

Introduction
The excitement of clinical science was first made apparent to me through the somewhat theatrical medium of the medical ‘grand round’. In the 1980s, at both Hammersmith Hospital and Oxford, where I undertook postgraduate training, the format was in its heyday. A junior doctor would present the history and clinical and investigative features of a patient with a puzzling illness. This was followed by a scholarly dissection of the key issues by the presenter, after which the chair and a distinguished audience would subject the arguments to forensic dissection. Not infrequently, ongoing research in laboratories within the hospital had actually generated new knowledge directly relevant to the patient and the audience would hear of these discoveries for the first time. It was a powerful vehicle for conveying the thrill of clinical science. It was, moreover, a form of discourse that William Harvey would have understood well. In endowing this lecture, he stipulated that the orator should ‘exhort the Fellows and Members of this College to search and study out the secrets of nature by way of experiment’.1 In the 17th century, non-invasive forms of medical science were limited in scope. Anatomical dissection was the principal tool and, of necessity, the only patients who could be subjected to that form of examination were those no longer alive. In his notes for the Lumleian Lectures, Harvey reminded himself ‘Not to praise or dispraise other anatomists, for all did well, and there was some excuse even for those who are in error’.1 That kind exhortation might bring a wry smile to those who remember the Hammersmith Hospital rounds of the 1980s, where impassioned clashes between the giants of medicine gathered there made the atmosphere more ‘Grand Guignol’ than ‘grand round’.
So, in homage to the tradition of the grand rounds, I will start this lecture with a description of a patient, whom I will call M, a 43-year-old woman who attends the diabetes clinic at Addenbrooke’s Hospital. Having lost touch with us a few years ago, she was re-referred by her GP because she had blood glucose levels that were persistently high. When she came to see me, she smiled resignedly, saying ‘my insulin isn’t working again’. Over several visits, the truth about M’s diabetes gradually emerged. She had been obese from early childhood, leading to severe bullying at school. Having struggled with her obesity throughout her teens and early twenties, in her late twenties she suddenly lost a lot of weight, which delighted her. However, the weight loss was actually due to the fact that she had developed type 2 diabetes and was now losing a large number of calories in her urine. After several years of oral medication, she eventually needed insulin treatment to control her blood glucose. However, she noticed that whenever she controlled her diabetes tightly, her weight piled back on. So she solved this problem by having ‘holidays’; periods of many months when she would omit her insulin and ‘let her diabetes rip’ before having enough of the extreme fatigue, nocturia and thrush. Doctors telling her that she was developing the microvascular complications of sustained hyperglycaemia, including retinopathy, neuropathy and nephropathy, had little impact because these issues didn’t bother her in the moment and, like many people, she was too preoccupied with her immediate life stresses to think much about the future. If we cannot help M move from her current metabolic trajectory, there is a strong likelihood that she will become blind, dialysis dependent and/or lose a limb well before the normal retirement age. Although her type 2 diabetes occurred unusually early for a person of European descent, her story is not at all atypical in other ethnicities, many of whom suffer a disproportionately higher burden of diabetes than white Europeans. The International Diabetes Federation (IDF) predicts that the number of people with diabetes will rise from an estimated 415 million in 2015 to 642 million in 2040,2 mostly in settings without adequate resources to detect complications early and intervene to reduce later disability.
In order to fully understand the conditions that have led to M’s plight, we need to better understand the environmental drivers of the current obesity epidemic, of which she is but one of many millions of victims. But that will not be enough. We need to know what it is that made M so susceptible to becoming obese and at such a young age and why, while many people with lifelong severe obesity remain normoglycaemic, in her case just a couple of decades of over-nutrition has been enough to lead her to develop type 2 diabetes.
The causes of obesity
Why has obesity become more common?
Obesity is not a new disease. The Palaeolithic sculptor of the Venus of Willendorf (Fig 1), working about 25,000 years ago, cannot have rendered the shape of a very obese female so perfectly from imagination alone. Hippocrates himself reported that obese people more frequently died suddenly and recommended that ‘they should eat only once a day and take no baths and sleep on a hard bed and walk naked as long as possible’.3 In 1727, the British physician Thomas Short wrote ‘No age has seen more instances of corpulency than our own’.3 Nonetheless, it is clear that obesity has become considerably more common in the vast majority of developed and developing countries since the beginning of the last century, with a particular acceleration appearing to start around the 1980s in highly developed countries such as the USA.4
Fig 1.

Venus of Willendorf. Palaeolithic carving in oolitic limestone from about 25,000 BCE. Wellcome Library, London.
