

Abstract
CRISPR/Cas9 technology has transformed mouse genome editing with unprecedented precision, efficiency, and ease; however, the current practice of microinjecting CRISPR reagents into pronuclear-stage embryos remains rate-limiting. We thus developed CRISRP-EZ (CRISPR RNP Electroporation of Zygotes), an electroporation-based technology that outperforms pronuclear and cytoplasmic microinjection in efficiency, simplicity, cost, and throughput. In C57BL/6J and C57BL/6N mouse strains, CRISPR-EZ achieves 100% delivery of Cas9/sgRNA ribonucleoproteins (RNPs), facilitating indel mutations (insertions or deletions), exon deletions, point mutations, and small insertions. In a side-by-side comparison in the high-throughput KnockOut Mouse Project (KOMP) pipeline, CRISPR-EZ consistently outperformed microinjection. Here, we provide an optimized protocol covering single guide RNA (sgRNA) synthesis, embryo collection, RNP electroporation, mouse generation, and genotyping strategies. Using CRISPR-EZ, a graduate-level researcher with basic embryo manipulation skills can obtain genetically modified mice in 6 weeks. Altogether, CRISPR-EZ is a simple, economic, efficient, and high-throughput technology that is potentially applicable to other mammalian species.
Keywords: Mouse genome editing, CRISPR-Cas9, CRISPR‐EZ, CRISPR, Cas9, sgRNA, ribonucleoprotein, RNP, zygote, embryo, electroporation, zygote electroporation
Introduction
The advent of CRISPR/Cas9 technology has greatly simplified and expedited in vivo genome editing in mice1–3. Standard practice involves pronuclear and/or cytoplasmic microinjection of Cas9 mRNA/sgRNA into pronuclear-stage embryos4–8, but microinjection remains a rate-limiting step due to its slow, costly, and technically demanding nature. This procedure is often performed by experienced personnel at dedicated transgenic facilities whose technical expertise commands a premium rate. Furthermore, Cas9 mRNA/sgRNA microinjection is prone to suboptimal editing efficiency and undesired mosaicism9–11, as the Cas9 protein must first be translated, folded, and complexed with sgRNAs prior to editing12–15
To expedite genome editing in vivo, we developed CRISPR-EZ (CRISPR RNP Electroporation of Zygotes), an electroporation-based mouse genome engineering technology to overcome these limitations16. By delivering preassembled Cas9/sgRNA ribonucleoproteins (RNPs) into pronuclear-stage embryos, editing occurs rapidly and transiently, maximizing efficiency while minimizing mosaicism9–11. CRISPR-EZ completely bypasses microinjection by utilizing a series of electrical pulses to deliver RNPs with 100% efficiency. In both mouse strains tested, C57BL/6J and C57BL/6N, CRISPR-EZ vastly surpassed Cas9 mRNA/sgRNA microinjection in editing efficiency, at approximately one quarter of the cost. CRISPR-EZ has now been successfully utilized by seven independent labs (personal communications), generating mice with editing schemes ranging from simple indels and exon deletions via the Non-Homologous End Joining (NHEJ) pathway, to precise point mutations and small insertions via the Homology-Directed repair (HDR) pathway (Fig. 1a). Other electroporation-based protocols have been developed by independent groups, with efficiencies exceeding that of microinjection9,17,18. These methods employ costly or proprietary equipment setups and were only tested on a limited number of editing experiments. In contrast, CRISPR-EZ uses commonly available reagents and equipment, requires only basic embryo handling skills, and has been comprehensively tested across a wide range of targets and editing schemes, making our protocol readily amenable to many investigators. Altogether, CRISPR-EZ is a powerful tool that stands to replace microinjection as the standard mouse genome editing method for most common editing schemes.
Figure 1. Overview of CRISPR-EZ technology and workflow.
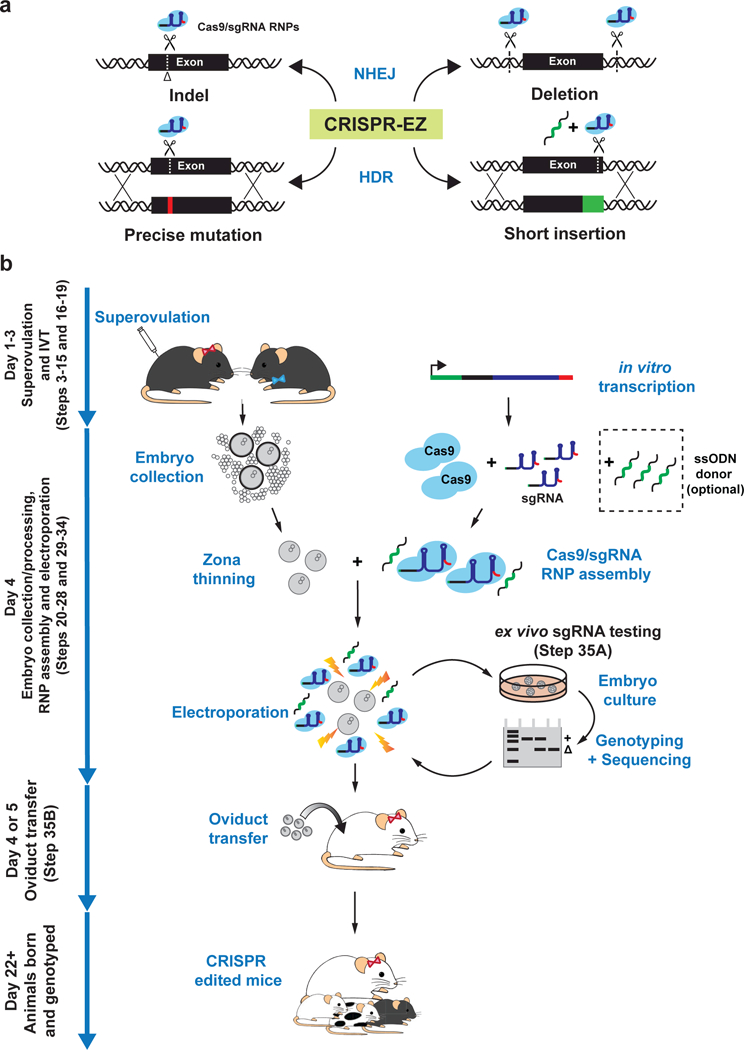
(a) An illustration of the most successful CRISPR-EZ editing strategies. A single sgRNA can be used to create a small indel via NHEJ repair or in conjunction with a ssODNs to create a precision mutation or a small insertion by HDR. Multiple sgRNAs can be used to engineer a genomic deletion by NHEJ repair. Design sgRNAs, HDR donor oligos, and editing validation assays prior to CRISPR-EZ experiments. (b) A graphic overview of CRISPR-EZ workflow. Day 1–3: ~4 week-old females are superovulated, first by PMSG injection, 46–48 hours later by hCG injection, before being housed with stud males for breeding. In parallel, sgRNAs are in vitro transcribed and purified. Day 4: Pronuclear stage embryos are collected and processed for electroporation, while Cas9/sgRNA complexes are assembled in vitro. Embryos (harvested at 0.5 dpc), Cas9/sgRNA RNPs, and optional ssODNs are combined in an electroporation cuvette and subjected to a series of electrical pulses. We recommend ex vivo validation of sgRNA editing efficiency in cultured morulae or blastocysts before generating edited mice. With a validated sgRNA design, electroporated embryos can be transferred to the oviduct of 0.5 dpc, pseudopregnant mothers to generate genetically engineered mice, which are then genotyped to confirm editing efficiency.
Protocol overview
Using CRISPR-EZ, one can quickly test sgRNAs directly in cultured mouse embryos in 1–2 weeks, and subsequently generate edited mice in one month (Fig. 1b). This protocol can be broken into 7 major stages, including sgRNA design (Steps 1–2), sgRNA synthesis (Steps 3–15), superovulation and mating (Steps 16–19), embryo culture, collection and processing (Steps 20–28), RNP assembly and electroporation (Steps 29–34), embryo culture and genotyping (Step 35A), and oviduct transfer (Step 35B). We have also included a video demonstrating all key procedures (Supplementary Video 1).
Development of protocol
Development of CRISPR-EZ was inspired by the reported success of delivering Cas9 mRNA/sgRNA into mouse and rat zygotes by electroporation19–22. However, the highly anionic properties of Cas9 mRNA may explain inefficient pronuclear delivery23. In contrast, CRISPR-EZ achieves 100% delivery by electroporating the relatively less anionic Cas9/sgRNA RNPs23,24. More importantly, preassembled Cas9/sgRNA RNPs initiate editing in embryos more rapidly than Cas9 mRNA, and their transient presence could reduce mosaicism and potential off-target effects9–11.
We developed CRISPR-EZ as a simple, high-throughput methodology that lowers the technical and financial barriers for mouse genome engineering. In a proof-of-principle editing experiment targeting the key pigment synthesis gene Tyrosinase (Tyr) (Fig. 2a, b, Supplementary Fig. 1a, b), we previously achieved 88% bi-allelic gene disruption and 42% HDR-mediated editing in C57BL/6J mice using a highly efficient sgRNA24. We also demonstrated that CRISPR-EZ could efficiently mediate a variety of editing schemes, including indels, precise mutations, exon deletions, and small insertions24 (Fig. 1a). We have since further optimized CRISPR-EZ electroporation conditions to achieve better editing efficiency without impacting embryo viability (Fig. 2c-i, Supplementary Table 1), and expanded its application to the widely used C57BL/6N mouse strain (Fig. 2d, h, Supplementary Table 1). Under these optimized conditions (30V, 6 pulses, 3 ms), we achieved 100% bi-allelic Tyr editing in both mouse strains, as shown by the albino coat in all mice generated (Fig. 2c, d, g, h), as well as 62.5% HDR efficiency in the C57BL/6J strain (Supplementary Table 1).
Figure 2. Optimization of CRISPR-EZ conditions for editing efficiency and embryo viability.
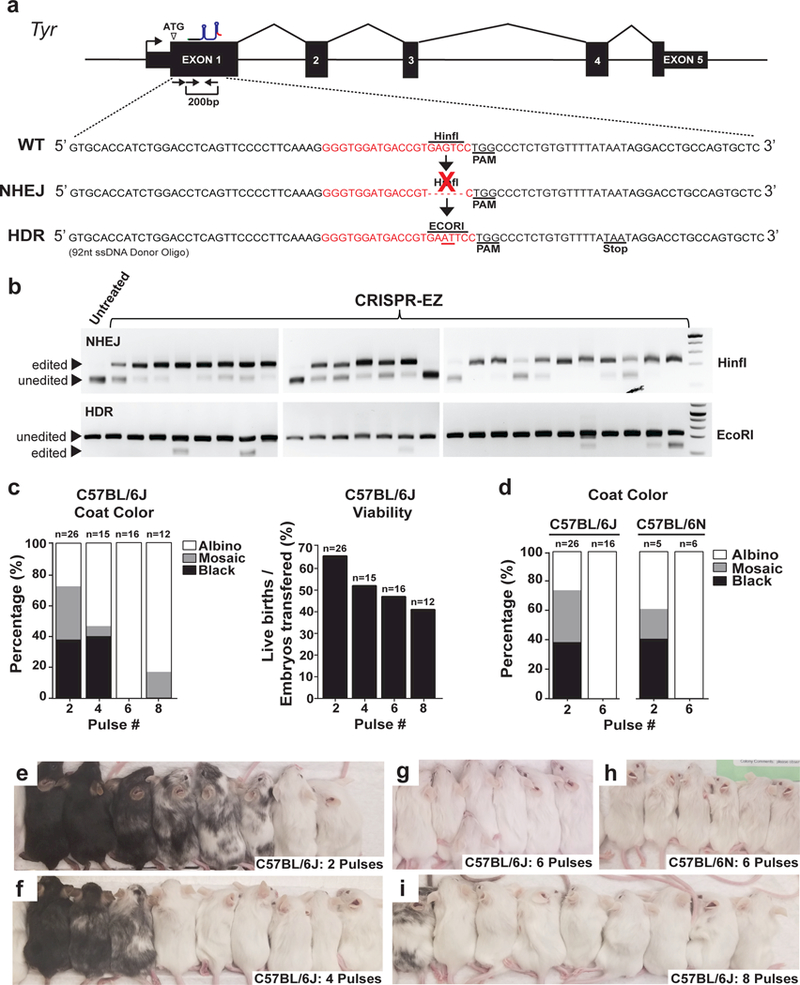
(a) A diagram illustrates the NHEJ and HDR editing strategies for exon 1 of the Tyr gene. A successful NHEJ editing ablates a HinfI site and disrupts Tyr gene function. A successful HDR editing replaces the HinfI site with an EcoRI site, introducing a frameshift mutation that abolishes Tyr gene function. (b) Representative RFLP results of Tyr edited mice indicate successful NHEJ editing (top) and HDR editing (bottom). (c) Since bi-allelic Tyr deficiency causes albinism in edited mice, the extent of albinism correlates the extent of Tyr editing that disrupts the genes function. Coat color (left) and viability (right) of C57B/6J edited mice generated from 2, 4, 6 or 8 pulse CRISPR-EZ conditions. Viability is defined as the percentage of live animals born out of total embryos transferred. The 6-pulse condition maximizes editing efficiency while minimally impacting pup viability. (d) Comparison of editing efficiency between C57B/6J and C57B/6N mouse strain using 2 or 6-pulse electroporation conditions. The 6-pulse CRISPR-EZ condition is equally effective in both strains. (e-i) Representative images are shown for the coat color of edited mice from experiments shown in (b-d). All animal procedures were approved by the Institutional Animal Care and Use Committee of UC Davis.
