

Beta-lactamase enzymes mediate the most common forms of gram-negative antibiotic resistance affecting clinical treatment. They also constitute an excellent model system for the difficult problem of understanding how allosteric mutations can augment catalytic activity of already-competent enzymes. Multiple allosteric mutations have been identified that alter catalytic activity or drug-resistance spectrum in class A beta lactamases, but predicting these in advance continues to be challenging. Here, we review computational techniques based on structure and/or molecular simulation to predict such mutations. Structure-based techniques have been particularly helpful in developing graph algorithms for analyzing critical residues in beta-lactamase function, while classical molecular simulation has recently shown the ability to prospectively predict allosteric mutations increasing beta-lactamase activity and drug resistance. These will ultimately achieve the greatest power when combined with simulation methods that model reactive chemistry to calculate activation free energies directly.
Beta lactamases provide the primary means of gram-negative bacterial resistance to the beta-lactam antibiotic classes that form a main part of the clinical armamentarium. Beta lactamase enzymes of classes (the focus of this work), C, and D hydrolyze beta-lactam antibiotics in a manner analogous to that of serine proteases: a serine in the active site mediates acylation and opening of the beta-lactam ring to form a covalent acyl intermediate; this is then resolved via nucleophilic attack by a water molecule to form deacylated, inactive product and regenerate the enzyme. Because beta lactamases are under such selective pressure—they also provide resistance against microbial toxins—a wide variety of functional mutations have been identified, either clinically or in laboratory evolution experiments [1–6]. Many of these mutations are allosteric in nature, yet predicting allosteric mutations prospectively has been challenging. The understanding required to make such predictions accurately would also yield deeper insight into the mechanisms of allosteric modulation in these well studied and clinically important enzymes.
Class A beta lactamases as model systems for allostery in well-structured proteins
Allostery can be broadly defined as perturbations to a molecule far from a binding or active site of interest that affect that site. These perturbations can take the form of allosteric ligands (other molecules) or allosteric mutations (changes to the reference molecule itself). Protein allostery is currently understood [7] in terms of shifts to the conformational ensemble P(x) to P’(x), where P(x) is used to denote the probability distribution of conformational states x across all position and velocity degrees of freedom. The simplest allosteric effect can be a structural change: in this case, the lowest free-energy state of an enzyme alters such that Pmax(x) ≠ P’max(x), thus changing likely conformation [8].Subtler shifts can manifest as population reweighting among minor(less populated) ensemble members [9] or emergence of a new minor population previously undetectable: in this case, Pmax(x) may not be altered (Fig. 1). We outline four broad motional regimes to classify protein allostery: intrinsically disordered proteins [10,11], highly flexible proteins [12–14], proteins that undergo substantial but well-defined conformational changes [15–20], and proteins that maintain one general tertiary structure and may undergo small conformational changes within that overall structure [21]. lass beta lactamases fall in the last category and are indeed relatively rigid. Therefore, one might expect them to be an easier target for predicting allosteric mutations. This, however, has been unexpectedly challenging, at least for predicting the relatively subtle changes to enzyme kinetics or substrate spectrum that are important for clinical resistance.
Figure 1. Two types of allosteric changes that can affect beta-lactamase function.
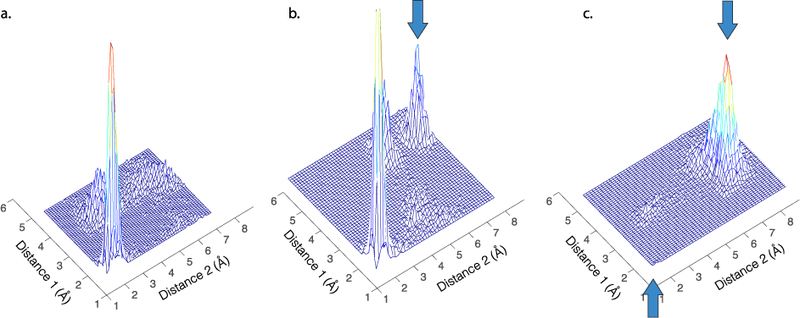
Allostery can either change the relative probabilities of minor states in the acyl-intermediate ensemble without altering the minimum-free-energy state (panels a, b) or change the minimum-free-energy state of a protein conformational ensemble (panel c). Both of these can alter enzyme function and drug resistance. Illustrative examples from active-site distance distributions of CTX-M9. Arrows indicate previously minor states that have greatly increased in probability or major states that have been abrogated. Simulations are of CTX-M9 S281A (a), S220R (b), and A99K (c) from [26]; distances plotted are the “oxyanion hole” hydrogen-bonding distances between the beta-lactam carbonyl oxygen and the backbone amide proton of Ser237 (distance 1) and that of Thr70 (distance 2).
