

Abstract
Studies have reported a positive correlation between elevated CD8+ T cells in the tumor microenvironment (TME) and good prognosis in cancer. However, the mechanisms linking T cell tumor-infiltration and tumor rejection are yet to be fully understood. The cells and factors of the TME facilitate tumor development in various ways. CD8+ T cell function is influenced by a number of factors, including CD8+ T cell trafficking and localization into tumor sites; as well as CD8+ T cell growth and differentiation. This review highlights recent literature as well as currently evolving concepts regarding the fates of CD8+ T cells in the TME from three different aspects CD8+ T cell trafficking, differentiation and function. A thorough understanding of factors contributing to the fates of CD8+ T cells will allow researchers to develop new strategies and improve on already existing strategies to facilitate CD8+ T cell mediated anti-tumor function, impede T cell dysfunction and modulate the TME into a less immunosuppressive TME.
Keywords: CD8+ T cell fates, Tumor microenvironment, CD8+ T cell differentiation, CD8+ T cell trafficking, T cell exhaustion
1. Introduction
The anti-tumor function of CD8+ T cells is highly dependent on two crucial factors: firstly, by CD8+ T cell differentiation; and secondly by the infiltration of CD8+ T cells into the tumor site which occurs by trafficking or transporting CD8+ T cells into the tumor microenvironment (TME) [1]. Researchers have linked elevated levels of cytotoxic CD8+ T cells in the TME with positive anti-tumor effects in breast [2], colorectal, glioblastoma [3], and cervical cancers [4]. Thus one can positively link elevated cytotoxic CD8+ T cells in the TME with a good prognosis in various cancer types.
The TME, more especially that of solid tumors, hinders CD8+ T cell trafficking and function due to a number of effects from chemokine secretions, abnormal tumor angiogenesis [5,6], and the activation of inhibitory checkpoint pathways [7]. Following tumor infiltration, naïve CD8+ T cells are differentiated into effector CD8+ T cells and further differentiated and activated into cytotoxic and memory CD8+ T cells in order to perform their targeted functions at the tumor site [1,[8], [9], [10]]. Cytotoxic CD8+ T cells classically secrete cytotoxic cytokines and perform tumor lytic functions upon initial encounter with foreign agents. After the initial encounter with antigens, memory CD8+ T cells remain in various areas in order to perform their specialized functions [11]. CD8+ T cell differentiation is dependent on the formation of the antigenic peptide–major histocompatibility complex (MHC). Furthermore, differentiation is influenced by cytokine production and costimulatory signals from antigen presenting cells (APCs); the secretion of extracellular cytokines [12]; and metabolic, epigenetic and transcriptional factors [13,14]. In this review we explore CD8+ T cell trafficking, differentiation and function to further understand the factors ultimately influencing the fate of CD8+ T cells in the TME.
2. The Tumor Microenvironment
The TME is well represented by cells of the innate and adaptive immune systems which contribute to tumor development and immune evasion. The TME results from an interaction between tumorigenesis and an individual's responses to tumorigenesis [15]. The TME is formed by interactions between tumor cells, immune cells, and cancer associated stromal cells however, there may be other factors that have not yet been identified. In addition, the TME is comprised of non-cellular components such as cytokines, chemokines and other factors released or created by the extracellular matrix [16].
The TME exhibits immunosuppressive features in the presence of tumor cells and immune cells, secreting cytokines, i.e. transforming growth factor beta (TGF-β), forkhead box O3 (FOXO3), cyclooxygenase-2 (COX-2), vascular endothelial growth factor (VEGF), serum-derived factor 1 (SDF-1), interleukins IL-6, IL-10, and others [17,18]. The TME may activate immunosuppressive cells, contributing to immune evasion in various ways. In this review, we discuss the following cells of the TME: cancer associated fibroblasts, tumor associated macrophages, myeloid-derived suppressor cells, regulatory T cells, effector T cells, and tumor cells.
Cancer associated fibroblasts (CAFs) alter the stromal structure producing a conducive environment for the growth of tumor cells [19,15]. Qiao and colleagues reported elevated levels of IL-6 secreted by CAFs in esophageal squamous cell carcinoma cells, to be positively correlated with chemoresistance and worse overall survival [20]. The presence of CAFs cause both physical and chemical changes in the TME which are immunosuppressive in nature.
Tumor associated macrophages (TAMs) are the term given to describe macrophages at the tumor site. Macrophages are capable of differentiating into various phenotypes. M1-polarized macrophages are classically activated by IFN-γ combined with lipopolysaccharide or tumor necrosis factor (TNF). M2-polarized macrophages are alternatively activated by interleukins IL-4, IL-10 and IL-13. M1-polarized macrophages characteristically secrete pro-inflammatory cytokines and promote inflammation, whereas M2-polarized macrophages promote tumor growth and metastasis [21,22]. TAMs closely resemble M2-polarized macrophages in function. TAMs prevent T cell proliferation and activation, by the secretion of restrictive chemokines IL-10, prostaglandins, TGF-β or reactive oxygen species (ROS) [15,23]. In mouse models, elevated TAMs have been reported to promote tumor growth. Additionally, elevated TAMs have been correlated with poor prognosis in human cancers. TAMs have been reported to promote carcinogenesis and metastasis by promoting the formation of new blood vessels and inhibiting CD8+ T cell infiltration and subsequent function, therefore preventing T cell facilitated adaptive immune responses [22]. TAMs secrete chemokines that impede the anti-tumor function of CD8+ T cells. In addition, TAMs play a marked role in angiogenesis. Therefore, identifying ways in which to decrease the level and function of TAMs in the TME may be successful therapeutic targets, as well as suitable adjuncts in cancer immunotherapy. Macrophages are adaptable, depending on the micro-environment in which they are located, which makes them valuable reprogramming targets for cancer immunotherapy. Reprogramming TAMs and M2-polorized macrophages into non-tumor promoting, tumor inhibiting M1-polorized macrophages is promising as a therapeutic strategy in cancer immunotherapy.
Myeloid- derived suppressor cells (MDSCs) suppress the function of natural killer cells (NK) cells and T cells by producing cytokines such as IL-6, IL-10, TGF-β and Arginase-1, an enzyme facilitating the production of superoxides, ROS and nitric oxide (NO). MDSCs are recruited to tumor cells by the production of the aforementioned suppressive cytokines and block the functionality of dendritic cells (DCs). Current data reports that MDSCs suppress both the adaptive and innate immune systems. Additionally, MDSCs promote metastasis and the formation of new blood vessels which are vital steps in sustaining carcinogenesis [[24], [25], [26], [27]]. MDSCs contribute towards T cell dysfunction and inhibit T cell activation and expansion. Additionally, MDSCs reduce IL-12 secretion, thus subsequently reducing the infiltration of CD8+ T cells in the TME. Reportedly, the immunosuppressive effect of MDSCs was altered when treated with IL-12, the percentage of MDSCs was reduced and the number of activated CD8+ T cells in the TME was increased [28]. MDSCs impair T cell trafficking through downregulating the expression of CD62L on CD4 and CD8+ T cells. CD62L is a L-selectin significant in facilitating the attachment of circulating CD8+ T cells to endothelial cells of secondary lymphoid organs, thus facilitating CD8+ T cell migration into tumor site [27]. In addition, MDSCs have been reported to promote the production of reactive nitrogen species (RNS), which leads to chemokine nitration, and the inhibition of CD8+ T cell trafficking, in both mouse and human cancers [5,27,29,30]. Therapeutic strategies preventing chemokine nitration as well as those decreasing MDSC accumulation at tumor sites are of investigative importance as adjuncts to improving adoptive T cell immunotherapies.
Regulatory T (Treg) cells are considered immunosuppressive cells of the TME and are obtainable from four various sources. Firstly, by migration from lymphatic or circulatory systems to the TME; Secondly, by differentiation resulting from the suppression of APCs by molecules in the TME; Thirdly, by expansion as a result of DC stimulation; Lastly, by the conversion of effector T cells into Treg cells occurring in the presence of TGF-β. Treg cells inhibit the specialization and function of APCs, decreasing the interactions between APCs and T cells and subsequently inhibiting effector T cell function [31]. Moreover, Treg cells suppress the effectivity of NK cells. Therefore, Treg cells are capable of preventing anti-tumor immunity by diminishing responses from both the adaptive and innate immune systems [32]. Curiel et al. discovered that human Treg cells function to prevent the cytotoxic effect of T cells in vivo, thus positively contributing to tumor growth [33]. Therefore, impeding Treg cell migration, differentiation, expansion, conversion as well as function within the TME may show positive results in improving anti-tumor immunity, from both adaptive and innate immune systems.
Effector T cells dynamically respond to antigenic peptides presented by MHC on the surface of diseased or foreign cells in the presence of co-stimulatory or co-inhibitory molecules. Helper, memory and cytotoxic T cells are all considered to be effector T cells. Helper T cells, CD4 T cells, assist cytotoxic T cells, and other immune cells to perform effector functions. Memory T cells remain circulating in the immune system to respond rapidly and pronouncedly upon subsequent exposure to pathogens or may remain in pathogenic tissue as in the case of resident memory T (TRM) cells [11,33,34]. Cytotoxic T cells in the TME may produce interleukins IL-2, IL-12 and interferon gamma (IFN-γ), which promote cytotoxic functions of CD8+ T cells through the production of TNF-related apoptosis-inducing ligands (TRAILs); ROS [35] and perforin [36].
Tumor cells express co-inhibitory receptors such as programmed death ligand-1 (PD-L1) and CD80 that interact with inhibitory molecules programmed death-1 (PD-1) and cytotoxic T lymphocyte antigen-4 (CTLA-4) expressed by CD8+ T cells. These interactions may inhibit CD8+ T cell activation and function [37,38]. Tumor cells may also express ectonucleotidase CD39 and CD73 on their surface, which have been reported to facilitate the metabolism of extracellular adenosine triphosphate (ATP) or adenosine diphosphate (ADP) to adenosine monophosphate (AMP) and finally into adenosine. Adenosine is considered an immunosuppressant, inhibiting the trafficking and function of CD8+ T cells at the tumor site [39,40]. Indoleamine 2,3-dioxygenase (IDO) is an enzyme produced by tumor cells that functions to convert tryptophan into kynurenine in the TME. Tryptophan is needed for the proliferation and activation of T cells, therefore in the presence of kynurenine T cell proliferation and activation are inhibited. In addition, kynurenine facilitates the production of Treg cells in the TME [41]. Lactate is produced by tumor cells as a metabolic by-product of anaerobic glycolysis and lactate production inhibits CD8+ T cell activation. Reportedly, high levels of lactate correlated with poor progression and distant metastasis [42]. Therefore, tumor cells may express certain receptors and secrete certain enzymes and metabolites that may facilitate T cell dysfunction and immunosuppression.
3. Trafficking and Localization of CD8+ T Cells
The function of T cells is dependent on their interaction with tumor cells and or other cells of the TME [43]. Naïve T cells are produced in the bone marrow. Naïve T cells are transported from the bone marrow; to the thymus where they undergo further development; to lymph nodes where they are activated and specialized; and through the circulation to reach target organs where they perform specialized functions [44,45]. In the thymus naïve T cells travel through the thymic cortex and medulla, where they undergo various developmental changes such as negative and positive selection which lead to their maturation. In the thymus medulla, these T cells begin to express their classification proteins CD4+ or CD8+. Mature T cells enter secondary lymphoid organs such as lymph nodes and spleen where they undergo cell division and multiplication. The lymph nodes are the site where foreign antigens are identified and taken up by APCs (mainly DCs) and presented to the appropriate T cell receptor (TCR) on T cells, signaling the TCR and leading to T cell activation [46,1]. The mature activated T cells exit the lymph nodes to reach the thoracic duct, circulatory system and finally reach target tissues where they may be localized to perform effector functions [7,47,48].
