

Abstract
The gene CDC73 (previously known as HRPT2) encodes the protein parafibromin. Biallelic mutation of CDC73 is strongly associated with malignancy in parathyroid tumors. Heterozygous germline mutations cause hyperparathyroidism jaw tumor syndrome,which is associated with a high life-time risk of parathyroid carcinoma. Therefore loss of parafibromin expression by immunohistochemistry may triage genetic testing for hyperparathyroidism jaw tumor syndrome and be associated with malignant behavior in atypical parathyroid tumors. We share our experience that parafibromin-negative parathyroid tumors show distinctive morphology. We searched our institutional database for parathyroid tumors demonstrating complete loss of nuclear expression of parafibromin with internal positive controls. Forty-three parafibromin-negative tumors from 40 (5.1%) of 789 patients undergoing immunohistochemistry were identified. Thirty-three (77%) were external consultation cases; the estimated incidence in unselected tumors was 0.19%. Sixteen (37.2%) fulfilled World Health Organization 2017 criteria for parathyroid carcinoma and 63% had serum calcium greater than 3mmol/L. One of 27 (3.7%) noninvasive but parafibromin-negative tumors subsequently metastasized. Parafibromin-negative patients were younger (mean, 36 vs. 63 y; P<0.001) and had larger tumors (mean, 3.04 vs. 0.62 g; P<0.001). Not all patients had full testing, but 26 patients had pathogenic CDC73 mutation/deletions confirmed in tumor (n=23) and/or germline (n=16). Parafibromin-negative tumors demonstrated distinctive morphology including extensive sheet-like rather than acinar growth, eosinophilic cytoplasm, nuclear enlargement with distinctive coarse chromatin, perinuclear cytoplasmic clearing, a prominent arborizing vasculature, and, frequently, a thick capsule. Microcystic change was found in 21 (48.8%). In conclusion, there are previously unrecognized morphologic clues to parafibromin loss/CDC73 mutation in parathyroid tumors which, given the association with malignancy and syndromic disease, are important to recognize.
Key Words: parathyroid, parathyroid carcinoma, CDC73, parafibromin, hyperparathyroidism jaw tumor syndrome
CDC73 (previously known as HRPT2) is a 17 exon, 133 kb gene that maps to 1q31.2 and encodes the protein parafibromin.1–3 CDC73 functions as a genuine tumor suppressor gene that is involved in the regulation of p53 and also as a component of the human PAF1 complex, which controls RNA polymerase II–mediated transcription.4–6 Pathogenic germline mutations in CDC73 are recognized as the cause of the autosomal-dominant hyperparathyroidism jaw tumor (HPT-JT) syndrome7,8—a rare hereditary tumor syndrome characterized by parathyroid neoplasms and unusual bony lesions of the mandible and maxilla, usually classified as ossifying fibromas.6,8,9
In HPT-JT syndrome, hyperparathyroidism commonly occurs at a young age with median first onset between the third and fourth decade (range, 7 to 65 y).6,9 In addition to its association with early onset hyperparathyroidism, HPT-JT syndrome is particularly important to recognize because of its strong association with parathyroid carcinoma. Parathyroid carcinoma, rare in all other circumstances, has been reported to account for 15% to 37.5% of hyperparathyroidism in HPT-JT patients, and the life-time risk of parathyroid carcinoma in patients with HPT-JT syndrome has been estimated to be as high as 15%.6,8–10 For these reasons, it has been recommended that patients with confirmed or suspected HPT-JT syndrome should be on surveillance programs beginning at age 5 to 10 years.10 Furthermore, the possibility of HPT-JT syndrome should be considered in all young patients with HPT.8–10
Biallelic CDC73 mutation/inactivation is also strongly associated with parathyroid carcinoma occurring outside the setting of HPT-JT syndrome.2,11 When the diagnosis is restricted to carcinomas, which not only fulfill histologic criteria for malignancy but also demonstrate biological evidence of malignant behavior (ie, recurrence or metastasis), the combined results of several studies indicate that the CDC73 mutation occurs in up to 77% of parathyroid carcinomas but in <1% of unselected apparently benign parathyroid adenomas.2,11–15 Furthermore, up to 20% of patients with apparently sporadic parathyroid carcinoma will be shown to have germline mutations in CDC73 and therefore have previously unrecognized HPT-JT syndrome.1,13,14 It has therefore been recommended that all patients with parathyroid carcinoma should be offered genetic testing for HPT-JT syndrome.8–10,16
Loss of immunohistochemical expression of parafibromin has been used as a marker of biallelic CDC73 mutation inactivation in parathyroid tumors.1,10,12,17–24 However, parafibromin immunohistochemistry (IHC) is not without controversies and difficulties. Different scoring systems for interpretation are in use and several groups have reported that parafibromin IHC can be a technically demanding or difficult antibody to deploy in the routine clinical setting—summarized in Gill.2 A fair reading of the literature indicates that, although some groups have found parafibromin IHC to be useful both in the differential diagnosis of atypical parathyroid tumors and as a maker of underlying CDC73 mutation,1,18–24 others found parafibromin IHC technically difficult to perform and interpret.25–27 Furthermore, parafibromin IHC is not widely available.
Given these difficulties with IHC, it would be particularly beneficial if morphologic clues to the presence of CDC73 mutation could be identified. As early adopters and proponents of parafibromin IHC,1,2,21 we now present our prospective experience of parafibromin IHC in the routine clinical setting. We seek to share our finding that parafibromin-deficient (HPT-JT type, CDC73 mutated) parathyroid tumors demonstrate distinctive but previously unrecognized morphologic features that can be used to triage IHC or, if parafibromin is locally unavailable or considered unreliable, molecular testing. Further we seek to address current controversies and uncertainties, including criteria for interpretation of parafibromin IHC, clinical associations of parafibromin-deficient tumors, and whether noninvasive but parafibromin-deficient/CDC73-mutated parathyroid tumors (which would be considered benign under the World Health Organization [WHO] 2017 classification) have metastatic potential.
