

ABSTRACT
Haemophagocytic lymphohistiocytosis is a rare inflammatory condition. It can present in adult general medical patients and is a challenging diagnostic conundrum. This article provides an overview of the pathophysiology and clinical presentation of the syndrome for the general physician who will be rarely confronted with this problem but will have to act promptly when the situation arises. Treatment is also briefly discussed, although this usually occurs in a specialist setting after the diagnosis has been established.
KEYWORDS: haemophagocytic lymphohistiocytosis, haemophagocytic syndrome, macrophage activation syndrome
Introduction
Haemophagocytic lymphohistiocytosis (HLH) is a rare syndrome, which can be difficult to recognise and diagnose. It could be expected that the general physician may only see one or two cases in their entire career. Despite the rarity of the condition, it is crucial that it is diagnosed quickly so that correct treatment can be given. The HLH patient often presents non-specifically unwell to the general physician with no significant medical history of note. Some patients may have pyrexia of unknown origin, already had an exhaustive battery of investigations and an overarching diagnosis is elusive. In medical literature, HLH has several other names: haemophagocytic syndrome or macrophage activation syndrome (MAS) when it occurs in conjunction with autoimmune/rheumatological disease.1,2
Pathology
HLH is caused by a pathological hyperactivation of macrophages (and T-cells) that engulf normal haematopoietic cells in the bone marrow, liver and spleen (Fig 1). This can be caused by genetic defects (primary HLH) or by acquired factors (secondary HLH/MAS).
Fig 1.
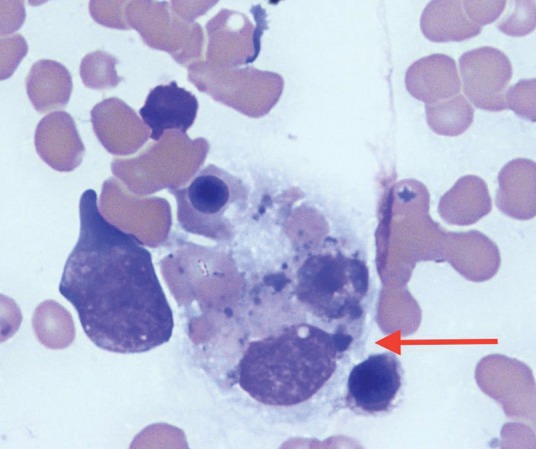
Bone marrow aspirate showing a macrophage (arrow) engulfing erythrocytes and an erythroblast. x40 magnification light microscopy.
Primary HLH
This is caused by exceedingly rare defects in genes involved in T-cell and natural killer (NK) cell function.3,4 As a result, there is loss of the ability to kill infected cells and antigen presenting cells, which leads to uncontrolled macrophage activity and high levels of inflammatory cytokines that further drive the pathological process. Causative gene mutations include PRF1, UNC13D, STX11 and STXBP2.4 This usually presents in childhood, although it is well recognised that primary HLH can occur in adults.5
Secondary HLH
This is the acquired form of the disease where there is a driving factor (such as infection) with no inborn genetic defect. In secondary HLH, an immune stimulus causes the uncontrolled macrophage activation.
MAS
MAS is a term used to describe HLH where it is found in conjunction with underlying rheumatological diseases. These diseases include systemic lupus erythematosus and systemic onset juvenile idiopathic arthritis.
Prognosis
Left untreated the syndrome results in irreversible organ damage and is often lethal. Therefore early recognition is key and initiation of appropriate treatment mandatory. In adults, prognosis is often poor with one large series showing a median survival of 2.1 months and an overall survival of 34% at 42 months.6 This is because of critical organ dysfunction and illness at time of diagnosis, but also because of the large number of patients with underlying malignant disease precipitating HLH, which may often confer a poor prognosis. In the paediatric population, overall survival is around 60%. Excellent results have been reported using reduced intensity condition haematopoietic stem cell transplantation (HSCT), with an overall survival of 92% reported for transplant patients.7,8 This will hopefully translate to improving survival in adults in the future.