The development, differentiation and location of memory precursor T cells are rather different from that of effector T cells. Lymph nodes house central memory T (TCM) cells which are reactivated upon secondary encounter with an antigen. Effector memory T (TEM) cells circulate through tissues and have cytotoxic properties. Resident memory T (TRM) cells persist in infection or tumor sites and do not recirculate in the circulatory system [11].
T cell trafficking is facilitated by the secretion of effector chemokines, lytic granules and other dynamic changes that occur in the circulatory system [1,49]. Dynamic associations occur between T cells and endothelial cells (ECs), which are guided by chemokine and chemokine receptor interactions, as well as adhesion molecules such as selectins and integrins. T cells in the circulatory system need to firstly bind themselves to the ECs, move along ECs towards the tumor site, firmly grip to the endothelial wall, and lastly bridge through the endothelial wall of the blood vessel in order to enter the infection or tumor site [12,50]. T cell trafficking is influenced by several factors: the matching of chemokine receptors on T cells with chemokines secreted in the TME; aberrant vasculature and endothelial anergy; and tumor immunosuppressive mechanisms of the cells of the TME as well as checkpoint pathways of the TME [12] (Fig. 1). These are further discussed in this review.
Fig. 1.

Factors affecting CD8+ T cell trafficking and localization. CD8+ T cells in the peripheral blood are recruited or trafficked locally into the TME in response to the binding of various secreted chemokines (CCL3, CXCL9, CXCL10, CXCL11, CCL4, CCL5, CCL20, and CXCL16) and their corresponding receptors expressed on tumor cells and stromal cells of the TME. CD8+ T cell trafficking is positively influenced by the interaction between adhesion molecules (ICAM-1, VCAM-1) expressed by endothelial cells and their corresponding receptors which are expressed on the surface of CD8+ T cells. The successful binding of the chemokine ligand fractalkine and CX3CR1 facilitates CD8+ T cell trafficking. The interaction between sphingosine-1-phosphate (S1P) and its corresponding receptors importantly facilitate CD8+ T cell trafficking. The above mentioned receptor-ligand interactions are marked with a solid line and arrow because they promote CD8 T cell trafficking. The binding of endothelin-1 to ETB, its corresponding receptor on endothelial cells, reduces ICAM-1 expression, thus preventing T cell adhesion and subsequently preventing CD8+ T cell trafficking. The chemokine-chemokine receptor interaction between CCL2 and CCR2 inhibits CD8+ T cell trafficking. The later mentioned interactions between endothelin-1 and ETB as well as CCL2 and CCR2 are marked as dotted lines because their interactions inhibit CD8+ T cell trafficking.
3.1. Matching of Chemokine Receptors and Chemokines
In order for CD8+ T cells to be successfully trafficked into the tumor site, certain chemokine receptors on the T cells should interact with the corresponding chemokines that are secreted by tumor cells and other cells in the TME [11]. Reportedly, CXCR3 is expressed on activated CD8+ T cells in breast and colorectal cancers and additionally in melanoma. CXCR3 attracts CXCL9, CXCL10, and CXCL11 chemokines which are highly secreted by tumor cells in a number of solid cancers [12,50,51,52]. Conversely, certain tumors may express low levels of CXCL9, CXCL10, and CXCL11 which in turn results in decreased infiltration of CD8+ T cells into the TME. Furthermore, both CCL5 and CXCL10 have reportedly been upregulated in esophageal cancer, positively correlating with high expressions of CD8 and Gzmb, markers of CD8+ T cells, and also elevated levels of CD8+ T cells within the tumor site. These correlations confirm that both CCL5 and CXCL10 are involved in improving CD8+ T cell trafficking and localization in esophageal tumor sites [53,54]. Matsumura and colleagues observed mice deficient of CXCR6, the receptor for CXCL16 is lowly expressed on the surface of T cells; and discovered that these mice exhibited decreased T cell migration, poor anti-tumor immunity and prognosis in breast cancer [55]. The expression of CCL4 was observed to be directly proportional to the expression of CD8, Gzmb and the infiltration of CD8+ T cells [55,56]. CCR5 is the common receptor for CCL4 and CCL5; CCL20 binds to its receptor CCR6; and CCL3 to its receptor CCR1 [56]. Reportedly CCL2, one of the most common chemokines in the TME of various cancers, is secreted in response to the production of ROS and RNS by MDSCs in the TME [29]. High levels of CCL2 expression and its subsequent binding to CCR2 is positively correlated with the infiltration of TAMs and negatively correlated with CD8+ T cell infiltration in the TME [29].
The successful binding of CX3CR1 and its corresponding ligand fractalkine has been reported to positively influence the recruitment CD8+ T cells in cancer. Elevated levels of fractalkine have been reported in various cancer types and through interacting with its receptor it promotes the recruitment of Th1 cells, helper T cells, and subsequently improves CD8+ T cell function. Fractalkine has also been reported to be correlated with reduced tumor growth. Further investigations of this interaction may allow scientists to capitalize on directing CD8+ T cells in and out of the TME [57,58]. The interaction between sphingosine-1-phosphates (S1Ps) and their corresponding receptors play an important role in the trafficking and activation of CD8+ T cell trafficking as well as determining the differentiation of CD8+ T cell subsets. SIP levels are most elevated in the circulatory system, followed by the lymphatic system and the least levels are in other tissues, this SIP concentration gradient guides CD8+ T cells from the blood to the lymph and to target tissues, thus facilitating CD8+ T cell trafficking. S1P signaling has been reported to reduce the number of circulating naïve and TCM cells in the circulatory system while enhancing circulating TEM cells which are short-lived cytotoxic memory cells. Thorough investigations into SIP signaling and its effect on TME modulation are warranted, as this may help researchers in further understanding the recruitment and exclusion of CD8+ T cell subsets in the blood, lymph and target tissues [59].
3.2. Aberrant Vasculature and Endothelial Anergy
The growth of all cells is highly dependent on their supply of nutrients consequently, angiogenesis has been stated as one of the hallmarks to cancer development [60]. New blood vessels are rapidly produced within the TME. These blood vessels lack the necessary qualities to transport nutrients and metabolites in and out of the TME and lack the necessary integrity, strength and flexibility in order to function optimally for the cells in the TME [7]. The ECs forming the blood vessels of the TME may present with little to no, poorly attached pericytes. In the absence of pericytes, blood vessels are abnormally leaky, failing to optimally transport blood in and out of the TME, thus resulting in decreased migration of CD8+ T cells to the tumor site [5]. As a result, these blood vessels fail to sustain their structural integrity, leaking their content, and resulting in an acidotic, hypoxic TME [7].
As angiogenesis continues, tumor cells continue to increase the production of VEGF, which is negatively correlated with the production of adhesion molecules: ICAM-1; ICAM-2; VCAM-1 and CD34 on ECs. ICAM-1 and VCAM-1 bind to their antigens LFA-1 and VLA-4 respectively on the surface of T cells to stimulate T cell trafficking. Additionally, the binding of endothelin-1 and ETB, its corresponding receptor on ECs, reduces ICAM-1 production and localization, thus suppressing T cell adhesion. The lack of adhesion molecules on ECs prevent CD8+ T cells from attaching to ECs, ultimately preventing CD8+ T cell trafficking from the circulatory system to the tumor site. At this stage, these dysfunctional ECs, devoid of adhesion molecules, are considered to be in a state of anergy, lacking the ability to react to CD8+ T cells [60,61,12]. Therefore, identifying mechanisms to prevent angiogenesis in cancer may result in maintaining or even improving CD8+ T cell migration to the tumor site as well as maintain a normal pH, both of which promote anti-tumor immunity of CD8+ T cells.
3.3. Immunosuppressive Cells and Checkpoint Pathways of the TME
Cells and factors of the TME have an immunosuppressive effect on tumor infiltrated CD8+ T cells and have been explored at the beginning of this review. Immune cells express checkpoint proteins, which bind to corresponding ligands presented on tumor cells, leading to the activation of a co-inhibitory checkpoint pathways and inhibiting CD8+ T cell function and tumor cellular death [7]. PD-1 is an immune checkpoint protein broadly expressed on T and B cells, monocytes, DCs and NK cells; the expression of which is directed by the release of quite a few cytokines namely IFN-γ, interleukins IL-1, IL-7, IL-15, and IL-21 [62]. PD-L1 is a co-inhibitory molecule and the main corresponding receptor ligand that binds to PD-1. PD-L1 is expressed on the surface of many tumor cell types, immune cells and stromal cells of the TME [37,38]. Likewise, PD-L1 expression is directed by the presence of different cytokines, primarily IFN-γ [63], which has been deemed responsible for the expression of PD-L1 expression on the cells of the TME [37,38,63,64].
4. CD8+ T Cell Differentiation and Function
The activation of CD8+ T cells is a 3-step process that follows T cell priming. It includes the interaction between TCR and the antigenic peptide–MHC complex; the delivery of a costimulatory or co-inhibitory signal from DCs; and lastly, the stimulation from extracellular cytokines [65] (Fig. 2). Once CD8+ T cells have been successfully activated, they differentiate into what is known as an early effector CD8+ T cell state. This is followed by the differentiation of CD8+ T cells into memory precursor as well as terminal effector CD8+ T cells (Fig. 3).
Fig. 2.
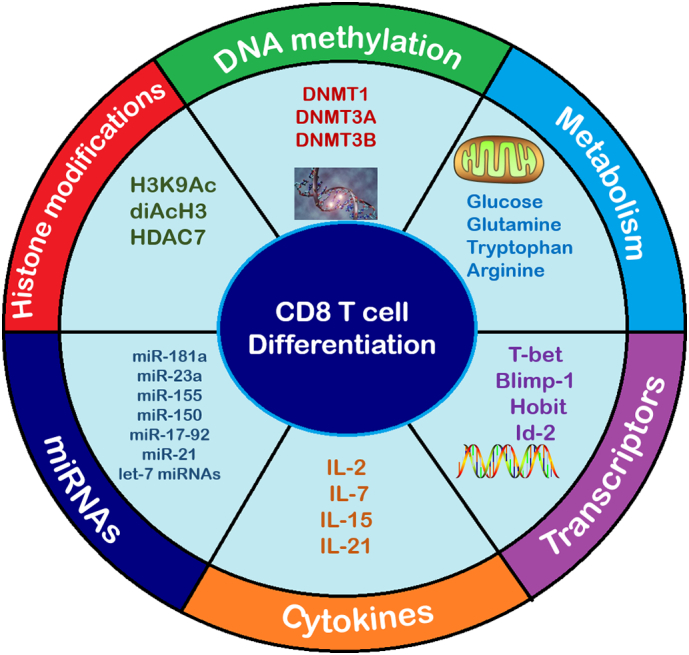
Factors promoting CD8+ T cell differentiation. CD8+ T cell differentiation and function is promoted by various factors and cellular changes. Metabolic factors such as glucose and amino acids (Glutamine, Tryptophan and Arginine) are used as primary or secondary sources of energy for CD8+ T cell differentiation and function. Transcription factors T-bet, Blimp-1, Hobit and ID2 promote CD8+ T cell differentiation. The presence of cytokines IL-2, IL-7, IL-15 and IL-21 promote CD8+ T cell activation and differentiation. miRNAs such as miR-181a, miR-23a, miR-155, miR-150, miR-17-92, miR-21 and let-7 miRNAs facilitate CD8+ T cell differentiation. Epigenetic changes such as certain histone modifications (H3K9Ac, diAcH3 and HDAC7) and DNA methylations (DNMT1, DNMT3A and DNMT3B) have been identified to facilitate CD8+ T cell differentiation.
Fig. 3.

