

Abstract
Recent progress in coarse-grained (CG) molecular modeling and simulation has facilitated an influx of computational studies on biological macromolecules and their complexes. Given the large separation of length- and time-scales that dictate macromolecular biophysics, CG modeling and simulation are well-suited to bridge the microscopic and mesoscopic or macroscopic details observed from all-atom molecular simulations and experiments, respectively. In this review, we first summarize recent innovations in the development of CG models, which broadly include structure-based, knowledge-based, and dynamics-based approaches. We then discuss recent applications of different classes of CG models to explore various macromolecular complexes. Finally, we conclude with an outlook for the future in this ever-growing field of biomolecular modeling.
Introduction
Many biological processes rely on macromolecules to serve as building blocks for large-scale complexes, exemplified by viruses, ribosomes, and cytoskeletal filaments [1–3]. These so-called macromolecular complexes often contain many copies of the same macromolecule that collectively aggregate through non-covalent interactions into ordered and functional suprastructures [4]. Furthermore, the lifecycles of these complexes are inherently dynamic, in which configurational transitions act as regulatory signals [5].
Since molecular phenomena at the scale of individual macromolecules translates into emergent and collective macroscopic behavior, it is clear that a fundamental understanding of these intriguing biophysical complexes requires a hierarchical approach. Recent advances in experimental techniques now offer multifaceted insights into these systems. For example, ensemble-averaged atomic structures can be resolved at high-resolution using X-ray crystallography or cryo-electron microscopy [6,7]. For dynamic information, one may use fluorescence techniques [8,9] or nuclear magnetic resonance (NMR) spectroscopy [10,11], albeit with lower spatial resolution. To complement these experimental approaches, theorists may attempt to leverage all-atom (AA) molecular dynamics (MD) simulations to access dynamical phenomena with molecular-scale resolution [12].
Within the space of MD simulation techniques, coarse-grained (CG) modeling and simulation are particularly attractive for the study of macromolecular complexes. By design, CG models are reduced representations of AA models that aim to retain the essential molecular aspects for the biophysical system of interest. As a result, CG simulations have three primary benefits in comparison to AA simulations. First, these models enable simulations of large, biologically-relevant systems by virtue of the reduced number of particles. Second, the removal of highly-fluctuating atomic degrees of freedom facilitates faster configurational sampling, as both larger integration time steps can be used while the underlying free energy surface is smoother. Hence, the combination of these first two benefits may facilitate observation of interesting collective behavior. Finally, the construction of useful CG models grants tacit insight into molecular features (i.e., from CG mappings) and interactions (i.e., from CG parameterizations) that are essential for biophysical function. For these reasons, CG simulations may provide insights and perspectives that would otherwise be inaccessible from AA molecular simulations, which has indeed driven the continued use and development of CG models and methods.
In this review, we summarize recent advances in the development and application of CG models for studying the structure and dynamics of biological macromolecules. In particular, we focus on highly CG models for proteins and nucleic acids, i.e., per-residue resolutions or coarser, which enable large-scale simulations of CG macromolecular complexes. Interested readers may refer to other in-depth reviews that discuss related topics, including higher-resolution CG models and ultra-low-resolution phenomenological models for proteins, membranes, carbohydrates and nucleic acids [13–17]. We first broadly describe the most common methodologies for CG modeling that have been developed over the last five years, with our intention to expand upon previous reviews on this subject, e.g., references [18,19]. We then survey recent uses of these models for macromolecular complexes, for which CG models have provided new insights. We conclude with a brief summary and discussion on the future outlook of the field.
Coarse-Grained Modeling of Large Biomolecules
As CG models have grown in both popularity and utility, so have the strategies for generating CG models of macromolecular complexes. Here, we focus on low-resolution models that have been used extensively; specifically, we refer to CG models that resolve proteins (nucleic acids) at the per-residue (per-backbone/sidechain) level or coarser. All procedures for CG modeling must answer two questions: how do we define the correspondence between atomic and CG degrees of freedom (i.e., mapping) and how do we define the effective interactions between CG sites (i.e., energetics)? Within our scope, we further classify three methodological avenues that have been used to approach these questions. We denote these as structure-based, knowledge-based, and dynamics-based approaches, which are each described below and schematically shown in Figure 1.
Figure 1.
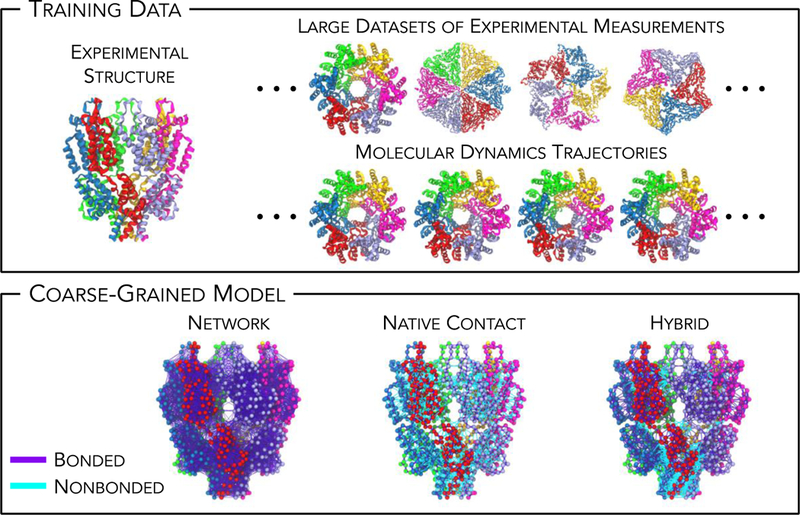
Different avenues of approach for model development of highly coarse-grained (CG) macromolecules, which are broadly classified into structure-based, knowledge-based, and dynamics-based strategies. Here, the capsid and spacer peptide 1 (CA-SP1) domains of the human immunodeficiency virus (HIV) protein is used as a representative example. (Top) Each model class relies on different training datasets and methods; for example, an experimental structure (PDB 5L93), an extended dataset of experimental measurements (PDB 5L93, 3ZX8, 3J37 and 6BVF), or statistics from an all-atom molecular dynamics trajectory (initial structures from PDB 5L93), respectively. (Bottom) Resultant models can be classified as network-based and native-contact-based models (or hybrids thereof); bonded (nonbonded) interactions are depicted by solid purple (cyan) lines while CG sites (per residue) are depicted as spheres.