Clinical features and diagnosis
Diagnosis of this condition is often challenging and part of this is because of the rarity of the disease. It will be invariably underestimated at the early stage because of the lack of specific laboratory and clinical features pointing towards the diagnosis. The presence of a fever with pancytopenia or lymphadenopathy may prompt a diagnosis (often in an expeditious way) of a haematological malignancy but the differential diagnosis would also include HLH. HLH is one of the diagnostic possibilities in the undifferentiated sick adult patient who is febrile and develops cytopenias with a raised ferritin level. This is often in the context of acute kidney injury, non-specific coagulopathy and abnormal liver function tests. The dilemma in these patients is often teasing out the abnormalities in the blood count, liver function, clotting screen and trying to differentiate these from the non-specific abnormalities seen in very sick patients who have had multiple insults from sepsis and drugs (perhaps with disseminated intravascular coagulation) from actual cases of HLH. These patients will often have had extensive investigations for fever (perhaps with no cause found) and treatment with multiple antibiotics for presumed infection. By this stage, they may well have been in hospital for quite some time, seen multiple teams and possibly be becoming sick enough to need support in the intensive care unit. Non-specific markers such as the C-reactive protein, erythrocyte sedimentation rate and lactate dehydrogenase may be raised. To summarise, these patients are sometimes the mystery patients with pyrexia of unknown origin that elude diagnosis. Readily available and cheap clinical and basic laboratory pointers that may help the general physician recognise and make the diagnosis of HLH include the presence (or constellation) of cytopenias, fever, splenomegaly, hypofibrinogenaemia, raised ferritin and raised triglycerides. Recognition of these features would then prompt more specialised investigations, including soluble CD25, NK cell activity studies, tissue biopsy and genetic tests.
After the possibility of HLH has been raised, the diagnostic criteria (Box 1) should be examined and specialist tests requested. In the UK, the specialist tests listed (soluble CD25 and NK cell activity studies) can be performed by reference laboratories. The local haematology team should usually be involved in performing a bone marrow biopsy and help with confirming or refuting a diagnosis. It is crucial to note that absence of haemophagocytosis in the bone marrow does not exclude the diagnosis. Other tissue may be obtained to demonstrate haemophagocytosis (eg lymph node or liver biopsy).
Box 1.
HLH-2004 diagnostic criteria3
| Molecular diagnosis known to cause HLH or meeting 5 out of the following 8 criteria |
|
HLH= haemophagocytic lymphohistiocytosis
Compared to other haematological emergencies, in HLH there is no rapid specific and objective confirmatory test (such as immunophenotyping) available but the diagnostic criteria are, in part, research tests that will not be available within a clinically useful time frame to help with the initial treatment decision. A variety of conditions, such as protracted septicaemia, will meet four out of eight criteria already (eg fever, splenomegaly, ferritin 500–2,000 ng/mL and low fibrinogen) and it can be a leap of faith to assess whether or not the degree of haemophagocytosis observed in bone marrow (which again can be seen in critically ill patients) is sufficiently severe to cross the diagnostic boundary and to initiate immunosuppressive therapy combined with chemotherapy in a patient with presumed septicaemia.
The other diagnostic difficulty is not only establishing diagnosis but then finding the underlying cause (Box 2). Primary HLH can be tested for in specialist immunology laboratories but is exceedingly rare. Secondary HLH needs to have a triggering disease identified. These triggers usually fall into the broad categories of infection, autoimmune or malignant. Evaluation therefore needs to take into account the family history (for primary HLH), travel and infection history (for infectious diseases) and laboratory tests (to help identify infections), as well as histology and radiology to look for malignancy. This is again where bone marrow or lymph node biopsy may be crucial in identifying a malignant diagnosis driving HLH.
Box 2.