Factors influencing T cell activation. The formation of the antigenic peptide–MHC-1 complex is a necessary process governing CD8+ T cell activation. Naïve CD8+ T cells may subsequently be activated through their interaction with molecules and cytokines facilitating the activation of their pathways. CD27, CD28, CD69, CD95 and OX40 are costimulatory molecules expressed on CD8+ T cells. The activation of CD8+ T cells is facilitated when any of these costimulatory molecules bind to their respective ligands on APCs. Hypoglycemia and hypoxia lead to increased signaling of peroxisome proliferator-activated receptor (PPAR)-α, facilitating energy generation through fatty acid catabolism and prioritizing CD8+ T cell activation. Th17 cell, a helper T cell, also facilitates CD8+ T cell activation. Naïve CD8+ T cells may be inactivated by the presence of immunosuppressive cells or factors of the TME; interactions with tumor cells; and effects from interactions with other APCs. TAMs secrete TGF-β, IL-10 and ROS to inhibit T cell activation. MDSCs secrete Arg1, iNOS and ROS to inhibit CD8+ T cell activation. Tumor cells expressing PD-L1 or CD80 receptors may bind to corresponding molecules PD-1 and CTLA-4 respectively, causing CD8+ T cell inactivation. Tumor cells may express CD39 and CD73 on their surface facilitating the metabolism of extracellular ATP into AMP and finally into adenosine which inhibits CD8+ T cell activation. Tryptophan is converted into kynurenine through the function of IDO which is produced by the tumor cell. Kynurenine inhibits CD8+ T cell activation. Tumor cells metabolize glucose into lactate and lactate production inhibits CD8+ T cell activation. APCs may express the B7S1 molecule which inhibits CD8+ T cell activation, upon binding to its receptor B7S1R.
4.1. Interaction between TCR and the Antigenic Peptide–MHC Complex
The TCR serves as a point of interaction for the antigenic peptide–MHC complex, otherwise known as the MHC-peptide-TCR, as well as antigens on APCs [33,34]. Naïve T cells are activated by professional APCs, DCs. This is considered a crucial step in T cell activation and antigenic specificity [47]. However, DCs, may fail to identify, and present foreign antigens to T cells, thus preventing CD8+ T cells from interacting with and being stimulated by the antigen [66]. The adaptive immune response hinges on effective antigen identification, presentation and interaction with the CD8+ T cell. Improving the aforementioned factors, within the context of the TME, will facilitate CD8+ T cell differentiation as well as CD8+ T cell function.
4.2. Co-Stimulation or Co-inhibition Signals between the DC and the T Cell
DCs deliver costimulatory or co-inhibitory signals to T cells in order to facilitate CD8+ T cell proliferation, cytokine production and to enhance effector CD8+ T cell function. Effector CD8+ T cell function is promoted by the stimulation of costimulatory cytokines and inhibited by the stimulation of inhibitory cytokines. T cell hypo-responsiveness may result from the chronic stimulation from inhibitory cytokines [67]. The activation of antigen-specific T cells will only be successful upon the reception of a costimulatory signal from a DC. In the absence of a costimulatory signal the T cells remain inactive.
CD69, CD95 and OX40 are costimulatory molecules commonly expressed on CD8+ T cells that bind to corresponding ligands that are expressed on DCs. The successful binding of these costimulatory molecules to their respective ligands leads to the activation of CD8+ T cells, thereby promoting tumor cell death [55,12,56]. CD69 activation stimulates the influx of calcium ions (Ca2+) and the activation of extracellular kinases ERK1/2 there-by facilitating CD8+ T cell proliferation. In addition, CD69 activation stimulates the secretion of IL-2 and IFN-γ which further promote the cytotoxic function of CD8+ T cells [68]. CD95 activation has been reported to facilitate CD8+ T cell growth, differentiation and migration. CD95L, the ligand of CD95 is expressed on DCs. The CD95/CD95L interaction promotes the differentiation of naïve CD8+ T cells into memory and cytotoxic CD8+ T cells. Elevated CD95L expressing DCs are correlated with improved anti-tumor immunity [69]. OX40 is expressed by antigen specific CD8+ T cells, and this expression may continue up to 72 h after antigen encounter. OX40L is the corresponding ligand of OX40 that is expressed on the surface of DCs. The expression of OX40 on CD8+ T cells has been reported to be correlated with an increase in CD8+ T cells with memory phenotype. OX40 agonists have been reported to improve the cytotoxic functions of CD8+ T cells by successfully overcoming CD8+ T cell tolerance to antigens or hypo-responsiveness, which usually occurs as a result of chronic stimulation. OX40 agonists have successfully been used to improve the immunological responses of certain metastatic cancer patients [70,71]. CD27 and CD28 are costimulatory molecules of the tumor necosis receptor super family and immunoglobulin family respectively, expressed by proliferating CD8+ T cells. The interaction between these receptors and their respective ligands CD80/CD86 and CD70/TNFSF7 stimulate CD8+ T cell activation [72].
Co-inhibitory signals are triggered by the successful interaction between co-inhibitory molecules and their ligands e.g., PD-1/PD-L1 or PD-1/PD-L2 interaction. This interaction results in the downregulation of effector cytokines produced by CD4+ and CD8+ T cells [[73], [74], [75]], therefore inhibiting the subsequent effector functions of these T cells. This downregulation of effector T cell function is correlated with the enhanced development and function of Treg cells in such a TME. Cytotoxic T lymphocyte antigen-4 (CTLA-4), an inhibitory receptor mainly expressed on the surface of T cells, primarily binds to ligand CD80 to inhibit T cell activation and IL-2 function. Furthermore, the PD-1/PDL1 and CTLA-4/CD80 interactions inhibit the Akt pathway [76]. Immune checkpoint inhibitors (ICIs) are used to block PD-1/PD-L1, CTLA-4/CD80 and similar co-inhibitory interactions [65]. ICIs have been found to be effective in treating different cancers as monotherapy and have also been effective as adjuncts to adoptive T cell and chimeric antigen receptor (CAR)-T cell therapies. These blockade immunotherapies upregulate effector CD8+ T cell responses, thus resulting in positive clinical responses through the formation, stimulation and development of tumor specific cytotoxic CD8+ T cells [77]. Interestingly, CD28 and CTLA-4 share a common ligand, CD80. CD28 promotes CD8+ T cell activation while CTLA-4 impedes CD8+ T cell activation, thus the CD28/CTLA-4 pathway may either have a costimulatory or co-inhibitory effect on CD8+ T cell activation. It has been hypothesized that specific, successful inhibitors of the CD28/CTLA-4 pathway will be able to diminish the possible suppressive aspects of the CD28/CTLA-4 pathway while improving the costimulatory signals to ultimately improve CD8+ T cell differentiation and funtion [72]. The presence of PD-1 expression on CD8+ T cells and PD-L1 expression on cancer cells warrants the use of ICIs as a mode of cancer immunotherapy. However, there are still various challenges and limitations to PD-1/PD-L1 therapies, these are further discussed later in this review.
DCs, may exhibit different immune functions depending on their level of maturation. Mature DCs stimulate Th1 cell production and subsequent effector CD8+ T cell function; and immature DCs stimulate Treg cell production through the secretion of IL-10, favoring immunosuppression. TGF-β and IL-6 are secreted by DCs to produce Th17 cells, which transport DCs to lymph nodes thus facilitating CD8+ T cell production and activation [78,79], and have been reported to enhance DC recruitment into the TME. Additionally, these helper T cells facilitate the production, differentiation and function of tumor specific CD8+ T cells in vivo by continuously facilitating CD8+ T cell IFN-γ production, delaying T cell exhaustion, and promoting the activation of CD8+ T cells [80]. Therefore, DCs facilitate tumor evasion by playing an important role in moderating the levels of T cell subsets through the secretion of immunosuppressive factors [47]. Additionally, DCs may produce indoleamine 2,3-dioxygenase (IDO) an enzyme preventing effector CD8+ T cell growth and multiplication, thus favoring the suppression of the immune system and tumor evasion [81]. DCs may also produce high levels of inducible costimulator ligand (ICOS-L) which facilitates CD4+ T cell specialization and IL-10 secretion, which in turn stimulate Treg cell production, and subsequent immunosuppression. DCs have been reported to secrete IL-15 and IL-21 and these positively influence the activation of effector CD8+ T cells. Chemokines may negatively or positively facilitate CD8+ T cell migration in the TME [12]. Therefore, regulating the secretion of certain chemokines is vital in improving CD8+ T cell infiltration. Combining chemokine based immunotherapies with anti-TAM and anti-MDSC therapies could significantly improve CD8+ T cell trafficking and function as well as modulate the TME into a less immunosuppressive TME.
4.3. Stimulation from Extracellular Cytokines
The expression of cytokines IL-2, IL-7, IL-15, and IL-21 promote cytokine-driven CD8+ T cell activation and further enhance CD8+ T cell expansion. Chemokines may be produced by DCs either having a costimulatory or co-inhibitory influence on CD8+ T cell migration and function. The MHC-peptide-TCR interaction is an advanced interaction occurring at the immunological synapse (IS) that may last for several hours in the presence of molecules and recruited chemokines that strengthen the interaction. This interaction is maintained in order to promote prolonged signaling, CD8+ T cell growth and production. CCL2, CCL5, CCL19, CCL21 CXCL12 are characteristic chemokines involved in stimulating CD8+ T cell growth and production. Reportedly, CD8+ T cell stimulation via TCR and CXCR4, increased the expression of activation markers CD69, CD25, and CD154, leading to increased T cell growth and production; as well as the production of IL-2, INF-γ, and IL-4 [82]. It was also discovered that CCL21 showed a higher binding affinity than CCL19 to their ligand, leading to an increase in CD8+ T cell growth, CD69 and CD25 expression, and the secretions of IL-2 and INF-γ [83,54,84]. The intricacies elucidating the full effects that cytokines have on CD8+ T cell differentiation and function is only partially understood.
4.4. Cellular Metabolic Changes
Tumor cells undergo various cellular and metabolic changes, allowing them to outcompete with immune cells for available nutrients in the TME. This is recognized as one of the hallmarks to cancer development discovered by Hanahan & Weinberg [85]. The growth and development of new, proliferating cells is highly dependent on the presence of oxygen and cellular nutrients, mainly glucose [37]. Tumor cells are able to obtain nutrients from alternative sources such as amino acids [86,87], acetate and fatty acids [88,89], especially in hypoxic and hypoglycemic environments [37]. Different cells of the TME, including T cell subtypes have different metabolic requirements enabling them to perform their various functions [90]. Not only do tumor cells deplete the TME of nutrients, but they additionally produce signaling and metabolic by-products that prevent CD8+ T cell function. An amalgamation of metabolic processes result in the production and accumulation of adenosine in the TME [91]. Ectonucleotidases, CD39 and CD73, are expressed on Treg, B and tumor cells, they facilitate the metabolism of extracellular ATP or ADP into AMP and finally into adenosine within a hypoxic TME [39]. Elevated adenosine levels in the TME have a negative correlation with CD8+ T cell function, and a positive correlation with Treg cell production, thus encouraging immune evasion [39,92,93]. Warburg and colleagues reported that tumor cells break down pyruvate into lactate in the presence and absence of oxygen, undergoing aerobic glycolysis, in order to obtain energy in a hypoglycemic environment. This Warburg effect proves that tumor cells are able to adapt and continue to proliferate even in the presence of limited nutrients [94]. Lactate, a by-product of glycolysis, is produced in a hypoxic TME and may be used by the cells of the TME as an alternative source of energy in oxidative phosphorylation (OXPHOS). Additionally, lactate accumulation may lead to lactic acid formation, promoting chronic inflammation [95], inhibiting CD8+ T cell function [96] and facilitating the migration of immunosuppressive cells into the TME [95]. The accumulation of metabolic by-products, such as adenosine and lactate, in the TME encourage immune evasion. Therefore, facilitating the downregulation of these metabolites in the TME may be conducive as combination therapies in cancer immunotherapy.