METHODS
We searched the computerized database of the Department of Anatomical Pathology, Royal North Shore Hospital, Sydney, Australia, for all parathyroid tumors that had undergone parafibromin IHC from its introduction into routine clinical practice in 20061 to 30 June 2017. Although there were no firm departmental policies and the decision to perform parafibromin IHC was left up to the discretion of the reporting pathologists, during this period there were a variety of relative indications for parafibromin IHC. These included suspected or confirmed parathyroid carcinoma, atypical parathyroid adenomas, multiple parathyroid adenomas, recurrent parathyroid tumors, hyperparathyroidism at a young age, a family history not explained by other syndromes, extreme hypercalcemia, large size, or the operative impression of the tumor being adherent to adjacent tissue.
Throughout this period we used the same mouse monoclonal anti-parafibromin antibody—clone 2H1 (cat. no.: SC-33638, Santa Cruz, CA).1,17 To achieve positive staining in internal control tissue, various dilutions, detection systems, and antigen retrieval protocols were required. The precise conditions varied during the 12-year period of the study, and, often, several attempts at different titers were required to achieve an interpretable result for individual cases. As an indication, our current default approach is to use the primary antibody at a dilution of 1 in 400, with heat-induced antigen retrieval in an alkaline solution for 40 minutes at 97°C, and, thereafter, adjust the titers if the slides are not interpretable. All cases initially reported as negative at the time of primary reporting were reviewed for this study to confirm this interpretation, or underwent repeat IHC.
Negative (abnormal) staining for parafibromin was defined as complete loss of nuclear expression in all neoplastic cells with internal positive controls in non-neoplastic tissue throughout the section (illustrated in Fig. 1).1 Cases with focal loss of staining in only some neoplastic cells were considered positive. If cases were negative but there was no staining in internal positive controls, the staining was considered noninformative and was repeated with different conditions, as described above until an interpretable result was achieved. Cytoplasmic staining was considered nonspecific and disregarded.
FIGURE 1.
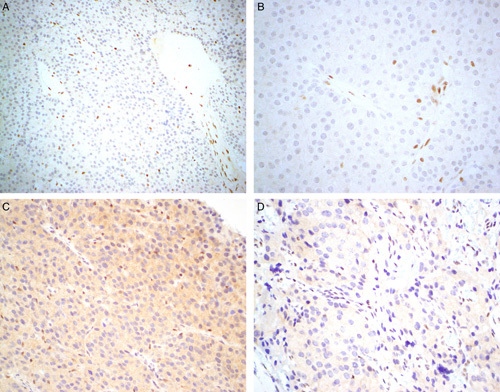
We emphasize that parafibromin can be a difficult stain to perform and interpret and often different conditions are required to achieve a workable result. This figure shows parafibromin IHC from the same tumor performed on the same block initially at primary diagnosis (A, B) and repeated 8 years later for this study (C, D). When first performed, all non-neoplastic cells are completely negative with crisp nuclear staining in internal positive controls (A, B). C, When repeated on archived material, a greater concentration of primary antibody was required to achieve expression in internal positive controls resulting in nonspecific cytoplasmic staining but still completely absent nuclear staining in neoplastic cells. D, The internal controls are weaker in some areas on repeat staining but still positive. Parafibromin IHC, original magnifications A) 200x, B) and C) 400x, D) 600x.
All parafibromin-negative cases underwent morphologic review of all available hematoxylin and eosin–stained sections. Cases were classified as either adenoma or carcinoma using the criteria outlined in the fourth edition of WHO 2017 classification of endocrine neoplasia.28 That is, the diagnosis of parathyroid carcinoma was restricted to tumors with evidence of unequivocal invasive growth involving adjacent structures, including the thyroid and soft tissues, blood vessels, or perineural spaces, or to those tumors with documented metastases. Specific morphologic features including invasive growth, vascular space invasion, prominent nucleoli, a thick capsule, cystic change, mitotic count per 10 hpf (2 mm2), sheet-like rather than acinar growth pattern, eosinophilic cell type, perinuclear cytoplasmic clearing, a prominent or hemangiopericytomatous vascular pattern, coagulative necrosis, cytologic atypia, and multinucleation were also specifically sought and recorded. Long-term clinical follow-up was sought for all cases by reviewing medical records and contacting the original referring pathologists and surgeons. We then developed a control cohort of truly unselected parathyroid tumors that excluded consultation cases by searching the anatomic pathology database for all parathyroid tumors undergoing routine surgical pathology reporting in our department from the calendar year 2012 to 2015.
Parafibromin-negative parathyroid tumors underwent Sanger sequencing on archived formalin-fixed paraffin-embedded (FFPE) neoplastic tissue. This testing was performed blinded to the results of germline mutation testing undertaken during routine clinical care. Briefly, neoplastic areas were macrodissected, and genomic DNA was extracted using the QIAmp DNA FFPE Tissue Kit (Qiagen), according to the manufacturer’s instructions. Primers were designed using Primer 3, using the NCBI gene reference, NG_012691.1 (primer sequences and conditions available upon request). To identify point mutations, 17 exons of CDC73 were amplified by polymerase chain reaction using MangoTaq polymerase (Bioline). Polymerase chain reaction products were purified using Wizard SV Gel and PCR Clean-up system (Promega) according to the manufacturer’s instructions. Amplified samples were sequenced using forward and reverse primers by Sanger sequencing (service provided by Australian Genome Research Facility, Sydney, Australia). Mutation nomenclature was according to accession number NM_024529.4 with numbering starting at the A of the ATG translation initiation codon.
If no mutations were identified by sequencing, samples underwent multiplex ligation-dependent probe amplification (MLPA) studies using SALSA P466-A1 Probemix (MRC-Holland), according to the manufacturer’s instructions. The relative peak area (RPA)—that is, the individual peak area in relation to the total of all peak areas in the sample—was normalized against the RPA of the control samples for each exon and reference genes included in the probe. A reduction of 40% to 50% in the ratio of RPA for the exon was considered to indicate deletion. This study was approved by the Northern Sydney Local Health District Human Research Ethics Committee.