We have performed side-by-side comparisons of CRISPR-EZ versus microinjection targeting the Tyr gene in the C57BL/6J strain24, as well as the Sh3rf2 gene in the C57BL/6N strain (Fig. 3a, b, c, d). In both cases, CRISPR-EZ achieved markedly greater editing efficiency24 (Fig. 3d). We then comprehensively compared 21 CRISPR-EZ and 27 microinjection-based knockout experiments integrated within the KnockOut Mouse Project (KOMP) pipeline, as part of their ongoing effort to knockout all protein-coding genes in mice. Remarkably, CRISPR-EZ exhibited on average ~3-fold increased editing efficiency over microinjection in C57BL/6N mice, while retaining comparable live birth rates (Fig. 3e, g, Supplementary Fig 2,3, Supplementary Table 2 and 3). Importantly, CRISPR-EZ efficiently generated edited founders for multiple genes that were refractory to editing by microinjection (Fig. 3f, Supplementary Fig. 3, Supplementary Tables 2 and 3). Based on these findings, the Mouse Biology Program at UC-Davis, a major operation center for KOMP, has adopted CRISPR-EZ as their primary method for knockout mouse generation. Altogether, CRISPR-EZ constitutes an economic, high-throughput, and highly efficient mouse genome engineering technology that stands to replace microinjection for a variety of editing strategies.
Figure 3. Comparing editing efficiency and viability of CRISPR-EZ and microinjection.
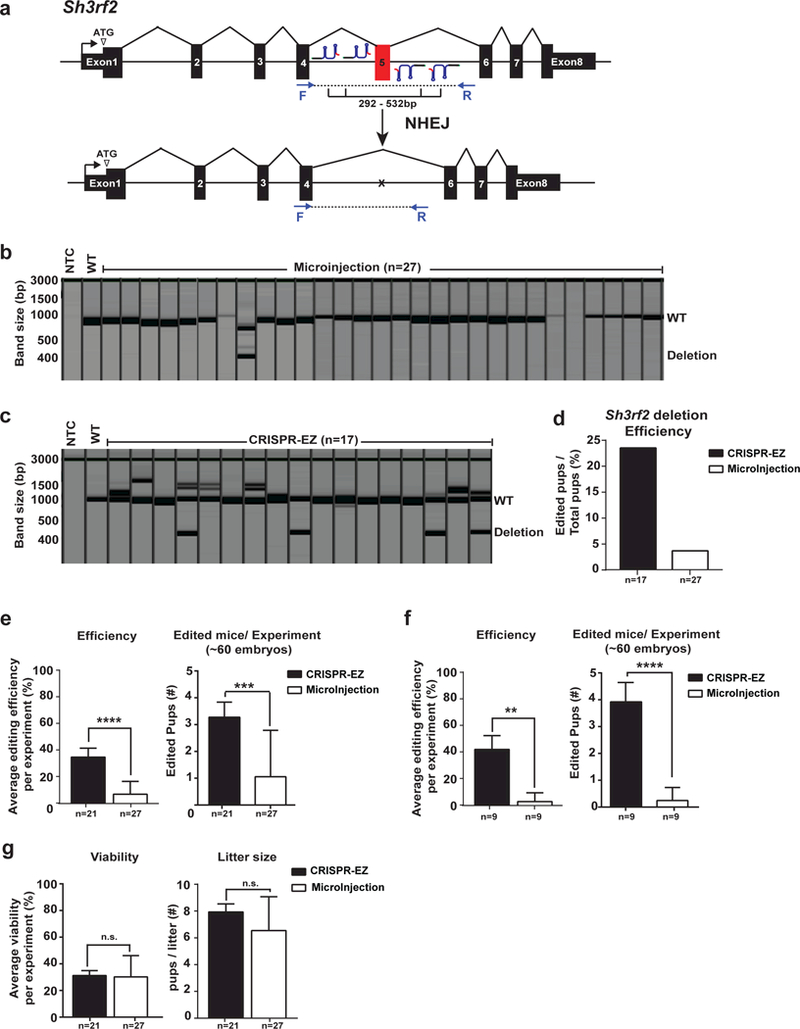
(a) A schematic diagram illustrates the strategy to employ two pairs of sgRNAs to mediate the deletion of Sh3rf2 exon 5. (b,c) QIAxcel fragment analysis images show PCR genotyping results of all mice generated through microinjection (b) and CRISPR-EZ (c) in C57BL/6N mice, demonstrating a greater editing efficiency in CRISPR-EZ experiments. (NTC= non-template control, WT=wildtype) (d) Deletion efficiency of Sh3rf2 exon 5 is shown for a side-by-side comparison between CRISPR-EZ and microinjection. (e) In a high throughput pipeline, up to two pairs of sgRNAs (4 sgRNAs) were designed to mediate exon deletion in C57BL/6N mice. Average editing efficiency is calculated across 21 CRISPR-EZ experiments or 27 microinjection experiments. CRISPR-EZ outperformed microinjection in editing efficiency (left), generating an average of 3 founder animals when starting with ~60 embryos for electroporation (right). (f) In a direct comparison, gene knockout experiments were carried out in C57BL/6N mice using either CRISPR-EZ (n=9) or microinjection (n=9), using identical sgRNA designs. CRISPR-EZ outperformed microinjection in editing efficiency (left), generating an average of 4 founder animals when starting with ~60 embryos for electroporation. (g) No obvious differences are observed for animal viability (left) or litter size (right) between CRISPR-EZ and microinjection. Data are means ± SD across all genes. ** P < 0.01; *** P < 0.001; **** P < 0.0001; n.s., not significant (All P-values were calculated on a basis of an unpaired one-tailed Student’s t test). All animal procedures were approved by the Institutional Animal Care and Use Committee of UC Davis.
Application of Protocol
CRISPR-EZ is broadly applicable to a wide range of mouse genome editing schemes for biomedical research. So far, CRISPR-EZ has been successfully employed in 34 editing projects across seven independent labs, generating edited mice with genomic deletions (n=28), indel mutations (n=3), and point mutations (n=3). Thus, CRISPR-EZ can replace microinjection for common editing schemes and multiple mouse strains.
CRISPR-EZ also provides a cost-effective means to test sgRNA designs in vivo. Despite recent advancements in sgRNA prediction algorithms, empirical testing remains necessary before in vivo experiments are attempted. Due to the high cost of microinjection, sgRNAs are frequently screened using immortalized or transformed mouse cell lines, whose editing efficiencies may differ from that of the embryo25. Using CRISPR-EZ, investigators can rapidly screen sgRNAs by directly genotyping electroporated embryos after culturing to the morula stage, thereby increasing confidence in obtaining the desired edited mice.
CRISPR-EZ may be a powerful tool for generating compound mouse genetic models to study complex genetic interactions or highly redundant gene families. Traditionally, mice harboring multiple edited alleles are generated through lengthy breeding of individually edited mice. With the advent of Cas9/sgRNA microinjection, it became possible to simultaneously edit multiple loci26, and CRISPR-EZ makes this even more accessible and efficient. Such approaches could be particularly useful when engineering multiple genetically linked loci, which are virtually impossible to obtain through breeding.
Given the similarities in preimplantation development among some mammalian species, CRISPR-EZ could potentially edit a much wider range of model organisms than the tested mouse strains. Notably, highly efficient porcine genome editing was recently demonstrated using a similar strategy27. The amenability of a specific mammalian zygote to CRISPR-EZ depends on its innate sensitivity to experimental manipulation, as well as the physical and biochemical properties of its zona pellucida and cellular membrane. Optimization will be necessary for each additional mouse strain or mammalian species. Given its high efficiency, CRISPR-EZ may be particularly suited for editing organisms whose embryos are scarce or difficult to obtain.
Comparison with other methods
The current de facto standard for mouse genome editing is microinjection of CRISPR reagents into pronuclear-stage embryos. Edited mice are routinely generated by microinjection of Cas9 mRNA/sgRNAs, and more recently, of Cas9/sgRNA RNPs28–30. It has been reported that Cas9/sgRNA RNPs are superior to Cas9 mRNA/sgRNAs owing to their immediate editing activity upon delivery12,14,15,31,32. In spite of its popularity, microinjection possesses several caveats, including high cost, low throughput, and technical difficulty, all of which are addressed by CRISPR-EZ. In our studies, CRISPR-EZ outperforms microinjection of Cas9 mRNA/sgRNAs for mouse genome engineering. Similar studies have demonstrated better mouse editing efficiency by CRISPR-EZ when compared with that of Cas9 RNP microinjection33.
While CRISPR-EZ efficiently performs a variety of simple editing schemes, its potential for more complex genome engineering, such as the insertion of fluorescent tags or conditional alleles, remains unclear, as we have only demonstrated electroporation of ssODN donors up to 162 nt24. Complex HDR-mediated editing schemes may require alternative approaches such as genetic engineering of embryonic stem cells (ESCs), microinjection of large double-stranded donors, or more recently, microinjection of synthesized long ssODNs34–37. As engineered ESCs can be clonally expanded and validated prior to mouse generation, this classic approach is still the preferred method for most sophisticated editing schemes.
In recent years, multiple electroporation-based methods have been utilized to deliver Cas9 mRNA into mouse zygotes under both ex vivo and in utero conditions19–21. However, the reported efficiencies for these approaches were generally suboptimal (~10% for indels, for example21) compared to CRISPR-EZ, possibly due to inefficient Cas9 mRNA delivery and/or delayed Cas9 expression in the embryo38,39. Several other groups also reported success in mouse genome engineering using zygote electroporation of Cas9 RNPs9,18,40. However, their specific experimental conditions were only tested on a limited number of editing experiments (no more than 1–2 targets per editing scheme were reported), and the variability across different targets, editing schemes and mouse strains, as well as high-throughput scalability were not clearly addressed9,18,40. Using CRISPR-EZ, we clearly demonstrated a 100% Cas9 RNP delivery in the zygotes of two different mouse inbred strains. We also demonstrated the effectiveness, robustness and reliability of CRISPR-EZ in a high-throughput pipeline across 21 different knockout targets (Fig. 3e, g, Supplementary Figs. 2 and 3, Supplementary Tables 2 and 3), showing an average of 35% editing success rate among the liveborn pups. For NHEJ-mediated deletion schemes, CRISPR-EZ typically uses 60 embryos to generate ~3 edited founders in most experiments (Fig. 3e). This editing efficiency and robustness significantly exceeds that of microinjection-based technology in the same editing schemes (Figs. 2 and 3); other Cas9 RNP electroporation protocols have not reported this level of robustness in a high throughput knockout pipeline. Finally, CRISPR-EZ is significantly more cost-effective compared to other Cas9 mRNA or Cas9 RNP electroporation methodologies, owing to its high editing efficiency, its technical simplicity, and its use of inexpensive, off-the-shelf equipment and consumables. Thus, CRISPR-EZ offers significant advantages over other microinjection-based and electroporation-based technologies, and can be employed as a standard technology for mouse editing experiments to generate indels, deletions, point mutations and small insertions.
Level of expertise required
CRISPR-EZ requires basic mouse embryo manipulation skills and molecular biology training. To successfully perform CRISPR-EZ in cultured mouse embryos, researchers will perform PCR, in vitro transcription, RNA purification, and RNP assembly to generate functional Cas9/sgRNA complexes, and then perform surgeries to collect pronuclear-stage embryos from superovulated females for RNP electroporation. To test editing strategies, the researchers will culture pronuclear-stage embryos to morula for genotyping analysis. A graduate student could master these skills after 2–3 months of training. To generate edited mice, researchers will transfer electroporated embryos into the oviducts of pseudopregnant mothers. This technically demanding procedure requires survival surgery and fine embryo manipulation skills and can also be learned by a graduate level researcher but will require considerably more hands-on experience than routine embryo manipulation. Researchers can utilize the service of a transgenic facility or acquire training to perform oviduct transfer independently. We refer researchers to “Manipulating the Mouse Genome” by Dr. András Nagy, if more experimental details are desired for this procedure41.
Limitations
CRISPR-EZ is well suited for generating indels and exon deletions through the NHEJ pathway (Figs. 2 and 3, Supplementary Figs. 1–3). We demonstrated that CRISPR-EZ facilitates deletions up to ~2.6 kb in length (Supplementary Fig. 2a, Supplementary Table 3), which greatly exceeds the average length of a protein-coding exon (~170 bp)42. However, deletion efficiency is inversely correlated with deletion size (Supplementary Fig. 1c), such that engineering very large deletions could present a challenge, albeit this caveat is not unique to CRISPR-EZ.
Cas9/sgRNA RNPs can be co-electroporated with ssODNs to mediate precise sequence replacement via HDR. Using CRISPR-EZ, we successfully inserted a V5 tag (42 bp) into the endogenous sox2 locus by co-electroporating a 162 nt ssODN24. Given the difficulty of synthesizing longer ssODNs, we have not yet investigated the maximal oligo length for delivery by CRISPR-EZ. A recent study has reported complex HDR-mediated editing in mice upon microinjection of Cas9/sgRNA RNPs along with long ssODN donors (~1000 nt)37. Hence, utilizing long ssODNs in CRISPR-EZ, if possible, would provide a promising strategy for complex genome editing. Currently, ESC genome editing and microinjection remain the preferred methodologies for complex mouse genome engineering.
While CRISPR/Cas9 has helped define an era of genome editing, concerns persist regarding the extent of Cas9 off-target effects43–45. We have not yet performed whole genome sequencing of edited mice generated by CRISPR-EZ or microinjection, and it is unclear if they differ in the extent of off-target effects. Engineered Cas9 variants may improve target specificity46,47, which have yet to be tested in the CRISPR-EZ system. Ultimately, one should exercise caution when interpreting the phenotype of CRISPR edited mice. To unambiguously demonstrate a causal relationship between genome modifications and the observed mouse phenotype, and to mitigate the concern of off-target artifacts, researchers may generate multiple edited mouse lines using independent sgRNA designs, and perform sufficient backcrossing to generate congenic mouse strains. When engineering mutant mice using classic ESC approaches, mutant phenotype is often validated using multiple ESC lines. Similar criteria are applied to transgenic mouse lines, where multiple founders are analyzed to definitively conclude the phenotype caused by the transgene. CRISPR editing in mice, either generated by CRISPR-EZ or by microinjection, may need to be subjected to the same level of scrutiny.