4.4.1. Hypoxia
Hypoxia in tumors affects the metabolism of tumor cells and has been characterized with a reduced response to radiotherapy and chemotherapy [91,39,92]. Tumors progressing in a hypoxic TME elicit a number of responses to promote tumor evasion namely, the facilitation of angiogenesis; and the formation of hypoxia inducible factors (HIFs), preventing antigen and TCR interaction [97]. Angiogenesis in the TME results in abnormally formed blood vessels which are prone to collapse, resulting in elevated interstitial pressure, and altered osmotic gradients, perpetuating a hypoxic TME [60,61]. Tumor cells may also increase the formation of HIFs which drive the expression of glucose transporter genes, enzymes required for glycolysis, and nitrogen oxide (NO) [95,96]. HIF-1α, facilitates the upregulation of nitrogen oxide synthase (iNOS) and Arginase-I, therefore facilitating the synthesis of NO and L-Arginine respectively [97]; and the expression of oxygen containing molecules NOX2 and ROS in the TME [98]. These are involved in limiting T cell growth, activation, differentiation, and ultimately favor the death of effector CD8+ T cells and immunosuppression [99].
The interaction between antigen and TCR is prevented in a hypoxic environment [97]. Additionally, CD8+ T cell function may be influenced by hypoxia in various ways, depending on the current level of CD8+ T cell differentiation [100]. Caldwell and colleagues studied the effect that different oxygen levels had on T cell growth at various levels of T cell differentiation. They observed the cytotoxic function of CD8+ T cells under different hypoxic environments. It was interestingly illustrated that the sustained function of cytotoxic CD8+ T cells is not purely oxygen dependent. The CD8+ T cells produced in a 2.5% oxygenated environment had a higher surface density of CD25+, thus improving their cytotoxic ability over those produced in the 20% oxygenated environment, which were merely higher in expression of CD25+, without an increased surface density. CD8+ T cells adapt in response to a hypoxic TME. Caldwell and colleagues further discovered that hypoxic environments may either impede or amplify CD8+ T cell function however, the mechanisms underlying this phenomenon warrant further investigation. In addition, further investigations need to be conducted in assessing the possible effects that cell-cell interactions and adhesions may have on cultured cells and their responses in various hypoxic environments. These effects may account for the contrasting possible effects that hypoxia may have on cytotoxic CD8+ T cells [101].
4.4.2. Hypoglycaemia
Immature or naïve T cells are able to obtain their metabolic functions through aerobic glycolysis, however when nutrients are limited they may obtain metabolic function through OXPHOS [93,95]. Both normal and tumor cells may contend for glucose by increasing their expression of glucose transporter receptor 1 (GLUT-1) [95], the receptor for the GLUT-1 gene expressed on the surface of most cancer cell types and also on T cells [93], but tumor cells outcompete T cells for glucose. Additionally, in a hypoxic environment, tumor cells may activate genes that regulate glycolysis and glucose transport, this may also occur as a stimulation from oncogenic signaling. Reportedly, high expressions of GLUT-1 are negatively correlated with CD8+ T cell influx into the TME [95] as well as patient prognosis in pancreatic adenocarcinoma [95], lung, ovarian [102], endometrial, breast and other cancers [103]. CD8+ T cells function optimally in a normal to high glucose environment, and may transit into an inactive state, known as T cell anergy and eventually undergo apoptosis in a hypoglycemic environment [104].
CD8+ T cells may use amino acids, l-Glutamine, l-Arginine and tryptophan as alternative sources of energy. Activated CD8+ T cells undergo aerobic glycolysis and glutaminolysis to promote the production of effector cytokines, INF-γ and IL-2 [91] and other growth factors facilitating effector CD8+ T cell clonal expansion and function [98,105]. IDO converts tryptophan in the TME into kynurenine, which interacts with Aryl hydrocarbon Receptor (AhR) and promotes Treg cell production in inflammation and cancer. Elevated IDO levels in tumors are correlated with poor patient prognosis [106,107].
Zhang and colleagues reported that CD8+ T cells may increasingly use fatty acids directly as a source of energy in a hypoxic hypoglycemic environment [108]. Fatty acids are used in the production of amino acids, and may contribute to the formation of effector factors of CD8+ T cells. Additionally, fatty acids may be converted into Acetyl-CoA, to facilitate the production of GADP, a vital enzyme involved in energy production through the Krebs cycle and glycolysis; and facilitating an increase in the production of INF-γ, thus improving effector functions of CD8+ T cells [109]. Additionally, in a hypoxic, hypoglycemic environment, CD8+ T cells metabolically breakdown fatty acids through increased signaling of peroxisome proliferator-activated receptor (PPAR)-α, a transcriptional factor, facilitating energy generation through fatty acid catabolism [108]. Fatty acid catabolism facilitates an increase in the production of INF-γ, thus improving effector functions of CD8+ T cells. This form of metabolism may be prioritized in a hypoxic hypoglycemic TME so as to improve CD8+ T cell mediated anti-tumor immunity.
Galectins are naturally occurring carbohydrate binding proteins that form a glycoprotein network with glycans in the extracellular matrix. They are secreted by macrophages, activated T cells as well as tumor cells and are elevated in several cancers [110,111]. Galectin-3 attaches to glycans in the TME such as VEGF-R2, promoting angiogenesis [112]; and the expression of immune cell surface markers such as CD8, TCR, LAG-3 and others, thus facilitating NK and T cell dysfunction [113,114]. In addition, galectin-3 attaches to glycosylated cytokines, i.e. IFN-γ, CXCL9 and CXCL10 decreasing their infiltration and the subsequent infiltration of CD8+ T cells into the TME. Gordon-Alonso et al. reported elevated IFN-γ, CXCL9 and CXCL10 levels in the TME as well as an improvement in T cell infiltration after galectin antagonist treatment. Galectin-3 inhibitor treatment was reported to improve T cell infiltration and activation furthermore, delaying tumor growth by improving IFN-γ levels. Thus proposing for the incorporation of galectin inhibitors in anti-tumor strategies [110].
4.4.3. Epigenetic and Transcriptional Factors
Various epigenetic, and transcriptional factors have been reported to control the differentiation and function of cytotoxic and memory CD8+ T cells, operating synergistically or contrastingly in regulating T cell differentiation [10,103], namely DNA methylation and histone modifications; as well as the functioning of T-bet and Eomesodermin (Eomes); B7 superfamily member 1 (B7S1); B lymphocyte-induced maturation protein 1 (Blimp-1) and Hobit; Forkhead box protein O1 (FoxO1), inhibitor of DNA-binding/ differentiation (ID) proteins and microRNAs (miRNAs).
DNA methylation is an epigenetic process facilitated by the function of the following DNA methyltransferases (DNMTs): DNMT1, DNMT3A, and DNMT3B [115]. Pathania and colleagues reported increased DNMT1 levels in breast cancer cells, highlighting that DNMT1 is crucial in the development of breast cancer stem cells. Furthermore, the elimination of DNMT1 in mice proved to limit breast cancer stem cell growth, therefore decreasing tumor progression and metastasis [116]. DNA methylation has been identified as an indicator of cancer aggressiveness. Similarly, DNA hypermethylation promotes carcinogenesis by decreasing gene expressions and transferring abnormal genetic sequences to subsequent dividing cells [36]. Alterations in DNA methylation, as seen in infection and carcinogenesis, are negatively correlated with the expression of effector factors for CD8+ T cell differentiation i.e. Gzmb, INF-γ. Interestingly, DNA methylation downregulates PD-1 expression on T cells [116,117]. DNMT1 has been identified as crucial for the differentiation and function of CD8+ T cells in viral infection, but not specifically in cancer. DNMT3A is expressed soon after CD8+ T cell activation and has been identified as a regulator of effector CD8+ T cell differentiation in the early stages of CD8+ T cell differentiation. DNMT3A favors the development of effector CD8+ T cells by impeding the development of memory CD8+ T cells. This effect of DNMT3A has been confirmed in both acute viral as well as cancer murine models [[118], [119], [120]]. Low dose DNA methyltransferase or DNA methylation inhibitors have proven to be successful therapies in hematologic cancers [36]. The above mentioned abnormal changes in the chromatin network or DNA may either facilitate CD8+ T cell differentiation and function, as in the case of DNMT3A expression. On the other hand, changes such as DNMT1 expression and DNA hypermethylation may promote carcinogenesis and metastasis.
Histone proteins undergo modifications allowing them to regulate genes of transcription in both normal and cancer cells. The modification of H3K9Ac results in the upregulation of genes responsible for cytotoxic and memory CD8+ T cell differentiation and function. G9a, another histone protein, interacts with DNMT3 to facilitate histone modifications in facilitating effector CD8+ T cell differentiation [121]. Additionally, di-acetylated histone H3 (diAcH3) has been identified as a marker of memory CD8+ T cells. HDAC7, a histone deacetylase has been identified as vital in facilitating TCR signaling and CD8+ T cell differentiation [122]. Thus, one can deduce that the genes acknowledged to control CD8+ T cell differentiation may be transcriptionally characterized by some form of histone modification [115,122].
T-bet and Eomes are T-box transcriptional factors that increase INF-γ stimulation by attaching to promoters that upregulate the stimulation of Gzmb, perforin and INF-γ. Additionally, they have been found to upregulate the expression of CXCR3 and CXCR4, thus positively facilitating effector CD8+ T cell differentiation. T-bet expression is stimulated by TCR signaling and is positively influenced by IL-2 and negatively influenced by mTOR signaling. Eomes expression is dependent on the presence of T-bet [108,10]. In contrast to Eomes, T-bet expression is most elevated during infection or inflammation during early stages of effector CD8+ T cell differentiation; and is least expressed once the infection or inflammation has cleared and memory CD8+ T cells are undergoing differentiation [10]. T-bet has been identified as an important prognostic marker in gastric cancer as well as an important marker in assessing therapeutic effects of immunotherapy [36,116]. CD8+ T cells expressing low levels of T-bet generally promote the production of memory precursor/ stem-cell like memory T (TSCM) cells which are long-lived pluripotent cells that are capable of differentiating into TCM and TEM cells. CD8+ T cells expressing elevated levels of T-bet are positively correlated with the production of short-lived effector CD8+ T cells and TEM cells. Reportedly, CD8+ T cells expressing elevated levels of Eomes are correlated with the production of TEM cells [123,124]. Li and colleagues observed successful elimination of established tumors after the adoptive transfer of T-bet and Eomes expressing CD8+ T cells [124]. The transcription factor IRF4 regulates the ratio of T-bet and Eomes expression on CD8+ T cells. The T-bet/ Eomes ratio is important in regulating the clearance of chronic viral infections such as lymphocytic choriomeningitis (LCMV). High levels of IRF4 maintain TCR signaling and slant the T-bet/ Eomes ratio in favor of T-bet, favoring the production of short-lived effector cells and antiviral function. In contrast, a T-bet/ Eomes ratio slanted in favor of Eomes promotes the production of memory precursor CD8+ T cells and reduces antiviral function [125]. As the immune response continues, the T-bet/ Eomes ratio favors the production of memory precursor cells. Therefore, T-bet and Eomes are essential transcription factors regulating the antiviral and antitumor functions of effector and memory CD8+ T cells. Therefore, targeting T-bet and Eomes could be of vital importance in encouraging CD8+ T cell mediated anti-tumor immunity, regulating CD8+ T cell differentiation and preventing T cell exhaustion.
B7 superfamily member 1 (B7S1) is expressed on APCs and various solid tumor cells. B7S1 is promoted by IL-10 and IL-6, and binds to its receptor B7S1R on the surface of CD8+ T cells to inhibit CD8+ T cell growth and cytokine secretions, thus negatively influencing CD8+ T cell trafficking and cytotoxic function. A high expression of B7S1 is a marker for poor patient prognosis in cancer. B7S1 upregulates Eomes, thus facilitating T cell dysfunction and exhaustion [122]. B7S1, T-bet and Eomes expression are closely correlated with each other, thus closely monitoring B7S1 targeted treatments together with T-bet and Eomes treatments is of beneficial importance in cancer immunotherapy.