RESULTS
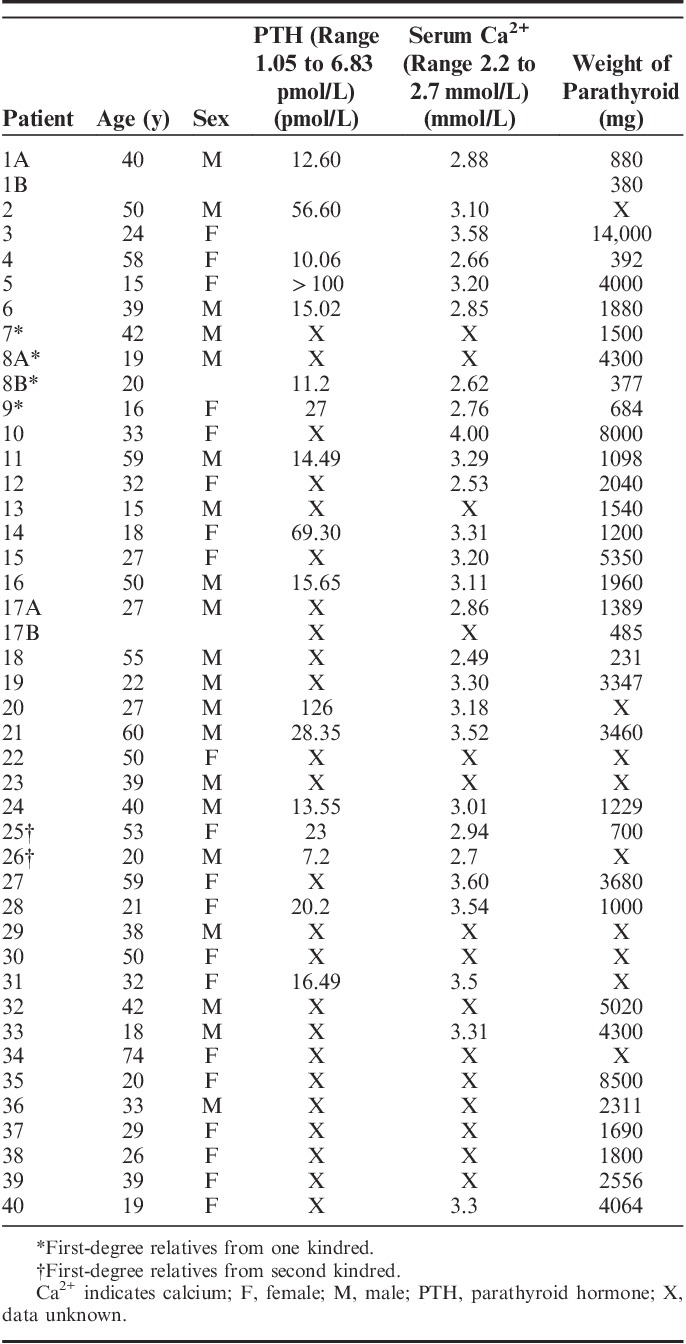
During the study period, a total of 815 parathyroid tumors from 789 patients underwent parafibromin IHC. IHC was confirmed negative in 43 tumors (5.2%) from 40 patients (5.1%) with material available for review. The clinical and demographic details of these patients, including the preoperative calcium and parathyroid hormone levels and gland weights, are presented in Table 1. There was a precisely equal sex distribution with 20 male patients and 20 female patients. The median age at diagnosis was 33 years, and the median weight was 1840 mg. The average serum calcium level was 3.12 mmol/L (range 2.49 to 4.0 mmol/L) with 17 of 27 (63%) patients having a preoperative serum calcium of greater than 3 mmol/L. Thirty-three (77%) of the IHC-negative cases were from external consultation cases.
TABLE 1.
Clinical Details of Parafibromin-negative Parathyroid Adenoma and Carcinoma
In the unselected control cohort excluding consultation cases from 2012 to 2015, there were 1055 patients of whom 69 (6.5%) underwent parafibromin IHC and only 2 were found to be parafibromin negative (0.19% incidence in a truly unselected cohort). There were several differences between the unselected cohort and the parafibromin-negative cohort. Eight hundred nineteen (78%) of the unselected control cohort were female patients versus 50% of the parafibromin-negative cohort (P<0.001). The mean age of the unselected control cohort was 63 years (range, 12 to 95 y) versus 36 years (range, 15 to 74 y) in the parafibromin-negative cohort (P<0.001). The mean weight of the tumors from the unselected control cohort was 620 mg (range, 58 to 13,800 mg) compared with 3036 mg (range, 231 to 14,000 mg) in the parafibromin-negative cohort (P<0.001). In univariate analysis, negative staining for parafibromin was significantly associated with younger age of onset, heavier weight, and male sex (all P<0.001). In a multivariate model including these factors, parafibromin negativity remained associated with age and weight (both P<0.001), but sex lost significance (P=0.187).
The morphologic features of the parafibromin-negative cases are presented in Table 2 and illustrated in Figures 2–5. Further whole-slide scanned images from all tumors are available for review at the Cancer Diagnosis and Pathology group website www.cancerdxpathology.org.au. Sixteen (37.2%) parafibromin-negative tumors fulfilled WHO 2017 criteria for carcinoma (Fig. 2). In addition to this strong association with malignancy, the parafibromin-negative tumors demonstrated consistent and distinctive morphology. The neoplastic cells displayed eosinophilic cytoplasm, which frequently lacked the granularity of usual parathyroid oxyphil cells. Another particularly distinctive feature was the very frequent presence of perinuclear cytoplasmic clearing imparting an almost koilocytic quality, which in some areas was reminiscent of chromophobe renal carcinoma. In most instances, there was quite prominent nuclear enlargement but, sometimes, with relatively preserved nuclear-to-cytoplasmic (N/C) ratios, giving a peculiar ancient quality to the atypia. In several cases, there were subclonal nodules, which demonstrated greater nuclear atypia, again sometimes with a relatively preserved N/C ratio.
TABLE 2.
Histologic Features of Parafibromin-negative Parathyroid Adenoma and Carcinoma

FIGURE 2.
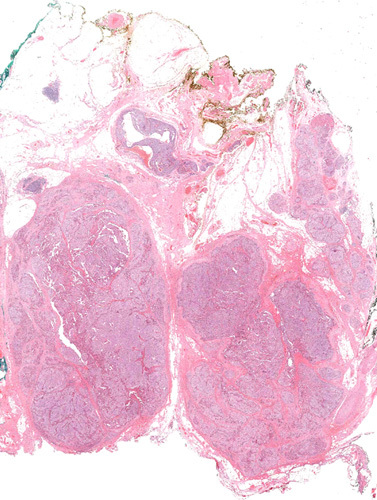
In accordance with the WHO 2017 classification the diagnosis of parathyroid carcinoma was restricted to cases that demonstrated unequivocal invasive growth. Hematoxylin and eosin, whole mount.