Structure-based approaches.
As perhaps the most prevalent CG approach in the literature, structure- based methods aim to leverage atomic-resolution experimental data on native structures to construct CG models. An underlying assumption in these models is that conservation of close contacts between residues that are observed in native structures are important for the functional dynamics of these biomolecules. Based on this intuition, mapping is typically prescribed such that each CG site represents a different residue, which, for example, may be based on C-α positions.
Two broad methods exist to describe the energetics of these CG models, i.e., the effective CG interactions. The first strategy is to use so-called network models in which the CG molecule is described by a graph of proximal masses that are connected by springs; by construction, the ground state predicted by network models yields the experimental structure. In the past, several network models have been proposed [20–23], which, when combined with spectral graph analysis techniques (e.g., normal mode or principal component analysis), provide considerable information on the conformational modes of macromolecules. The other strategy is to use so-called (off-lattice) Gō models [24,25], which instead describe native contacts using attractive, non-bonded interactions, while all other (i.e., non-native) interactions are assumed to be purely repulsive. However, in all of these earlier methods, the prescribed energy landscape is funneled such that only states that minimally frustrate the experimental topology are allowed [26]. Consequently, both network and Gō models have limited usability when large conformational transitions or changes in chemical environment (e.g., through mutations or ligand interactions) are of interest.
To address these deficiencies, variants of both network and native-contact models have recently been reported. The ability to investigate large-amplitude changes during conformational transitions, for example, has been enabled by network models [27,28] and Gō models [29–31] that utilize an energy landscape constructed from the mixture of single-state energetics from two or more different conformational states. However, it remains to be seen if physically-relevant transition states and associated pathways can be predicted by these methods, which warrants further investigation. Other proposed variants have suggested the need to go beyond simple harmonic (Lennard-Jones) interactions that are common in network (native-contact) models. For instance, algorithms inspired from graph theory [32,33] have been proposed as a means to differentiate network weights based on chemical fragments to ultimately improve accuracy. In alternative formulations, network bonds have been replaced by local density kernels for flexibility analysis [34]. Native-contact models have undergone a similar treatment. To represent implicit anisotropic interactions (e.g., due to the presence of side chains), which may only be active in certain configurations, virtual binding sites have been introduced [35,36]. The self-organizing polymer (SOP) model has also been suggested as a variant of Gō models with softer bonded and repulsive interactions. This approach ostensibly improves agreement with force-induced folding and unfolding behavior, and has shown empirical success for both proteins and RNA [37–39].
Importantly, many recent CG models have opted to hybridize both network-based and native-contact-based approaches [35,36,40–43]; for example, a network model could represent different intra-protein conformers, while a native-contact model represents inter-protein interfaces. Indeed, this type of approach is well-suited for studies of large-scale macromolecular complexes. However, several aspects of structure-based CG modeling remain open-ended. Mapping of CG sites, for example, is almost entirely decided by chemical intuition, and mostly at the level of C-α atoms. While systematic mapping methods for structure-based CG models are not as prevalent, some promising directions have recently been reported. For instance, graph decimation methods have been shown to generate a hierarchy of network model resolutions, which may be easily extended as a mapping operator [33]. In addition, the ability to discriminate phenomena based on physical chemistry principles remains a challenge. In the next section, we describe knowledge-based CG models which attempt to resolve some of these issues.
Knowledge-based approaches.
Broadly speaking, we define knowledge-based approaches as CG parameterization strategies that leverage the growing collection of different macromolecules (and their conformers) with solved experimental structures or measured macroscopic properties. Here, the goal is to design CG models with greater degrees of transferability and chemical specificity, i.e., a generalized model that can be used to independently describe any given macromolecular assembly of interest. Knowledge-based approaches have been used in the past to propose residue-specific pair interactions, such as in the well-known Miyazama-Jernigan potential [44,45]. Recently, there has been a resurgence of knowledge-based approaches, which have been timely given advances in macromolecular structure characterization and statistical methodologies.
Knowledge-based models that build upon aforementioned structure-based models have been proposed for both protein and nucleic acid CG models. The primary distinction between these methods is the choice of the target observable. For instance, parameter sets for network models have been introduced that delineate different inter-residue coupling forces to reproduce experimental Debye-Waller factors [46]. On the basis of statistical distributions of residue contacts observed in training sets of experimental structures, it is possible to formulate hybrid network/native-contact models that capture pairwise energies [47], vibrational entropies [42,43], relative entropies [48,49], and maintained contacts [50]. In the case of intrinsically disordered proteins, in which native states are largely unknown, large datasets of radius of gyrations were used to parameterize effective CG potentials. Similar approaches have been adopted for RNA, although it appears that more complex CG interactions are needed to also account for base-pair orientation [51,52].