Causes of secondary haemophagocytic lymphohistiocytosis
Infections:
|
Autoimmune diseases:
|
Malignant diseases:
|
Management principles
Treatment is aimed at controlling the over-reactive immune response and preventing irreversible organ damage. In general, the condition should be managed at a hospital with sufficient expertise in dealing with the condition, given the specialist nature of the treatment. Given the need for patients to proceed with HSCT in some cases, close liaison with a transplant centre is needed.
In 1994, the Histiocyte Society setup the first international treatment protocol for HLH: HLH-94.9 This involves an initial 8 weeks of therapy, at which point a disease assessment occurs, followed by a continuation phase, which may serve as a bridge to allogeneic HSCT. It has been found that etoposide (a topoisomerase II inhibitor) is an effective therapy for HLH and is the backbone of the HLH-94 protocol.7,9,10 The protocol was later updated to form HLH-2004 (Table 1).3
Table 1.
Summary of HLH-2004 treatment protocol.3 Supportive care with prophylactic co-trimoxazole, anti-virals (eg aciclovir), intravenous immunoglobulin (0.5 g/kg 4-weekly) and anti-fungals are also administered
| Week of treatment | Treatment |
|---|---|
| Initial treatment | |
| 1–2 | Dexamethasone 10 mg/m2 Etoposide 150 mg/m2 on day 1, 4, 8 and 11 Ciclosporin 3 mg/kg twice daily aiming for a trough level 200 mcg/L |
| 3–4 | Dexamethasone 5 mg/m2 Etoposide 150 mg/m2 weekly Ciclosporin 3 mg/kg twice daily aiming for a trough level 200 mcg/L Intrathecal methotrexate 12 mg with hydrocortisone 50 mg at week 3 (repeat weekly on week 4–6 if evidence of central nervouse system involvement) |
| 5–6 | Dexamethasone 2.5 mg/m2 Etoposide 150 mg/m2 weekly Ciclosporin 3 mg/kg twice daily aiming for a trough level 200 mcg/L |
| 7–8 | Dexamethasone 1.25 mg/m2 Etoposide 150 mg/m2 weekly Ciclosporin 3 mg/kg twice daily aiming for a trough level 200 mcg/L |
| Continuation therapy | |
| 9 onwards | Dexamethasone 10 mg/m2 for 3 days every 2 weeks Etoposide 150 mg/m2 every 2 weeks Ciclosporin 3 mg/kg twice daily aiming for a trough level 200 mcg/L |
Primary HLH
In primary HLH, the treatment is to follow HLH-2004 to get the pathological process into a remission. This is followed by an allogeneic HSCT aimed at replacing the defective immune system with a fully functional donor immune system. This procedure carries with it the complications, morbidity and mortality of both intensive chemotherapy and allogeneic HSCT (principally severe infections, relapse of the HLH and graft versus host disease).
Secondary HLH
There are three situations in which there may be a pathological driver identified as causing secondary HLH.
1. Infection driving secondary HLH
Treatment of the underlying infection is needed with close clinical observation (eg liposomal amphotericin if the driver is visceral leishmaniasis). If there is not a fast response (eg within 2 days) to the infection targeted therapy then formal HLH therapy containing etoposide should be considered.