As the mass of any object can only increase if the energy put into it is greater than the energy lost from it, then attention has obviously been paid to changes in the environment that acted to promote caloric ingestion and those that have acted to diminish the expenditure of energy. In brief, until about the past 50 years, never in the course of history have the majority of humans had such continuous access to sufficient ingestible calories at a time when they did not require a large expenditure of energy to acquire them. There are many powerful influences that together have acted to encourage caloric intake.
Ingestible calories are cheaper than ever before in human history. The percentage of household income spent on food has progressively decreased in all developed and developing countries over the past century, much of this decrease being attributable to advances in food production, packaging, refrigeration and transport.5 For instance, in the USA the proportion of household income spent on food fell from about 50% to about 12% between the late 19th and the late 20th centuries.5 In the UK, the average household spend on food and drink has also fallen, currently being 11.1% of all household expenditure.6 Ingestible calories have been made more desirable and attractive through food advertising, higher sugar and fat content, and a sophisticated industry that has cleverly used sensory science to encourage purchase and consumption of food. What is presented as a ‘standard’ portion has risen dramatically in size over a relatively short period.7 Calories are more ubiquitously and continually available. There are many more shops selling food,8 which is also now freely available in non-traditional outlets that are often open all hours. There has been a relative increase in the availability of cheap, processed, calorie-dense food, which tends to be less satiating owing to lower concentrations of protein, fibre and slowly digested carbohydrates than non-processed food, while there has been a growing price gap, with unprocessed foods with higher concentration of protein and fibre becoming consistently more expensive than processed foods high in sugar and fat.9
These changes have occurred at the same time as the requirements for the use of energy to support physical activity (at work or domestically) have diminished,10 and when the widespread increase in domestic central heating means that the demands for thermogenesis for maintenance of body temperature have decreased.11 It is not particularly surprising that a state where energy intake chronically exceeds energy expenditure is now commonplace.
An interesting idea that is emerging, but is as yet unproven, is that over-nutrition in early life somehow sets a long-term trajectory influencing levels of adult adiposity.12 Other hypotheses implicating factors such as environmental chemicals including antibiotics, perhaps acting through changes in the human microbiome, currently remain in the realms of speculation.13 Before any public health efforts based on these ideas are launched, such hypotheses will need to be tested rigorously and repeatedly with full consideration of the confounding effects of genetic background and other environmental and socio-economic influences.
Why are some people resistant to the obesogenic environment while some are susceptible?
In the face of these major environmental stresses, why is it that many people are still lean? It is possible, of course, that all of those who remain lean are making conscious choices to restrain their food intake and increase their expenditure to obtain the social, economic and health benefits that leanness brings. It is also possible that those who are resistant to the obesogenic environment are somehow different in their biological makeup. So, a somewhat tongue-in-cheek question is the following: are lean people ‘morally superior’ or are they ‘biologically different’? Of course, few things in biology are black and white and it is likely that both biological predisposition and conscious exertion of choice both impact on a person’s state of adiposity at any particular time.
One way of providing a quantitative estimate of the impact of inherent biological (genetically encoded) factors is to compare the degree of similarity between identical (monozygotic) twin pairs who share 100% of their genetic makeup, with non-identical (dizygotic) twin pairs who share only 50%. Both types of pair largely share similar pre- and postnatal environments. This type of study can lead to estimates of the influence that shared genes, shared environment and non-shared environment have on the inter-individual variance of any human trait. Multiple independent studies of this type have clearly shown that genetic factors dominate, with some studies reporting that up to as much as 70% of the population variance in body mass index (BMI) or related measures of adiposity is attributable to inherited factors.14 In a remarkable study, an American psychiatrist, Mickey Stunkard, studied Scandinavian twins who had been adopted by different families shortly after birth.15 In adult life, the adiposity of these adopted children was not at all correlated with the adiposity of their adoptive parents or the siblings who they grew up with. But it was remarkably strongly correlated with that of the twin they had been separated from. This was particularly striking (intra-pair correlation coefficient of about 0.7) when the twins raised apart were identical and less so when non-identical, where the correlation coefficient was around about 0.3. These estimates are supported by recent advances allowing single nucleotide polymorphism (SNP)-based heritability estimates among unrelated individuals with genome-wide genotyping. Such approaches also highlight a major contribution of common genetic variation underlying variation in BMI.16 Thus, a compelling and consistent body of data indicates that genetic factors play a major role in determining an individual’s position along the population distribution of adiposity.