FIGURE 5.
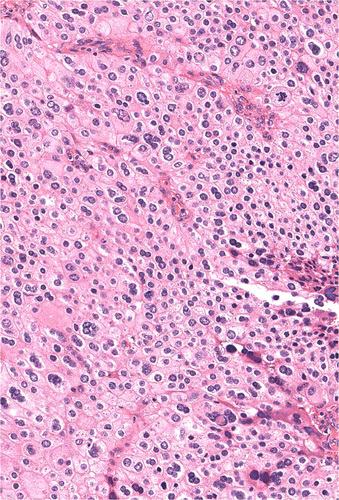
In some cases, both multinucleation and nuclear atypia were present. The nuclear atypia was sometimes associated with smudged chromatin and relatively preserved N/C ratios imparting an ancient quality. An atypical mitotic figure is noted in the upper right quadrant. Hematoxylin and eosin, original magnification 400x.
FIGURE 3.
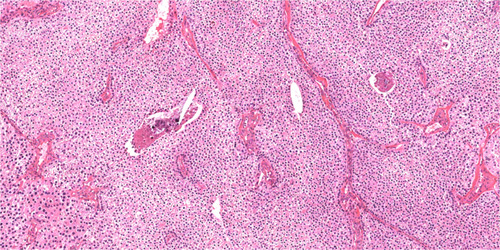
Parafibromin-deficient (HPT-JT type, CDC73 mutated) parathyroid tumors are characterized by cells with eosinophilic cytoplasm demonstrating a sheet-like growth pattern. At this magnification both the prominent arborizing vasculature and the distinctive perinuclear cytoplasmic clearing are also evident. Hematoxylin and eosin, original magnification 100x.
FIGURE 4.
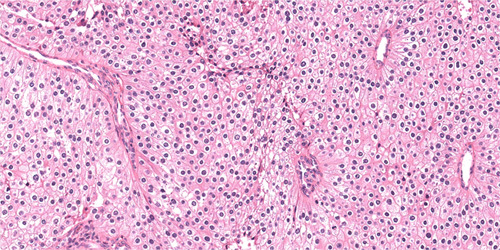
Parafibromin-deficient (HPT-JT type, CDC73 mutated) parathyroid tumors demonstrate eosinophilic cytoplasm, but these tumor cells usually lack the distinct cytoplasmic granularity of usual oxyphil cells and are notable for their relative nuclear enlargement and perinculear cytoplasmic clearing. Hematoxylin and eosin, original magnification 200x.
The chromatin was usually speckled, often, coarsely so, sometimes with prominent nucleoli. Rather than being arranged in an acinar architecture, the neoplastic cells usually grew in solid sheets. Microcystic change was not uncommon, being found in 21 (48.8%) cases; however, grossly evident macrocystic change was absent. In most cases, there was a conspicuous arborizing vasculature, at least focally. In a few cases, there were some prominent dilated vessels imparting a hemangiopericytomatous quality, which contributed to the microcystic appearance appreciated at low power. Some cases were associated with a thick capsule. Multinucleation was noted in 5 (12%) cases. Broad bands of fibrosis were rare.
The results of molecular testing are presented in Table 3. Of the 39 tumors from 38 patients that underwent molecular testing on FFPE tumor tissue, pathogenic CDC73 variants were confirmed in 23. Eight (34.8%) of these were large-scale deletions. Loss of heterozygosity was confirmed in 4 tumors, and, in an additional 2 tumors, (#6 and #23) putative “second-hit” somatic variants were identified that were absent in germline testing.
TABLE 3.
Mutation and Follow-up Status of Parafibromin-negative Patients

Of the 40 patients, 24 had some degree of germline mutation testing as part of their clinical care. In 16 (66.7%), pathogenic germline variants were identified, of which 2 were whole-gene deletions. Of note, 4 of the patients in whom no germline mutations were identified did not undergo MLPA, and 3 of these patients had large-scale deletions in their tumors. Therefore, germline deletions could not be excluded. In 3 tumors, somatic testing was not able to identify the known germline CDC73 variants because of incomplete coverage.
There were no tumors in which we could confidently exclude CDC73 mutation. One case (#4) had neither somatic nor germline testing performed, and, in 13 cases, somatic sequencing data had insufficient coverage in at least some exons. Therefore, a total of 26 of 40 (65%) patients with parafibromin-negative tumors had confirmed pathogenic somatic and/or germline CDC73 mutations, and, in the remaining 14 patients, there was insufficient coverage to exclude a pathogenic mutation.
The long-term follow-up of the parafibromin-negative patients is presented in Table 3—median duration of follow-up 26 months. One patient (2.5%) developed a jaw tumour with the typical characteristics of the type of ossifying fibroma reported in the setting of HPT-JT syndrome.6 Of the 16 patients who fulfilled WHO 2017 criteria for carcinoma at first presentation, subsequent recurrence with the properties of malignant disease rather than metachronous tumors, that is, infiltrative or destructive growth, vascular invasion, and distant metastasis, arose in 5 (31%) patients after intervals of up to 10 years. Several of the patients with histologically benign parathyroid adenoma developed recurrent disease that could be definitively attributed to metachronous tumors arising in a different parathyroid gland.
However, 1 patient, with a noninvasive primary tumor and therefore classified as adenoma under the WHO 2017 system, developed recurrent disease that seemed to represent unequivocal metastatic carcinoma in the absence of a second metachronous or synchronous tumor and is presented in detail. Patient 5 initially presented at the age of 15 with a serum calcium level of 3.2 mmol/L (normal range, 2.2 to 2.7 mmol/L) and a serum parathyroid hormone level of >100 pmol/L (normal range, 1.05 to 6.83 pmol/L). A right inferior parathyroid tumor weighing 4 g was removed. This tumor was fragmented upon receipt. The entire specimen was embedded in 7 blocks. It demonstrated typical features for a parafibromin-deficient tumor, including a solid growth pattern, eosinophilic cytoplasm, and some perinuclear cytoplasmic clearing. Although the fragmentation of the tumor made assessment of the interface with non-neoplastic tissue difficult (Fig. 6), there was no invasive growth evident. Of note, all slides from this initial tumor were reviewed by 3 pathologists (2 with subspecialty expertise in endocrine pathology) and reviewed again for this paper, and all reviewers agreed that there was no invasive growth and therefore the tumor was best classified as adenoma. The hypercalcemia resolved immediately after surgery, and, in fact, the patient developed hungry bone syndrome.