Experimental Design
Animal use authorization.
Live animals are used throughout this protocol. As a matter of caution and compliance, all appropriate authorizations must be acquired from institutional and/or federal regulatory bodies prior to performing this protocol. All mouse use, including but not limited to housing, breeding, production, sample collection for genotyping, and euthanasia, is in accordance with the Animal Welfare Act, the AVMA Guidelines on Euthanasia and are in compliance with the ILAR Guide for Care and Use of Laboratory Animals, and the UC Davis institutional animal care and use committee (IACUC) guidelines and policies. Our animal care and use protocol has been reviewed and approved by our IACUC for this project.
sgRNA design.
Designing optimal sgRNAs is crucial for successful editing, as efficiency varies depending on Cas9 sequence preference, target specificity, genome topology, and cell type48–50. Several publicly-available design programs can be used to determine initial sgRNA candidates. Some examples include but are not limited to: Sequence scan for CRISPR (http://crispr.dfci.harvard.edu/SSC/), Gene Perturbation Platform (http://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design), Chop-Chop (https://chopchop.rc.fas.harvard.edu/), CRISPR Design (http://crispr.mit.edu/). We recommend screening 2–3 sgRNA designs per target site directly in mouse embryos to ensure faithful in vivo editing (Fig. 1b). For exon deletions, we screen 2–3 sgRNAs for each cutting site and identify the paired sgRNA combination(s) with the best efficiency. Although we have had some success testing sgRNAs in vitro using Cas9-overexpressing mouse cancer cell lines, we have encountered cases where cell line results differ from those obtained from embryos (data not shown).
sgRNA synthesis.
We recommend a rapid and cost-effective cloning-free method to generate large quantities of high quality in vitro transcribed sgRNAs51 (Fig. 4). In this strategy, a unique oligo containing a T7 promoter and the guide sequence is annealed to a universal oligo containing the tracrRNA sequence, forming a PCR template that is amplified using two additional primers. The product of this reaction serves as the template for T7 in vitro transcription, which typically yields >100 µg of sgRNA.
Figure 4. Diagram illustrating a cloning-free strategy for sgRNA synthesis.
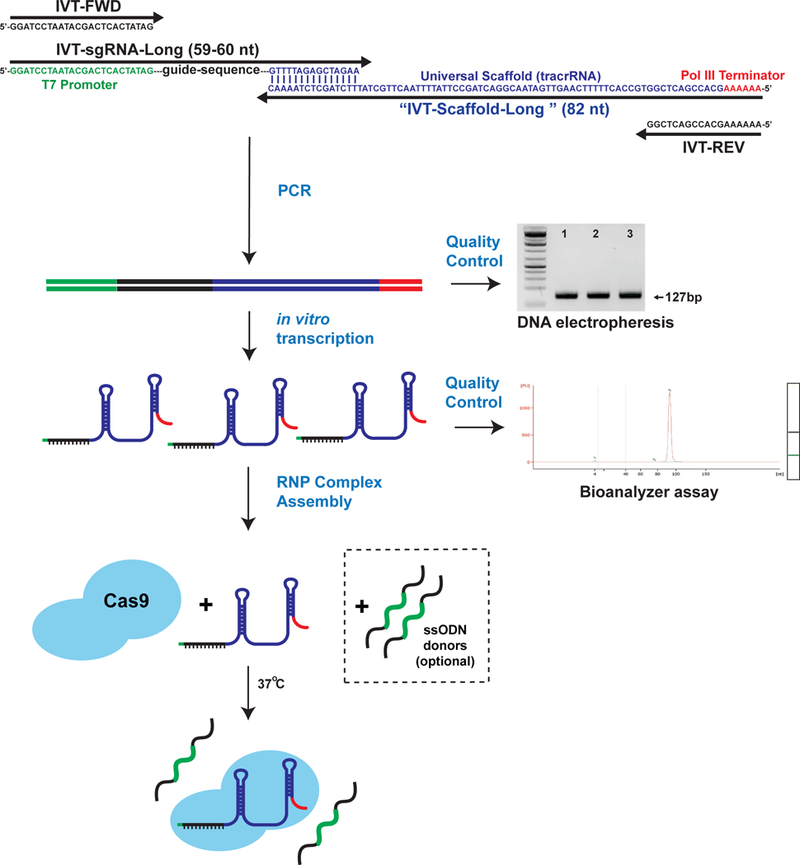
The sequences and purpose of each synthesized oligo are diagrammed to show how they function in this cloning free strategy. The components include a pair of PCR primers (black: IVT-FWD and IVT-REV), a common reverse template oligo (blue/red: IVT-Scaffold-Long), and an oligo containing a 5’ T7 promoter and a unique sgRNA sequence (green/black/blue: IVT-VAR-sgRNA). The DNA template for sgRNA synthesis is generated by a PCR reaction. The product of this reaction is a single 127 bp amplicon which should be confirmed by gel electrophoresis prior to continuing. Shown on the right is a representative DNA electrophoresis image of the PCR reaction. Subsequently, sgRNAs are synthesized by T7 in vitro transcription (IVT) and purified. Before moving forward, the quality and quantity of the newly synthesized sgRNA can be determined by submitting the sample for BioAnalyzer testing. A representative bioanalyzer trace of the IVT products is shown to the right. Recombinant Cas9 protein, purified sgRNA(s), and ssODN (optional) are assembled into RNPs in vitro by combining the components with a stabilizing buffer at 37°C for 10 minutes. The active RNP Complex is now ready for electroporation.
Superovulation and mating.
Mouse genome engineering requires large numbers of pronuclear-stage embryos. Consecutive injection of pregnant mare serum gonadotropin (PMSG) and human chorionic gonadotropin (hCG) is a well-established method to induce superovulation in rodents and generate large numbers of embryos (Fig. 5a-h). Both PMSG and hCG are functional analogs of gonadotropins normally produced in the anterior pituitary glands for natural sexual development and reproductive function52. Superovulated females are immediately housed with stud males to produce pronuclear-stage embryos, with a typical yield of 10–20 embryos per mouse. The number of females required for each CRISPR experiment depends on the specific experimental design. For NHEJ-mediated editing schemes, we usually electroporate ~30 embryos to test sgRNA efficiency and ~60 embryos to generate edited mice, but more complex editing schemes may necessitate additional embryos.
Figure 5. Overview of key superovulation and zygote collection procedures.
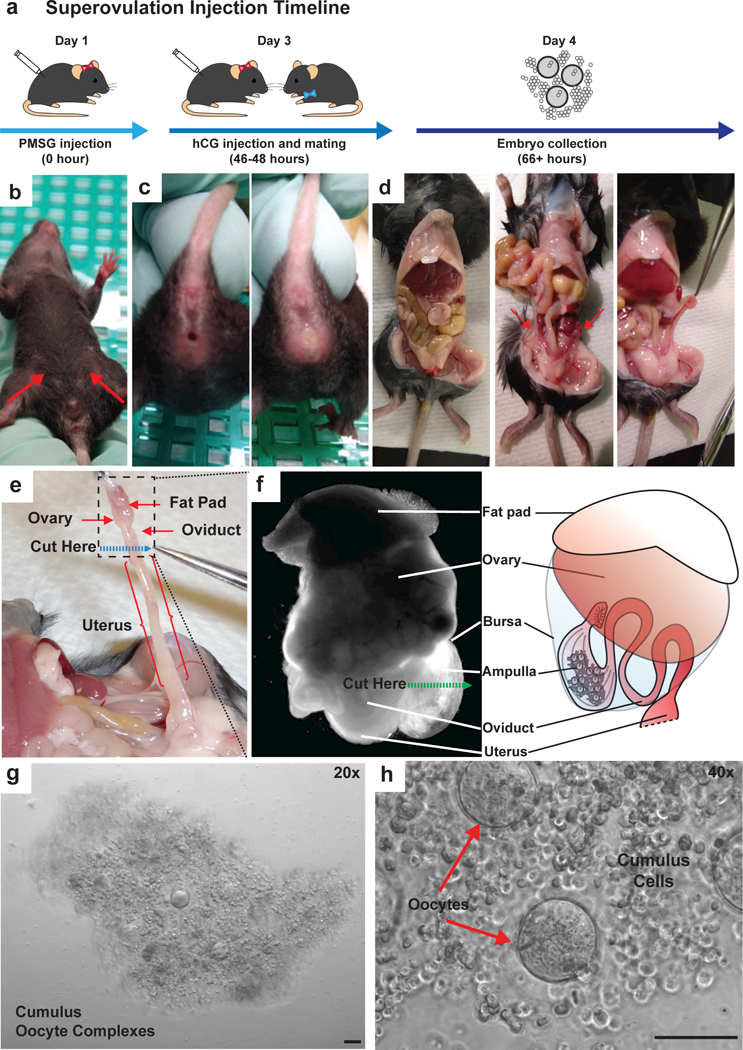
(a) A diagram illustrating the hormone injection, mating and embryo collection timeline for superovulation. (b) IP injection sites of PMSG (day 1) and hCG (day 3) are indicated in the abdominal region of a female mouse (red arrows). C. After hCG injection at day 3, superovulated females are paired 1:1 with stud males for breeding, and copulatory plugs are evident in successfully mated females in the following morning (day 4). An example of non-plugged female (c, left) and a plugged female (c, right) is shown. (d) An image shows the internal anatomy of a female dissected abdominal cavity (d. left). Intestines are placed to the side for easier visualization and manipulation. Ovaries are located behind the intestines, each sharing a fat pad with kidney (d, middle, red arrows). Fat pads are used as manipulation points to avoid damaging the ovaries or oviducts during extraction. (d, right). Fat pads are gently detached from the kidneys by trimming along the mesometrium. (e) The ovary and associated oviduct is dissected by cutting the oviduct/uterine junction (dashed blue line). The tissue in the dotted black square is isolated. (f) A bright field image of isolated ovary and oviduct under a stereoscope (left), next to a cartoon representation of the same structure (right). Cumulus oocyte complexes can be seen through the expanded ampulla membrane, and are released by nicking the ampulla. The location and direction of oviduct nicking procedure is indicated by a green dashed line. (g) A representative image of cumulus oocyte complexes released as a single intact mass from an individual ampulla. (h) A magnified segment of panel G shows the composition of the cumulus oocyte complex, with oocytes shown with red arrows and cumulus cells between oocytes. All scale bars: 100 μm.
Embryo collection and processing.
CRISPR-EZ is best performed on pronuclear-stage embryos, where the maternal and paternal pronuclei have yet to reach S-Phase or fuse (Supplementary Fig. 4). In practice, embryos collected from superovulated females contain both pronuclear-stage embryos and unfertilized oocytes. Researchers can use differential interference contrast (DIC) microscopy to select healthy pronuclear-stage embryos for CRISPR-EZ. Alternatively, overnight culture of electroporated embryos allows healthy embryos to develop into 2-cell embryos, which can be selected for oviduct transfer.
Pronuclear-stage embryos are surrounded by the zona pellucida, a glycoprotein layer that plays an essential role for spermatozoa binding, acrosome reaction, and fertilization53 (Fig. 6a). Partial zona erosion by transient exposure to Acid Tyrode’s (AT) solution dissociates the zona protein constituents and enhances RNP delivery (Fig. 6b). Zona dissolution is a critical step of the CRISPR-EZ protocol, which greatly facilitates the Cas9 RNP delivery under electroporation conditions with minimal detrimental effects on embryo viability. We empirically determined ~30% zona erosion as suitable for allowing efficient RNP delivery without affecting embryo viability (Fig. 6b), as prolonged Acid Tyrode’s treatment may impair preimplantation development and blastocyst hatching54.
Figure 6. An overview of key zygote processing procedures.
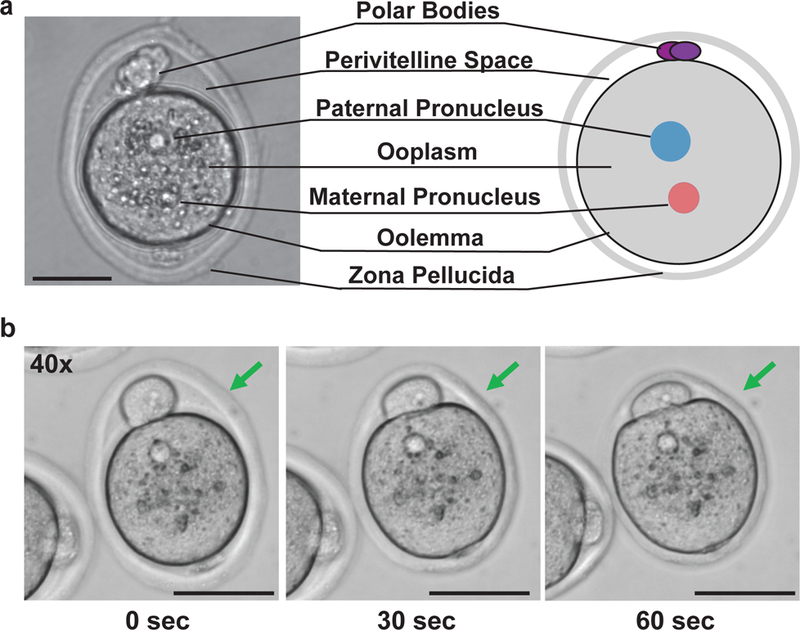
(a) Cartoon diagram illustrating various cellular structures of a pronuclear stage embryo (right), with a corresponding DIC image (left). (b) Representative images are shown for pronuclear stage embryos at various degrees of zona thinning upon treatment with Acid Tyrode’s solution: fully intact zona (left), properly thinned zona (middle), overly thinned zona showing collapse of the perivitelline space (right). Prolonged embryo to acidic conditions impairs embryo viability. Scale Bars: 50 μm.