A prime example of knowledge-based models has been for protein structure prediction and homology modeling, e.g., as evaluated by the Critical Assessment of Structure Prediction (CASP) experiments [53]. To this end, one consideration that requires further investigation is the transferability of current knowledge-based models. In practice, training sets are composed of related proteins, and it is unclear if models generated from one set (e.g., globular proteins) can be successful applied toward another set (e.g., intrinsically disordered proteins).
Another promising direction for knowledge-based models is to leverage Bayesian inference techniques, which have previously been applied to calibrate and select optimal force fields for MD simulations [54–56]. Within our scope, the question is how can we infer reference atomic distributions from CG data? Recently, an approach was reported that sought to determine optimal mappings and energetics of individual proteins by the use of a combination of experimental atomic models and low-resolution cryoEM density maps [57,58].
Dynamics-based approaches.
While the previous two approaches are largely dependent on experimental data, dynamics-based approaches instead use systematic algorithms to derive CG mappings and energetics based on molecular-level statistics from AA MD simulations. Mapping procedures, for instance, may determine the optimal clustering of atoms into CG sites to recapitulate the lowest-frequency collective motions [59,60] or to maximize the relative fluctuations between sites [61] for a given number of CG particles. Determination of the optimal number of CG sites is also possible based on scaling laws for the residual thermal fluctuations obtained from CG-mapped AA trajectories [62]. Similarly, methods to parameterize network models to recapitulate fluctuations from MD simulations have been reported [63,64], while generalized extensions of this model have been trained on ensembles of proteins with different force-fields [65,66]. However, while numerous strategies have been developed to systematically parameterize CG interactions [67–74], these methods have found limited use in CG modeling studies of macromolecular complexes. In part, the difficulty is in capturing all of the relevant physics, including many-body effects such as hydrophobicity, in CG models (e.g., due to the use of simple pairwise interactions). We note that this is one aspect of the general CG representability problem [75]. Recent work has demonstrated both the means and utility of increasingly expressive CG interactions, which include CG interactions that are based on order parameters [76,77], such as local densities [78,79]. Furthermore, the general “Ultra-CG” (UCG) machinery enables the systematic mixing of different CG interaction models, which may represent different physical (e.g., conformational changes, ligand binding) or chemical (e.g., hydrolysis, protonation) states [41,77,80,81]; these would be a natural extension to the aforementioned multi-state structure-based CG models. Another general challenge is that CG models based on statistical mechanics formulations may preclude transferability, given their inherent dependence on thermodynamic state [75]. However, empirical evidence on the utility of state-dependent CG potentials for improved transferability has begun to emerge [82,83], and careful analysis on the origins of this behavior may provide new insights into this problem. Finally, dynamics-based approaches can be augmented with knowledge of experimental observables, which has recently been proposed as an integrated framework to generate new classes of CG models [84].
Applications of Coarse-Graining for Macromolecular Complexes
Recent advances in CG models have resulted in new mechanistic insights into large macromolecular complexes and their polymeric or aggregated assemblies. For example, network models are amenable for the exploration of collective motions with large computational efficiency [27,85]. On the other hand, native-contact models have been particularly useful in studying dynamical processes during the lifecycle of macromolecular complexes [15]. Finally, CG models have found additional utility as part of so-called integrative modeling, in which information from different hierarchies of scale (e.g., a continuum from AA to CG descriptions) are leveraged to gain hierarchical insights [86]. Below, we highlight a few recent applications of CG models, e.g., as seen in Figure 2, within these three broad categories.
Figure 2.
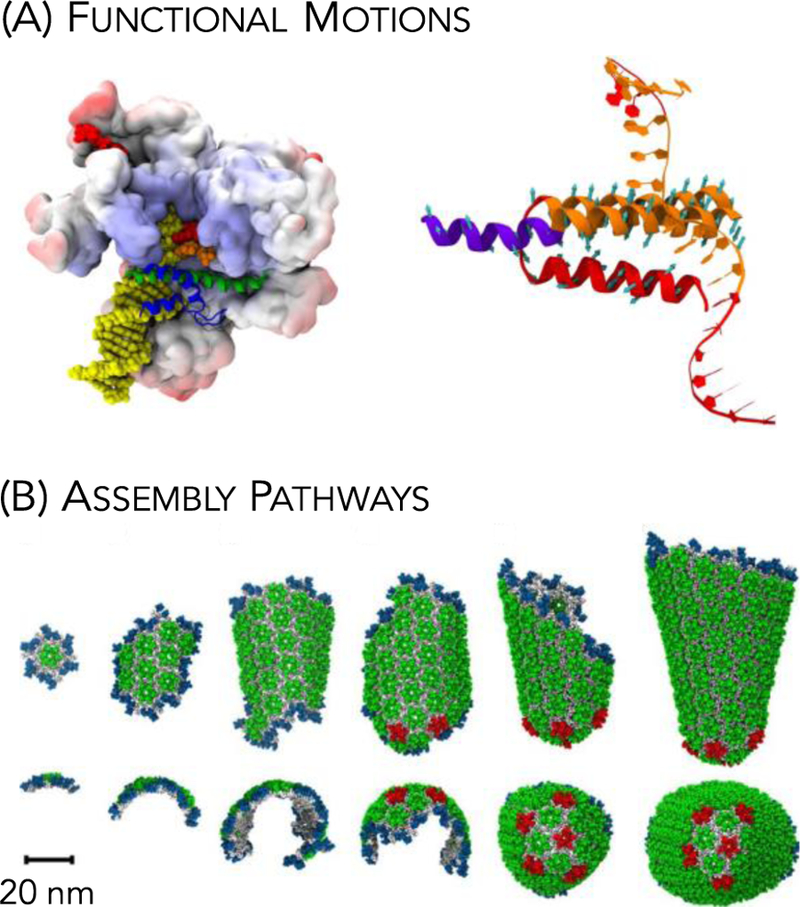
Two representative applications of coarse-grained modeling. (A) Schematic of the closed loop complex of RNA polymerase (colored surface) and RNA (yellow balls) [left] with large-scale collective motions indicated by the teal arrows along the bridge helix [right] (adapted from Ref. [34]). (B) Self-assembly pathway of the human immunodeficiency virus (HIV) CA capsid protein subunits in forming the mature capsid of infectious virions. Identified are CA hexamers (green), pentamers (red) and the growing edge of the conical capsid (blue) (adapted from Ref. [35]).