FIGURE 6.
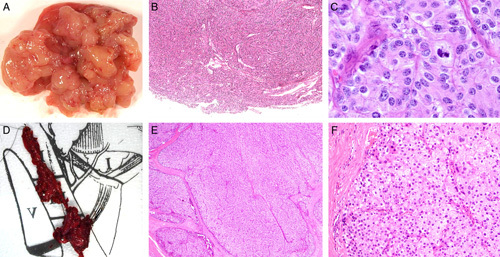
Patient 5’s case was initially classified as adenoma at first excision (A–C) but subsequently demonstrated unequivocal metastasis warranting classification as carcinoma (D–F). At initial resection, the tumor came out easily but was fragmented (A) making assessment of the interface with non-neoplastic tissue difficult. B, However, no unequivocal invasive growth was evident. C, At high power the primary tumor demonstrated cytologic features of parafibromin deficiency, including nuclear enlargement with relatively preserved N/C ratios and eosinophilic but not oxyphilic cytoplasm. D, At recurrence, the tumor was resected from the level 3/4 lymph nodes, effectively excluding seeding of benign disease from the previous operation. The recurrence demonstrated an unequivocal invasive growth pattern into soft tissue (E), but it still showed similar cytologic features to that seen in the original tumor (F). Hematoxylin and eosins, original magnifications. B) 100x, C) 400x, E) 100x , F) 200x.
Eight months later the patient developed recurrent hyperparathyroidism. She underwent repeat surgical exploration, and each of the left superior, left inferior, and right superior parathyroid glands were identified and confirmed on biopsy to be histologically normal and to demonstrate retained parafibromin expression. The central neck compartment and the site of the previously resected right inferior parathyroid were also cleared and lacked neoplastic tissue. The hyperparathyroidism did not resolve. Six months later, imaging revealed enlarged level 3/4 lymph nodes, and the patient underwent a modified right neck dissection during which 4 separate deposits of parafibromin-negative parathyroid carcinoma were identified at level 4—an area that was outside the previous operative fields. The hyperparathyroidism resolved but then recurred 14 months later when the patient underwent further resection of parathyroid carcinoma adherent to the recurrent laryngeal nerve and cricothyroid muscle in the central neck compartment. MLPA studies of FFPE tissue from the original tumor demonstrated deletion of exon 17. No germline mutation was identified on clinical sequencing of peripheral blood; however, germline tissue did not undergo MLPA studies, and therefore large-scale deletions could not be excluded. At last follow-up, 7 years after first presentation, she was disease free.
DISCUSSION
In this report we describe the previously unrecognized but distinctive morphology of parafibromin-deficient (HPT-JT type, CDC73 mutated/inactivated) parathyroid neoplasms and propose that they be considered a distinct subtype of parathyroid tumor. We accept that there were no firm department guidelines for which cases underwent parafibromin IHC and that therefore there may be an ascertainment bias toward a distinct morphology as we became more familiar with the stereotypical morphology of these cases. These tumors are characterized by a sheet-like growth of neoplastic cells. Although this sheet-like pattern is frequently interrupted by an arborizing vasculature, it contrasts with the acinar architecture that is found in the majority of parathyroid neoplasms. A particular feature of these tumors is their distinctive eosinophilic cytoplasm. Usually, at least in some areas, the eosinophilic cytoplasm characteristic of parafibromin-deficient tumors demonstrates a unique appearance that is different to and, with experience, may be distinguished from the very granular cytoplasm of the type of oxyphil cells that commonly occur elsewhere in parafibromin-deficient tumors, but are not uncommon in non-neoplastic parathyroid tissue and other parathyroid adenomas.29 Other features characteristic of parafibromin-negative parathyroid tumors include nuclear enlargement (sometimes with preserved N/C ratios and smudged or coarse chromatin) and distinctive perinuclear cytoplasmic clearing imparting an almost koilocytic-like appearance. Microcystic change or a thick hyalinized fibrous capsule are also commonly present, but these features are not uncommon in usual adenomas. To familiarize pathologists with these morphologic features, whole-slide scanned images from all tumors in this study are available for review at the Cancer Diagnosis and Pathology group website www.cancerdxpathology.org.au.
Clinical experience with parafibromin IHC is mixed.1,18–27 Our prospective experience of parafibromin IHC over the 12-year period of this study during which we performed IHC on 815 tumors is that, while it can be a technically difficult stain to perform and interpret, meaningful results can be achieved with care and time. The very high specificity of negative parafibromin IHC is supported by the confirmation of pathogenic germline or somatic mutations in 26 of 40 (65%) patients, with a high likelihood that there are further mutations that have been missed because of incomplete coverage, and the limited sensitivity of sequencing and MLPA on FFPE tissue. Evidence for the suboptimal sensitivity of sequencing using FFPE tissue includes the fact that we did not identify the known germline mutation in the somatic testing of 3 patients with confirmed HPT-JT syndrome despite the fact that we did not microdissect the tissue.
However, we fully accept the limitations of parafibromin IHC, and our experience is that a diagnostic result cannot always be obtained. In a significant number of cases, we had to repeat the stain with different concentrations and antigen retrieval conditions to achieve an interpretable result—that is, positive staining in non-neoplastic internal controls as well as negative staining in neoplastic cells. We commonly find that, in larger tumors, which were otherwise positive for parafibromin, toward the center of specimens, there is focal nonspecific loss of nuclear expression in neoplastic cells associated with decreased or absent expression in internal positive controls—a pattern of staining we still interpret as positive. All the cases were prospectively identified as parafibromin negative (and confirmed on repeat testing); therefore age in paraffin blocks does not appear to be a significant confounding factor, particularly if attention is payed to the need for internal positive controls.