I have, I confess, created a somewhat false dichotomy, implying that adiposity could only be influenced by ‘conscious choice’ or ‘biological factors’. This is over-simplistic. A wealth of evidence is emerging that demonstrates the powerful impact of environmental factors which exploit the fact that, as recently summarised by Theresa Marteau and colleagues, ‘much human behaviour is automatic, cued by environmental stimuli, resulting in actions that are largely unaccompanied by conscious reflection’.17
What physiological processes are these genes likely to be impacting on?
As genes act through physical rather than metaphysical means, it behoves us to ask whether and how body fat stores are regulated by the organism, and what are the signals used to govern such regulatory processes? In this regard, what we now know about human biology is indebted to decades of careful and insightful work in animal models, largely rodents. This work has revealed the existence of two broad classes of signal that influence appetite and energy expenditure; namely, longer-term signals that convey information about stored energy reserve (mostly triglyceride in adipose tissue) and shorter-term signals that provide information about the amount and composition of ingested macronutrients.
Pathways that sense nutritional stores
Experiments in rodents undertaken in the 1940s and 1950s by scientists such as Adolph,18 Kennedy,19 Hetherington20 and Anand21 first established that nutritional stores were under some sort of homeostatic control and that a functioning hypothalamus was a necessary component of that process. The latter had in fact been first suggested by clinical reports at the turn of the 19th and 20th centuries by German physicians such as Frohlich, Mohr and Babinski.3
Arguably, the modern era of obesity science began in 1959 with a publication by GR Hervey, from the Medical Research Council Department of Experimental Medicine in Cambridge,22 which demonstrated the existence of a circulating factor influencing eating behaviour and body fat stores. Hervey created a partial surgical union of skin and subcutaneous tissues between pairs of genetically identical rats, establishing a permanent circulatory connection between the two across which blood-borne signals could equilibrate. One member of each parabiotic pair was then subjected to a hypothalamic lesion that made it overeat and become obese. Remarkably, those rats parabiosed to such a partner rapidly reduced their food intake and became markedly undernourished.
In Hervey’s own words:
the experiments demonstrate what in a normal single animal would constitute a feedback system. The results also imply that the information reaches the hypothalamus in some form which is capable of crossing through the parabiotic union.22
In the 1970s, studying genetically obese strains of mice previously described by the Jackson Laboratories,23,24 Coleman used the technique of parabiosis to prove that the ob/ob mouse was lacking a blood-borne signal to which the db/db mouse was completely unresponsive.25,26
It took, however, a further 20 years and the power of murine molecular genetics before Friedman and colleagues at the Rockefeller University in New York discovered that this blood-borne signal was a cytokine-like hormone, which they termed leptin. This new hormone is produced exclusively in fat cells and its secretion is proportionate to increases in adipose tissue mass.27,28 Leptin interacts with receptors in the hypothalamus and elsewhere in the brain to reduce food intake and increase energy expenditure; thus, remarkably, this most unprepossessing of cell types, the adipocyte, turned out to be an ‘intelligent’ member of the neuroendocrine signalling community, participating directly in providing information to the brain about the state of nutrient stores. Leptin plays a major (perhaps its principal) role in the signalling of states of nutritional depletion to the brain, with many – if not all – of the neuroendocrine responses to starvation being critically dependent on the brain sensing a decrement in circulating leptin.29 The discovery of leptin was a major milestone in the history of physiology, and the work of Friedman and Coleman (sadly now deceased) that led to its discovery has been widely recognised by major international prizes.