B lymphocyte-induced maturation protein 1 (Blimp-1), a transcriptional factor produced by effector CD8+ T cells, inhibits T cell transcription [10]. Additionally, Blimp-1 downregulates PD-1 expression on T cells, further confirming that it negatively influences effector CD8+ T cell differentiation. Another transcriptional factor produced by CD8+ T cells is BCL-6 and its production is negatively correlated to that of Blimp-1 [108,115]. CD8+ T cells producing Blimp-1 in excess facilitate the differentiation of memory CD8+ T cells. Reportedly, Blimp-1 expression was elevated in T cells of patients with acute myeloid leukaemia (AML), and it was identified as a valuable biomarker in monitoring the treatments of AML. Blipm-1 positively encourages the transcriptional expression of ICIs on T cell surfaces, therefore significantly encouraging T cell exhaustion in both infection and cancer [126]. Naïve CD8+ T cells differentiate into short-lived effector CD8+ T cells with cytotoxic functions; or memory precursor effector CD8+ T cells. These memory precursor CD8+ T cells are capable of developing into long-lived CD8+ T cells such as TEM, TCM and TRM. Short-lived effector CD8+ T cells have been reported to express T-bet and Blimp-1 in comparison to memory precursor CD8+ T cells which have been reported to express Eomes.
Hobit, the homolog for Blipmp-1, is primarily expressed on effector CD8+ T cells of humans. Hobit has been identified as an important transcriptional factor expressed on short-lived effector CD8+ T cells that regulates the fate of effector CD8+ T cells similar to T-bet. However, further investigations need to be conducted in order to clearly identify the mechanisms and possible synergistic effects that these transcriptional factors may have on the fates of effector CD8+ T cells [127,128]. The mechanisms in which Blimp-1 and Hobit control effector CD8+ T cell function is not fully understood. Further investigations need to be conducted in order to clearly identify these mechanisms as well as possible synergistic effects that Blimp-1, Hobit, T-bet and Eomes may have on each other as well as on regulating the fate of memory and effector CD8+ T cells. These investigations could be used to improve T cell exhaustion targeted therapies and may be promising in reversing T cell exhaustion.
FoxO1 is a transcriptional factor, essential in the differentiation of memory CD8+ T cells. It is known to work in combination with other transcriptional factors in facilitating the chronic, long-term functions of memory CD8+ T cells, their differentiation and continued existence in infection [121] as well as in cancer. Targeting FoxO1 through deletion has resulted in a decrease in IL-12 production, the downregulation of mTORC1 and encouraging the production of memory CD8+ T cells, thus promoting CD8+ T cell survival [129].
A positive correlation has been identified between the overexpression of certain ID transcription genes and perpetuating metastasis in various cancers. The overexpression of ID2 has been identified in high metastatic tumor cells wherein ID2 promotes further metastasis as opposed to low metastatic tumor cells which impede metastasis through the decreased expression of Semaphorin 3F, an inhibitor of metastasis [130]. In addition, ID2 has been identified as an essential factor in increasing effector CD8+ T cell count during the acute onset of an infection and ID3 has been identified as a facilitator for the continued existence of TSCM cells [36].
microRNAs (miRNAs), control gene transcription of antigen-specific tumor infiltrating lymphocytes (TILs), as well as other cells. The expression of certain miRNAs have been reported to be elevated in effector CD8+ T cells. Additionally, miRNAs are vital in the production and function of immune cells such as MDSCs, DCs, and macrophages [131]. miRNAs have been reported as major regulators of gene and protein expression, immune checkpoints and signaling pathways, which all have an impact on CD8+ T cell differentiation and function [132]. miRNA profiling studies have identified either a downregulation or upregulation in the expression of certain miRNAs on various CD8+ T cell subsets during CD8+ T cell differentiation. It has been proposed that the efficacy of T cell-based therapies may be enhanced by incorporating miRNAs in TCR- and CAR-T cell therapies thus, enhancing CD8+ T cell mediated anti-tumor immunity through the regulation of miRNAs and their associated genes in T cell engineering [133].
miRNA26a and miRNA101, immunosuppressive miRNAs, reduce the production of EZH2, which in high levels facilitates anti-tumor immunity through enhancing effector cytokine production, function and improving patient prognosis. Thus the elevation of these miRNAs ultimately leads to T cell dysfunction [133]. Reportedly, the effects of miRNA139 and miRNA342 expression prevent Eomes and perforin production, and subsequently impede cytotoxic enhancing factors. miRNA150 is broadly expressed in TILs [134], limiting IL-2 function, thus negatively impacting on effector CD8+ T cell development [135]. Contrastingly in a later study, Smith and colleagues discovered miRNA150 to be essential in enhancing effector CD8+ T cell production and differentiation [136]. It was reported that the lack of miRNA150 positively correlates with the production and differentiation of memory CD8+ T cells [137]. Immunosuppressive miRNAs negatively correlate with the differentiation and function of cytotoxic CD8+ T cells, impacting poorly on patient prognosis in cancer.
miRNA17–92 enhances CD8+ T cell differentiation through stimulation of the PI3K/AKT/mTOR pathway and the inhibition of PTEN. The inhibition of miRNA17–92 has been identified to favor the differentiation of memory CD8+ T cells and elevated miRNA17–92 and miRNA181 have been identified in facilitating CD8+ T cell differentiation especially at its onset, following antigen stimulation [138]. Both miRNA17–92 and miRNA181 expression are negatively correlated with let-7 expression, which is characterized by a decrease in effector CD8+ T cells [139]. Reportedly, miRNA155 is an enhancer of effector CD8+ T cell differentiation in infections and cancer alike [140] and miRNA23a inhibits Blimp-1 therefore inhibiting the differentiation of effector CD8+ T cells [54]. Elevated miRNA21 levels have been reported in effector CD8+ T cells, which in turn facilitate T cell activation and differentiation; and decreased levels in naïve and memory CD8+ T cells [141]. miRNAs may be immunostimulatory in function, facilitating effector CD8+ T cell differentiation and function and may be considered as markers reflecting positive CD8+ T cell anti-tumor immunity.
4.4.4. Signaling Pathways
Signaling pathways interact with each other to highly influence the growth and survival of healthy and unhealthy cells [142]. The phosphatidylinositol-3-kinase (PI3K)/Akt signaling pathway is fundamental in continuing cellular existence in a nutrient low, hypoxic and acidic TME. Mammalian target of rapamycin (mTOR) obtains stimuli from nutrients, growth factors and other cellular stimuli to facilitate the downregulation of effectors necessary for protein synthesis, thus inhibiting CD8+ T cellular growth, development and metabolism [104,143]. Gene alterations and dysfunctions in solid and haematological tumors influence the activation of these pathways, facilitating cell growth and proliferation; improving tumor cell survival; and or facilitating metastasis, angiogenesis and therapeutic resistance [142]. Additionally, CD8+ T cell metabolism is preserved through the equilibrium between the stimulation of the adenosine monophosphate-activated protein (AMPK) and mTOR complex 1 (mTORC1) pathways. mTORC1 is essential in the differentiation of CD8+ T cells and the suppression of mTORC1 has been reported to attenuate the differentiation of memory CD8+ T cells. In addition, mTORC1 activation is achieved through TCR signaling and is maintained by the uptake of amino acids, which is achieved by the function of amino acid transporters. ASCT2, a transporter of glutamine and leucine, stimulates the uptake of these amino acids into CD8+ T cells, triggering the mTORC1 signaling pathway and promoting the differentiation of effector CD8+ T cells [96]. On the other hand, increased extracellular levels of potassium ions (K+) have been reported in the TME and these ions inhibit the activation of the AKT-mTORC1 signaling pathway. As a result of the concentration gradient and in the presence of the enzyme protein phosphatase 2A (PP2A), K+ ions move into CD8+ T cells and the mTORC1 signaling pathway is inhibited [144,145]. The stimulation of the PI3K/Akt/mTOR signaling pathway promotes tumor cellular proliferation within a nutrient deficient TME; yet also impeding the production of T cell effector factors.
The Wnt/β-catenin; STAT3 and SMAD; and the Notch signaling pathways have been identified as additional pathways regulating T cell growth, trafficking, differentiation, activation and ultimately function [146]. Upon activation, the Wnt/β-catenin pathway impedes the expression of Eomes, a chief CD8+ T cell regulator necessary for the production of INF-γ, therefore impeding effector CD8+ T cell differentiation. Consequently, the induction of Wnt/β-catenin by TWS119 has been known to enhance the promotion of TSCM cells [146], which are capable of re-establishing themselves, promoting the differentiation of TCM and TEM cells which re-circulate in the blood and are reactivated upon re-encounter with an antigen [147]. However, the activation of the Wnt/β-catenin pathway has been positively correlated with carcinogenesis and metastasis. It is an important oncogenic pathway that leads to tumor evasion. A negative correlation between increased β-catenin expression and the infiltration of CD8+ T cells at tumor sites has been identified. In addition, increased β-catenin expression is correlated with the migration, function and survival of Treg cells [148]. Activation of Wnt/β-catenin pathway may have contrasting effects on tumor immunity, therefore immunomodulation through this pathway is considered contentious. Highly specific inhibitors would have to target certain aspects of the pathway while amplifying other aspects of the pathway in order to improve effector CD8+ T cell mediated anti-tumor immunity.
STAT3 and SMAD activation is initiated by mature T cells and is further maintained by the secretion of IL-21. Additionally, IL-6, IL-10, TGF-β, VEGF and other inhibitory cytokines trigger STAT3 signaling, impede effector CD8+ T cell differentiation and exhaustion, favoring the production of TSCM cells. The production of TSCM cells is enhanced by the overexpression of ID transcriptional genes as well as STAT3 signaling in mature CD8+ T cells. In addition, STAT3 signaling triggered by oncogenes facilitates the expansion of MDSCs and Treg cells [147]. STAT3 controls the expression of transcriptional factors Eomes, Blimp-1, BCL-6 thus protecting the survival of memory precursors, rather than cytotoxic CD8+ T cells.
The Notch signaling pathway determines Eomes expression [149]. In vitro studies have associated Notch signaling with the production of effector genes and markers of cytotoxic CD8+ T cells, namely Gzmb, perforin, INF-γ, and T-bet. Furthermore, Notch expresses a vital factor forming the receptor for IL-2, which additionally facilitates the PI3K/Akt/mTOR signaling pathway [142,150]. The Notch pathway favors T cell differentiation towards the production of TSCM cells which are highly proliferative, self -renewing, pluripotent cells that display superior anti-tumor efficiency than TCM and terminal effector CD8+ T cells. In addition, TSCM cells respond rapidly to antigens upon re-exposure, are resistant to apoptosis, and are highly cytotoxic to cancer cells [151,152]. Therefore, developing specific and non-specific techniques to modulate Notch signaling may enhance the production and development of TSCM cells with cytotoxic abilities. Targeting the Notch pathway may possibly by-pass CD8+ T cell dysfunction by replenishing the supply of TSCM cells in the TME. Thus, immunomodulation of the Notch signaling pathway is of vital importance in enhancing adoptive T cell therapies.
5. T Cell Dysfunction
T cell anergy, exhaustion, senescence and stemness are all considered as different types of T cell dysfunction [153]. T cell anergy is an inactive state in which T cells produce low levels of IL-2 resulting in incomplete effector T cell differentiation. Exhausted T cells are characterized as effector T cells that produce reduced levels of effector cytokines, express co-inhibitory receptors on their surface, exhibit low effector functions and are resilient to being converted back into functional effector T cells. T cell senescence is characterized by decreased CD28 expression, thus preventing T cell costimulation, leading to the loss of genes at chromosomal ends and subsequently the cessation of the cell cycle [131,154]. T cell stemness is the ability that TSCM cells have in re-establishing themselves and facilitating the differentiation of TCM cells and TEM cells. T cell stemness is a desired type of T cell dysfunction [153].