The main challenges in predicting beta-lactamase allosteric effects involve detecting subtle changes to the active-site conformational ensemble and correlating those to conformational fluctuations elsewhere in the protein. Since many class beta lactamases have kcat values ranging from 0.1 s−1 to 1000 s−1 [22–24], catalytically permissive conformations of the enzyme could easily be a small portion (e.g. 1%) of the overall equilibrium population without this equilibrium forming the major barrier for catalysis. However, such equilibria can still affect kcat by reducing the steady-state concentration of enzyme-substrate or enzyme-intermediate complex in an active form [25,26].The chemical changes controlling substrate spectrum and resulting drug resistance are also relatively subtle. It is thus more difficult to predict how allosteric changes affect which beta-lactam drugs a given enzyme will hydrolyze, particularly gain or loss of catalytic ability for different beta-lactam drug classes e.g. penicillins, cephalosporins, carbapenems, etc. This effectively becomes a problem of predicting the allosteric coupling between changes elsewhere in the protein, the functional groups on the drug, and the catalytic geometry.
A number of studies have characterized these interactions retrospectively, using either drug-resistant clinical isolates or resistance mutations identified by laboratory evolution studies and characterizing the location and catalytic effects of mutations [1,27–29]. Apart from the many mutations in the active site that affect class A beta-lactamase catalysis, the omega loop has been identified as having a particular effect on catalysis, even when the mutations lie in portions of the loop not contacting substrate [30–34]. Intriguingly, a number of other allosteric mutations have been identified that are challenging either to rationalize or particularly to have predicted in advance.
Here, we discuss three broad sets of approaches to predicting allosteric effects on the resistance spectrum and enzymatic activity of class A beta lactamases: structure-based methods, methods based on classical molecular dynamics, and methods that explicitly represent reactive chemistry. While some of the methods we discuss include sequence phylogenetic data as well, methods primarily sequence-based in nature are not discussed here.
Structure-based methods for predicting allostery in beta lactamases
Most structure-based methods that have been applied to beta lactamases utilize some sort of network analysis: spatial connectivity, rigid-body analysis, or elastic network models. The fundamental idea behind network analyses of allostery, whether structure-based or otherwise, is that some sort of pairwise convolution holds for residue-residue interactions: if f(i,j) and g(j,k) describe the interaction of residues i with j and j with k, then f * g describes the interaction of I with k via j. Different statistical measurements and assumptions can be used to build such pairwise edges, different convolution operators applied, and different approaches can be used to analyze the resulting networks for allosteric interactions, but this is the fundamental underlying principle. One such approach has constructed proximity-based networks constructed from crystal structures and analyzed them via a centrality criterion [35] to identify “critical” residues involved in long-range interactions in the class A beta-lactamase TEM-1 [36]. Another approach used rigid-body network analysis on different class A beta lactamase structures to predict long-distance coupling matrices for each enzyme, noting similarities in long-distance coupling and overall enzyme rigidity between phylogenetically and functionally related enzymes [37]. However, since functionally important conformational substates throughout the catalytic cycle of class beta lactamases differ in their hydrogen-bonding patterns at the active site, methods based on a single structure may not capture all the relevant network interactions required to predict allosteric effects. This can be addressed both by multi-structure methods and by methods that first estimate a conformational ensemble and then analyze interactions, as described below.
A third approach uses a perturbation measure, the dynamic flexibility index, that measures the change in protein flexibility upon single deletion of each residue from an empirical interaction network. The dynamic flexibility index spans structure-based and classical molecular-dynamics-based approaches, as it was initially developed using structure-based elastic network models as the underlying network [38] and then applied to TEM-1 and reconstructed ancestral beta lactamases using a residue-residue covariance matrix calculated from temperature replica-exchange simulations [39]. This application of the dynamic flexibility index to analyze molecular dynamics simulation data reports on the nearby and distant effects of perturbation at a given residue site and yields predictions of the effects specifically on conformational dynamics of the enzyme. Proximity-based networks, rigidity-based networks, and elastic network models have all been used to develop and apply different network and graph-theoretic analyses to beta lactamases; systematic prospective testing would help assess the current fidelity of their predictions and identify areas for further improvement.