Etoposide has been shown to be very effective in HLH driven by Epstein-Barr virus (EBV) infection and therefore should be introduced as the first-line treatment for this indication.10 Rituximab, a murine-derived monoclonal anti-CD20 antibody used to treat B-cell lymphomas and autoimmune disease, has also been reported to have efficacy in treating HLH caused by EBV.11 Rituximab effectively targets B-cells where EBV resides. Serology may be unreliable in the immunocompromised patient because of a lack of antibody response, although a finding of IgM antibodies to EBV would be highly suggestive of primary EBV infection as a precipitant cause of HLH and prompt treatment. We therefore would check for circulating EBV in the blood using polymerase chain reaction (PCR). The question of whether the finding of EBV in the blood PCR with IgG on the EBV serology (suggesting historical rather than primary infection) would suggest HLH caused by EBV re-activation is a difficult question. EBV re-activation may be seen in immunocompromised states and a search for other underlying precipitants, such as malignancy, would still have to occur prior to fully attributing the HLH to EBV re-activation. EBV re-activation in the context of a patient with fatal HLH and underlying Hodgkin lymphoma has been previously reported, reflecting the co-existence that it may have with other precipitants.12
2. Malignancy driving HLH
Beginning disease-specific chemotherapy should depend upon the malignancy diagnosed. Many treatment protocols within haemato-oncology contain etoposide and these treatments may be preferred, considering the known efficacy of etoposide in HLH. Full treatment following the HLH-2004 protocol would begin only if the patient was refractory to initial treatment with disease-specific chemotherapy. HLH may also occur as a complication of HSCT in patients with a prior history of malignancy (usually haematological) and pre-treated and conditioned with chemotherapy. This is a rare and often fatal situation for which very little management consensus exists.
3. MAS
Treatment is initially with steroids as well as specific treatment of the underlying rheumatological disease precipitating the MAS.
Idiopathic HLH
Sometimes a precipitating cause for HLH may not be obvious (eg in adults).
It can be hard for clinicians to commit a patient to HLH therapy given the following three factors:
other possible diagnoses (eg sepsis with some of the non-specific features of HLH, such as fever commonly seen)
the lack of a driving factor for HLH to support the overall diagnosis
committing the patient to several toxic therapies, while already cytopenic and the use of etoposide, when they may already be critically unwell with multi-organ dysfunction.
These factors, as well as the vague presenting features of HLH, mean delayed diagnosis, which commonly leads to delayed treatment. If HLH is a viable diagnosis, however, then the diagnostic work-up should be promptly completed and HLH therapy started without further delay. This approach will often involve intensive care physicians, haematologists, infectious disease physicians and rheumatologists to ensure that a comprehensive work-up and plan has been made.
In the authors’ experience, the diagnosis driving HLH may arise at a slightly later date after treatment with the HLH-2004 protocol has commenced. Two case examples may high light these difficulties:
A 70-year-old male was admitted to the intensive care unit with a diagnosis of HLH established without precipitating cause, despite extensive work-up. The bone marrow aspirate revealed reactive changes and haemophagocytosis. HLH-2004 protocol was started and subsequently the bone marrow trephine biopsy result showed diffuse large B-cell lymphoma. There had been no lymphoma detected on the bone marrow aspirate, computed tomography of the neck to pelvis or on examination of the skin. Treatment was therefore switched to R-CHOP chemotherapy (rituximab, cyclophosphamide, vincristine and prednisolone) for lymphoma.
A 63-year-old male was admitted to the intensive care unit with HLH. The bone marrow aspirate revealed haemophagocytosis and the HLH-2004 protocol was commenced. However, the bone marrow PCR for Leishmania was later found to be positive and a diagnosis of visceral leishmaniasis was established. Treatment with liposomal amphotericin B was then commenced.
In both these cases, there was extensive investigation for rheumatological, malignant and infectious triggers. Both the cases emphasise the dilemmas that occur between starting HLH therapy when an underlying cause has not been established (with outstanding investigations still pending) and the clinical urgency of the situation. Both patients had similar, non-specific presentations with fever and progressive pancytopenias, as well as the haematological and biochemical features of HLH. It is also of note that, with very similar patient presentations, until the bone marrow biopsy results were reported, there were no distinguishing features between a malignant and infectious trigger. This only adds to the diagnostic conundrum facing the clinician and makes the comprehensive search for a precipitant crucial.
It is unlikely that commencing HLH therapy would jeopardise the care of the patient where an underlying cause was subsequently found for two reasons. The first is that HLH therapy will provide overall control of the inflammatory nature of the syndrome in lieu of a precipitant being found; the second reason is that HLH therapy may contain some form of treatment for the underlying cause (eg steroids and etoposide are well recognised lymphoma therapy).