Much subsequent science has been stimulated by the exploration of the downstream cellular and molecular pathways mediating these effects. The arcuate nucleus in the mediobasal hypothalamus is a major site for leptin action.30 There, it acts on two key populations of neurons whose profile of neuropeptide expression is unique to this tiny and highly specific anatomical site. Neurons expressing pro-opiomelanocortin (POMC) are stimulated by leptin, and the products of the POMC gene, such as alpha and beta melanocyte-stimulating hormone (MSH), are powerful suppressors of appetite and stimulate energy expenditure through activation of the sympathetic nervous system.31,32 Reciprocally, a population of neurons expressing neuropeptide Y (NPY) and agouti-related peptide (AGRP) are rapidly switched off by leptin,33 consistent with the fact that NPY is a powerful appetite stimulant and AGRP is an antagonist of MSH peptides.34 The power of these tiny populations of neurons (there are around 800 AGRP-expressing neurons in a murine brain that contains about 75 million neurons) is illustrated by the remarkable finding that an adult mouse in which the AGRP-positive neurons are acutely ablated through a transgenic toxin approach will continue to behave otherwise normally but ceases eating completely and will, if permitted, starve itself to death.35 It has recently been recognised that hypothalamic neurons concerned with leptin sensing and body weight regulation have particular electrophysiological properties that make them uniquely suited to the integration of incoming hormonal and neural signals over the medium to long term.36
Pathways that sense ingested nutrients and alter meal termination
While the leptin–melanocortin signalling system informs the brain about long-term energy stores in adipose tissue, the brain also receives much more acute information about the size and macronutrient composition of individual meals. Such information is integrated with the longer-term adipostatic signals to influence the process of satiety, leading to meal termination. Cholecystokinin (CCK) was the classic postprandial hormone involved in satiety. Rodents given exogenous CCK eat smaller meals, but compensate by eating more frequently.37 A growing body of research into cells of the entero-endocrine system, which are stimulated to release their contents by the presence of nutrients in the gastrointestinal tract, has demonstrated how important signals such as those derived from glucagon-like peptide 1 (GLP1), peptide YY (PYY) and ghrelin are in mediating the sense of postprandial satiety.38–40 More recently, advances are being made in identifying signals that influence macronutrient choice. Thus, the liver-derived hormone FGF21 is potently induced by high carbohydrate loads and, acting through the hypothalamus, powerfully reduces preference for ingestion of carbohydrate versus other macronutrients.41
The human dimension
The discovery of the leptin–melanocortin system in the mid-1990s generated great excitement, but the relevance of this system to human physiology and/or disease remained unknown. As a young endocrinologist, I had paid little attention to the field of body weight regulation, considering obesity (including my own!) to be largely the cosmetically unfortunate result of poor lifestyle choices. This changed in the early 1990s when, in my regular endocrine clinic, I saw a woman with a complex poly-endocrine syndrome, one feature of which was severe obesity from early childhood. We worked out that she was failing to cleave several hormonal precursors into mature peptide hormones42 and eventually went on to pinpoint her disorder as being due to biallelic mutations in the neuroendocrine-specific subtilisin-like pro-protein convertase PCSK1, the first disorder of pro-protein processing to be described in a metazoan organism.43
It seemed implausible to me that her obesity would be unrelated to her underlying genetic lesion. The corollary of this was that human beings could become severely obese owing to mutations in a single specific gene. Around the time of the first report of leptin, we heard of two severely obese and hyperphagic British first cousins from a consanguineous family of Pakistani origin. The human genomic sequence of leptin was not yet known, so we initially obtained subcutaneous adipose biopsies and sequenced the complementary DNA (cDNA) of these children. However, using the radiolabel- and gel-based methodologies of the day, we didn’t see a mutation. A year or so later, Sadaf Farooqi joined the lab as a clinical research fellow and, as her first foray into laboratory work, I suggested that she use newly validated assays for circulating leptin to examine the plasma of these children. There was none detectable. We pulled out the old sequencing films and, with a mix of excitement and horror, realised that at one point in the sequence in the normal control there was a tightly bunched run of six guanine nucleotides, but in the affected children there were only five. We had missed this the first time around. The children were homozygotes for a frameshift mutation that would truncate the leptin molecule.44
Having only ever studied two obese pedigrees, we had found the genetic cause in both, representing the first humans for whom a clear single gene explanation for obesity had been established.45 So, human beings could indeed become obese for purely biological reasons. How common was this? We reckoned that if a phenotype is due to a highly penetrant genetic mutation, it is likely to occur early in life and be severe, so we focused our attention on severe childhood obesity. Over the next few years we discovered several other genes, mutations in which cause obesity, including MC4R46 simultaneously with Froguel,47TRKB48 and BDNF.49 The first mutations in POMC,50 leptin receptor51 and SIM152 were reported by others. Our efforts in genetic discovery in childhood-onset obesity have continued to the present day under the scientific leadership of Sadaf Farooqi.53,54
How did these genetic mutations lead to chronic energy imbalance? We carried out detailed phenotypic evaluation of children carrying these mutant genes. This clearly showed that the main driver for their obesity was a markedly increased appetite. Affected children had increased hunger and reduced duration of satiety, leading to increased food-seeking behaviour and diminished meal-related satiation.48,55,56 This has been, in some cases, accompanied by a small reduction in energy expenditure per unit lean mass.53,57 Thus, through the discovery and study of human genetic disorders, we established that a state of human obesity could be produced by a purely genetic route, with those mutations leading to a change in eating behaviour through impairing the function of brain circuits concerned with sensing nutrient stores. Therefore, these forms of obesity were not so much ‘metabolic’ as ‘neurobehavioural’, with the defective gene resulting in an aberrantly strong drive for what is, after all, a necessary human behaviour.