Methods that use classical molecular dynamics to predict allostery
If allostery is best expressed as shifts in the protein conformational ensemble, classical molecular dynamics provides the means to estimate the native and allosterically perturbed ensembles directly. While methods based on this approach can sample conformational equilibria involving states too rare or too transient to detect experimentally, two main limitations apply: since classical molecular dynamics generates simulated single-molecule trajectories in phase space to sample the often-rough free energy landscapes corresponding to protein conformational ensembles, sufficient sampling is required to accurately estimate the perturbation P’(x) for each mutation considered. Second, classical molecular dynamics does not treat chemical reactions directly, so each chemical state modeled must be considered separately. In recent years, two major improvements have extended the reach of classical molecular dynamics in accurately predicting allosteric effects on beta lactamase function: building long-timescale kinetic models from many individual simulations and coupling large-scale simulations of mutants to machine-learning analysis of how allosteric effects manifest in the enzyme active site.
Markov State Models can be constructed from molecular dynamics trajectories to yield kinetic models of conformational equilibria and dynamics [40–46]. They require extensive simulation to construct and are sensitive to choice of structural similarity metric, but they provide statistical estimates that can be used to analyze mechanistic pathways, identify important conformational states, and predict experimental observables in a quantitative fashion. These approaches have been used to predict the conformational equilibria of TEM-1 and several mutational variants, which were then leveraged in a hybrid docking strategy to improve prediction of antibiotic binding and implicitly KM. The Markov State Models generated in this process were then queried to explain the mutational effects on KM and predict additional mutants to achieve desired perturbations [47,48]. These results demonstrate both the specific predictive ability of Markov State Model approaches for ligand binding and their potential for broader use in predicting allosteric mutations to beta lactamases.
An alternate approach utilizes many molecular dynamics simulations of many beta-lactamase mutants to predict conformational equilibria directly. This forgoes the additional information from Markov State Model analysis but side-steps the challenges in constructing structural metrics that discriminate between subtly different active-site conformations affecting catalysis. This approach has been applied to the rapidly spreading class beta-lactamase CTX-M9. In this work, 125 point mutants were simulated and the top ones tested experimentally, yielding increased bacterial minimum inhibitory concentration(MIC) and enzymatic kcat for three of the top five mutants computationally predicted to have improved enzymatic function [26]. This study used a different approach from the TEM-1 study [47] in that the chemical state simulated was the acylenzyme intermediate rather than the enzyme-substrate complex, in an attempt to predict the effects of conformational equilibria on kcat (and in particular on the deacylation reaction that is rate-limiting for CTX-M9 and the substrates examined). Perhaps most interestingly, after confirming these predictions, positional mutual information feature selection and statistical decision trees were used to identify the active-site atoms that shift positional equilibria in response to allosteric mutation. Network analysis of pairwise positional mutual information [49] can also identify critical interactions and residues for mutation in a fashion analogous to structure-based or phylogenetically-based methods but in this case based on conformational dynamics. This approach was tested and prospectively validated using alanine mutagenesis for CTX-M9 [50]. Graph centrality algorithms such as used for structure-based analyses [35] can also be applied to positional mutual information graphs (Fig. 2). A simulation and feature-selection analysis was also performed, using machine learning on molecular dynamics simulations to predict off-pathway conformations in the class carbapenemase KPC-2 [51]. These approaches offer the potential to directly identify how allosteric changes alter active-site conformational equilibria and thus construct statistical tools to address a central question of allosteric modulation. With increasing computational power, such tools becoming increasingly viable solutions.
Figure 2. Network analyses of enzyme motions identify critical residues for allostery.
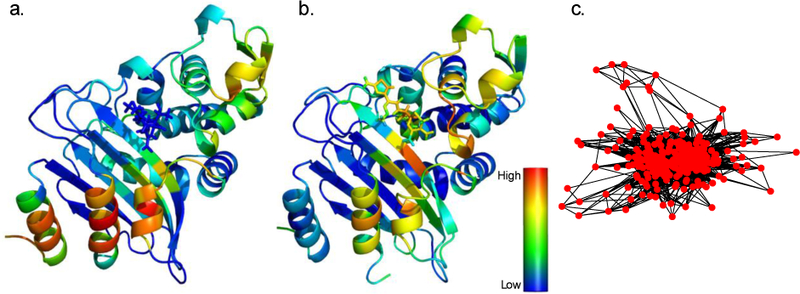
Rendered here are network centrality scores for the CTX-M9 beta lactamase based on positional mutual information in molecular dynamics simulations [50]. Coloring in panel (a) is based on network eigencentrality of the CTX-M9:cefotaxime acylenzyme complex, while coloring in panel (b) is based on the same complex with meropenem. The residue-residue interaction graph used to calculate (a) is rendered in panel (c). Network-based analyses have identified residues predicted to be either closely coupled to active site residues or broadly critical to beta-lactamase structure and dynamics. Edges in these networks can derive from both structure-based and dynamics-based methods; this rendering uses dynamics-based methods (symmetric uncertainty calculated from sitional mutual information). The symmetric uncertainty matrix is thresholded at 0.01 for graph rendering, although centrality is calculated on the full graph. Several important allosteric mutants score highly using this analysis, but such combinations of positional mutual information and network centrality await systematic experimental confirmation.