After the initial treatment phase of idiopathic HLH, using the HLH-2004 protocol, a decision would need to be made about continuation of therapy, stopping treatment or proceeding to allogeneic HSCT. Primary or relapsing HLH would be an indication for HSCT. Secondary HLH, where remission and disease control cannot be secured by treatment of the underlying disease, would warrant consideration of HSCT. In some high-risk malignant haematological diseases, HSCT may be needed to try and secure long-term remission, irrespective of the presentation with HLH.
Conclusion
The first step in diagnosing a condition is to suspect it in the differential diagnosis. HLH should be suspected in sick medical patients who exhibit the clinical and laboratory features discussed in this article. Symptoms will usually be fever and cytopenias with a raised ferritin level. Diagnosis will often occur in a collaborative way between haematologists, rheumatologists and infectious diseases teams. Underlying causes (eg genetic, infectious or malignant) should be investigated. Treatment of the condition then depends upon the identified cause and should be in a centre with sufficient expertise in managing HLH. Although this is a rare diagnosis, it is important to be aware of it as treatment of the condition is specific and HLH is also a significant differential diagnosis in pyrexia of unknown origin.
Conflicts of interest
MB has received speaking honorarium from GlaxoSmithKline.
References
- 1.Jordan MB. Allen CE. Weitzman S. Filipovich AH. McClain KL. How I treat haemophagocytic lymphohistiocytosis. Blood. 2011;118:4041–52. doi: 10.1182/blood-2011-03-278127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lehmberg K. Ehl S. Diagnostic evaluation of patients with suspected haemophagocytic lymphohistiocytosis. Br J Haematol. 2013;160:275–87. doi: 10.1111/bjh.12138. [DOI] [PubMed] [Google Scholar]
- 3.Henter JI. Horne A. Arico M, et al. HLH-2004: diagnostic and theraputic guidelines for haemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 4.Cetica V. Pende D. Griffiths GM. Arico M. Molecular basis of familial hemophagocytic lymphohistiocytosis. Haematologica. 2010;95:538–41. doi: 10.3324/haematol.2009.019562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sieni E. Cetica V. Piccin A, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS One. 2012;7:e44649. doi: 10.1371/journal.pone.0044649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parikh SA. Kapoor P. Letendre L. Kumar S. Wolanskyj AP. Prognostic Factors and Outcomes of Adults With Hemophagocytic Lymphohistiocytosis. Mayo Clin Proc. 2014;89:484–92. doi: 10.1016/j.mayocp.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 7.Trottestam H. Horne A. Arico M. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long term results of the HLH-94 protocol. Blood. 2011;118:4577–84. doi: 10.1182/blood-2011-06-356261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marsh RA. Vaughn G. Kim M, et al. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood. 2010;116:5824–31. doi: 10.1182/blood-2010-04-282392. [DOI] [PubMed] [Google Scholar]
- 9.Henter JI. Samuelsson-Horne A. Arico M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100:2367–73. doi: 10.1182/blood-2002-01-0172. [DOI] [PubMed] [Google Scholar]
- 10.Imashuku S. Kuriyama K. Sakai R. Treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis (EBV-HLH) in young adults: a report from the HLH study center. Med Pediatr Oncol. 2003;41:103–9. doi: 10.1002/mpo.10314. [DOI] [PubMed] [Google Scholar]
- 11.Chellapandian D. Das R. Zelley K, et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol. 2013;162:376–82. doi: 10.1111/bjh.12386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasselblom S. Linde A. Ridell B. Hodgkin’s lymphoma, Epstein-Barr virus reactivation and fatal haemophagocytic syndrome. J Intern Med. 2004;255:289–95. doi: 10.1046/j.0954-6820.2003.01249.x. [DOI] [PubMed] [Google Scholar]