We note that different criteria have been used for parafibromin interpretation by different groups. We1,21 and others18,20,24 require completely absent nuclear expression of parafibromin in all neoplastic cells in the presence of an internal positive control in non-neoplastic tissue—an approach highly analogous to that commonly used for DNA mismatch repair IHC interpretation in colorectal carcinoma.30 In contrast, some other groups consider focal loss of parafibromin expression as sufficient to indicate negative staining.19,22,23 This study confirms the high specificity of our approach to interpretation but was not intended or designed to assess sensitivity. Indeed, we have previously reported that, although truncation or large-scale deletions of CDC73 are consistently associated with completely negative parafibromin expression, some pathogenic point mutations may be associated with retained positive expression for parafibromin using our criteria.1,21 The reported incidence of large-scale germline deletions of CDC73 is up to 35%,31 similar to our finding of 8 of 23 (34.8%) in the somatic testing in our series. We emphasize that large-scale deletions are usually not detected by sequencing alone, and several of the patients thought to lack germline CDC73 mutations in this and previous studies did not have MLPA studies performed, and therefore may have had unrecognized deletions.
There is an extensive literature linking CDC73 mutations with parathyroid carcinoma.2,11–15,32 However, the role of CDC73 mutation as a marker of malignancy in atypical parathyroid tumors remains controversial. This may be because mutation testing is not readily available, and some groups have found parafibromin IHC unreliable.2,25–27,33 However, we also believe it is partially because the accuracy of parafibromin IHC is commonly compared with morphologic criteria as a gold standard, despite evidence that morphology is an imperfect predictor of biologically malignant behavior. It can be argued that parathyroid carcinoma is both “overdiagnosed” and “underdiagnosed.” That is, the majority of cases diagnosed as carcinoma on the basis of histologic criteria alone do not recur, and the diagnosis of carcinoma is frequently not made by histologic criteria until metastasis/recurrence has occurred.2,34–37 It is therefore worth noting that 16 of the 40 (40%) patients with tumors that fulfilled WHO criteria for malignancy demonstrated complete loss of parafibromin staining in their tumors and, most importantly, in 5 (31%) of these patients, the tumor went on to demonstrate unequivocal malignant behavior. In contrast, although parafibromin IHC was not performed in all tumors, loss of expression was found in only 2 of 1055 (<0.2%) of truly unselected parathyroid tumors.
When confronted with an atypical parathyroid tumor that does not fulfill WHO 2017 criteria for malignancy, we have taken the pragmatic approach that tumors lacking immunohistochemical evidence of CDC73 mutation/inactivation are very unlikely to recur and can be safely followed-up similarly to benign adenomas.33 Of course, this is useful in the great majority of cases primarily because CDC73 mutation and recurrence after resection of an atypical adenoma are both rare events. However, it leaves the very real difficulty of how to classify parafibromin-negative parathyroid tumors that do not fulfill WHO criteria for malignancy. We now believe that these are best classified simply as “parafibromin-deficient (HPT-JT type, CDC73 mutated) parathyroid tumors” and considered a distinct entity.
There are isolated case reports of CDC73 mutated but noninvasive and therefore “histologically benign” parathyroid tumors that have subsequently metastasized.38 Some of these events may be attributable to metastasis from other unrecognized synchronous or metachronous primary tumors.6 Our experience with patient 5 in this report, where every possible effort was made to exclude a second primary tumor, adds another case of a noninvasive parathyroid tumor associated with CDC73 mutation which subsequently behaved in a malignant manner. This supports our approach that parafibromin-deficient parathyroid tumors have some metastatic potential.2 However, it is clear that the metastatic potential is very low in the absence of invasive growth. When care was taken to exclude second primary tumors, none of the 26 other parafibromin deficient tumors that did not fulfill WHO criteria for carcinoma behaved aggressively at long-term follow-up. Therefore, although we believe that recurrence and metastasis may occur in parafibromin-deficient tumors lacking WHO 2017 criteria for malignancy, we emphasize that this is an unusual event. Indeed, the risk of metastasis in noninvasive parafibromin-deficient tumors is probably even lower than the 3.7% estimated in our study, given that it was subject to a selection bias toward identifying such patients (as recurrence/metastasis would be considered an indication for parafibromin IHC).
In our experience, much more common than metastasis from a noninvasive parafibromin-deficient tumor are cases where malignant features were only recognized on pathologic review of parafibromin-deficient tumors that subsequently behaved in a malignant manner.39 Because of the very low risk of metastasis from noninvasive tumors we do not recommend altering the WHO criteria for parathyroid carcinoma to include all parafibromin-deficient tumors. However, we would recommend that particular care should be taken in examining all parafibromin-deficient tumors, perhaps with a lower threshold for diagnosing invasive growth, and therefore parathyroid carcinoma, in equivocal cases.
Patients with confirmed HPT-JT syndrome have a very high risk of recurrence in the same or other glands, which is at least 25%, but which may occur in the majority of patients at follow-up extending to 30 years.6,8,9,38,40–43 Therefore, the diagnosis of parafibromin-deficient (HPT-JT type) parathyroid tumors should precipitate long-term follow-up for the possibility of late recurrence or metachronous disease, whether or not WHO criteria for malignancy are met. That is, simply identifying these unique tumors not as benign or atypical adenomas but as parafibromin-deficient (HPT-JT type, CDC73 mutated) parathyroid tumors should be sufficient to justify long-term follow-up and adequately convey the risk of recurrence, which is usually attributable to metachronous disease in patients with germline mutations and only very rarely to true metastasis from non-invasive tumors. Confirmation of germline CDC73 mutation in such patients also allows for cascade testing of family members, to identify carriers for whom screening for parathyroid disease is then appropriate.1,10,12,17–24,44–48
In conclusion, we report that parafibromin-deficient (HPT-JT type, CDC73 mutated) parathyroid tumors demonstrate distinctive morphologic features including sheet-like rather than acinar architecture, eosinophilic (but not very oxyphilic) cytoplasm, nuclear enlargement with distinctive chromatin and perinuclear cytoplasmic clearing, a prominent arborizing vasculature sometimes with a hemangiopericytomatous quality, and, frequently, a thick capsule. Other important clues to the diagnosis include a younger age of onset (mean, 36 vs. 63 y; P<0.001), a larger size (mean, 3.04 vs. 0.62 g; P<0.001) and more marked hypercalcemia (average 3.12 mmol/L in our series). Although parafibromin IHC can be technically difficult to perform and interpret, recognition of these tumors is important because of their strong association with malignancy (rare in all other circumstances) and HPT-JT syndrome. If a parafibromin-deficient (HPT-JT type) parathyroid tumor is identified, we recommend particularly careful assessment for malignancy by conventional morphologic criteria and genetic testing by both sequencing and MLPA for germline CDC73 mutation/deletion—present in at least 40% of patients in our series but probably more. For noninvasive parafibromin-deficient (HPT-JT type, CDC73 mutated) tumors, we still recommend long-term follow-up, primarily because of the risk of metachronous disease but also because of the risk of aggressive behavior in a small minority of cases.