Methods that include reactive chemistry
Because beta lactamase function involves substrate hydrolysis, it is natural to hypothesize that explicitly considering the reactive steps in this process would improve prediction of allosteric effects on catalysis. Common techniques for modeling reactions catalyzed by enzymes include quantum-mechanical cluster calculations, mixed-resolution quantum mechanical / molecular mechanics (QM/MM) simulation, and semi-empirical methods such as empirical valence bond theory (EVB) [52–54]. Each of these has been applied to beta-lactamase function, although most studies have concentrated on catalytic mechanism, mechanism of inhibition, or active-site mutations rather than allostery [55–57]. An intriguing exception is a comparative QM/MM study of different class A beta lactamases that retrospectively predicts carbapenemase activity based on crystal structure and simulation [58]. The same approach was also used to distinguish between efficient and inefficient inhibition of class beta lactamases by clavulanic acid [55]. This work does not explicitly treat allostery, but it nevertheless suggests both that QM/MM approaches have predictive power for hydrolytic activity on key clinically used antibiotics and that, given sufficient sampling, these approaches could be extended to treat allostery. The challenge, of course, is that QM/MM, QM-cluster, and EVB simulations are all more computationally expensive than the structure-based or classical molecular dynamics methods described above, so combinations of these methods may be most effective in making accurate yet tractable predictions.
New experimental methods that potentiate computational analysis of allostery
Several recent experimental developments offer exciting new opportunities to integrate experimental and computational measurements in order better to understand allosteric modulation of beta-lactamase activity. New tools to analyze conformational heterogeneity in crystallographic datasets combined with room-temperature and multi-temperature crystallography have helped detect allosteric changes and changes to the conformational ensemble in other enzymes [59–62]; the resolving power of such approaches is limited to a few The authors declare no conflict of interest. conformations but is nonetheless a substantial increase on a single-conformation model. Such analyses provide experimental measurements directly comparable to computational predictions of allosterically induced shifts to beta-lactamase conformational ensembles. Second, the phenomenon of epistasis (which we use here to mean when an identical mutation has different effects in different genetic backgrounds) has long challenged a general understanding of how beta-lactamase function is controlled [34, 63–65]. Recent deep-mutational-scanning experiments on TEM-1 measure the fitness of thousands of point mutations on genetic backgrounds that sequentially vary by one mutation each [4, 66]. This approach allows quantification of epistatic fitness effects: it demonstrates that epistasis does indeed play a complicating role here: the effect of single mutations is not additive, but it provides quantitative measures that can be used to inform computational analyses of allosteric effects and conformational ensembles to achieve a deeper understanding of the physical underpinnings for these genetic fitness effects.
Conclusions
Despite substantial successes in analyzing and predicting allostery in class A beta lactamases, the ultimate goal of predicting allosteric mutations that selectively add or remove resistance to a single antibiotic or a single class of antibiotics has not yet been achieved. This goal is admittedly more ambitious than many allosteric prediction tasks, as augmenting the activity of naturally occurring enzymes for their target substrates is more challenging than disrupting activity. The high degree of epistasis that is observed experimentally further complicates this task and makes computational prediction all the more useful, as exhaustive experimental exploration becomes more challenging when mutations are non-additive. Ultimately, prospective prediction of allosteric mutations that add or remove drug resistance in beta lactamases will likely require a combination of approaches (Fig. 3). These could include first performing machine learning on deep mutational scanning datasets and phylogenetic data to narrow the space of potential mutations, then classical molecular dynamics in conjunction with some of the analytic methods described here to sample conformation space, and finally QM/MM or semi-empirical methods to model catalysis itself. This would permit analyses that could capture changes to both conformational equilibria and reactive chemistry over a large number of relevant mutations and enable a better quantitative understanding of allostery.