Large-scale functional motions.
Conformational transitions within macromolecular complexes have been explored using variants of both network and native-contact models. For example, flexibility analysis has revealed the collective motions that contribute to catalysis and translocation of RNA polymerase [34] (see Figure 2A) and to mechanical phenotypes of amyloid fibrils with disease expression [87]. Highly CG multi-state network models have also successfully recapitulated transition paths between the large-scale structural transitions in ribosomal complexes between the initiation and elongation states [33]. Finally, multi-state native-contact models have recently revealed the mechanism of action of motor protein motility, including kinesin [30] and myosin VI [29].
Dynamic assembly/disassembly pathways.
A key attribute of macromolecular complexes is their dynamic ability to assemble from their macromolecular constituents with high fidelity and then disassemble, to recycle, release, or replace components based on regulatory signals. Viruses are a quintessential example as viral cargo must be packaged and transported from host cells and released into newly infected cells. To this end, native-contact models have revealed hierarchical assembly modes during the replication cycle of HIV-1 (see Figure 2B) [35,36,88,89]. Similarly, dynamic de-polymerization responses of microtubules due to either mechanical forces or severing enzymes have been explored using native-contact models [90,91]. Most recently, UCG models of actin filaments under different hydrolysis states have revealed cooperativity effects that regulate polymerization of actin subunits [41].
Role of CG in hierarchical and integrative modeling.
The final aspect of CG modeling that we highlight is its integration in hierarchical modeling frameworks, in which a separation of length-and/or time-scales in the biophysical system of interest requires multiple resolutions of data that can span AA models to experimental data. For instance, CG models can accelerate the sampling of AA statistics. One such example is a multi-resolution approach known as the Adaptive Resolution Scheme (AdResS), which simultaneously models a region of interest in atomic detail within an environment of CG representation. An example would be ligand recognition in lysozymes [92]. An alternative approach is to explicitly use distributions from CG simulations to initiate AA simulations via back-mapping procedures [93] or to construct biased simulations that enhance AA configurational sampling [94]. In the opposite direction, continuum or mesoscopic fields can be applied to CG simulations. For instance, hydrodynamic flow fields, which can affect macromolecule relaxation and aggregation, have been integrated into CG MD simulations using the Lattice Boltzmann technique [95–97].
Conclusions and Future Outlook
Driven by a desire to understand the hierarchical nature of macromolecular complexes and their assemblies with molecular fidelity, the field of coarse-grained (CG) modeling and simulation continues to rapidly evolve. Here, we present recent advances in methodology and application of highly CG models for proteins and nucleic acids. Interestingly, previously classified boundaries between CG models, which broadly contain structure-based, knowledge-based, and dynamics-based approaches, appear to be melding into a unified nexus that blends these various philosophies. Nonetheless, we emphasize two main challenges to be considered for future advances. The first is that many aspects of CG modeling remain heavily reliant on either intuition or arbitrary modeling decisions. The second is that many CG models are inherently prescriptive, thereby limiting their ability to predict new phenomena. To address these issues, we anticipate that new methodological advances to generate CG mappings and energetics, especially those related to statistical inference approaches or based on a deeper understanding of systematic coarse-graining theory, will greatly expand the utility of CG modeling and simulation.
Highlights.
Coarse-grained models are reduced representations of all-atom models that aim to retain the essential molecular aspects for the biophysical system of interest.
Coarse-grained simulations may provide insights and perspectives that would otherwise be inaccessible from all-atom molecular simulations.
Coarse-grained simulations bridge insights between microscopic and mesoscopic or macroscopic phenomena.
Coarse-grained modeling strategies are discriminated on the basis of their use of experimental structural data, large datasets of experimental observables, or molecular simulations.
Advances in methodology have enabled models to more expressively capture conformational changes in macromolecular complexes.
Coarse-grained simulations have been used to elucidate hierarchical and dynamical behavior in complex macromolecular systems.
Acknowledgements
This work was supported in part by the National Institute of General Medical Sciences of the National Institutes of Health (grant P50-GM082545) and the National Science Foundation (grant CHE-1465248). Support from the Ruth L. Kirschstein National Research Service Award Postdoctoral Fellowship is gratefully acknowledged by A.J.P (grant F32-GM125218).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Disclosures
The authors declare no conflict of interest.