RNP assembly and electroporation.
Processed pronuclear-stage embryos are mixed with the in vitro assembled Cas9/sgRNA RNPs and subjected to a series of electrical pulses that transiently permeabilize the zona and cell membrane. For mouse embryos, constant, low-voltage, repeated electrical pulses with a long duration (Square Wave Form) offers the best electroporation conditions to achieve efficient RNP delivery with minimal impact on embryo viability55. In short, the first electrical pulse opens pores in the zona and embryo cell membrane, while subsequent pulses maintain pore integrity and can presumably electrophorese the RNP molecules through the cytoplasm56. Our optimal electroporation condition is empirically determined as six 3-ms pulses at 30 volts for a pool of 35–100 embryos, which consistently achieves 100% Cas9 RNP delivery efficiency24 (Fig. 2c,d).
Embryo culture and genotyping.
One of the key benefits of CRISPR-EZ is its capacity to rapidly test sgRNAs ex vivo in mouse embryos prior to generating live animals by first culturing electroporated embryos until the morula stage of preimplantation development (Fig. 1b). Depending on the experimental design, editing events can be validated in individual mouse morula by various methods, including restriction fragment length polymorphism (RFLP)57, T7 endonuclease digestion58, and PCR-based analysis. The exact sequence modification is then validated by sequencing. The same genotyping strategies can also be applied to mice using tail DNA.
RFLP is a powerful genotyping strategy when an endogenous restriction enzyme recognition site is predicted to be altered by editing, such that unedited, partially edited (heterozygous or mosaic editing), and bi-allelic edited embryos can be distinguished and quantified. We present an example RFLP assay for our Tyr editing scheme scheme (Fig. 2a. b).
When an appropriate restriction enzyme site is absent from the predicted target site, a T7 endonuclease assay can be employed to estimate the sgRNA efficiency. T7 endonuclease selectively cleaves DNA heteroduplexes that form after edited and unedited DNA is heat denatured and reannealed. However, as a single embryo lacks sufficient DNA heterogeneity to form heteroduplexes, multiple embryos (we recommend >8 embryos) must be pooled for one T7 reaction. While more versatile than RLFP, the T7 endonuclease assay provides less precise information.
A PCR-based genotyping strategy is often employed for CRISPR-mediated exon deletions. We typically design a PCR strategy to amplify both the deleted and unedited sequence. The relative abundance of PCR product from the predicted deletion allele versus the unedited allele provides an estimation of the extent of editing.
Oviduct transfer.
Live animals can be generated from electroporated embryos by performing oviduct transfer into pseudopregnant CD1 females (Fig. 7a-j). Electroporated embryos are typically implanted into the ampulla of pseudopregnant females either immediately following electroporation, or at the 2-cell stage 24 hours later. If pronuclear-stage embryos can be pre-screened by DIC microscopy, we recommend same-day transfer, as this minimizes in vitro culture time and increases overall viability. Oviduct transfer is technically demanding but achievable with sufficient training. At first, researchers will likely require the expertise of a highly trained technician from a transgenic facility.
Figure 7. An overview of key procedures for oviduct transfer.
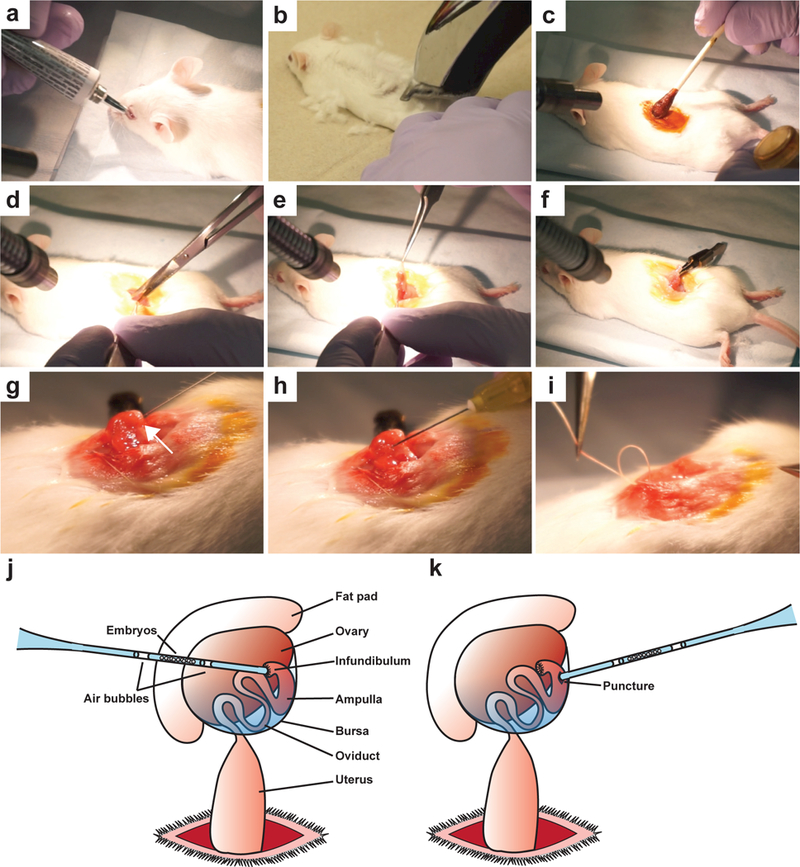
(a) Following anesthetic cocktail administration for anesthesia, artificial tears ointment is applied to the eyes of pseudopregnant mice to prevent drying as mice are unable to close their eyes under anesthetics. (b) Shave the hair over the surgical area of the mice, along the midline of the back near the last rib. (c) Apply betadine by cotton tipped applicator to the surgical area followed by two 70% EtOH treatments using a cotton tipped applicator. (d) Using a pair of small scissors, make a small longitudinal incision along the midline at the level of the last rib and carefully position the skin to expose the body wall. Make another small incision through the peritoneal cavity above the ovary. (e) Using blunt fine forceps, grasp the fat pad and gently pull out the ovary and upper part of the uterus from the incision site. (f) Clip the fat pad with serrefine forceps and lay down the ovary over the middle of the back. (g) Using a 30G x ½” needle, puncture a small hole near the ampulla, proximal to the ovary (white arrow). (h) Insert the glass transfer pipet with loaded embryos into the hole, and gently blow the embryos into the oviduct. (i) After gently returning the organs with blunt forceps, sew the body cavity closed using sutures. (j) Cartoon diagram illustrating the anatomy of the fat pad, ovary, oviduct, and uterus, as well as the proper location of the embryo transfer into the ampulla. The glass transfer needle is preloaded as follows: M2+BSA media, an air bubble, M2+BSA media with embryos, an air bubble, M2+BSA media. Successful transfer is indicated by the appearance of both air bubbles within the oviduct. (k) Cartoon diagram illustrating an alternative method to introduce embryo into the oviduct. A small opening is made between the infundibulum and ampulla using a 30G needle, and transferred embryos will settle in the ampulla.
MATERIALS
REAGENTS
sgRNA synthesis:
Phusion high fidelity DNA polymerase (New England Biolabs, cat. no. M0530)
HiScribe T7 high yield RNA synthesis kit (New England Biolabs, cat. no. E2040)
DNase I, RNase-free (New England Biolabs, cat. no. M0303)
SeraMag Speedbeads (GE Healthcare, cat. no. 65152105050250)
-
100% Ethanol (EtOH, Koptec, cat. no. V1016)
CAUTION Flammable: store in appropriate storage container. Eye irritant: wear safety glasses.
Nuclease free water (Ambion, cat. no. AM9937, molecular biology grade)
1 M Tris-HCl pH 7.4 (Sigma, cat. no. T2663–1L, molecular biology grade)
0.5 M EDTA (Sigma, cat. no. 03690–100ML, molecular biology Grade)
Oligos for sgRNA synthesis, Donor Oligo and PCR Primers for genotyping (Table 1, Integrated DNA Technologies, custom DNA oligonucleotides)
Table 1.
Oligo sequences for sgRNA synthesis, donor oligo and PCR primers for genotyping
| Name | Sequence | Concentration |
|---|---|---|
| IVT-SGRNA- LONG |
5’-GGATCCTAATACGACTCACTATAG-−−20nt guide sequence— GTTTTAGAGCTAGAA |
1 µM |
| IVT- SCAFFOLD- LONG |
5’-AAAAAAGCACCGACTCGGTGCCACTTTTT CAAGTTGATAACGGACTAGCCTTATTTTAAC TTGCTATTTCTAGCTCTAAAAC |
1 µM |
| IVT-FWD | 5’-GGATCCTAATACGACTCACTATAG | 100 µM |
| IVT-REV | 5’-AAAAAAGCACCGACTCGG | 100 µM |
| IVT-TYR-LONG | 5’-GGATCCTAATACGACTCACTATAGGGGTGGATGACCGTGAGTCCG TTTTAGAGCTAGAA |
1 µM |
| TYR-ECORI- DONOR |
5’- GTGCACCATCTGGACCTCAGTTCCCCTTCAAAGGGGTGGAT GACCGTGAATTCCTGGCCCTCTGTGTTTTATAATAGGACCTG CCAGTGCTC |
200 µM |
| sgTyr F1 | 5’-TCTTTTCGGAGACACTCAAATCA | 100 µM |
| sgTyr F2 | 5’-TCTGTACAATTTGGGCCCCC | 100 µM |
| sgTyr R1 | 5’-GCTTTCAGGCAGAGGTTCCT | 100 µM |
Superovulation and mating
-
3–8-month-old male mice and 3–5-week-old female mice (C57BL/6J (Jax 000664) or C57B/6N (Jax 005304)
CAUTION Experiments involving live mice must conform to appropriate institutional regulations. All animal experiments in this protocol were performed in accordance with the UC Davis institutional animal care and use committee (IACUC) guidelines and policies.
Pregnant mare serum gonadotropin (PMSG; ProspecBio, cat. no. HOR-272, lyophilized)
Human chorion gonadotropin (hCG; Millipore, cat. no. 230734, lyophilized)
Dulbecco’s phosphate buffered saline (DPBS, calcium and magnesium free; Gibco, cat. no. 14190–144)
Embryo collection and processing:
M2 medium (Zenith Biotech, cat. no. ZFM2–050)
Bovine serum albumin (BSA; Sigma, cat. no. A3311, embryo culture grade)
Hyaluronidase/M2 (Millipore, cat. no. MR-051-F)
Acid Tyrode’s solution (Sigma, cat. no. T1788, embryo culture grade)
RNP assembly and electroporation:
HEPES (Sigma, cat. no. H4034, cell culture grade)
Potassium chloride (KCl; Sigma, cat. no. P9333, molecular biology grade)
Magnesium chloride (MgCl2; Sigma, cat. no. M8266, anhydrous)
100% glycerol (Fisher, cat. no. BP229, molecular biology grade)
Tris (2-carboxyethyl) phosphine hydrochloride (TCEP; Sigma, cat. no. C4706)
OptiMEM reduced serum media (Thermo, cat. no. 31985062)
Cas9 protein (Alt-R™ S.p. Cas9 Nuclease 3NLS (IDT, cat. no. 1074181, 61 µM)
Embryo culture and genotyping
KSOM+AA medium (Potassium-supplemented simplex optimized medium plus amino acids; Zenith Biotech, cat. no. ZEKS-050)
Mineral oil (Millipore, cat. no. ES-005C, embryo grade)
Potassium chloride (KCl, Fisher, cat. no. P217–3, certified ACS grade)
1M Tris-HCl, pH 8.5 Solution. (Teknova, cat. no. T1085, molecular biology grade)
Ethylenediaminetetraacetic acid (EDTA; Sigma, cat. no. EDS-100G, anhydrous)
Magnesium chloride (MgCl2; Fisher, cat. no. M33–500 certified ACS grade)
Gelatin type B (Fisher, cat. no. G7–500, laboratory grade)
Nonidet P-40 substitute (NP-40; Sigma, cat. no. 74385)
Tween-20 (Sigma, cat. no. P7949–500, molecular biology grade)
Proteinase K (Fisher, cat. no. BP1700–100, molecular biology grade)
GoTaq (Promega, cat. no. M712)
HinfI (NEB, cat. no. R0155S, 10,000 units/ml)
EcoRI (NEB, cat. no. R3101S, 20,000 units/ml)
Additional restriction enzymes for RFLP (Optional, NEB)
LE agarose (BioExpress, cat. no. E-3120–500, analytical grade)
Tris Base (Fisher cat. no. 77–68-1, molecular biology grade)
Acetic acid (Fisher cat. no. 64–19-7, Glacial Certified ACS)
1 kb Plus DNA ladder (Thermo, cat. no.10787018)
-
EtBr (Ethidium Bromide 10 mg/ml; Thermo, cat. no. 15585011, Ultrapure)
CAUTION EtBr is considered to be a potent mutagen. Avoid skin, eye, mouth and upper respiratory exposure by wearing appropriate personal protective equipment. Non-Toxic alternatives can be used, such as Sybr-Safe (Thermo, cat. no. S33102)
Gel loading dye, 6x (Thermo, cat. no. R0611, 6X)
Oviduct transfer
-
7–9-week-old CD1 recipient female mice and 2–8-month-old vasectomized males (Charles River strain code 022, vasectomy code VASEX)
CAUTION Experiments involving live mice must conform to appropriate institutional regulations. All animal experiments in this protocol were performed in accordance with the UC Davis institutional animal care and use committee (IACUC) guidelines and policies.
Artificial tears ointment (Akorn, cat. no.17478–062-35)
Betadine (Purdue products, cat. no. 67618–150-01)
-
Buprenorphine hydrochloride injection CIII (Pfizer, cat. no. 00409–2012-32)
CAUTION Partial opioid agonist and Schedule III controlled substance. Obtain appropriate authorizations prior to use.