Figure 3. Proposed coupling of large-scale molecular dynamics, QM/MM, and statistical learning for insight into allostery.
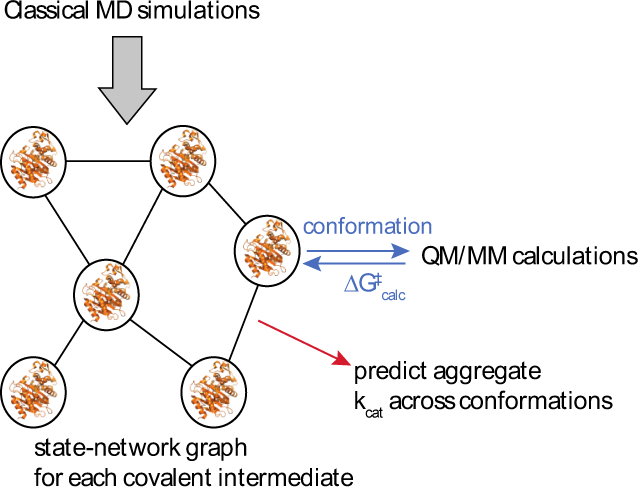
In this scheme, classical molecular dynamics simulations are used to construct a state network for each covalent intermediate in beta-lactamase hydrolysis. QM/MM calculations on conformations sampled from this state network are used to predict ∆G‡ for each conformation. These predictions are used both to guide lumping/splitting of the state network (to maintain uniform ∆G‡ within a state) and to calculate aggregate kcat across the states. This process could either be repeated across multiple enzyme mutants for retrospective analysis or used prospectively to guide targeted mutagenesis.
Highlights.
Class A beta lactamases demonstrate substantial allostery
Predicting allosteric mutations increasing drug resistance is important and hard
Structural analysis and classical MD have successfully identified key residues
QM/MM methods have successfully predicted catalytic activity
Combining these techniques, and new structural data, may yield robust prediction
Acknowledgements
The authors thank Jennifer Hays for many helpful discussions. We also thank Banu Ozkan and Marc van der Kamp for helpful comments. This work was funded by a National Institutes of Health fellowship [F31 GM113543] to G.A.C and grants from The Hartwell Foundation and the Knut and Alice Wallenberg Foundation to P.M.K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflict of interest.
References
- •1.Perez-Llarena FJ, Kerff F, Abian O, Mallo S, Fernandez MC, Galleni M, Sancho J, Bou G: Distant and new mutations in CTX-M-1 beta-lactamase affect cefotaxime hydrolysis. Antimicrob Agents Chemother 2011, 55:4361–4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mehta SC, Rice K, Palzkill T: Natural Variants of the KPC-2 Carbapenemase have Evolved Increased Catalytic Efficiency for Ceftazidime Hydrolysis at the Cost of Enzyme Stability. PLoS Pathog 2015, 11:e1004949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haidar G, Clancy CJ, Shields RK, Hao B, Cheng S, Nguyen MH: Mutations in blaKPC-3hat Confer Ceftazidime-Avibactam Resistance Encode Novel KPC-3 Variants That Function as Extended-Spectrum beta-Lactamases. Antimicrob Agents Chemother 2017, 61. [DOI] [PMC free article] [PubMed]
- 4.Firnberg E, Labonte JW, Gray JJ, Ostermeier M: A comprehensive, high-resolution map of a gene’s fitness landscape. Mol Biol Evol 2014, 31:1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deng Z, Huang W, Bakkalbasi E, Brown NG, Adamski CJ, Rice K, Muzny D, Gibbs RA, Palzkill T: Deep sequencing of systematic combinatorial libraries reveals beta-lactamase sequence constraints at high resolution. J Mol Biol 2012, 424:150–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacquier H, Birgy A, Le Nagard H, Mechulam Y, Schmitt E, Glodt J, Bercot B, Petit E, Poulain J, Barnaud G, et al. : Capturing the mutational landscape of the beta-lactamase TEM-1. Proc Natl Acad Sci U S A 2013, 110:13067–13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••7.Motlagh HN, Wrabl JO, Li J, Hilser VJ: The ensemble nature of allostery. Nature 2014, 508:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monod J, Wyman J, Changeux JP: On the Nature of llosteric Transitions: A Plausible Model. J Mol Biol 1965, 12:88–118. [DOI] [PubMed] [Google Scholar]
- 9.Cooper A, Dryden DT: Allostery without conformational change. A plausible model. Eur Biophys J 1984, 11:103–109. [DOI] [PubMed] [Google Scholar]
- 10.Hilser VJ,Thompson EB:Intrinsicdisorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci U S 2007, 104:8311–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferreon AC, Ferreon JC, Wright PE, Deniz AA: Modulation of allostery by protein intrinsic disorder. Nature 2013, 498:390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuxreiter M, Tompa P, Simon I: Local structural disorder imparts plasticity on linear motifs. Bioinformatics 2007, 23:950–956. [DOI] [PubMed] [Google Scholar]