Footnotes
Conflicts of Interest and Source of Funding: The authors have disclosed that they have no significant relationships with, or financial interest in, any commercial companies pertaining to this article.
REFERENCES
- 1.Gill AJ, Clarkson A, Gimm O, et al. Loss of nuclear expression of parafibromin distinguishes parathyroid carcinomas and hyperparathyroidsim-jaw tumor (HPT-JT) syndrome related adenomas from sporadic parathyroid adenomas and hyperplasias. Am J Surg Pathol. 2006;30:1140–1149. [DOI] [PubMed] [Google Scholar]
- 2.Gill AJ. Understanding the genetic basis of parathyroid carcinoma. Endocr Pathol. 2014;25:30–34. [DOI] [PubMed] [Google Scholar]
- 3.Hahn MA, Marsh DJ. Identification of a functional bipartite localization signal in the tumor suppressor parafibromin. Oncogene. 2005;15:6241–6248. [DOI] [PubMed] [Google Scholar]
- 4.Jo JH, Chung TM, Youn H, et al. Cytoplasmic parafibromin/hCdc73 targets and destabilizes p53 mRNA to control p53-mediated apoptosis. Nat Commun. 2014;12:5433. [DOI] [PubMed] [Google Scholar]
- 5.Xu Y, Bernecky C, Lee CT, et al. Architecture of the RNA polymerase II-Paf1C-TFIIS transcription elongation complex. Nat Commun. 2017;8:15741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lloyd RV, Arnold A, Gill A, et al. Lloyd RV, Osamura RY, Klöppel G, Rosai J. Hyperparathyroidism jaw tumour syndrome. WHO Classification of Tumours of Endocrine Organs, 4th ed Lyon: IARC Press; 2017:255–256. [Google Scholar]
- 7.Carpten JD, Robbins CM, Villablanca A, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet. 2002;32:676–680. [DOI] [PubMed] [Google Scholar]
- 8.van der Tuin K, Tops CMJ, Adank MA, et al. CDC73-related disorders: clinical manifestatons and case detection in primary hyperparathyroidism. J Clin Endocrinol Metab. 2017;102:4534–4454. [DOI] [PubMed] [Google Scholar]
- 9.Iacobone M, Carnaille B, Palazzo FF, et al. Hereditary hyperparathyroidism—a consensus report of the European Society of Endocrine Surgeons (ESES). Langenbecks Arch Surg. 2015;400:867–886. [DOI] [PubMed] [Google Scholar]
- 10.Wasserman JD, Tomlinson GE, Druker H, et al. Multiple endocrine neoplasia and hyperparathyroid-jaw tumor syndromes: clinical features, genetics, and surveillance recommendations in childhood. Clin Cancer Res. 2017;23:e123–e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu W, McPherson JR, Stevenson M, et al. Whole-exome sequencing studies of parathyroid carcinomas reveal novel PRUNE2 mutations, distinctive mutational spectra related to APOBEC-catalyzed DNA mutagenesis and mutational enrichment in kinases associated with cell migration and invasion. J Clin Endocrinol Metab. 2015;100:E360–E364. [DOI] [PubMed] [Google Scholar]
- 12.Howell VM, Haven CJ, Kahnoski K, et al. HRPT2mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet. 2003;40:657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shattuck T, Stiina V, Obara T, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med. 2003;349:1722–1729. [DOI] [PubMed] [Google Scholar]
- 14.Cetani F, Pardi E, Borsari S, et al. Genetic analyses of HRPT2 gene in primary hyperparathyroidism: germline and somatic mutations in familial and sporadic parathyroid tumours. J Clin Endocinol Metab. 2004;89:5583–5591. [DOI] [PubMed] [Google Scholar]
- 15.Krebs L, Shattuck TM, Arnold A. HRPT mutation analysis of typical sporadic parathyroid adenomas. J Clin Endocinol Metab. 2005;90:5015–5017. [DOI] [PubMed] [Google Scholar]
- 16.Weinstein LS, Simonds WF. Perspective: HRPT2, a marker of parathyroid cancer. N Engl J Med. 2003;349:1691–1692. [DOI] [PubMed] [Google Scholar]
- 17.Tan M-H, Morrison C, Wang P, et al. Loss of parafibromin immunoreactivity is a distinguishing feature of parathyroid carcinoma. Clin Cancer Res. 2004;10:6629–6637. [DOI] [PubMed] [Google Scholar]
- 18.Witteveen JE, Hambdy NA, Dekkers OM, et al. Downregulation of CASR expression and global loss of parafibromin staining are strong negative determinants of prognosis in parathyroid carcinoma. Mod Pathol. 2011;24:688–697. [DOI] [PubMed] [Google Scholar]
- 19.Juhlin CC, Villablanca A, Sandelin K, et al. Parafibromin immunoreactivity: its use as an additional diagnostic marker for parathyroid tumor classification. Endocr Relat Cancer. 2007;14:501–512. [DOI] [PubMed] [Google Scholar]
- 20.Fernandez-Ranvier GG, Khanafshar E, Tacha D, et al. Defining a molecular phenotype for benign and malignant parathyroid tumors. Cancer. 2009;115:334–344. [DOI] [PubMed] [Google Scholar]
- 21.Howell VM, Gill AJ, Clarkson A, et al. Accuracy of combined protein gene product 9.5 and parafibromin markers for immunohistochemical diagnosis of parathyroid carcinoma. J Clin Endocrinol Metab. 2009;94:434–441. [DOI] [PubMed] [Google Scholar]
- 22.Kim HK, Oh YL, Kim SH, et al. Parafibromin immunohistochemical staining to differentiate parathyroid carcinoma from parathyroid adenoma. Head Neck. 2012;34:201–206. [DOI] [PubMed] [Google Scholar]