References
- 1.ZhangW, CaoS,Martin J L,MuellerJ D,ManskyL M:Morphology and ultrastructure ofretrovirus particles. AIMS Biophysics 2015, 2:343–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de la Cruz J, Karbstein K, Woolford JL: Functions of Ribosomal Proteins in Assembly of Eukaryotic Ribosomes In Vivo. Annu. Rev. Biochem 2015, 84:93–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huber F, Schnauß J, Rönicke S, Rauch P, Müller K, Fütterer C, Käs J: Emergent complexity of the cytoskeleton: From single filaments to tissue. Adv. Phys 2013, 62:1–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williamson JR: Cooperativity in macromolecular assembly. Nat. Chem. Biol 2008, 4:458–465. [DOI] [PubMed] [Google Scholar]
- 5.Marsh JA, Teichmann SA: Structure, Dynamics, Assembly, and Evolution of Protein Complexes. Annu. Rev. Biochem 2015,84:551–575. [DOI] [PubMed] [Google Scholar]
- 6.Shi Y: A glimpse of structural biology through X-ray crystallography. Cell 2014, 159:995–1014. [DOI] [PubMed] [Google Scholar]
- 7.Henderson R: Overview and future of single particle electron cryomicroscopy. Arch. Biochem. Biophys 2015, 581:19–24. [DOI] [PubMed] [Google Scholar]
- 8.Sieben C, Douglass KM, Guichard P, Manley S: Super-resolution microscopy to decipher multi-molecular assemblies. Curr. Opin. Struct. Biol 2018, 49:169–176. [DOI] [PubMed] [Google Scholar]
- 9.Shashkova S, Leake MC: Single-molecule fluorescence microscopy review: shedding new light on old problems. Biosci.Rep 2017,37:BSR20170031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovermann M, Rogne P, Wolf-Watz M: Protein dynamics and function from solution state NMR spectroscopy. Q.Rev. Biophys 2016, 49:e6. [DOI] [PubMed] [Google Scholar]
- 11.Kleckner IR, Foster MP: An introduction to NMR-based approaches for measuring protein dynamics. Biochim. Biophys. Acta 2011, 1814:942–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perilla JR, Goh BC, Cassidy CK, Liu B, Bernardi RC, Rudack T, Yu H, Wu Z, Schulten K: Molecular dynamics simulations of large macromolecular complexes. Curr. Opin. Struct. Biol 2015, 31:64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kmiecik S, Gront D, Kolinski M, Wieteska L, Dawid AE, Kolinski A: Coarse-Grained Protein Models and Their Applications. Chem. Rev 2016, 116:7898–7936. [DOI] [PubMed] [Google Scholar]
- 14.Ingólfsson HI, Lopez CA, Uusitalo JJ, de Jong DH, Gopal SM, Periole X, Marrink SJ: The power of coarse graining in biomolecular simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci 2014, 4:225–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takada S, Kanada R, Tan C, Terakawa T, Li W, Kenzaki H: Modeling Structural Dynamics of Biomolecular Complexes by Coarse-Grained MolecularSimulations. Acc.Chem. Res 2015,48:3026–3035. [DOI] [PubMed] [Google Scholar]
- 16.Noid WG: Perspective: Coarse-grained models for biomolecular systems. J. Chem. Phys 2013, 139:090901. [DOI] [PubMed] [Google Scholar]
- 17.Simunovic M, Bassereau P, Voth GA: Organizing membrane-curving proteins: the emerging dynamical picture. Curr. Opin. Struct. Biol 2018, 51:99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saunders MG, Voth GA: Coarse-graining of multiprotein assemblies. Curr. Opin. Struct. Biol 2012, 22:144–150. [DOI] [PubMed] [Google Scholar]
- 19.Takada S: Coarse-grained molecular simulations of large biomolecules. Curr. Opin. Struct.Biol 2012, 22:130–137. [DOI] [PubMed] [Google Scholar]
- 20.Tirion MM: Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett 1996, 77:1905–1908. [DOI] [PubMed] [Google Scholar]
- 21.Haliloglu T, Bahar I, Erman B: Gaussian dynamics of folded proteins. Phys. Rev. Lett 1997, 79:3090–3093. [Google Scholar]
- 22.Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, Bahar I: Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J 2001, 80:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin T-L, Song G: Generalized spring tensor models for protein fluctuation dynamics and conformation changes. BMC Struct. Biol 2010, 10:S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gō N: Theoretical Studies of Protein Folding. Annu. Rev. Biophys. Bioeng 1983, 12:183–210. [DOI] [PubMed] [Google Scholar]
- 25.Clementi C, Nymeyer H, Onuchic JN: Topological and energetic factors: What determines the structural details of the transition state ensemble and ‘en-route’ intermediates for protein folding? An investigation for small globular proteins. J. Mol. Biol 2000, 298:937–953. [DOI] [PubMed] [Google Scholar]
- 26.Nymeyer H, Garcia AE, Onuchic JN: Folding funnels and frustration in off-lattice minimalist protein landscapes. Proc. Natl. Acad. Sci. USA 1998, 95:5921–5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng W, Wen H: A survey of coarse-grained methods for modeling protein conformational transitions. Curr. Opin. Struct. Biol 2017, 42:24–30. [DOI] [PubMed] [Google Scholar]
- 28.Das A, Gur M, Cheng MH, Jo S, Bahar I, Roux B: Exploring the Conformational Transitions of Biomolecular Systems Using a Simple Two-State Anisotropic Network Model. PLoS Comp. Biol 2014, 10:e1003521.[*] This paper introduces a simple yet efficient method to approximate large conformational transition pathways using two-state anisotropic network models.