- 13.Fox DA, Larsson P, Lo RH, Kroncke BM, Kasson PM, Columbus L: Structure of the Neisserial outer membrane protein Opa60: loop flexibility essential to receptor recognition and bacterial engulfment. J Am Chem Soc 2014, 136:9938–9946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wrabl JO, Gu J, Liu T, Schrank TP, Whitten ST, Hilser VJ: The role of protein conformational fluctuations in allostery, function, and evolution. Biophys Chem 2011, 159:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Popovych N, Sun S, Ebright RH, Kalodimos CG: Dynamically driven protein allostery. Nat Struct Mol Biol 2006, 13:831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baldwin J, Chothia C: Haemoglobin: the structural changes related to ligand binding and its allosteric mechanism. J Mol Biol 1979, 129:175–220. [DOI] [PubMed] [Google Scholar]
- 17.Ogawa S, Mcconnell HM: Spin-Label Study of Hemoglobin Conformations in Solution. Proceedings of the National Academy of Sciences of the United States of America 1967, 58:19.-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nussinov R, Tsai CJ: Allostery in disease and in drug discovery. Cell 2013, 153:293–305. [DOI] [PubMed] [Google Scholar]
- 19.Schrank TP, Bolen DW, Hilser VJ: Rational modulation of conformational fluctuations in adenylate kinase reveals a local unfolding mechanism for allostery and functional adaptation in proteins. Proc Natl Acad Sci U S A 2009, 106:16984–16989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doshi U, Holliday MJ, Eisenmesser EZ, Hamelberg D: Dynamical network of residue-residue contacts reveals coupled allosteric effects in recognition, catalysis, and mutation. Proc Natl Acad Sci U S A 2016, 113:4735–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui Q, Karplus M: Allostery and cooperativity revisited. Protein Sci 2008, 17:1295–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yigit H, Queenan AM, Anderson GJ, Domenech Sanchez A,Biddle JW, Steward CD, Alberti S, Bush K, Tenover FC: Novel carbapenem-hydrolyzing beta-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob Agents Chemother 2001, 45:1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venkatachalam KV, Huang W, LaRocco M, Palzkill T: Characterization of TEM-1 beta-lactamase mutants from positions 238 to 241 with increased catalytic efficiency for ceftazidime. J Biol Chem 1994, 269:23444–23450. [PubMed] [Google Scholar]
- 24.Delmas J, Robin F, Carvalho F, Mongaret C, Bonnet R: Prediction of the evolution of ceftazidime resistance in extended-spectrum beta-lactamase CTX-M-9. Antimicrob Agents Chemother 2006, 50:731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waley SG: The kinetics of substrate-induced inactivation. Biochem J 1991, 279 ( Pt 1):87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••26.Latallo MJ, Cortina GA, Faham S, Nakamoto RK, Kasson PM: Predicting allosteric mutants that increase activity of a major antibiotic resistance enzyme. Chem Sci 2017, 8:6484–6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zaccolo M, Gherardi E: The effect of high-frequency random mutagenesis on in vitro protein evolution: a study on TEM-1 beta - lactamase. J Mol Biol 1999, 285:775–783. [DOI] [PubMed] [Google Scholar]
- 28.He D, Chiou J, Zeng Z, Chan EW, Liu JH, Chen S: Comparative Characterization of CTX-M-64 and CTX-M-14 Provides Insights into the Structure and Catalytic Activity of the CTX-M Class of Enzymes. Antimicrob Agents Chemother 2016, 60:6084–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Minasov G, Shoichet BK: Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J Mol Biol 2002, 320:85–95. [DOI] [PubMed] [Google Scholar]