- 23.Wang O, Wang C, Nie M, et al. Novel HRPT2/CDC73 gene mutations and loss of expression of parafibromin in Chinese patients with clinically sporadic parathyroid carcinomas. PLoS One. 2012;7:e45567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ozolins A, Narbuts Z, Vanags A, et al. Evaluation of malignant parathyroid tumours in two European cohorts of patients with sporadic primary hyperparathyroidism. Langenbecks Arch Surg. 2016;401:943–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mangray S, Delellis RA. Parafibromin in the diagnosis of parathyroid carcinoma. Adv Anatomic Pathol. 2007;14:299–301. [Google Scholar]
- 26.Quinn CE, Healy J, Lebastchi AH, et al. Modern experience with aggressive parathyroid tumors in a high-volume. New England referral center. J Am Coll Surg. 2015;220:1054–1062. [DOI] [PubMed] [Google Scholar]
- 27.Delellis RA. Challenging lesions in the differential diagnosis of endocrine tumors: parathyroid carcinoma. Endocr Pathol. 2008;19:221–225. [DOI] [PubMed] [Google Scholar]
- 28.De Lellis RA, Arnold A, Bilezikian JP, et al. Lloyd RV, Osamura RY, Klöppel G, Rosai J. Parathyroid carcinoma. WHO Classification of Tumours of Endocrine Organs, 4th ed Lyon: IARC Press; 2017:147–152. [Google Scholar]
- 29.Howson P, Kruijff S, Aniss A, et al. Oxyphil cell parathyroid adenomas causing primary hyperparathyroidism: a clinico-pathological correlation. Endocr Pathol. 2015;26:250–254. [DOI] [PubMed] [Google Scholar]
- 30.Hall G, Clarkson A, Shi A, et al. Immunohistochemistry for PMS2 and MSH6 alone can replace a four antibody panel for mismatch repair deficiency screening in colorectal adenocarcinoma. Pathology. 2010;42:409–413. [DOI] [PubMed] [Google Scholar]
- 31.Bricaire L, Odou MF, Cardot-Bauters C, et al. Frequent large germline HRPT2 deletions in a French National cohort of patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 2013;98:E403–E408. [DOI] [PubMed] [Google Scholar]
- 32.Guarnieri V, Battista C, Muscarella LA, et al. CDC73 mutations and parafibromin immunohistochemistry in parathyroid tumors: clinical correlations in a single-centre patient cohort. Cell Oncol (Dordr). 2012;35:411–422. [DOI] [PubMed] [Google Scholar]
- 33.Krujiff S, Sidhu SB, Sywak MS, et al. Negative parafibromin staining predicts malignant behaviour in atypical parathyroid adenomas. Ann Surg Oncol. 2014;21:426–433. [DOI] [PubMed] [Google Scholar]
- 34.Fernandez-Ranvier GG, Khanafshar E, Jensen K, et al. Parathyroid carcinoma, atypical parathyroid adenoma, or parathyromatosis? Cancer. 2007;110:255–264. [DOI] [PubMed] [Google Scholar]
- 35.Sandelin K, Tullgreen O, Farnebo LO. Clinical course of metastatic parathyroid carcinoma. World J Surg. 1994;18:594–598. [DOI] [PubMed] [Google Scholar]
- 36.Ippolito G, Palazzo FF, Sebag F, et al. Intraoperative diagnosis and treatment of parathyroid cancer and atypical parathyroid adenoma. Br J Surg. 2007;94:566–670. [DOI] [PubMed] [Google Scholar]
- 37.Ryhanen EM, Leiojon H, Mesto S, et al. A nationwide study on parathyroid carcinoma. Acta Oncol. 2017;56:991–1003. [DOI] [PubMed] [Google Scholar]
- 38.Sarquis MS, Silveira LG, Pimenta FJ, et al. Familial hyperparathyroidism: surgical outcome after 30 years of follow-up in three families with germline HRPT2 mutations. Surgery. 2008;143:630–640. [DOI] [PubMed] [Google Scholar]
- 39.Lim S, Elston MS, Gill AJ, et al. Metastatic parathyroid carcinoma initially misdiagnosed as parathyroid adenoma—the role of parafibromin in increasing diagnostic accuracy. Intern Med J. 2011;41:695–699. [DOI] [PubMed] [Google Scholar]
- 40.Marsh DJ, Hahn MA, Howell VM, et al. Molecular diagnosis of primary hyperparathyroidism in familial cancer syndromes. Expert Opin Med Diagn. 2007;1:377–392. [DOI] [PubMed] [Google Scholar]
- 41.Iacobone M, Masi G, Barzon L, et al. Hyperparathyroidism-jaw tumor syndrome: a report of three large kindred. Langenbecks Arch Surg. 2009;394:817–825. [DOI] [PubMed] [Google Scholar]
- 42.Mehta A, Patel D, Rosenberg A, et al. Hyperparathyroidism-jaw tumor syndrome: results of operative management. Surgery. 2014;156:1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silveira LG, Dias EP, Marinho BC, et al. HRPT2-related familial isolated hyperparathyroidism: could molecular studies direct the surgical approach? Arq Bras Endocrinol Metabol. 2008;52:1211–1220. [DOI] [PubMed] [Google Scholar]
- 44.Juhlin CC, Nilsson IL, Johansson K, et al. Parafibromin and APC as screening markers for malignant potential in atypical parathyroid adenomas. Endocr Pathol. 2010;21:166–177. [DOI] [PubMed] [Google Scholar]
- 45.Erovic BM, Harris L, Jamali M, et al. Biomarkers of parathyroid carcinoma. Endocr Pathol. 2012;23:221–231. [DOI] [PubMed] [Google Scholar]
- 46.Hu Y, Liao Q, Cao S, et al. Diagnostic performance of parafibromin immunohistochemical staining for sporadic parathyroid carcinoma: a meta-analysis. Endocrine. 2016;54:612–619. [DOI] [PubMed] [Google Scholar]
- 47.Kumari N, Chaudhary N, Pradhan R, et al. Role of histological criteria and immunohistochemical markers in predicting risk of malignancy in parathyroid neoplasms. Endocr Pathol. 2016;27:87–96. [DOI] [PubMed] [Google Scholar]
- 48.Wilhelm SM, Wang TS, Ruan DT, et al. The American Association of Endocrine Surgeons Guidelines for definitive management of primary hyperparathyroidism. JAMA Surg. 2016;151:959–968. [DOI] [PubMed] [Google Scholar]