Chronic T cell stimulation results from prolonged illness or inflammation, due to injury, infections or cancer. Chronic T cell stimulation re-exposes the TCR to antigens and thus weakens the TCR response to antigens; inhibiting T cell growth, effector cytokine production and cytotoxic function of CD8+ T cells [61,131,154]. Exhausted CD8+ T cells express high levels of co-inhibitory receptors on their surface, such as PD-1, CTLA-4, lymphocyte activation gene 3 protein (LAG-3), and T-cell immunoglobulin domain and mucin domain protein 3 (TIM-3) [131]. As T cells develop into exhausted T cells they lose their ability to produce various immunostimulatory cytokines. At an early phase of T cell exhaustion, exhausted T cells lose their production of IL-2, leading to their loss of cytotoxic function. TNF-α production is lost at an intermediate phase of T cell exhaustion, and the loss in IFN-γ and Gzmb production occurs at an advanced phase of T cell exhaustion [149,155]. The increase in expressions of co-inhibitory receptors on exhausted T cells as well as the decrease in secretions of immunostimulatory cytokines results in decreased CD8+ T cell growth, differentiation and trafficking.
Insufficient T cell trafficking and T cell exhaustion may contribute to resistance to ICI therapies. In order to address these common challenges and limitations in the therapeutic efficacy of ICI therapies, ICI therapies are being explored in combination with other therapies [99]. Successful results have been reported in combining anti-PD-1/PD-L1 therapies with mainstream chemotherapy in gastric [156] and colorectal cancer [157]. Similarly, positive results have been reported in combining anti-PD-1/PD-L1 with radiation therapies in melanoma [158]. Preventing and reversing T cell exhaustion are promising strategies in targeting the challenges and limitations of ICI therapies. Kamphorst and colleagues investigated various strategies in order to improve the efficacy of PD-1 therapies. They identified the important role that the CD28/B7 costimulatory pathways play in reversing CD8+ T cell exhaustion. Stimulation of this costimulatory pathway improved the efficacy of PD-1 blockade therapy in cancer as well as chronic viral infection. They further suggested that future investigations should explore the use of CD28 as a predictive biomarker for possible PD-1 blockade sensitive patients [159].
The effector function of T cells is positively correlated with the differentiation of CD8+ T cells. Investigators have observed the sustained survival of TCM cells as opposed to effector CD8+ T cells in vivo. In addition, it was deduced that co-infusing CD4+ T cells with CD8+ T cells during adoptive cell therapy improved the efficacy of CD8+ T cells in vivo. The efficacy of the CD8+ T cells was enhanced by the supportive aid of the CD4+ T cells. Similarly, researchers have highlighted that the infusion of memory or TSCM cells may improve the efficacy of CAR-T cell therapy as we know it. This would address T cell exhaustion which usually occurs in the conventional approach of infusing terminally differentiated effector CD8+ T cells in adoptive cell and CAR-T cell therapies [160].
During infection, illness or cancer, foreign antigens are identified by CD8+ T cells, in order to facilitate antigen presentation and to initiate the immune response. This concept is based on self-nonself discrimination. Foreign antigens express molecular signatures that make them easily identifiable. Tumor cells do not always express these foreign antigens however; they may exhibit an overexpression of tumor associated antigens (TAAs) which are lowly expressed in normal tissues. Many TAAs have been identified as promising targets for cytotoxic CD8+ T cells which are tumor specific in their function. However, the expression of TAAs in normal tissues may trigger mechanisms that encourage the production of T cells with TCRs of reduced sensitivity. This process is termed central or peripheral tolerance. On the other hand, in their attempt to perform their tumor specific function, highly sensitive cytotoxic CD8+ T cells may successfully destroy tumor cells and normal cells. Tumor-specific neoantigens are alternative antigens expressed exclusively on the surface of tumor cells. Neoantigen-specific CD8+ T cells are able to identify, specifically target and destroy these tumor cells without leading to toxicity or the destruction of normal cells [161,162]. It has been postulated that utilizing neoantigen vaccines together with ICI or other combination therapies may activate neoantigen-specific CD8+ T cell expansion and responses. Tumor specific CD8+ T cells may have improved anti-tumor function in various cancer types [162,163]. The specificity of neoantigens make them ideal targets for tumor specific CD8+ T cell antitumor immunity.
6. Conclusion
The anti-tumor functions of CD8+ T cells in the TME are dependent on antigen presentation; successful T cell priming, trafficking, differentiation and function. Naïve CD8+ T cells migrate from the bone marrow and secondary lymphoid organs; differentiate from naïve CD8+ T cells to effector CD8+ T cells and further differentiate into memory and cytotoxic cells. Cytotoxic CD8+ T cells enter the tumor site to perform their function and memory CD8+ T cells may either enter the tumor site as TRM cells or recirculate in the blood to perform their various functions as TCM cells. In theory, once cytotoxic CD8+ T cells enter the tumor site they should be able to destroy tumor cells however, the cells and factors of the TME provide an immunosuppressive environment preventing the function of CD8+ T cells [164]. Various forms of cancer immunotherapy have proven to be effective in re-establishing and promoting CD8+ T cell anti-tumor immunity however, many of these therapies have a short-lived and or limited effectivity due to the immunosuppressive effects of the TME. Combination therapies focusing on upregulating and improving effector CD8+ T cell function as well as those impeding immunosuppressive effects of the TME are of increasing vital significance in cancer immunotherapy. In addition, preventing and reversing T cell exhaustion and enhancing the stem-cell like properties of CD8+ T cells are promising mechanisms to improve CD8+ T cell proliferation and survival within the TME, enhance anti-tumor function, as well as valuably improve adjunct modifications to adoptive T cell therapies in future.
Conflict of Interest
The authors declare that they have no competing interests.
Acknowledgments
This study was supported by the grants from the National Natural Science Foundation of China (81771781), the National Key Research and Development Program of China (2016YFC1303501) and Major Science and Technology Projects of Henan Province (1611003101000).
References
- 1.Nolz J.C. Molecular mechanisms of CD8+ T cell trafficking and localization. Cell Mol Life Sci. 2016;72:2461–2473. doi: 10.1007/s00018-015-1835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim P.S., Ahmed R. Features of responding T cells in cancer and chronic infection. Curr Opin Immunol. 2010;22:223–230. doi: 10.1016/j.coi.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kmiecik J. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. 2013;264:71–83. doi: 10.1016/j.jneuroim.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 4.Piersma S.J. High number of intraepithelial CD8+ tumor-infiltrating lymphocytes is associated with the absence of lymph node metastases in patients with large early-stage cervical cancer. Cancer Res. 2007;67:354–361. doi: 10.1158/0008-5472.CAN-06-3388. [DOI] [PubMed] [Google Scholar]
- 5.Bellone M., Calcinotto A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front Oncol. 2013;3:1–15. doi: 10.3389/fonc.2013.00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung A., Lee J., Ferrera N. Targeting the tumour vasculature: insights from physiological angiogenesis. Nat Rev Cancer. 2010;10:505–514. doi: 10.1038/nrc2868. [DOI] [PubMed] [Google Scholar]
- 7.Oelkrug C., Ramage J.M. Enhancement of T cell recruitment and infiltration into tumours. Clin Exp Immunol. 2014;178:1–8. doi: 10.1111/cei.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harty J.T., Badovinac V.P. Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol. 2008;8:107–119. doi: 10.1038/nri2251. [DOI] [PubMed] [Google Scholar]
- 9.Butler N.S., Nolz J.C., Harty J.T. Immunologic considerations for generating memory CD8 T cells through vaccination. Cell Microbiol. 2011;13:925–933. doi: 10.1111/j.1462-5822.2011.01594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaech S.M., Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mami-Chouaib F. Resident memory T cells. Crit. Comp. Tumor Immunol. 2018:1–10. [Google Scholar]
- 12.Slaney C.Y., Kershaw M.H., Darcy P.K. Trafficking of T cells into tumors. Cancer Res. 2014;74:7168–7175. doi: 10.1158/0008-5472.CAN-14-2458. [DOI] [PubMed] [Google Scholar]
- 13.Cairns R.A., Harris I.S., Mak T.W. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 14.Mellor A.L., Munn D.H. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol. 2008;8:74–80. doi: 10.1038/nri2233. [DOI] [PubMed] [Google Scholar]
- 15.Whiteside T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904–5912. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mbeunkui L., Johann D.J. Cancer and the tumor microenvironment: a review of an essential relationship. Cancer Chemother Pharmacol. 2009;63:571–582. doi: 10.1007/s00280-008-0881-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Becker C., Hald M. Immune-suppressive properties of the tumor microenvironment. Cancer Immunol Immunother. 2013;62:1137–1148. doi: 10.1007/s00262-013-1434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanahan D., Coussens L.M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 19.Zhou L., Yang K., Wickett R.R., Zhang Y. Dermal fibroblasts induce cell cycle arrest and block epithelial–mesenchymal transition to inhibit the early stage melanoma development. Cancer Med. 2016;5:1566–1579. doi: 10.1002/cam4.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiao Y. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene. 2018;37:873–883. doi: 10.1038/onc.2017.387. [DOI] [PubMed] [Google Scholar]
- 21.Wang F. CD163+CD14+ macrophages, a potential immune biomarker for malignant pleural effusion. Cancer Immunol Immunother. 2015;64:965–976. doi: 10.1007/s00262-015-1701-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang L., Zhang Y. Tumor-associated macrophages, potential targets for cancer treatment. Biomark Res. 2017;5:1–6. doi: 10.1186/s40364-017-0106-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez F.O., Sica A., Mantovani A., Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 24.Filipazzi P., Huber V., Rivoltini L. Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol Immunother. 2012;61:255–263. doi: 10.1007/s00262-011-1161-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chandra D., Gravekamp C. Myeloid-derived suppressor cells Cellular missiles to target tumors. Oncoimunology. 2014;2 doi: 10.4161/onci.26967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brandau S., Moses K., Lang S. The kinship of neutrophils and granulocytic myeloid-derived suppressor cells in cancer: cousins, siblings or twins? Semin Cancer Biol. 2013;23:171–182. doi: 10.1016/j.semcancer.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 27.Hanson E.M., Clements V.K., Sinha P., Ilkovitch D., Ostrand-Rosenberg S. Myeloid-derived suppressor cells down-regulate L-Selectin expression on CD4+ and CD8+ t cells. J Immunol. 2009;183:937–944. doi: 10.4049/jimmunol.0804253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steding C.E. The role of interleukin-12 on modulating myeloid-derived suppressor cells, increasing overall survival and reducing metastasis. Immunology. 2011;133:221–238. doi: 10.1111/j.1365-2567.2011.03429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Molon B. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. 2011;208:1949–1962. doi: 10.1084/jem.20101956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruger A.M. How to measure the immunosuppressive activity of MDSC: assays, problems and potential solutions. Cancer Immunol Immunother. 2018 doi: 10.1007/s00262-018-2170-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–308. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 32.Ghiringhelli F. CD4+ CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. 2005;202:1075–1085. doi: 10.1084/jem.20051511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curiel T.J. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 34.Andersen M.H., Schrama D., Thor Straten P., Becker J.C. Cytotoxic T Cells. J Invest Dermatol. 2006;126:32–41. doi: 10.1038/sj.jid.5700001. [DOI] [PubMed] [Google Scholar]