- 30.Vakulenko SB, Toth M, aibi P, Mobashery S, Lerner SA: Effects of Asp-179 mutations in TEMpUC19 beta-lactamase on susceptibility to beta-lactams. Antimicrob Agents Chemother 1995, 39:1878–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levitt PS, Papp-Wallace KM, Taracila MA, Hujer AM, Winkler ML, Smith KM, Xu Y, Harris ME, Bonomo RA: xploring the role of a conserved class A residue in the Omega-Loop of KPC-2 beta-lactamase: a mechanism for ceftazidime hydrolysis. J Biol Chem 2012, 287:31783–31793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yi H, hoi JM, Hwang J, Prati F, Cao TP, Lee SH, Kim HS: High adaptability of the omega loop underlies the substrate-spectrum-extension evolution of a class A beta-lactamase, PenL. Sci Rep 2016, 6:36527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saves I, Burlet-Schiltz O, Maveyraud L, Samama JP, Prome JC, Masson JM: Mass spectral kinetic study of acylation and deacylation during the hydrolysis of penicillins and cefotaxime by beta-lactamase TEM-1 and the G238S mutant. Biochemistry 1995, 34:11660–11667. [DOI] [PubMed] [Google Scholar]
- •34.Dellus-Gur E, Elias M, Caselli E, Prati F, Salverda ML, de Visser JA, Fraser JS, Tawfik DS: Negative Epistasis and Evolvability in TEM-1 beta-Lactamase--The Thin Line between an Enzyme’s Conformational Freedom and Disorder. J Mol Biol 2015, 427:2396–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freeman LC: A Set of Measures of Centrality Based on Betweenness. Sociometry 1977, 40:35–41. [Google Scholar]
- 36.Cusack MP, Thibert B, Bredesen DE, Del Rio G: Efficient identification of critical residues based only on protein structure by network analysis. PLoS One 2007, 2:e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verma D, Jacobs DJ, Livesay DR: Variations within class-beta-lactamase physiochemical properties reflect evolutionary and environmental patterns, but not antibiotic specificity. PLoS Comput Biol 2013, 9:e1003155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nevin Gerek Z, Kumar S, Banu Ozkan S: Structural dynamics flexibility informs function and evolution at a proteome scale. Evol Appl 2013, 6:423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••39.Zou TS, Risso VA, Gavira JA, Sanchez-Ruiz JM, Ozkan SB: Evolution of Conformational Dynamics Determines the Conversion of a Promiscuous Generalist into a Specialist Enzyme. Molecular Biology and Evolution 2015, 32:132–143. [DOI] [PubMed] [Google Scholar]
- 40.Singhal N, Snow CD, Pande VS: Using path sampling to build better Markovian state models: Predicting the folding rate and mechanism of a tryptophan zipper beta hairpin. Journal of Chemical Physics 2004, 121:415–425. [DOI] [PubMed] [Google Scholar]
- 41.Swope WC, Pitera JW, Suits F: Describing protein folding kinetics by molecular dynamics simulations. 1. Theory. Journal of Physical Chemistry B 2004, 108:6571–6581. [Google Scholar]
- 42.Chodera JD, Swope WC, Pitera JW, Dill KA: Long-time protein folding dynamics from short-time molecular dynamics simulations. Multiscale Modeling & Simulation 2006, 5:1214–1226. [Google Scholar]
- 43.Noe F, Horenko I, Schutte C, Smith JC: Hierarchical analysis of conformational dynamics in Biomolecules: transition networks of metastable states. J Chem Phys 2007, 126:155102. [DOI] [PubMed] [Google Scholar]
- 44.Huisinga W, Schutte C, Stuart AM: Extracting macroscopic stochastic dynamics: Model problems. Communications on Pure and Applied Mathematics 2003, 56:234–269. [Google Scholar]
- 45.Roblitz S, Weber M: Fuzzy spectral clustering by PCCA plus : application to Markov state models and data classification. Advances in Data Analysis and Classification 2013, 7:147–179. [Google Scholar]
- 46.Scherer MK, Trendelkamp-Schroer B, Paul F, Prez-Hernndez G, Hoffmann M, Plattner N, Wehmeyer C, Prinz J-H, No: PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models. J. Chem. Theory Comput 2015, 11:5525–5542. [DOI] [PubMed] [Google Scholar]
- ••47.Hart KM, Ho CMW, Dutta S, Gross ML, Bowman GR: Modelling proteins’ hidden conformations to predict antibiotic resistance. Nature Communications 2016, 7. [DOI] [PMC free article] [PubMed]