- 29.Mugnai ML, Thirumalai D: Kinematics of the lever arm swing in myosin VI. Proc. Natl. Acad. Sci. USA 2017,114:E4389–E4398.[*] The mechanism of myosin VI motility is revealed through a multi-state self-organizing polymer model that captures the conformational transitions associated with the power stroke.
- 30.Zhang Z, Thirumalai D: Dissecting the kinematics of the kinesin step. Structure 2012, 20:628–640. [DOI] [PubMed] [Google Scholar]
- 31.Knott M, Best RB: Discriminating binding mechanisms of an intrinsically disordered protein via a multi-state coarse-grained model. J. Chem. Phys 2014, 140:175102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bramer D, Wei GW: Multiscale weighted colored graphs for protein flexibility and rigidity analysis. J. Chem. Phys 2018, 148:054103. [DOI] [PubMed] [Google Scholar]
- 33.Koehl P, Poitevin F, Navaza R, Delarue MH: The renormalization group and its applications to generating coarse-grained models of large biological molecular systems. J. Chem. Theory Comput 2017, 13:1424–1438.[*] This paper and Ref. 62 describe approaches to identify hierarchies of coarse-grained models for macromolecules based on renormalization group theory.
- 34.Opron K, Xia K, Burton Z, Wei GW: Flexibility-rigidity index for protein-nucleic acid flexibility and fluctuation analysis. J. Comput. Chem 2016, 37:1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grime JMA, Dama JF, Ganser-Pornillos BK, Woodward CL, Jensen GJ, Yaeger MJ, Voth GA: Coarse--grained simulation reveals key features of HIV--1 capsid self-assembly. Nat. Commun 2016, 7:11568.[*] Ultra-coarse-grained models of viral capsid proteins reveal the importance of inert crowder and protein conformational sampling for robust self-assembly into mature HIV cores.
- 36.Pak AJ, Grime JMA, Sengupta P, Chen AK, Durumeric AEP, Srivastava A, Yeager M, Briggs JAG, Lippincott-Schwartz J, Voth GA: Immature HIV-1 lattice assembly dynamics are regulated by scaffolding from nucleic acid and the plasma membrane. Proc. Natl. Acad. Sci. USA 2017, 114:E10056–E10065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hyeon C, Dima RI, Thirumalai D: Pathways and Kinetic Barriers in Mechanical Unfolding and Refolding of RNA and Proteins. Structure 2006, 14:1633–1645. [DOI] [PubMed] [Google Scholar]
- 38.Hyeon C, Lorimer GH, Thirumalai D: Dynamics of allosteric transitions in GroEL. Proc. Natl. Acad. Sci. USA 2006, 103:18939–18944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JY, Iverson TM, Dima RI: Molecular investigations into the mechanics of actin in different nucleotide states. J. Phys. Chem. B 2011, 115:186–195. [DOI] [PubMed] [Google Scholar]
- 40.Sfriso P, Hospital A, Emperador A, Orozco M: Exploration of conformational transition pathways from coarse-grained simulations. Bioinformatics 2013, 29:1980–1986. [DOI] [PubMed] [Google Scholar]
- 41.Katkar HH, Davtyan A, Durumeric AEP, Hocky GM, Schramm AC, Cruz EMDL, Voth GA: Insights into the cooperative nature of ATP hydrolysis in actin filaments. bioRxiv 2018, 10.1101/319558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frappier V, Najmanovich RJ: A Coarse-Grained Elastic Network Atom Contact Model and Its Use in the Simulation of Protein Dynamics and the Prediction of the Effect of Mutations. PLoS Comp. Biol 2014, 10:e1003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mailhot O, Frappier V, Major F, Najmanovich R: The Elastic Network Contact Model applied to RNA: enhanced accuracy for conformational space prediction. bioRxiv 2017: 10.1101/198531. [DOI]
- 44.Miyazawa S, Jernigan RL: Estimation of effective interresidue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules 1985, 18:534–552. [Google Scholar]
- 45.Miyazawa S, Jernigan RL: Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation andthreading. J. Mol. Biol 1996, 256:623–644. [DOI] [PubMed] [Google Scholar]
- 46.Kaynak BT, Findik D, Doruker P: RESPEC Incorporates Residue Specificity and Ligand Effect into Elastic Network Model. J. Phys. Chem. B 2018, 122:5347–5355. [DOI] [PubMed] [Google Scholar]
- 47.Hamacher K, McCammon JA: Computing the amino acid specificity of fluctuations in biomolecular systems. J. Chem. Theory Comput 2006, 2:873–878. [DOI] [PubMed] [Google Scholar]
- 48.Sankar K, Jia K, Jernigan RL: Knowledge-based entropies improve the identification of native protein structures. Proc. Natl. Acad. Sci. USA 2017, 114:2928–2933.[*] This paper trains a knowledge-based free-energy function, in which entropies are determined based on changes to close contacts, to discriminate native protein states.