- 35.McClory S. Evidence for a step-wise program of T cell development within the human tonsil. J Clin Invest. 2012;122:1403–1415. doi: 10.1172/JCI46125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wongtrakoongate P. Epigenetic therapy of cancer stem and progenitor cells by targeting DNA methylation machineries. World J Stem Cells. 2015;7:137–148. doi: 10.4252/wjsc.v7.i1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson K.G., Stromnes I.M., Greenberg P.D. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell. 2017;31:311–325. doi: 10.1016/j.ccell.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merelli B., Massi D., Cattaneo L., Mandalà M. Targeting the PD1/PD-L1 axis in melanoma: Biological rationale, clinical challenges and opportunities. Crit Rev Oncol Hematol. 2014;89:140–165. doi: 10.1016/j.critrevonc.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 39.Li J. CD39/ CD73 up-regulation on myeloid-derived suppressor cells via TGF- β -mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology. 2017;6 doi: 10.1080/2162402X.2017.1320011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi L. Adenosine signaling: next checkpoint for gastric cancer immunotherapy? Int Immunopharmacol. 2018;63:58–65. doi: 10.1016/j.intimp.2018.07.023. [DOI] [PubMed] [Google Scholar]
- 41.Gholami M.D. Exhaustion of T lymphocytes in the tumor microenvironment: significance and effective mechanism. Cell Immunol. 2017;322:1–14. doi: 10.1016/j.cellimm.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 42.Brand A. LDHA-Associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016;24:657–671. doi: 10.1016/j.cmet.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 43.Hodge G. Lung cancer is associated with decreased expression of perforin, granzyme B and interferon (IFN)-γ by infiltrating lung tissue T cells, natural killer (NK) T-like and NK cells. Clin Exp Immuology. 2014;178:79–85. doi: 10.1111/cei.12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Permanyer M., Bosnjak B., Forster R. Dendritic cells, T cells and lymphatics: dialogues in migration and beyond. Curr Opin Immunol. 2018;53:173–179. doi: 10.1016/j.coi.2018.05.004. [DOI] [PubMed] [Google Scholar]
- 45.Randolph G.J., Ivanov S., Zinselmeyer B.H., Scallan J.P. The lymphatic system: Integral roles in immunity. Annu Rev Immunol. 2017;35:31–52. doi: 10.1146/annurev-immunol-041015-055354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.James K.D., Jenkinson W.E., Anderson G. T-cell egress from the thymus: should I stay or should I go? J Leukoc Biol. 2017;104:275–284. doi: 10.1002/JLB.1MR1217-496R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bandola-Simon J., Roche P.A. Dysfunction of antigen processing and presentation by dendritic cells in cancer. Mol Immunol. 2018 doi: 10.1016/j.molimm.2018.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abbas A.K., Lichtman A.H., Pillai S. Cell Mol Immunol. 2007:397–417. [Google Scholar]
- 49.De La Cruz-Merino L. Immune microenvironment in colorectal cancer: a new hallmark to change old paradigms. Clin Dev Immunol 2011. 2011;2011 doi: 10.1155/2011/174149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cham C.M., Driessens G., O'Keefe J.P., Gajewski T.F. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. 2008;38:2438–2450. doi: 10.1002/eji.200838289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masopust D., Schenkel J.M. The integration of T cell migration, differentiation and function. Nat Rev Immunol. 2013;13:309–320. doi: 10.1038/nri3442. [DOI] [PubMed] [Google Scholar]
- 52.Harlin H. Chemokine expression in melnma metastasiswith CD8+ T-cell recruitment. Cancer Res. 2009;69:1–18. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mulligan A.M. Tumoral lymphocytic infiltration and expression of the chemokine CXCL10 in breast cancers from the Ontario Familial Breast Cancer Registry. Clin Cancer Res. 2013;19:336–346. doi: 10.1158/1078-0432.CCR-11-3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu J. Local production of the chemokines CCL5 and CXCL10 attracts CD8+ T lymphocytes into esophageal squamous cell carcinoma. Oncotarget. 2015;6:24978–26489. doi: 10.18632/oncotarget.4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matsumura S. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. 2008;181:3099–3107. doi: 10.4049/jimmunol.181.5.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laufer J.M., Legler D.F. Beyond migration — Chemokines in lymphocyte priming, differentiation, and modulating effector functions. J Leukoc Biol. 2018;1:1–12. doi: 10.1002/JLB.2MR1217-494R. [DOI] [PubMed] [Google Scholar]
- 57.Ferretti E., Pistoia V., Corcione A. Role of fractalkine/ CX3CL1 and its receptor in the pathogenesis of inflammatory and malignant diseases with emphasis on B cell malignancies. Mediators Inflamm. 2014;2014(480941) doi: 10.1155/2014/480941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Siddiqui I., Erreni M., Van Brakel M., Debets R., Allavena P. Enhanced recruitment of genetically modified CX3CR1-positive human T cells into fractalkine/CX3CL1 expressing tumors: Importance of the chemokine gradient. J Immunother Cancer. 2016;4:1–12. doi: 10.1186/s40425-016-0125-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garris C.S., Blaho V.A., Hla T., Han M.H. Sphingosine-1-phosphate receptor 1 signalling in T cells: trafficking and beyond. Immunology. 2014;142:347–353. doi: 10.1111/imm.12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nolz J.C., Starbeck-Miller G.R., Harty J.T. Naive, effector and memory CD8 T cell trafficking: parallels and distinctions. Immunotherapy. 2011;3:1223–1233. doi: 10.2217/imt.11.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paulsen M., Janssen O. Pro- and anti-apoptotic CD95 signalling in T cells. Cell Commun Signal. 2011;9 doi: 10.1186/1478-811X-9-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 63.Kinter A.L. The common y- chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol. 2008;181:6738–6746. doi: 10.4049/jimmunol.181.10.6738. [DOI] [PubMed] [Google Scholar]
- 64.Huang A. A human programmed death-ligand 1-expressing mouse tumor model for evaluating the therapeutic efficacy of anti-human PD-L1 antibodies. Nat Sci Rep. 2017;7:1–9. doi: 10.1038/srep42687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang Y., Li Y., Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015;6:1–9. doi: 10.1038/cddis.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Finn O., Immuno-Oncology J. Understanding the function and dysfunction of the immune system in cancer. Ann Oncol. 2012;23:86–89. doi: 10.1093/annonc/mds256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Obst R. The timing of T cell priming and cycling. Front Immunol. 2015;6:1–10. doi: 10.3389/fimmu.2015.00563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.González-Amaro R., Cortés J.R., Sánchez-Madrid F., Martín P. Is CD69 an effective brake to control inflammatory diseases? Trends Mol Med. 2013;19:625–632. doi: 10.1016/j.molmed.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu J.Y. CTL- vs T reg lymphocyte-attracting chemokines , CCL4 and CCL20 , are strong reciprocal predictive markers for survival of patients with oesophageal squamous cell carcinoma. Br J Cancer. 2015;113:747–755. doi: 10.1038/bjc.2015.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Linch S.N., McNamara M.J., Redmond W.L. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:1–14. doi: 10.3389/fonc.2015.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lei F. Regulation of A1 by OX40 contributes to CD8+T cell survival and anti-tumor activity. PLoS One. 2013;8:257–264. doi: 10.1371/journal.pone.0070635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Crepeau R.M., Ford M.L. Challenges and opportunities in targeting the CD28/CTLA-4 pathway in transplantation and autoimmunity. Expert Opin Biol Ther. 2017;17:1001–1012. doi: 10.1080/14712598.2017.1333595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen L., Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384–3391. doi: 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shin D., Ribas A. The evolution of checkpoint blockade as a cancer therapy: what's here, what's next. Curr Opin Immunol. 2015;33:23–35. doi: 10.1016/j.coi.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 75.Zou W., Wolchok J., Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016 doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kamphorst A.O., Ahmed R. Manipulating the PD-1 pathway to improve immunity. Curr Opin Immunol. 2013;25 doi: 10.1016/j.coi.2013.03.003. 381±8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ma W., Gilligan B.M., Yuan J., Li T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J Hematol Oncol. 2016;9:47. doi: 10.1186/s13045-016-0277-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maj T., Wei S., Welling T., Zou W. T cells and costimulation in cancer. Cancer J. 2013;19:473–482. doi: 10.1097/PPO.0000000000000002. [DOI] [PubMed] [Google Scholar]
- 79.Faget J. ICOS ligand expression on plasmacytoid dendritic cells supports breast cancer progression by promoting the accumulation of immunosuppressive CD4+ T cells. Cancer Res. 2012;72:6130–6141. doi: 10.1158/0008-5472.CAN-12-2409. [DOI] [PubMed] [Google Scholar]
- 80.Zhu J., Yamane H., Paul W.E. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martin-Orozco N. Th17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31:787–798. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gollmer K. CCL21 mediates CD4+ T-cell costimulation via a DOCK2/Rac-dependent pathway. Blood. 2009;114:580–588. doi: 10.1182/blood-2009-01-200923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ito T. Plasmacytoid dendritic cells prime IL-10–producing T regulatory cells by inducible costimulator ligand. J Exp Med. 2007;204:105–115. doi: 10.1084/jem.20061660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Riella L.V., Murakami N. Co-inhibitory pathways and their importance in immune regulation. Transplantation. 2014;98:3–14. doi: 10.1097/TP.0000000000000169. [DOI] [PubMed] [Google Scholar]
- 85.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 86.Fan J. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. 2013;9:1–11. doi: 10.1038/msb.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mocellin S., Bronte V., Nitti D. Nitric oxide, a double edged sword in cancer biology: searching for therapeutic opportunities. Med Res Rev. 2007;27:317–352. doi: 10.1002/med.20092. [DOI] [PubMed] [Google Scholar]
- 88.O'Flanagan C.H., Bowers L.W., Hursting S.D. A weighty problem: Metabolic perturbations and the obesity-cancer link. Horm Mol Biol Clin Invest. 2015;23:47–57. doi: 10.1515/hmbci-2015-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nieman K.M. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498–1503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kouidhi S., Elgaaied A.B., Chouaib S. Impact of metabolism on T-cell differentiation and function and cross talk with tumor microenvironment. Front Immunol. 2017;8:1–13. doi: 10.3389/fimmu.2017.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Clayton A., Al-Taei S., Webber J., Mason M.D., Tabi Z. Cancer eosomes express CD39 and CD73, which suppress T cells through adenosine production. J Immunol. 2011;187:676–683. doi: 10.4049/jimmunol.1003884. [DOI] [PubMed] [Google Scholar]
- 92.Deaglio S. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Simoni Y. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018 doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 94.Warburg B.Y.O., Wind F. The metabolism of tumors in the Body. J Gen Physiol. 1926;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cohen R. Targeting cancer cell metabolism in pancreatic adenocarcinoma. Oncotarget. 2015;6:16832–16847. doi: 10.18632/oncotarget.4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Beckermann K.E., Dudzinski S.O., Rathmell J.C. Dysfunctional T cell metabolism in the tumor microenvironment. Cytokine Growth Factor Rev. 2017;35:7–14. doi: 10.1016/j.cytogfr.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim H. Engineering human tumor-specific cytotoxic T cells to function in a hypoxic environment. Mol Ther. 2008;16:599–606. doi: 10.1038/sj.mt.6300391. [DOI] [PubMed] [Google Scholar]
- 98.Corzo C.A. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Song M., Chen X., Wang L., Zhang Y. Future of anti-PD-1 / PD-L1 applications: Combinations with other therapeutic regimens. Chin J Cancer Res. 2018;4:157–172. doi: 10.21147/j.issn.1000-9604.2018.02.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xu Y. Glycolysis determines dichotomous regulation of T cell subsets in hypoxia. J Clin Invest. 2016;126:2678–2688. doi: 10.1172/JCI85834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Caldwell C.C. Differential Effects of Physiologically Relevant Hypoxic Conditions on T Lymphocyte Development and Effector Functions. J Immunol. 2001;167:6140–6149. doi: 10.4049/jimmunol.167.11.6140. [DOI] [PubMed] [Google Scholar]
- 102.Cho H., Lee Y.S., Kim J., Chung J.Y., Kim J.H. Overexpression of glucose transporter-1 (GLUT-1) predicts poor prognosis in epithelial ovarian cancer. Cancer Invest. 2013;31:607–615. doi: 10.3109/07357907.2013.849722. [DOI] [PubMed] [Google Scholar]
