Driven Macrophage Glycolytic Metabolism Is a Crucial Component of Innate Antiviral Defense. J Immunol 197, 2880–2890. [DOI] [PubMed] [Google Scholar]
- Kamitani T, Kito K, Nguyen HP, and Yeh ET (1997). Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J Biol Chem 272, 28557–28562. [DOI] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, and Akira S (2005). IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6, 981–988. [DOI] [PubMed] [Google Scholar]
- Lachmandas E, Boutens L, Ratter JM, Hijmans A, Hooiveld GJ, Joosten LA, Rodenburg RJ, Fransen JA, Houtkooper RH, van Crevel R, et al. (2016). Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat Microbiol 2, 16246. [DOI] [PubMed] [Google Scholar]
- Lazarus MB, Nam Y, Jiang J, Sliz P, and Walker S (2011). Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 469, 564–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y, Wen H, Yu Y, Taxman DJ, Zhang L, Widman DG, Swanson KV, Wen KW, Damania B, Moore CB, et al. (2012). The Mitochondrial Proteins NLRX1 and TUFM Form a Complex that Regulates Type I Interferon and Autophagy. Immunity 36, 933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ZG, and Walker S (2016). The Biochemistry of O-GlcNAc Transferase: Which Functions Make It Essential in Mammalian Cells? Annu Rev Biochem 85, 631–657. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang Z, Li L, Gong W, Lazenby AJ, Swanson BJ, Herring LE, Asara JM, Singer JD, and Wen H (2017). Myeloid-derived cullin 3 promotes STAT3 phosphorylation by inhibiting OGT expression and protects against intestinal inflammation. J Exp Med 214, 1093–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G, Wang P, Zhao K, Hou J, Wang X, et al. (2017a). The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat Immunol 18, 214–224. [DOI] [PubMed] [Google Scholar]
- Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, Di Conza G, Cheng WC, Chou CH, Vavakova M, et al. (2017b). alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 18, 985–994. [DOI] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, and Chen ZJ (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- Martinez-Fleites C, Macauley MS, He Y, Shen DL, Vocadlo DJ, and Davies GJ (2008). Structure of an O-GlcNAc transferase homolog provides insight into intracellular glycosylation. Nat Struct Mol Biol 15, 764–765. [DOI] [PubMed] [Google Scholar]
- Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, and Tschopp J (2005). Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172. [DOI] [PubMed] [Google Scholar]
- Moore CB, and Ting JP (2008). Regulation of mitochondrial antiviral signaling pathways. Immunity 28, 735–739. [DOI] [PubMed] [Google Scholar]
- O’Neill LA, and Pearce EJ (2016). Immunometabolism governs dendritic cell and macrophage function. J Exp Med 213, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman M, Janetzko J, Fan C, Duveau DY, Tan ZW, Thomas CJ, and Walker S (2015). A small molecule that inhibits OGT activity in cells. ACS Chem Biol 10, 1392–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak S, Borodkin VS, Albarbarawi O, Campbell DG, Ibrahim A, and van Aalten DM (2012). O-GlcNAcylation of TAB1 modulates TAK1-mediated cytokine release. EMBO J 31, 1394–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz S, Vilasco M, Arguello M, Sun Q, Lacoste J, Nguyen TL, Zhao T, Shestakova EA, Zaari S, Bibeau-Poirier A, et al. (2009). Ubiquitin-regulated recruitment of IkappaB kinase epsilon to the MAVS interferon signaling adapter. Mol Cell Biol 29, 3401–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan P, Clark PM, Mason DE, Peters EC, Hsieh-Wilson LC, and Baltimore D (2013). Activation of the transcriptional function of the NF-kappaB protein c-Rel by O-GlcNAc glycosylation. Sci Signal 6, ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, Cascante M, and Bosca L (2010). Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 185, 605–614. [DOI] [PubMed] [Google Scholar]
- Roers A, Hiller B, and Hornung V (2016). Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity 44, 739–754. [DOI] [PubMed] [Google Scholar]
- Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, and Fitzgerald KA (2005). The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol 175, 5260–5268. [DOI] [PubMed] [Google Scholar]
- Sanchez EL, and Lagunoff M (2015). Viral activation of cellular metabolism. Virology 479-480, 609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, and Chen ZJ (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. [DOI] [PubMed] [Google Scholar]
- Shafi R, Iyer SP, Ellies LG, O’Donnell N, Marek KW, Chui D, Hart GW, and Marth JD (2000). The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci U S A 97, 5735–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, King RJ, Abrego J, Goode GD, Dasgupta A, et al. (2017). MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 32, 392. [DOI] [PubMed] [Google Scholar]
- Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, and Chen ZJ (2006). The specific and essential role of MAVS in antiviral innate immune responses. Immunity 24, 633–642. [DOI] [PubMed] [Google Scholar]
- Wang A, Huen SC, Luan HH, Yu S, Zhang C, Gallezot JD, Booth CJ, and Medzhitov R (2016). Opposing Effects of Fasting Metabolism on Tissue Tolerance in Bacterial and Viral Inflammation. Cell 166, 1512–1525 e1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, and Thompson CB (2010). Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell 40, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, and Chen ZJ (2014). Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol 32, 461–488. [DOI] [PubMed] [Google Scholar]
- Xia J, Mandal R, Sinelnikov IV, Broadhurst D, and Wishart DS (2012). MetaboAnalyst 2.0--a comprehensive server for metabolomic data analysis. Nucleic acids research 40, W127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, and Shu HB (2005). VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 19, 727–740. [DOI] [PubMed] [Google Scholar]
- Yang WH, Park SY, Nam HW, Kim do H, Kang JG, Kang ES, Kim YS, Lee HC, Kim KS, and Cho JW (2008). NFkappaB activation is associated with its O-GlcNAcylation state under hyperglycemic conditions. Proc Natl Acad Sci U S A 105, 17345–17350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, and Qian K (2017). Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 18, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Zhang F, and Kudlow JE (2002). Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repression. Cell 110, 69–80. [DOI] [PubMed] [Google Scholar]
- You F, Sun H, Zhou X, Sun W, Liang S, Zhai Z, and Jiang Z (2009). PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat Immunol 10, 1300–1308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.