- 49.Sankar K, Mishra SK, Jernigan RL: Comparisons of Protein Dynamics from Experimental Structure Ensembles, Molecular Dynamics Ensembles, and Coarse-Grained Elastic Network Models. J. Phys. Chem. B 2018, 122:5409–5417. [DOI] [PubMed] [Google Scholar]
- 50.Putz I, Brock O: Elastic network model oflearnedmaintained contacts to predict protein motion. PLoS ONE 2017, 12:e0183889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poblete S, Bottaro S, Bussi G: A nucleobase-centered coarse-grained representation for structure prediction of RNA motifs. Nucleic Acids Res 2018, 46:1674–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Villada-Balbuena M, Carbajal-Tinoco MD: One-bead coarse-grained model for RNA dynamics. J. Chem. Phys 2017, 146:045101. [DOI] [PubMed] [Google Scholar]
- 53.Moult J, Fidelis K, Kryshtafovych A, Schwede T, Tramontano A: Critical assessment of methods of protein structure prediction (CASP)—Round XII. Proteins: Struct. Funct. Bioinform 2018, 86:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ge Y, Voelz VA: Model Selection Using BICePs: A Bayesian Approach for Force Field Validation and Parameterization. J. Phys. Chem. B 2018, 122:5610–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Farrell K, Oden JT, Faghihi D: A Bayesian framework for adaptive selection, calibration, and validation of coarse-grained models of atomistic systems. J. Comput. Phys 2015, 295:189–208. [Google Scholar]
- 56.Schöberl M, Zabaras N, Koutsourelakis PS: Predictive coarse-graining. J. Comput. Phys 2017, 333:49–77. [Google Scholar]
- 57.Chen YL, Habeck M: Data-driven coarse graining of large biomolecular structures. PLoS ONE 2017, 12:e0183057.[*] This paper introduces a Bayesian inference approach to develop coarse-grained mappings and interactions based on experimental atomic models or electron density maps.
- 58.Habeck M: Bayesian Modeling of Biomolecular Assemblies with Cryo-EM Maps. Frontiers in Molecular Biosciences 2017, 4:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Z, Lu L, Noid WG, Krishna V, Pfaendtner J, Voth GA: A systematic methodology for defining coarse-grained sites in large biomolecules. Biophys. J 2008, 95:5073–5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Z, Voth GA: Coarse-grained representations of large biomolecular complexes from low-resolution structural data. J. Chem. Theory Comput 2010, 6:2990–3002. [DOI] [PubMed] [Google Scholar]
- 61.Li M, Zhang JZ, Xia F: Constructing Optimal Coarse-Grained Sites of Huge Biomolecules by Fluctuation Maximization. J. Chem. Theory Comput 2016, 12:2091–2100. [DOI] [PubMed] [Google Scholar]
- 62.Sinitskiy AV, Saunders MG, Voth GA: Optimal number of coarse-grained sites in different components of large biomolecular complexes. J. Phys. Chem. B 2012, 116:8363–8374.[*] See Ref. 33.
- 63.Lyman E, Pfaendtner J, Voth GA: Systematic multiscale parameterization of heterogeneous elastic network models of proteins. Biophys. J 2008, 95:4183–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moritsugu K, Kurkal-Siebert V, Smith JC: REACH coarse-grained normal mode analysis of protein dimer interaction dynamics. Biophys. J 2009, 97:1158–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Orellana L, Rueda M, Ferrer-Costa C, Lopez-Blanco J, Chacón P, Orozco M: Approaching Elastic Network Models to Molecular Dynamics Flexibility. J. Chem. Theory Comput 2010, 6:2910–2923. [DOI] [PubMed] [Google Scholar]
- 66.Orellana L, Yoluk O, Carrillo O, Orozco M, Lindahl E: Prediction and validation of protein intermediate states from structurally rich ensembles and coarse-grained simulations. Nat. Commun 2016, 7:12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Noid WG, Chu JW, Ayton GS, Krishna V, Izvekov S, Voth GA, Das A, Andersen HC: The multiscale coarse-graining method. I. A rigorous bridge between atomistic and coarse-grained models. J. Chem. Phys 2008, 128:244144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Noid WG, Liu P, Wang Y, Chu J-W, Ayton GS, Izvekov S, Andersen HC, Voth GA: The multiscale coarse-graining method. II. Numerical implementation for coarse-grained molecular models. J. Chem. Phys 2008, 128:244115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Izvekov S,Voth GA: A multiscale coarse-graining method for biomolecular systems. J. Phys. Chem. B 2005, 109:2469–2473. [DOI] [PubMed] [Google Scholar]
- 70.Rudzinski JF, Noid WG: Bottom-up coarse-graining of peptide ensembles and helix-coil transitions. J. Chem. Theory Comput 2015, 11:1278–1291. [DOI] [PubMed] [Google Scholar]
- 71.Shell MS: The relative entropy is fundamental to multiscale and inverse thermodynamic problems. J. Chem. Phys 2008, 129:144108. [DOI] [PubMed] [Google Scholar]
- 72.Carmichael SP, Shell MS: A New Multiscale Algorithm and Its Application to Coarse-Grained Peptide Models for Self-Assembly. J. Phys. Chem. B 2012, 116:8383–8393. [DOI] [PubMed] [Google Scholar]
- 73.Savelyev A, Papoian GA: Chemically accurate coarse graining of double-stranded DNA. Proc. Natl. Acad. Sci. USA 2010, 107:20340–20345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rudzinski JF, Noid WG: Coarse-graining entropy, forces, and structures. J. Chem. Phys 2011, 135:214101.[*] This paper provides an elegant analysis that compares systematic coarse-graining methods based on a general information measure.