- 103.Krzeslak A. Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol Oncol Res. 2012;18:721–728. doi: 10.1007/s12253-012-9500-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Siska P.J., Rathmell J.C. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015;36:1–8. doi: 10.1016/j.it.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rodriguez P.C., Ochoa A.C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: Mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–191. doi: 10.1111/j.1600-065X.2008.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ottensmeier C.H. Upregulated glucose metabolism correlates inversely with CD8+ T-cell infiltration and survival in squamous cell carcinoma. Cancer Res. 2016;76:4136–4148. doi: 10.1158/0008-5472.CAN-15-3121. [DOI] [PubMed] [Google Scholar]
- 107.Davis-Yadley A.H., Abbott A.M., Pimiento J.M., Chen D.T., Malafa M.P. Increased expression of the glucose transporter type 1 gene is associated with worse overall survival in resected pancreatic adenocarcinoma. Pancreas. 2016;45:974–979. doi: 10.1097/MPA.0000000000000580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang Y. Enhancing CD8 + T cell fatty acid catabolism within a metabolically challenging tumor microenvironment icreases the efficacy of melanoma immunotherapy. Cancer Cell. 2017;32:377–391. doi: 10.1016/j.ccell.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lu K. Expression and clinical significance of glucose transporter-1 in pancreatic cancer. Oncol Lett. 2016;12:243–249. doi: 10.3892/ol.2016.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gordon-Alonso M., Hirsch T., Wildmann C., van der Bruggen P. Galectin-3 captures interferon-gamma in the tumor matrix reducing chemokine gradient production and T-cell tumor infiltration. Nat Commun. 2017;8:1–15. doi: 10.1038/s41467-017-00925-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gordon-Alonso M., Demotte N., van der Bruggen P. Sugars boost exhausted tumor- infiltrating lymphocytes by counteracting immunosuppressive activities of galectins. Oncoimmunology. 2014;3:2–4. doi: 10.4161/onci.28783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Markowska A.I., Jefferies K.C., Panjwani N. Galectin-3 protein modulates cell surface expression and activation of vascular endothelial growth factor receptor 2 in human endothelial cells. J Biol Chem. 2011;286:29913–29921. doi: 10.1074/jbc.M111.226423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Demotte N. A Galectin-3 ligand corrects the impaired function of human CD4 and CD8 tumor-infiltrating lymphocytes and favors tumor rejection in mice. Cancer Res. 2010;70:7476–7489. doi: 10.1158/0008-5472.CAN-10-0761. [DOI] [PubMed] [Google Scholar]
- 114.Kouo T. Galectin-3 shapes antitumor immune responses by suppressing CD8 + T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunonolgy Res. 2015;3:412–423. doi: 10.1158/2326-6066.CIR-14-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Henning A.N., Roychoudhuri R., Restifo N.P. Epigenetic control of CD8+ T cell differentiation. Nat Rev Immunol. 2018 doi: 10.1038/nri.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pathania R. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat Commun. 2015;6:706–721. doi: 10.1038/ncomms7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Balmer M.L. Memory CD8+ T cells require increased concentrations of acetate induced by stress for optimal function. Immunity. 2016;44:1312–1324. doi: 10.1016/j.immuni.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 118.Ladle B.H. De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8 + T-cell fate decisions following activation. Proc Natl Acad Sci U S A. 2016;113:10631–10636. doi: 10.1073/pnas.1524490113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang Z. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature. 2018;554:387–391. doi: 10.1038/nature25477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Youngblood B. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature. 2018;552:404–409. doi: 10.1038/nature25144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gray S.M., Kaech S.M., Staron M.M. The interface between transcriptional and epigenetic control of effector and memory CD8+ T-cell differentiation. Immunol Rev. 2014;261:157–168. doi: 10.1111/imr.12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kasler H.G. Histone deacetylase 7 regulates cell survival and TCR signaling in CD4/CD8 double-positive thymocytes. J Immunol. 2011;186:4782–4793. doi: 10.4049/jimmunol.1001179. [DOI] [PubMed] [Google Scholar]
- 123.McLane L. Different localizations of T-bet and Eomes in CD8 T-cell memory populations. J Immunol. 2013;190:3207–3215. doi: 10.4049/jimmunol.1201556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li G. T-Bet and Eomes regulate the balance between the effector/central memory T cells versus memory stem like T cells. PLoS One. 2013;8:1–10. doi: 10.1371/journal.pone.0067401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nayar R. IRF4 regulates the ratio of T-Bet to eomesodermin in CD8+T cells responding to persistent LCMV infection. PLoS One. 2015;10:1–20. doi: 10.1371/journal.pone.0144826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhu L. Blimp-1 impairs T cell function via upregulation of TIGIT and PD-1 in patients with acute myeloid leukemia. J Hematol Oncol. 2017;10:1–13. doi: 10.1186/s13045-017-0486-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Braun J., Frentsch M., Thiel A. Hobit and human effector T-cell differentiation: the beginning of a long journey. Eur J Immunol. 2015;45:2762–2765. doi: 10.1002/eji.201545959. [DOI] [PubMed] [Google Scholar]
- 128.Amsen D., van Gisbergen K.P.J.M., Hombrink P., van Lier R.A.W. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol. 2018;19:538–546. doi: 10.1038/s41590-018-0114-2. [DOI] [PubMed] [Google Scholar]
- 129.Rao R.R., Li Q., Bupp M.R.G., Shrikant P.A. Transcription factor Foxo1 represses T-bet mediated effector functions and promotes memory CD8+ T cell differentiation. Immunity. 2012;36:374–387. doi: 10.1016/j.immuni.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Coma S. Id2 promotes tumor cell migration and invasion through transcriptional repression of semaphorin 3F. Cancer Res. 2011;70:3823–3832. doi: 10.1158/0008-5472.CAN-09-3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen S., Zhang Y., Kuzel T.M., Zhang B. Regulating tumor myeloid-derived suppressor cells by microRNAs. Cancer cell Microenviron. 2015;2:1–6. doi: 10.14800/ccm.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yang C.Y. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8 + T cell subsets. Nat Immunol. 2011;12:1221–1230. doi: 10.1038/ni.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhao E. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol. 2016;17:95–103. doi: 10.1038/ni.3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xu Y. Higher numbers of T-Bet + tumor- infiltrating lymphocytes associate with better survival in human epithelial ovarian cancer. Cell Physiol Biochem. 2017;41:475–483. doi: 10.1159/000456600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Trifari S. MicroRNA-directed program of cytotoxic CD8+ T-cell differentiation. Proc Natl Acad Sci U S A. 2013;110:18608–18613. doi: 10.1073/pnas.1317191110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Smith N.L., Wissink E.M., Grimson A., Rudd B.D. miR-150 regulates differentiation and cytolytic effector function in CD8 + T cells. Sci Rep. 2015;5:1–11. doi: 10.1038/srep16399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chen Z. miR-150 regulates memory CD8 T cell differentiation via c-Myb. Cell Rep. 2017;20:2584–2597. doi: 10.1016/j.celrep.2017.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shin H.M. Epigenetic modifications induced by blimp-1 regulate CD8+ T cell memory progression during acute virus infection. Immunity. 2013;39:661–675. doi: 10.1016/j.immuni.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Khan A.A., Penny L.A., Yuzefpolskiy Y., Sarkar S., Kalia V. MicroRNA-17~92 regulates effector and memory CD8 T-cell fates by modulating proliferation in response to infections. Blood. 2013;121:4473–4483. doi: 10.1182/blood-2012-06-435412. [DOI] [PubMed] [Google Scholar]
- 140.Dudda J.C. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity. 2013;38:742–753. doi: 10.1016/j.immuni.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ji Y., Hocker J.D., Gattinoni L. Enhancing adoptive T cell immunotherapy with microRNA therapeutics. Semin Immunol. 2016;28:45–53. doi: 10.1016/j.smim.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Porta C., Paglino C., Mosca A. Targetting PI3K/Akt/Mtor signalling in cancer. Front Oncol. 2014;4 doi: 10.3389/fonc.2014.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Murray I.A., Patterson A.D., Perdew G.H. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer. 2014;14:801–814. doi: 10.1038/nrc3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zeng H., Chi H. mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr Opin Immunol. 2017;46:103–111. doi: 10.1016/j.coi.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Eil R. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nat Publ Gr. 2016;537:539–543. doi: 10.1038/nature19364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gattinoni L. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15:808–813. doi: 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gattinoni L., Klebanoff C.A., Restifo N.P. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12:671–684. doi: 10.1038/nrc3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pai S.G. Wnt/ beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol. 2017;10:1–12. doi: 10.1186/s13045-017-0471-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Backer R.A. A central role for notch in effector CD8+ T cell differentiation. Nat Immunol. 2014;15:1143–1151. doi: 10.1038/ni.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wells A.C. Modulation of let-7 miRNAs controls the differentiation of effector CD8 T cells. Elife. 2017;6:1–22. doi: 10.7554/eLife.26398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kondo T. Notch-mediated conversion of activated T cells into stem cell memory-like T cells for adoptive immunotherapy. Nat Commun. 2017;8:1–14. doi: 10.1038/ncomms15338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tsukumo S., Yasutomo K., Osborne B.A. Regulation of CD8+ T cells and antitumor immunity by notch signaling. Front Immunol. 2018;9:1–7. doi: 10.3389/fimmu.2018.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Crespo J., Haoyu S., Welling T.H., Tian Z., Zou W. T cell anergy, exhaustion, senescence and stemness in the tumour microenvironment. Curr Opin Immunol. 2013;25:214–221. doi: 10.1016/j.coi.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bartel D., Micrornas P. Target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cho O.H. Notch regulates cytolytic effector function in CD8+ T cells. J Immunol. 2015;182:3380–3389. doi: 10.4049/jimmunol.0802598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fuchs C.S. Safety and efficacy of Pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer Phase 2 clinical KEYNOTE-059 Trial. JAMA Oncol. 2018;06510:1–8. doi: 10.1001/jamaoncol.2018.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Shahda S. A phase II study of pembrolizumab in combination with mFOLFOX6 for patients with advanced colorectal cancer. J Clin Oncol. 2017;35:3541. [Google Scholar]
- 158.Haymaker C.L. Metastatic melanoma patient had a complete response with clonal expansion after whole brain radiation and PD-1 blockade. Cancer Immunol Res. 2017;5:100–106. doi: 10.1158/2326-6066.CIR-16-0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kamphorst A.O. Rescue of exhausted CD8 T cells by PD-1 targeted therapies is CD28-dependent. Science. 2017;355:1423–1427. doi: 10.1126/science.aaf0683. (80-.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Golubovskaya V., Wu L. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers (Basel) 2016;8 doi: 10.3390/cancers8030036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lu Y., Robbins P.F. Seminars in Immunology Cancer immunotherapy targeting neoantigens. Semin Immunol. 2016;28:22–27. doi: 10.1016/j.smim.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zou W., Wolchok J.D., Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers and combinations. Sci Transl Med. 2016;8 doi: 10.1126/scitranslmed.aad7118. 328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Schumacher T., Schreiber R. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. (80-.) [DOI] [PubMed] [Google Scholar]
- 164.Nouri-Shirazi M. Dendritic cells capture killed tumor cells and present their antigens to elicit tumor-specific immune responses. J Immunol. 2000;165:3797–3803. doi: 10.4049/jimmunol.165.7.3797. [DOI] [PubMed] [Google Scholar]