Ketamine (VetOne, cat. no. 501072, 100 mg/ml)
-
Xylazine (Akorn, cat. no. 59399011020, 100mg/ml)
CAUTION Seek institutional approval prior to acquiring and using anesthetics.
EQUIPMENT
sgRNA synthesis:
RNase-free 1.5 ml microcentrifuge tubes (VWR, cat. no. 20170–333)
RNase-free 8-well PCR strip tube. (VWR, cat. no. 82006–606)
Thermalcycler (Biorad S1000 or equivalent)
Gel electrophoresis apparatus (Biorad 1704489EDU or equivalent)
Gel doc imager XR+ system (Biorad 1708195 or equivalent)
Magnetic stand (Invitrogen, cat. no. 12321D)
Nanodrop ND-2000-US (Thermo, or equivalent spectrophotometer)
Agilent Bioanalyzer 2100 (Optional, typically a facility owned equipment)
Superovulation and mating:
-
26 G x ½” needle (BD, cat. no. 305111)
CAUTION Sharps hazard. Please dispose of needles in sharps container.
1 ml syringe (BD, cat. no. 309659, without needle, latex free)
Embryo collection and processing:
Stereomicroscope (Nikon SMZ-U or equivalent)
0.22 µm PVDF syringe filter (Millipore, cat. no. SLGV033RB)
0.45 µm PVDF syringe filter (Millipore, cat. no. SLHV033RB)
15-inch rubber aspirator tube assembly (Sigma, cat. no. A5177)
Capillary tubes (Sigma, cat. no. P0674)
CAUTION Sharps hazard. Dispose of used needles in sharps container.
60 mm tissue culture dish (Greiner Bio-One, cat. no. 628–160)
RNP assembly and electroporation:
0.1 cm gap electroporation cuvette (Biorad, cat. no. 1652089)
Electroporator (Biorad Gene Pulser XCell, cat. no. 1652660, including CE and PC modules)
Embryo culture and genotyping
35 mm tissue culture dish (Greiner Bio-One, cat. no. 627–160)
Cell culture incubator (Thermo, Napco Series 8000 DH or equivalent)
Oviduct transfer:
Warm stage/platform (C & A Scientific XH-2001 or equivalent)
Weighing scale (OHAUS, cat. no. LS200)
Fine dissection forceps (Roboz, cat. no. RS-4976)
-
Dumont straight forceps (Fine Science Tools, cat. no. 11252–23)
CAUTION Sharps hazard. Dispose of needles in sharps container.
Dissection scissors (Roboz, cat. no. RS-5960)
Serrefine forceps (Fine Science Tools, cat. no. 18050–28)
-
Bead Sterilizer (Fine Science Tools, cat no. 18000–45)
CAUTION Sterilizer could reach extremely high temperatures.
-
30 G x ½” needle (BD, cat. no. 305106)
CAUTION Sharps hazard. Dispose of needles in sharps container.
Hair trimmer (Andis: model no. D-4)
Cotton tipped applicator (Medline, cat. no. MDS202000Z)
Wound clip (Braintree Scientific, cat. no. EZC CS)
Wound clip applier (Braintree Scientific, cat. no. EZC APL)
-
Absorbable Suture (CP Medical, cat. no. 421A)
CAUTION Sharps hazard. Dispose of needles in sharps container.
REAGENT SETUP
Superovulation hormones:
Reconstitute lyophilized hormones by adding pre-chilled DPBS directly to the glass bottles containing PMSG or hCG powders. Mix by gentle inversion and pipetting to ensure full reconstitution. Prepare 50 IU/ml stock solutions for PMSG and hCG. Dispense 600 µl aliquots into pre-chilled 1.5 ml centrifuge tubes, and immediately flash freeze by submerging in a liquid nitrogen bath or a dry ice/ethanol bath. PMSG and hCG stocks can be stored at −80°C for up to 1 year.
CRITICAL
All reagents and vessels are pre-chilled on ice, as hormones should be kept cold throughout preparation with minimal exposure to ambient temperature.
Mice for superovulation:
Injections for superovulation should be timed such that female mice are ~4 weeks old at the time of mating. Females up to 5 weeks can be used but females older than 5 weeks do not respond as well to superovulation. In our experience, reliable male studs are 3–8-month-old. Replace males if they fail to produce copulatory plugs after 3 consecutive mating attempts. After each mating, males should be rested for at least 4 or more days to recover sperm count.
CRITICAL
In order to ensure fertilization success, we test the plugging rate of stud males with reproductively mature females (8+ weeks). We consider males to have proven fertility if they leave copulatory plugs after three consecutive matings. Optimal superovulation conditions vary by mouse strains, and may need to be empirically determined. The described procedures are tested for C57BL/6J and C57BL/6N mice.
Cas9 protein:
After first thaw, make 4 µl or 8 µl Cas9 protein aliquots for single or double use, respectively. We recommend testing new lots of protein against previous lots in embryos to ensure efficacy. Avoid freeze thaw and store in −80°C freezer for up to 10 weeks.
Embryo culture media (KSOM+AA+BSA):
Supplement KSOM+AA medium with BSA to a final concentration of 1 mg/ml, and sterilize through a 0.22 μm PVDF syringe filter. Make 1 ml aliquots and store at 4 °C for up to 1 week once BSA is added.
Embryo manipulation media (M2+BSA):
Supplement M2 media with BSA to a final concentration of 4 mg/ml, and sterilize through a 0.22 μm PVDF syringe filter. Make 1 ml aliquots and store at 4 °C for up to 1 week. M2 without BSA can be stored for up to 4 months at 4 °C.
Ethanol, 80% (vol/vol):
Combine 8 ml of 100% ethanol with 2 ml nuclease free water. Store at room temperature (20–25°C), indefinitely.
Ethanol, 70% (vol/vol):
Combine 7 ml of 100% ethanol with 3 ml nuclease free water. Store at room temperature, indefinitely.
TE Buffer (10 mM Tris-HCl, 1 mM EDTA, pH 7.4):
Prepare TE buffer by combining 10 mL of 1 M Tris-HCL (pH 7.4) with 2 mL 0.5 M EDTA and 988 mL of nuclease free water. TE Buffer can be stored at room temperature, indefinitely.
RNP Buffer for Cas9 RNP assembly:
Prepare RNP buffer stock by combining the reagents listed in the table below. Prepare 500 µl aliquots and store at −20 °C for up to 6 months. Immediately prior to RNP assembly, dilute 100 mM TCEP 1:20 in RNP buffer to make complete RNP buffer (by adding 1 µl of 100 mM TCEP to 19 µl of RNP buffer). This complete reagent is single use and should not be stored.
CRITICAL
TCEP acts as a reducing agent and prevents Cas9 aggregation. TCEP is unstable in phosphate buffers at neutral pH and is therefore added just prior to complex formation. 100mM TCEP should be stored as single-use, 5 µl aliquots at −20 °C indefinitely.
Embryo Lysis Buffer for genotyping:
Prepare Embryo Lysis Buffer by combining the reagents listed in the table below, and store as 500 µl aliquots at −20 °C for up to 6 months. Prior to embryo lysis, dilute 20 mg/ml Proteinase K 1:100 in Embryo Lysis Buffer to a final concentration of 0.2 mg/ml (by adding 5 µl of 20 mg/ml Proteinase K to 495 µl Embryo Lysis Buffer). This complete reagent is single use and should not be stored.
Pseudopregnant female mice for oviduct transfer:
7–9-week-old female CD1 mice are mated to 2–8-month-old vasectomized males. Pseudopregnant CD1 females are identified by the presence of copulatory plugs the next morning.
CRITICAL STEP
Vasectomized males should be tested for infertility at least once by pairing with a reproductively mature female (8+ weeks), checking presence of copulatory plug, and confirming absence of sired litter. Replace males if they fail to produce copulatory plugs after 3 consecutive mating attempts.
Anesthetic cocktail preparation:
Dilute Ketamine (stock 100 mg/ml) and Xylazine (stock 100 mg/ml) by aseptically combining 1 ml stock Ketamine with 0.1 ml stock and 8.9 ml of sterile water. Store at 4 °C until the expiration of either component. Check manufacturer information for expiration dates.
EQUIPMENT SETUP
Mouth pipet for embryo manipulation:
For multiple CRISPR-EZ procedures, mouse embryos are manipulated using a mouth pipet loaded with a glass transfer pipet (Supplementary Fig. 5). Glass transfer pipets are produced by pulling glass capillary tubes over an open flame. An ideal glass transfer pipet possesses a 4–5 cm tip of near uniform diameter, with an opening of approximately 5x the diameter of an oocyte. To make a mouth pipet, first cut the 15-inch rubber aspirator tube in half and connect both new ends to a 0.45 µm PVDF syringe filter to prevent contamination from the researcher. Next, insert a glass transfer pipet into the clear pipet holder.
Embryo culture plates:
For prolonged embryo culture, prepare culture plates by even distributing 5–6 20 µl droplets of KSOM+AA+BSA media into a 35-mm cell culture plate (Supplementary Fig. 6). Carefully overlay with 2–3 ml of mineral oil using a serological pipet, and equilibrate by pre-incubating for at least 4 hours in a water jacketed, 5% CO2 incubator at 37°C and 95% humidity. CRITICAL As KSOM is a sodium bicarbonate buffered solution, pH is extremely sensitive to air exposure. Mineral oil serves to buffer against temperature and pH fluctuations outside the incubator59.
Embryo manipulation plates: For embryo manipulation, collection, treatment, and washes, prepare M2+BSA medium plates immediately prior to use by evenly distributing droplets of 40 µl M2 + 4 mg/ml BSA on 60 mm plates. As M2 is a HEPES buffered solution, it is refractory to pH fluctuations and therefore does not require a mineral oil overlay (Supplementary Fig. 7).
Embryo workstation:
For optimal expediency, we recommend establishing an embryo workstation containing the following equipment, tools and supplies within easy reach: stereomicroscope, electroporator, CO2 incubator, embryo culture/manipulation plates, hand pipets, mouth pipets, and dissection tools. Arrange these items such that dissected mouse oviducts can be immediately processed under the stereomicroscope for embryo collection. Pronuclear-stage embryos are rapidly isolated, pulsed with the electroporator, and placed into the incubator, with minimal exposure of embryos to ambient temperature.
PROCEDURE:
sgRNA design ● TIMING 1 day; 1–2 hours hands on
-
1.
Candidate sgRNAs can be selected from several widely used online algorithms. To engineer indels, point mutations or small insertions, we recommend selecting 2–3 candidate sgRNAs for the target cut site based on algorithm score criteria. To engineer genomic deletions, design a pair of sgRNAs to flank the region to be excised, using one of the commonly used algorithms for sgRNA design. Some publicly-available design programs include but are not limited to: Sequence scan for CRISPR (http://crispr.dfci.harvard.edu/SSC/), Gene Perturbation Platform (http://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design), Chop-Chop (https://chopchop.rc.fas.harvard.edu/), CRISPR Design (http://crispr.mit.edu/).
CRITICAL STEP
Although it remains unclear how extensive Cas9 off-target activity is in edited mice, it is important to avoid sgRNA designs with highly possible off-target sites. Not all algorithms provide off-target analysis on predicted sgRNAs. It is necessary, in some cases, to perform BLAT analyses on the selected sgRNA sequences to manually identify potential off-target sites. In our experience, the most successful sgRNAs are often highly ranked across multiple algorithms.
sgRNA synthesis ● TIMING 2 days; 3 hours hands on
-
3.
Generate the DNA template for sgRNA in vitro transcription through PCR by preparing the following reaction mixture in sterile RNase-free 8-well PCR strip tube. Clean up is not required after the PCR reaction.
Place the PCR reaction in a thermal cycler using the following cycling conditions:
-
4.
Confirm the presence of template by running 5 µl of PCR product combined with 1 µl of 6x DNA loading dye on a 2% (wt/vol) agarose gel. Expected product size will be 127bp (Fig. 4).
? TROUBLESHOOTING
PAUSE POINT
The completed PCR reaction can be stored at −20°C until needed.
-
5.
For T7 in vitro transcription, follow manufacturer’s instructions. Briefly, prepare the following reaction mixture in an RNase-free 8-well PCR strip tube, and mix the reagents by gentle pipetting. All components except DNA template (generated in Step 3) are provided by the NEB HiScribe T7 high yield RNA synthesis kit.
CRITICAL STEP
The in vitro transcription reaction is highly sensitive to RNase contamination. It is essential to ensure an RNase-free work environment by use of RNase-free reagents and supplies.
-
6.
Incubate the in vitro transcription reaction mixture for >18 hours at 37°C in a thermal cycler.
-
7.
Add 1 µl of RNase-free DNase I (2 units/reaction), and incubate for 20 minutes at room temperature (20–25°C) to remove DNA template.
-
8.
Add 129 µl 100% ethanol to the reaction to facilitate subsequent small RNA binding to the magnetic SeraMag Speedbeads for sgRNA purification.
-
9.
Thoroughly vortex SeraMag Speedbeads for 10 seconds to resuspend. Add 100 µl bead mixture to the 150 µl in vitro transcription reaction. Gently pipet the reaction 10 times to mix, and incubate for 5 minutes at room temperature.
-
10.
Place the RNA+bead reaction on the magnetic stand, and wait for 5 min to allow beads to pellet towards the magnet.
-
11.
Discard supernatant using a pipet. Add 200 µl of 80% (vol/vol) EtOH and wash the beads by gently pipetting for 5 times. Incubate for 5 minutes on the magnetic stand to allow beads to re-pellet.
-
12.
Repeat the wash step (Step 10), then air dry for 5–10 minutes.
-
13.
Elute RNA with 20 µl of nuclease-free water, pipet to mix 10 times, and incubate for 2 minutes at room temperature.
-
14.
Place the reaction tube on the magnetic stand for 5 minutes at room temperature. Using a pipet, carefully transfer the RNA-containing supernatant to an RNase-free 1.5 ml microcentrifuge tube.
-
15.