- 75.Wagner JW, Dama JF, Durumeric AEP, Voth GA: On the representability problem and the physical meaning of coarse-grained models. J. Chem. Phys 2016, 145:044108–044108. [DOI] [PubMed] [Google Scholar]
- 76.Wagner JW, Dannenhoffer-Lafage T, Jin J, Voth GA: Extending the range and physical accuracy of coarse-grained models: Order parameter dependent interactions. J. Chem. Phys 2017, 147:044113. [DOI] [PubMed] [Google Scholar]
- 77.Dama JF, Jin J, Voth GA: The Theory of Ultra-Coarse-Graining. 3. Coarse-grained Sites with Rapid Local Equilibrium of Internal States. J. Chem. Theory Comput 2017, 13:1010–1022. [DOI] [PubMed] [Google Scholar]
- 78.Delyser MR, Noid WG: Extending pressure-matching to inhomogeneous systems via local-density potentials. J. Chem. Phys 2017, 147:134111. [DOI] [PubMed] [Google Scholar]
- 79.Sanyal T, Shell MS: Coarse-grained models using local-density potentials optimized with the relative entropy: Application to implicit solvation. J. Chem. Phys 2016, 145:034109. [DOI] [PubMed] [Google Scholar]
- 80.Dama JF, Sinitskiy AV, McCullagh M, Weare J, Roux Bt, Dinner AR, Voth GA: The theory of ultra-coarse-graining. 1. General principles. J. Chem. Theory Comput 2013, 9:2466–2480.[*] This paper outlines the ultra-coarse-graining framework, which is a systematic approach to enhance CG model expressiveness through the use of embedded internal states.
- 81.Davtyan A, Dama JF, Sinitskiy AV, Voth GA: The theory of ultra-coarse-graining. 2. Numerical implementation. J. Chem. Theory Comput 2014, 10:5265–5275. [DOI] [PubMed] [Google Scholar]
- 82.Sanyal T, Shell MS: Transferable Coarse-Grained Models of Liquid-Liquid Equilibrium Using Local Density Potentials Optimized with the Relative Entropy. J. Phys. Chem. B 2018, 122:5678–5693. [DOI] [PubMed] [Google Scholar]
- 83.Jin J, Voth GA: Ultra-Coarse-Grained Models Allow for an Accurate and Transferable Treatment of Interfacial Systems. J. Chem. Theory Comput 2018, 14:2180–2197. [DOI] [PubMed] [Google Scholar]
- 84.Chen J, Chen J, Pinamonti G, Clementi C: Learning Effective Molecular Models from Experimental Observables. J. Chem. Theory Comput 2018, 14:3849–3858. [DOI] [PubMed] [Google Scholar]
- 85.López-Blanco JR, Chacón P: New generation of elastic network models. Curr. Opin. Struct. Biol 2016, 37:46–53. [DOI] [PubMed] [Google Scholar]
- 86.Joseph AP, Polles G, Alber F, Topf M: Integrative modelling of cellular assemblies. Curr. Opin. Struct. Biol 2017, 46:102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yoon G, Kwak J, Kim JI, Na S, Eom K: Mechanical characterization of amyloid fibrils using coarse-grained normal mode analysis. Adv. Funct. Mater 2011, 21:3454–3463. [Google Scholar]
- 88.Grime JMA, Voth GA: Early stages of the HIV-1 capsid protein lattice formation. Biophys. J 2012, 103:1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qiao X, Jeon J, Weber J, Zhu F, Chen B: Mechanism of polymorphism and curvature of HIV capsid assemblies probed by 3D simulations with a novel coarse grain model. Biochim. Biophys. Acta 2015, 1850:2353–2367. [DOI] [PubMed] [Google Scholar]
- 90.Theisen KE, Desai NJ, Volski AM, Dima RI: Mechanics of severing for large microtubule complexes revealed by coarse-grained simulations. J.Chem. Phys 2013, 139:121926. [DOI] [PubMed] [Google Scholar]
- 91.Theisen KE, Zhmurov A, Newberry ME, Barsegov V, Dima RI: Multiscale modeling of the nanomechanics of microtubule protofilaments. J. Phys. Chem. B 2012, 116:8545–8555. [DOI] [PubMed] [Google Scholar]
- 92.Fogarty AC, Potestio R, Kremer K: A multi-resolution model to capture both global fluctuations of an enzyme and molecular recognition in the ligand-binding site. Proteins: Struct. Funct. Bioinform 2016, 84:1902–1913. [DOI] [PubMed] [Google Scholar]
- 93.Shimizu M, Takada S: Reconstruction of Atomistic Structures from Coarse-Grained Models for Protein-DNA Complexes. J. Chem. Theory Comput 2018, 14:1682–1694. [DOI] [PubMed] [Google Scholar]
- 94.Hocky GM, Dannenhoffer-Lafage T, Voth GA: Coarse-Grained Directed Simulation. J. Chem. Theory Comput 2017, 13:4593–4603.[*] This paper describes a method to minimally bias atomistic simulations toward coarse-grained observables, e.g., to describe a protein as if it were embedded within a multi-protein complex.
- 95.Chiricotto M, Melchionna S, Derreumaux P, Sterpone F: Hydrodynamic effects on β-amyloid (16–22) peptide aggregation. J. Chem. Phys 2016, 145:035102. [DOI] [PubMed] [Google Scholar]
- 96.Sterpone F, Derreumaux P, Melchionna S: Molecular Mechanism of Protein Unfolding under Shear: A Lattice Boltzmann Molecular Dynamics Study. J. Phys. Chem. B 2018, 122:1573–1579. [DOI] [PubMed] [Google Scholar]
- 97.Sterpone F, Derreumaux P, Melchionna S: Protein simulations in fluids: Coupling the OPEP coarse-grained force field with hydrodynamics. J. Chem. Theory Comput 2015, 11:1843–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]