- 48.Bowman GR, Bolin ER, Hart KM, Maguire BC, Marqusee S: Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proc Natl Acad Sci U S 2015, 112:2734–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kasson PM, Ensign DL, Pande VS: Combining Molecular Dynamics with Bayesian Analysis To Predict and Evaluate Ligand-Binding Mutations in Influenza Hemagglutinin. J Am Chem Soc 2009, 131:1338–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cortina GA, Kasson PM: Excess positional mutual information predicts both local and allosteric mutations affecting beta lactamase drug resistance. Bioinformatics 2016, 32:3420–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •51.Cortina GA, Hays JM, Kasson PM: Conformational Intermediate That Controls KPC-2 Catalysis and Beta-Lactam Drug Resistance. ACS Catalysis 2018, 8:2741–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kamerlin SCL, Warshel A: The empirical valence bond model: theory and applications. WileyInterdisciplinary Reviews-Computational Molecular Science 2011, 1:30–45. [Google Scholar]
- 53.van der Kamp MW, Mulholland AJ: Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry 2013, 52:2708–2728. [DOI] [PubMed] [Google Scholar]
- 54.Siegbahn PE, Himo F: Recent developments of the quantum chemical cluster approach for modeling enzyme reactions. J Biol InorgChem 2009,14:643–651. [DOI] [PubMed] [Google Scholar]
- •55.Fritz RA, Alzate-Morales JH, Spencer J, Mulholland AJ, van der Kamp MW: Multiscale Simulations of Clavulanate Inhibition Identify the Reactive Complex in Class A beta-Lactamases and Predict the Efficiency of Inhibition. Biochemistry 2018, 57:3560–3563. [DOI] [PubMed] [Google Scholar]
- 56.Meroueh SO, Fisher JF, Schlegel HB, Mobashery S: Ab initio QM/MM study of class A beta-lactamase acylation: dual participation of Glu166 and Lys73 in a concerted base promotion of Ser70. J Am Chem Soc 2005, 127:15397–15407. [DOI] [PubMed] [Google Scholar]
- 57.Langan PS, Vandavasi VG, Cooper SJ, Weiss KL, Ginell SL, Parks JM, oates L: Substrate Binding Induces Conformational Changes in a Class β-lactamase That Prime It for Catalysis. ACS Catalysis 2018, 8:2428–2437. [Google Scholar]
- ••58.Chudyk EI, Limb MA, Jones C, Spencer J, van der Kamp MW, Mulholland AJ: QM/MM simulations as an assay for carbapenemase activity in class A beta-lactamases. Chem Commun (Camb) 2014, 50:14736–14739. [DOI] [PubMed] [Google Scholar]
- •59.Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T: Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc Natl Acad Sci U S A 2011, 108:16247–16252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Keedy DA, Fraser JS, van den Bedem H: Exposing Hidden Alternative Backbone Accessing ConformationsinX-rayrystallography Using qFit. PLoS Comput Biol 2015, 11:e1004507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Acker TM, Gable JE, Bohn MF, Jaishankar P, Thompson MC, Fraser JS, Renslo AR, Craik CS: Allosteric Inhibitors, Crystallography, and Comparative Analysis Reveal Network of Coordinated Movement across Human Herpesvirus Proteases. J Am Chem Soc 2017, 139:11650–11653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••62.Keedy DA, Hill ZB, Biel J, Kang E, Rettenmaier TJ, Brandao-Neto J, Pearce NM, von Delft F, Wells JA, Fraser JS: An expanded allosteric network in PTP1B by multitemperature crystallography, fragment screening, and covalent tethering. Elife 2018, 7. [DOI] [PMC free article] [PubMed]
- •63.Bershtein S, Segal M, Bekerman R, Tokuriki N, Tawfik DS: Robustness-epistasis link shapes the fitness landscape of a randomly drifting protein. Nature 2006, 444:929–932. [DOI] [PubMed] [Google Scholar]
- 64.Weinreich DM, Delaney NF, DePristo MA, Hartl DL: Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 2006, 312:111–114. [DOI] [PubMed] [Google Scholar]
- •65.Knies JL, Cai F, Weinreich DM: Enzyme Efficiency but Not Thermostability Drives Cefotaxime Resistance Evolution in TEM-1 beta-Lactamase. Mol Biol Evol 2017, 34:1040–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••66.Steinberg B, Ostermeier M: Shifting Fitness and Epistatic Landscapes Reflect Trade-offs along an Evolutionary Pathway. J Mol Biol 2016, 428:2730–2743. [DOI] [PubMed] [Google Scholar]