Determine the quality and quantity of sgRNA preparation using a bioanalyser and spectrophotometer, respectively. Alternatively, run 2 µl of sgRNAs on an RNase-free 2% (wt/vol) agarose gel to check quality.
?TROUBLESHOOTING
PAUSE POINT
Purified sgRNAs can be stored for up to a year at −80°C. To minimize freeze-thaw cycles, store sgRNAs as 5 µl aliquots.
Superovulation and mating ● TIMING 3 days; 2 hours hands on
-
16.
Determine the number of females required for superovulation prior to the experiment, based on the assumption of obtaining 10–20 embryos per female. For most editing strategies, we aim to electroporate ~30 embryos per condition for testing sgRNA design in embryos, and ~60 embryos per condition for generating edited mice. Hence, 2–3 superovulated females are sufficient for testing sgRNA designs in embryos, and 4–5 recipient females are sufficient for one mouse editing experiment. It is prudent to include some extra embryos to allow for procedural embryo loss, embryo lethality and unfertilized oocytes.
-
17.
Follow the hormone injection schedule for superovulation (Fig. 5a) by administering 5 IU PMSG (100 µl) to ~4-week-old female mice through intraperitoneal (IP) injection at 2:00–3:00 PM on day 1. To perform IP injection, restrain the mouse by grasping behind its neck with thumb and forefinger, while securing the tail between pinky and ring finger. Invert the mouse to expose the lower abdomen, and with a 26-gauge syringe, inject at a 45° angle between two visible nipples on either side of the abdomen (Fig. 5b).
-
18.
Approximately 46–48 hours after PMSG injection (1:00–3:00 PM on day 3), administer 5 IU hCG (100 µl) to the same female mice by IP injection to induce ovulation.
CRITICAL STEP
Perform IP injections within 30 minutes of thawing either PMSG or hCG, as hormones rapidly lose activity at room temperature.
?TROUBLESHOOTING
-
19.
After hCG injection, pair each hormone stimulated female with one 3–8 month old stud male with proven fertility in a mating cage.
Embryo collection and processing ● TIMING 2 hours
-
20.
The morning after hCG IP injection (66–68 hours after PMSG, 7:00–9:00 AM on day 4), collect females with copulation plugs for embryo collection. Typically, 60% of superovulated females display copulation plugs, an indication of a successful mating (Fig. 5c).
CRITICAL STEP
We provide a detailed video for all experimental procedures described following this Step (Supplementary Video 1).
?TROUBLESHOOTING
-
21.
Euthanize females by CO2 asphyxiation and/or additional measures according to institutional guidelines. Lay the animals on their backs and spray the abdominal area thoroughly with 70% ethanol. Using dissection scissors, open the abdominal cavity and locate the ovaries on both sides (Fig. 5d). Surgically isolate the ovaries with attached oviducts and place each ovary/oviduct in a 50 µl droplet of M2+BSA media in an embryo manipulation plate (Fig. 5e, f). We typically array 9 droplets of M2+BSA media for oviduct dissection around the perimeter of a 60 mm dish, leaving one droplet in the center for embryo collection (Supplementary Fig.7).
-
22.
Under a stereomicroscope, nick the ampulla of the oviduct using dissection forceps (Fig. 5f), releasing the cumulus oocyte complex containing oocytes or pronuclear stage embryos surrounded by supporting cumulus cells (Fig. 5g, h, Supplementary Video 1).
-
23.
Transfer all dissected cumulus oocyte complexes to a single 50 µl droplet of M2+BSA media using a hand pipet (Fig. 5g, h).
-
24.
Prepare a 60-mm plate with a 100 µL droplet of Hyaluronidase/M2, and transfer the cumulus oocyte complexes to the Hyaluronidase/M2 droplet using a pipet with as little carryover as manageable. Gently pipet the cumulus oocyte complexes up and down until the majority of the embryos are dissociated from the cumulus cells. In our experience, this typically takes ~1 minute, but the length of treatment depends on the number of cumulus oocyte complexes collected. An additional 50 µL of Hyaluronidase/M2 can be added to this droplet if cumulus cell dissociation does not occur within 2 minutes.
CRITICAL STEP
Embryos should not be left in Hyaluronidase/M2 for longer than 5 minutes.
-
25.
Hyaluronidase treatment releases embryos from the cumulus oocyte complex. Transfer embryos to a new 50 µl M2+BSA droplet using a mouth pipet to dilute active Hyaluronidase and wash away surrounding cumulus cells.
-
26.
Pass the embryos by mouth pipet through 3–6 additional 50 µl M2+BSA droplets until the cumulus cells are completely removed. If cumulus cells remain, additional washes are necessary, which have minimal impact on embryo viability. At this point, embryo morphology and quality can be assessed by visual inspection under stereomicroscope (Fig. 6a). The status of fertilization can be assessed by checking for the presence of both the male and female pronuclei using a DIC capable microscope.
PAUSE POINT
Embryos can be temporarily maintained for no more than 30 minutes in a 50 µl M2+BSA droplet in a water jacketed, 5% CO2 incubator at 37°C and 95% humidity.
CRITICAL STEP
Embryos are extremely sensitive to culture conditions, including temperature, CO2 level, and media conditions. Be sure to test incubator conditions and check media expiration dates once a month to ensure successful ex vivo embryo culture.
?TROUBLESHOOTING
-
27.
Mildly erode the zona pellucida by brief embryo exposure to Acid Tyrode’s solution. Using a mouth pipet, transfer the embryos to a droplet of Acid Tyrode’s solution in a 60 mm plate. Observe the embryos under a stereomicroscope, until the zona pellucida is ~30% eroded. This usually occurs at approximately 30–40 seconds (Fig. 6b, Supplementary Video 1).
CRITICAL STEP:
The timing of Acid Tyrode’s treatment is crucial, as prolonged exposure can completely dissolve the zona pellucida and compromise embryo viability. Expect some heterogeneity in the extent of zona erosion among the embryos.
?TROUBLESHOOTING
-
28.
Using a mouth pipet, swiftly pass the embryos sequentially through at least four 50 µl droplets of M2+BSA to dilute out the Acid Tyrode’s solution. Obtain a final embryo count to confirm sufficient numbers for experimental conditions. It is not uncommon to lose up to 5% of embryos during these manipulations steps.
PAUSE POINT
Embryos can be temporarily stored for no more than 30 minutes in a 50 µl M2+BSA droplet in a water jacketed, 5% CO2 incubator at 37°C with 95% humidity.
CRITICAL STEP:
As the exact timing of fertilization is unknown, and embryos from multiple females are combined, it is important to realize that the embryos will be within a few hours of each other on a development timeline and this heterogeneity should not be misinterpreted as a developmental delay or consequence of manipulation. Some embryos will fail to be fertilized (oocytes) or develop and do not progress beyond the 1–2 Cell stages.
RNP assembly and electroporation ● TIMING 1 hour
-
29.
Immediately following embryo processing, prepare Cas9 RNP complex reaction mixtures. To assemble Cas9/sgRNA RNP complex for NHEJ mediated indel, use option A; to engineer HDR mediated editing, use option B; to make defined deletions, use option C.
A. For NHEJ-mediated indels
-
i)
Combine the following reagents at room temperature in a sterile, RNAse free 8-well PCR strip.
-
ii)
Incubate the reaction mixture in a thermal cycler at 37°C for 10 minutes to allow complex formation.
B. For HDR mediated editing
-
i)
Combine the following reagents at room temperature in a sterile, RNAse free 8-well PCR strip.
-
ii)
Incubate the reaction mixture in a thermal cycler at 37°C for 10 minutes to allow complex formation.
C. For engineering deletions using paired sgRNAs
-
i)
Combine the following reagents at room temperature in a sterile, RNAse free 8-well PCR strip.
-
ii)
Incubate the reaction mixture in a thermal cycler at 37°C for 10 minutes to allow complex formation.
PAUSE POINT
Assembled RNP complex made using option A,B or C can be stored at room temperature for up to 2 hours.
-
30.
Transfer the processed embryos from Step 28 through two 50 µl droplets of OptiMEM to dilute out the M2+BSA in preparation for electroporation. CRITICAL STEP: It is important to remove BSA, as it provides undesired resistance during embryo electroporation.
-
31.
Transfer 30 (for in vitro testing) or 60 (for animal derivation) embryos to a 10 µl droplet of OptiMEM. Add 10 µl pre-assembled Cas9/sgRNA RNP complex (from Step 29) to this droplet and mix thoroughly by pipetting up and down 10 times, until the embryo/RNP mixture is homogenous by visual inspection under stereomicroscope.
CRITICAL STEP
The Cas9/sgRNA RNP mixture has a thicker viscosity than OptiMEM due to the presence of glycerol. The embryo/RNP mixture must be pipetted gently to reach homogeneity.
-
32.
Transfer the entire 20 µl embryo/RNP mixture to a 0.1 cm gap electroporation cuvette, avoid making bubbles, and electroporate embryos in a Biorad Gene Pulser XCell apparatus with a square wave protocol and the following parameters:
?TROUBLESHOOTING
-
33.
Recover electroporated embryos by flushing the cuvette at least three times with 50 µl KSOM+AA+BSA media using a pipet, transferring and combining each wash into a 60 mm plate. Avoid introducing air bubbles during this procedure. It is optional to repeat this procedure to maximize the recovery of electroporated embryos. We typically recover >95% of the originally loaded embryos.
-
34.
Using a mouth pipet, transfer the recovered embryos to one droplet of KSOM+AA+BSA media in an equilibrated embryo culture plate. This serves to dilute the remaining RNP/OptiMEM mixture. Then transfer the embryos to a second droplet in the same plate for subsequent embryo culture (Step 35A) or in preparation for oviduct transfer (Step 35B).
PAUSE POINT
Embryos can be cultured for up to four days in the equilibrated embryo culture plate while in a water jacketed, 5% CO2 incubator at 37°C with 95% humidity.
CRITICAL STEP
Prior to use, the embryo culture plate containing KSOM+AA+BSA media overlaid with mineral oil that and is equilibrated by pre-incubation in a cell incubator for at least 4 hours is prepared according to Supplementary Fig. 6.
-
35.
At this point, embryos can either be cultured to the morula stage for embryo genotyping (option A) or transferred to a recipient female oviduct for the generation of CRISPR edited mice (option B). When attempting this protocol for the first time, subject the embryos to ex vivo culture conditions according to option A to test sgRNA editing efficiency. Once the results from the ex vivo testing conditions are adequate, one can transfer electroporated embryos to generate mice using option B.
(A). Embryo culture and genotyping ● TIMING 4–5 days; 4–6 hours hands on
-
i.
The following day, under a stereoscope, transfer healthy 2-cell embryos to a fresh droplet of KSOM+AA+BSA, and culture to morula/blastocyst embryos in an equilibrated embryo culture plate while in a water jacketed, 5% CO2 incubator at 37°C with 95% humidity. Typically, pronuclear stage embryos develop to morula after ~72 hours of culture, and to blastocysts after ~84 hours of culture.
-
ii.
Wash morula/blastocyst embryos by sequentially passing them through two droplets of DPBS using a mouth pipet.
-
iii.
Using a pipet set to 1 µl, transfer each individual embryo into one well of an 8-well PCR strip containing 10 µl of Embryo Lysis Buffer + Proteinase K (0.2 mg/ml).
-
iv.
Lyse the embryos to extract DNA by heating the samples in a thermal cycler at 55°C for 4 hours, followed by a 10-minute incubation at 95°C to inactivate Proteinase K.
PAUSE POINT
Embryo lysates can be stored at 4°C for 2–3 days or −20°C for up to 2 weeks before genotyping analysis. Avoid multiple freeze-thaws.
CRITICAL STEP
Embryo genotyping assays vary depending on the exact editing scheme. Provided are procedures to assess NHEJ-or HDR-mediated editing for the Tyr editing scheme using RFLP. As this workflow is highly optimized and reproducible, we strongly recommend performing the Tyr editing experiment as a positive control in CRISPR-EZ experiments, particularly for the first-time users.
-
v.
Perform PCR #1 of a nested PCR to amplify DNA fragments flanking the targeted site from small amounts of DNA from crude single-embryo lysates (from Step 35Aiv). Prepare the first PCR reaction mix as described:
Place the PCR reaction in a thermal cycler using the following cycling conditions:
PAUSE POINT
PCR product can be stored at 4°C for 2–3 days or −20°C for up to 2 weeks before genotyping analysis. Avoid multiple freeze-thaws.
-
vi.
Perform PCR #2 of a nested PCR reaction to generate a highly specific PCR product using 2 µl of 1:10 diluted first PCR reaction as the template. Dilution of the first PCR product serves to reduce primer carryover in the nested PCR. Prepare the nested PCR reaction mix as described:
Place the PCR reaction in a thermal cycler using the following cycling conditions:
CRITICAL STEP
Annealing temperature depends on primer sequences. This particular reaction is showing sgTyr conditions. We typically achieve >90% reliable PCR reactions when amplifying products up to 500 bp from individual embryos.
?TROUBLESHOOTING
-
vii.
To genotype NHEJ-mediated Tyr indel mutations, digest 10 µl of the nested PCR products from Step 35Avi with 10 units of HinfI in a 20 µl reaction and incubate for 4 hours at 37°C. Run digestion products on a 2% (wt/vol) agarose gel, using non-digested PCR product as a loading control for 30 minutes at 135 volts. There is sufficient loading dye carry over from the previous PCR reaction.
-
viii.
To genotype HDR-mediated Tyr small sequence substitution, digest 10 µl of the nested PCR products from Step 35Avi with 20 units of EcoRI in a 20 µl reaction and incubate for 4 hours at 37°C. Run the digestion products on a 2% (wt/vol) agarose gel, using non-digested PCR product as a loading control for 30 minutes at 135 volts. There is sufficient loading dye carry over from the previous PCR reaction.
CRITICAL STEP
In any HDR-mediated editing, both HDR and NHEJ editing events can occur, which are not mutually exclusive within the same embryo. We recommend to perform both NHEJ and HDR RFLP analyses to determine the genotype of each embryo in a HDR editing experiment.
(B). Oviduct Transfer ● TIMING 19 Days; 2–3 hours hands on
-
i.
On the day before the embryo transfer, prepare 5 pseudopregnant recipient females by combining 5 females with 5 vasectomized males, so that each mating cage has a single male and female mouse.
CRITICAL STEP
Typically, 60 embryos are electroporated, from which 40+ viable embryos are expected to be recovered, which can be transferred to 2x CD1 pseudopregnant mothers. If more embryos are available for transfer, be sure to prepare additional mating cages for pseudopregnant females.
-
ii.
On the morning of the embryo transfer, prepare for survival surgery by sterilizing all surgical tools with 70% ethanol and placing into a bead sterilizer until use. Prepare an empty mouse cage by placing it on a warm stage/platform set at 37 °C.
-
iii.
On the same morning, identify 0.5 days post coitum (dpc) females with visible copulatory plugs. Weigh each pseudo-pregnant female and IP inject 0.1 ml of anesthetic cocktail for every 10g of body weight (dosage of Ketamine at 100 mg/kg and Xylazine at 10mg/kg) using a 1 ml syringe attached to a 26G x ½” needle.
CRITICAL STEP
Once administered, mice typically succumb to anesthetics within 1–2 minutes which lasts for 60–120 minutes. Mice should be monitored after experiment according to institutional guidelines. Ketamine over dosage may cause death. Repeat injections should be avoided if at all possible.
-
iv.
After one minute, check if the mouse is fully anesthetized by pinching its toes with forceps. Once the mouse is unresponsive, place the anesthetized mouse under the stereomicroscope and apply artificial tears ointment to its eyes to prevent drying during surgery (Fig 7a). Using a hair trimmer, shave the hair over the surgical area, along the midline of the back near the last rib (Fig. 7b).
-
v.
To disinfect the surgical area, apply betadine by cotton tipped applicator, scrubbing from the center of the surgical site and radiating outwards to disinfect approximately a 2-inch square area (Fig. 7c). Subsequently wipe off with 70% (vol/vol) ethanol. Repeat this procedure twice to fully sterilize the surgical area (Supplementary Video 1).
-
vi.
Make a small longitudinal incision (≤1cm) parallel to the midline at the level of the last rib and slide the skin to expose the body wall. Pick up the body wall with forceps over the site of the ovary and make a small incision through the body wall (Fig. 7d).
CRITICAL STEP
Take precaution to avoid cutting blood vessels, as bleeding may obstruct the oviduct transfer and increase risk of surgical complications.
-
vii.
Identify the fat pad attached to the ovary, and using blunt fine forceps, pull it out along with the ovary and upper part of the uterus from the incision site (Fig. 7e). Clip the fat pad with serrefine forceps and lay down the ovary over the middle of the back (Fig. 7f).
-
viii.
Load the glass transfer pipet by drawing up M2+BSA medium about 1 cm up the pipet, then one small air bubble, then 10 embryos from Step 34 within a minimal amount of media, and finally another small air bubble (Fig. 7j).
CRITICAL STEP
Up to 20 embryos can be transferred into the oviducts of each pseudopregnant female (10 embryos per oviduct). Typically, 60 embryos are electroporated where 40+ viable embryos are expected to be recovered and transferred to 2 CD1 pseudopregnant mothers.
-
ix.
Insert the transfer pipette with loaded embryos into the infundibulum, and gently blow the embryos into the infundibulum of the oviduct until the two bubbles are seen in the ampulla to ensure successful transfer (Fig. 7g, j). Alternatively, if the infundibulum is too difficult to locate, use a 30 G x ½” needle to puncture a small hole upstream of the ampulla and insert the transfer needle into this newly made opening (Fig. 7h, k).
-
x.
Remove the transfer pipette and unclip the serrefine forceps. Using blunt forceps, gently place the fat pad, ovary, and uterus back into the peritoneal cavity, and sew up the body wall with absorbable sutures (Fig. 7i, Supplementary Video 1).
-
xi.
Transfer embryos into the other oviduct of the same mouse by sliding the skin over to expose the opposite side of the back, and repeat Steps 35Bvi-x.
-
xii.
Hold the skin together and close the opening with wound clips.
-
xiii.
For pain relief, deliver 0.1 ml of Buprenorphine to the mouse by subcutaneous injection into the lower abdominal region using a 26 G x ½” needle.
-
xiv.
Place the mouse into the pre-warmed empty cage and monitor for vital signs (e.g. respiratory rate) until awake. House mice in an IACUC facility until pups are born. For C57BL6/J mice, gestation period is typically 18–19 days.
CRITICAL STEP
Animals should be monitored daily for at least 10 days following surgery and additional analgesia can be given at the recommendation of the veterinarian.
After the gestational period, collect tail samples from the pups for genomic DNA extraction60, in accordance with institutional guidelines. Perform genotyping analysis to determine editing efficiency by repeating Steps 35Av-viii xv.
● TIMING
Steps 1–2, sgRNA design, 1 day; 1–2 hours hands on.
Steps 3–15, sgRNA synthesis, 2 days; 3 hours hands on.
Steps 16–19, Superovulation and mating, 3 days; 2 hours hands on.
Steps 20–28, Embryo collection and processing, 2 hours.
Steps 29–34, Complex formation and electroporation, 1 hour hands on.
Step 55A, Embryo culture and genotyping, 4–5 days; 4–6 hours hands on
Step 35B, Oviduct Transfer, 19 days; 2–3 hours hands on
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
Table 2.
Troubleshooting Advice
| Step | Problem | Possible Reason | Solution |
|---|---|---|---|
| 3 | Failed PCR amplification of DNA template for in vitro transcription |
Failed PCR reaction, DNase contamination. |
We recommend running 5 µl of the 50 µl reaction on a 2% (wt/vol) agarose gel to confirm PCR product size and PCR efficiency/specificity. Faint or smeared bands indicate failed PCR reactions. One can attempt the PCR genotyping again with optimized primer design and PCR reagents. |
| 15 | Failed sgRNA synthesis. |
Failed in vitro
transcription, RNase contamination. |
We recommend evaluating sgRNA quality using bioanalyzer analysis. Denaturing the sample by pre-heating at 70°C for 5 minutes before running on the bioanalyzer removes additional bands by denaturing any secondary structure (Supplementary Fig. 8). If sgRNA synthesis fails, repeat IVT with RNase-free reagents. |
| 18 | Failed superovulation |
Hormone leaks during IP injection, expiration of hormones. |
If hormone leaks during IP injection, re- inject a half dose of PMSG or hCG (~3 IU). When using a new batch of hormone, we recommend to perform a positive control by IP injection of validated hormones that works well in the past. |
| 20 | Suboptimal embryo yield |
Lack of plugged females. |
In our experience, roughly 50% of non- plugged females have fertilized embryos. We suggest collecting from the non- plugged females, with the anticipation to recover fewer fertilized embryos. |
| 26 | Embryos fail to develop after electroporation. |
Inappropriate embryo culture. |
Embryos are sensitive to perturbation of culture conditions, such as temperature, oxygen, and humidity. Calibrate temperature and CO2 level of your incubator on a monthly basis. KSOM+AA+BSA is sensitive to pH fluctuations when exposed to air. (Supplementary Fig. 6). Do not leave embryos in M2+BSA wash droplets at room temperature for longer than 5–10 minutes at a time (Supplementary Fig. 7). |
| 26 | Embryos fail to develop after electroporation. |
Embryos are lysed/inviable. |
Low embryo yield or poor-quality embryos can be caused by compromised hormones, poor injection technique, or improper hormone dosage. The vasculature of the ovary can provide an indication of proper superovulation—a red, highly vascularized ovary has undergone superovulation, while a pale ovary suggests unsuccessful superovulation. Embryos that do not develop or appear lysed could be due pipetting steps that are done too vigorously (Supplementary Fig. 9). If problem persists, replace hormones and ensure injections are administered in the appropriate time window (Fig 5b). |
| 27 | Embryos show low editing efficiency |
Insufficient Acid Tyrode’s treatment |
To determine optimal Acid Tyrode’s treatment duration, transfer 2–3 embryos to the droplet while viewing under a stereomicroscope. Determine how many seconds are required to completely dissolve the zona pellucida, then test again using half this amount of time. The optimal treatment duration is typically close to half the time required to completely dissolve the zona pellucida. On average, it takes about 1–2 minutes. If the Zona is still present after 2 minutes, replace aliquot and test on 3–4 more zygotes. |
| 32 | Low viability/low editing. |
Pulse number optimization. |
A certain balance can be struck when performing the electroporation. Fewer pulses have less impact on viability, but lower editing efficiency. More pulses tend to have higher editing but with a slight loss in viability. For complex genome editing schemes, we recommend trying different pulse numbers to enhance editing efficiency. |
| 32 | Low viability/low editing |
Electroporator Functionality |
If there are doubts whether the machine delivers the correct voltage at the correct intervals, an electrical engineer or the manufacturer technical services could be consulted to confirm the instrument in proper working order. |
| 35Avi | Failed genotyping PCR |
Inefficient lysis or lost embryo. |
Attempt PCR with robust primers to confirm template is present. It is also possible the embryo was lost during transfer to the microcentrifuge tube. Check for the presence of an embryo immediately prior to lysis. |
| 35Avi | All samples are edited |
Premature lysis. | Since embryos are used for this protocol, essentially only a few dozen molecules of template DNA are used per reaction. It is important to use appropriate non-template and water only controls to ensure the measured editing efficiency is accurate. |
ANTICIPATED RESULTS
Once the desired editing strategy is designed, our protocol typically yields >100 µg of sgRNA at >6000 ng/µl concentration for efficient Cas9/sgRNA RNP assembly. In both C57BL/6J and C57BL/6N mouse strains, one can achieve 100% embryo delivery of Cas9/sgRNA RNPs, with or without a ssODN (tested up to 162 nt in length). For NHEJ-mediated editing experiments, our superovulation protocol typically recovers 10–20 viable embryos per plugged female, ~80% of which are fertilized and viable, and ideal for electroporation. The editing efficiency ranges from 3% to 100% when engineering genomic deletions (21 independent experiments, 1275 zygotes electroporated, 249 pups born; Fig. 3e, g, Supplementary Fig 2,3, Supplementary Table 2, 3), from 50% to 100% when engineering indel mutations (3 independent experiments, 405 embryos electroporated, 97 pups born; Fig. 2c-i, Supplementary Table 1; data not shown), and from 14% to 63% when engineering precise small sequence replacements (3 independent experiments, 439 zygotes electroporated, 125 pups born; Supplementary Table 1; personal communications, Polina Lishko and Ellen Robey). Taken together, CRISPR-EZ outperforms microinjection in a variety of CRISPR genome engineering applications in mice.
Supplementary Material
| Component | Amount (ml) | Final concentration |
|---|---|---|
| 1 M HEPES, pH=7.5 | 1.0 | 100 mM |
| 3 M KCl | 2.5 | 750 mM |
| 1 M MgCl2 | 0.05 | 5 mM |
| 100% Glycerol | 5.0 | 50% (vol/vol) |
| Nuclease-free H2O | 0.95 |
| Component | Amount (ml) | Final concentration |
|---|---|---|
| 1 M KCl | 0.5 | 50 mM |
| 1 M Tris-HCl, pH=8.5 | 0.1 | 10 mM pH=8.5 |
| 1 M MgCl2 | 0.025 | 2.5 mM |
| Gelatin (powder) | 1 mg | 0.1 mg/ml |
| Nonidet P-40 | 0.045 | 0.45% (vol/vol) |
| 20% Tween 20 | 0.225 | 0.45% (vol/vol) |
| Nuclease-free H2O | To 10 ml | - |
| Component | Amount (µl) | Final Concentration |
|---|---|---|
| H2O | 35.5 | - |
| 5x Phusion HF Buffer | 10 | 1X |
| 10 mM dNTPs | 1 | 0.2 mM |
| IVT-FWD primer (1 µM) | 1 | 0.02 µM |
| IVT-REV primer (1 µM) | 1 | 0.02 µM |
| IVT-SGRNA-LONG oligo (100 µM) | 0.5 | 1 µM |
| IVT-SCAFFOLD-LONG oligo (100 µM) | 0.5 | 1 µM |
| Phusion HF (2 Units/µl) | 0.5 | 0.02 Units |
| Total | 50 | - |
| Cycle Number | Denature | Anneal | Extend |
|---|---|---|---|
| 1 | 95°C, 2 min | ||
| 2–31 | 95°C, 10 s | 72°C, 10 s | 72°C, 10 s |
| 32 | 72°C, 2 min |
| Component | Amount (µl ) | Final Concentration |
|---|---|---|
| 10x HiScribe Reaction Buffer | 2 | 1X |
| ATP (100 mM) | 2 | 10 mM |
| GTP (100 mM) | 2 | 10 mM |
| CTP (100 mM) | 2 | 10 mM |
| UTP (100 mM) | 2 | 10 mM |
| DNA template | 8 | (Step 3) |
| T7 RNA polymerase mix | 2 | - |
| Total | 20 | - |
| Component | Amount (µl) | Working Concentration |
|---|---|---|
| 5x RNP Buffer + TCEP | 2 | 1x |
| 61 µM Cas9 Protein | 1.31 | 8 µM |
| 2 µg/µl sgRNA (from Step 15) | 2 | 4 µg |
| TE Buffer pH 7.4 | 4.69 | - |
| Total | 10 | - |
| Component | Amount (µl) | Working Concentration |
|---|---|---|
| 5x RNP Buffer + TCEP | 2 | 1x |
| 61 µM Cas9 Protein | 1.31 | 8 µM |
| 2 µg/µl sgRNA (from Step 15) | 2 | 4 µg |
| TE Buffer pH 7.4 | 3.69 | - |
| 200 µM ssDNA oligo (optional) | 1 | 20 µM |
| Total | 10 | - |
| Component | Amount (µl) | Working Concentration |
|---|---|---|
| 5x RNP Buffer + TCEP | 2 | 1x |
| 61 µM Cas9 Protein | 2.62 | 16 µM |
| 2 µg/µl sgRNA 1 (from Step 15) | 2 | 4 µg |
| 2 µg/µl sgRNA 2 (from Step 15) | 2 | 4 µg |
| TE Buffer pH 7.4 | 1.38 | - |
| Total | 10 µl | - |
| Condition | Value |
|---|---|
| Voltage | 30 V |
| Pulses | 4–6 |
| Pulse Length | 3 ms |
| Pulse Interval | 100 ms |
| Cycle Number | Denature | Anneal | Extend |
|---|---|---|---|
| 1 | 95°C, 2 min | ||
| 2–31 | 95°C, 10 s | 60°C, 30 s | 72°C, 10 s |
| 32 | 72°C, 10 min |
| Cycle Number | Denature | Anneal | Extend |
|---|---|---|---|
| 1 | 95°C, 2 min | ||
| 2–31 | 95°C, 10 s | 60°C, 30 s | 72°C, 10 s |
| 32 | 72°C, 10 min |
ACKNOWLEDGMENTS
We thank A Lee, B Lee and for technical assistance. We thank C. Di Biagio for help in making the Protocol Video. We thank C Jeans and the QB3 Macrolab for purified Cas9 protein used during testing. We thank J Doudna, D Carrol, J Corn for stimulating discussions, and N Anchell, P Dao, K Jager, and D Young for zygote collection, electroporation, and embryo transfer. We thank Kristin N. Grimsrud for veterinary oversight. L.H. is supported by a HHMI faculty scholar award, a Bakar fellow award at UC-Berkeley, and several grants from the National Institutes of Health (NIH, R01GM114414, R01CA139067, 2R01CA139067, 1R21HD088885 and R21HD088885) and a research scholar award from American Cancer Society (ACS). AJM is supported by a F32 postdoctoral fellowship from NIH (CA192636–03). SC is supported by a CCRC predoctoral fellowship. JAW and KCL acknowledge the support from two NIH grants (UM1 OD003321 and U42 OD011175) and a grant from the American College of Laboratory Animal Medicine.
Footnotes
EDITORIAL SUMMARY: CRISRP-EZ (CRISPR RNP Electroporation of Zygotes) achieves 100% delivery of Cas9/sgRNA ribonucleoproteins (RNPs) by zygote electroporation, enabling efficient indels, exon deletions, point mutations, and small insertions in mouse genome, and outperforming microinjection-based methods.
TWEET: CRISPR-EZ employs zygote electroporation to deliver RNPs for efficient mouse genome editing
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA & Charpentier E A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science (80-. ) 337, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE & Church GM RNA-Guided Human Genome Engineering via Cas9. Science (80-. ) 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA & Zhang F Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–23 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F & Jaenisch R One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M, Li Y, Gao N, Wang L, Lu X, Zhao Y & Liu M Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol 31, 681–683 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Yasue A, Mitsui SN, Watanabe T, Sakuma T, Oyadomari S, Yamamoto T, Noji S, Mito T & Tanaka E Highly efficient targeted mutagenesis in one-cell mouse embryos mediated by the TALEN and CRISPR/Cas systems. Sci. Rep 4, 5705 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujii W, Kawasaki K, Sugiura K & Naito K Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res 41, e187–e187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang H, Wang H, Shivalila CS, Cheng AW, Shi L & Jaenisch R One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hashimoto M, Yamashita Y & Takemoto T Electroporation of Cas9 protein/sgRNA into early pronuclear zygotes generates non-mosaic mutants in the mouse. Dev. Biol 418, 1–9 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Tu Z, Yang W, Yan S, Yin A, Gao J, Liu X, Zheng Y, Zheng J, Li Z, Yang S, Li S, Guo X & Li X-J Promoting Cas9 degradation reduces mosaic mutations in non-human primate embryos. Sci. Rep 7, 42081 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aida T, Chiyo K, Usami T, Ishikubo H, Imahashi R, Wada Y, Tanaka KF, Sakuma T, Yamamoto T & Tanaka K Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol 16, 87 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung CJ, Zhang J, Trenchard E, Lloyd KC, West DB, Rosen B & de Jong PJ Efficient gene targeting in mouse zygotes mediated by CRISPR/Cas9-protein. Transgenic Res 26, 263–277 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, Sridharan M, Carte J, Chen W, Roark N, Ranganathan S, Ravinder N & Chesnut JD Rapid and Highly Efficient Mammalian Cell Engineering via Cas9 Protein Transfection. J. Biotechnol 208, 44–53 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Skarnes WC Is mouse embryonic stem cell technology obsolete? Genome Biol 16, 109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sung YH, Kim JM, Kim H-T, Lee J, Jeon J, Jin Y, Choi J-H, Ban YH, Ha S-J, Kim C-H, Lee H-W & Kim J-S Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Res 24, 125–31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen S, Lee B, Lee AY-F, Modzelewski AJ & He L Highly Efficient Mouse Genome Editing by CRISPR Ribonucleoprotein Electroporation of Zygotes. J. Biol. Chem 291, 14457–67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W, Kutny PM, Byers SL, Longstaff CJ, DaCosta MJ, Pang C, Zhang Y, Taft RA, Buaas FW & Wang H Delivery of Cas9 protein into mouse zygotes through a series of electroporation dramatically increased the efficiency of model creation. J. Genet. Genomics (2016). at <http://www.sciencedirect.com/science/article/pii/S1673852716300029> [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hur JK, Kim K, Been KW, Baek G, Ye S, Hur JW, Ryu S-M, Lee YS & Kim J-S Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat. Biotechnol 34, 807–808 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Qin W, Dion SL, Kutny PM, Zhang Y, Cheng AW, Jillette NL, Malhotra A, Geurts AM, Chen Y-G & Wang H Efficient CRISPR/Cas9-Mediated Genome Editing in Mice by Zygote Electroporation of Nuclease. Genetics 200, 423–430 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hashimoto M & Takemoto T Electroporation enables the efficient mRNA delivery into the mouse zygotes and facilitates CRISPR/Cas9-based genome editing. Sci. Rep 5, 11315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi G, Gurumurthy CB, Wada K, Miura H, Sato M & Ohtsuka M GONAD: Genome-editing via Oviductal Nucleic Acids Delivery system: a novel microinjection independent genome engineering method in mice. Sci. Rep 5, 11406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaneko T, Mashimo T, Zhang Y, Zernicka-Goetz M, Tokumasu D & Sakane Y Simple Genome Editing of Rodent Intact Embryos by Electroporation. PLoS One 10, e0142755 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen Z-Y & Liu DR Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol 33, 73–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen S, Lee B, Lee AY-F, Modzelewski AJ & He L Highly Efficient Mouse Genome Editing by CRISPR Ribonucleoprotein Electroporation of Zygotes. J. Biol. Chem 291, 14457–67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh P, Schimenti JC & Bolcun-Filas E A mouse geneticist’s practical guide to CRISPR applications. Genetics 199, 1–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F & Jaenisch R One-step generation of mice carrying mutations in multiple genes by CRISPR/cas-mediated genome engineering. Cell 153, 910–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanihara F et al. Somatic cell reprogramming-free generation of genetically modified pigs. Sci. Adv 2, e1600803 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakagawa Y, Sakuma T, Nishimichi N, Yokosaki Y, Yanaka N, Takeo T, Nakagata N & Yamamoto T Ultra-superovulation for the CRISPR-Cas9-mediated production of gene-knockout, single-amino-acid-substituted, and floxed mice. Biol. Open 5, 1142–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park K-E, Powell A, Sandmaier SES, Kim C-M, Mileham A, Donovan DM & Telugu BP Targeted gene knock-in by CRISPR/Cas ribonucleoproteins in porcine zygotes. Sci. Rep 7, 42458 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aida T, Chiyo K, Usami T, Ishikubo H, Imahashi R, Wada Y, Tanaka KF, Sakuma T, Yamamoto T & Tanaka K Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol 16, 87 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z, Lv J, Xie X, Chen Y, Li Y, Sun Y, Bai Y, Songyang Z, Ma W, Zhou C & Huang J CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 6, 363–372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeWitt MA, Corn JE & Carroll D Genome editing via delivery of Cas9 ribonucleoprotein. Methods 121–122, 9–15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willis BJ, Lloyd KCK & Wood JA High throughput electroporation of zygotes increases efficiency in the generation of genetically edited rodent models using CRISPR-Cas9. in Transgenic Animal Research Conference XI (2017). [Google Scholar]
- 34.Doetschman T, Gregg RG, Maeda N, Hooper ML, Melton DW, Thompson S & Smithies O Targetted correction of a mutant HPRT gene in mouse embryonic stem cells. Nature 330, 576–8 (1987). [DOI] [PubMed] [Google Scholar]
- 35.Doetschman T, Maedat N & Smithiestt O Targeted mutation of the Hprt gene in mouse embryonic stem cells (gene targeting/embryonic stem cells/homologous recombination/polymerase chain reaction amplification). Genetics 85, 8583–8587 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW & Zhang F Resource One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR / Cas-Mediated Genome Engineering. Cell 153, 910–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quadros RM et al. Easi-CRISPR: a robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol 18, 92 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Howe CC & Solter D Cytoplasmic and nuclear protein synthesis in preimplantation mouse embryos. J. Embryol. Exp. Morphol 52, 209–25 (1979). [PubMed] [Google Scholar]
- 39.CULLEN BR, EMIGHOLZ K & MONAHAN JJ Protein Patterns of Early Mouse Embryos During Development. Differentiation 17, 151–160 (1980). [DOI] [PubMed] [Google Scholar]
- 40.Wang W, Kutny PM, Byers SL, Longstaff CJ, DaCosta MJ, Pang C, Zhang Y, Taft RA, Buaas FW & Wang H Delivery of Cas9 Protein into Mouse Zygotes through a Series of Electroporation Dramatically Increases the Efficiency of Model Creation. J. Genet. Genomics 43, 319–327 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagy A, Gertenstein M, Vintersten K & Behringer R Manipulating the Mouse Embryo: A Laboratory Manual Third Edition. (Cold Spring Harbor Laboratory Press, 2003). doi: 10.1086/380032 [DOI] [Google Scholar]
- 42.Zhu L, Zhang Y, Zhang W, Yang S, Chen J-Q & Tian D Patterns of exon-intron architecture variation of genes in eukaryotic genomes. BMC Genomics 10, 47 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schaefer KA, Wu W-H, Colgan DF, Tsang SH, Bassuk AG & Mahajan VB Unexpected mutations after CRISPR–Cas9 editing in vivo. Nat. Methods 14, 547–548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakajima K, Kazuno A, Kelsoe J, Nakanishi M, Takumi T & Kato T Exome sequencing in the knockin mice generated using the CRISPR/Cas system. Sci. Rep 6, 34703 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iyer V, Shen B, Zhang W, Hodgkins A, Keane T, Huang X & Skarnes WC Off-target mutations are rare in Cas9-modified mice. Nat. Methods 12, 479–479 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX & Zhang F Rationally engineered Cas9 nucleases with improved specificity. Science (80-. ) 351, 84–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z & Joung JK High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moreno-Mateos MA, Vejnar CE, Beaudoin J-D, Fernandez JP, Mis EK, Khokha MK & Giraldez AJ CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 12, 982–988 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu H, Xiao T, Chen C-H, Li W, Meyer C, Wu Q, Wu D, Cong L, Zhang F, Liu JS, Brown M & Liu SX Sequence determinants of improved CRISPR sgRNA design. Genome Res. gr. 191452.115 (2015). at <http://genome.cshlp.org/content/early/2015/06/10/gr.191452.115> [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horlbeck MA, Witkowsky LB, Guglielmi B, Replogle JM, Gilbert LA, Villalta JE, Torigoe SE, Tijan R & Weissman JS Nucleosomes impede Cas9 access to DNA in vivo and in vitro. Elife 5, e12677 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin S, Staahl B, Alla RK & Doudna JA Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3, e04766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Godine JE, Chin WW & Habener JF alpha Subunit of rat pituitary glycoprotein hormones. Primary structure of the precursor determined from the nucleotide sequence of cloned cDNAs. J. Biol. Chem 257, 8368–71 (1982). [PubMed] [Google Scholar]
- 53.Wassarman PM Mammalian Fertilization: Molecular Aspects of Gamete Adhesion, Exocytosis, and Fusion. Cell 96, 175–183 (1999). [DOI] [PubMed] [Google Scholar]
- 54.Malter HE & Cohen J Blastocyst formation and hatching in vitro following zona drilling of mouse and human embryos. Gamete Res 24, 67–80 (1989). [DOI] [PubMed] [Google Scholar]
- 55.Saito T In vivo electroporation in the embryonic mouse central nervous system. Nat. Protoc 1, 1552–1558 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Escoffre J-M, Portet T, Wasungu L, Teissié J, Dean D & Rols M-P What is (Still not) Known of the Mechanism by Which Electroporation Mediates Gene Transfer and Expression in Cells and Tissues. Mol. Biotechnol 41, 286–295 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Urnov FD, Miller JC, Lee Y-L, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD & Holmes MC Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435, 646–651 (2005). [DOI] [PubMed] [Google Scholar]
- 58.Kim HJ, Lee HJ, Kim H, Cho SW & Kim J-S Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res 19, 1279–88 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morbeck DE & Leonard PH in Embryo Culture 325–331 (Humana Press, 2012). doi: 10.1007/978-1-61779-971-6_18 [DOI] [Google Scholar]
- 60.Wang Z & Storm DR Extraction of DNA from mouse tails. Biotechniques 41, 410, 412 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.