In one case, congenital deficiency of leptin (Fig 2), we have had the great privilege of witnessing the dramatic effects of correcting a genetic disorder by providing the missing gene product.56,58 In all cases, the obesity of congenital leptin deficiency is completely reversible with leptin therapy, no matter how long the deficiency has been present.59 Within a few days after initiation of leptin therapy, spontaneous food intake reduces by over 70% and this is accompanied by astonishing changes in higher brain centres, as exemplified by the blood-flow changes to an area of the brain such as the ventral striatum in response to the visual presentation of food-related stimuli. This occurs before there is any discernible change in weight.60 The implications of such studies, demonstrating the power of a simple secreted protein coming only from adipose tissue to exert such powerful influences on a higher brain centre, is profound and counters the notion that, as far as human obesity is concerned, we can readily separate (as Gordon Kennedy actually tried to do)19 ‘hypothalamic’ (subconscious, not under voluntary control) from ‘higher’ (conscious, wholly controllable by free will) elements.
Fig 2.
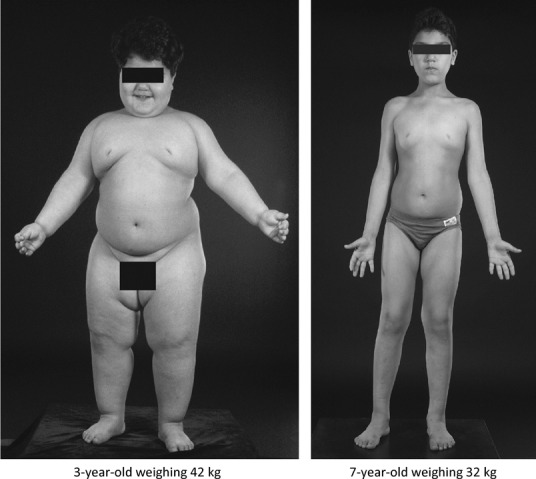
Child with congenital leptin deficiency before (aged 3 years) and after (aged 7 years) leptin therapy. Modified with permission from the Annual Review of Medicine 2005, volume 56, pp 443–58. © by Annual Reviews, www.annualreviews.org
The importance of postprandially released endocrine signals in the control of human food intake and energy balance has been demonstrated not so much through genetics as through pharmacological studies. The administration of a variety of these gut-derived peptides in a manner that mimics what would happen after a meal has been repeatedly shown to have major effects on eating behaviour in humans.38
Does this have any relevance to variation of adiposity in the general population and common forms of obesity?
Most obese humans do not harbour single mutations that, alone, cause their energy imbalance. However, adiposity is a highly heritable trait, so it should be possible to gain insights by looking in a hypothesis-free way at variation at the level of the whole genome in the broader population. Such studies, which represent a triumph of international collaboration, have been undertaken in a rigorous way so that the genetic determinants of adiposity have been systematically explored in hundreds of thousands of humans. Some scepticism has been expressed by the broader biomedical community about the value of these ‘genome-wide association study (GWAS)’ approaches, as it has often proven challenging to establish beyond reasonable doubt what bioactive molecule is actually mediating the change in physiology and/or disease risk. Obesity is an excellent example of how GWAS approaches can provide a very important ‘gestalt’ regarding the global genetic architecture of a complex trait. When one takes all the common genetic variants that reproducibly associate with measures of fatness in humans, and collectively look at the tissues and genes that these variants are most likely to be impacting, the result is remarkable. The vast majority of human variants influencing fatness impact on genes that are most highly expressed in the brain.61 Additionally, many of the variants are close to genes encoding highly neuronally expressed molecules in which loss of function mutations have previously been described to cause monogenic obesity (eg MC4R,46POMC50 and BDNF).49
The late Jane Wardle convincingly demonstrated that variants in these genes had a quantifiable impact on eating behaviour in normal children and, through those behaviours, significantly influenced a child’s risk of becoming obese.14 It can now be unequivocally stated that, across many populations, an individual’s susceptibility to be either obese or lean is powerfully influenced by the chance events associated with the inheritance of particular variants of genes that are functioning predominantly in the central nervous system. Despite these unarguable findings, it has been hard to shift the concept of ‘greed’ as a moral failing rather than a biological predisposition, and the increased appetite drive that predisposes many people to obesity is not infrequently linked in the public mind, and the popular press, with laziness and stupidity.
One markedly obese person who was neither lazy nor stupid was William Taft, the 27th president of the USA and the 10th chief justice of the US Supreme Court, the only person to hold both offices. At his heaviest, he weighed 340 lbs and had a BMI of over 46. Perusal of his diaries and those of members of his staff give a poignant picture of a man desperate to avoid the medical consequences of severe obesity, and making prodigious but only partially and intermittently successful efforts to control his weight.62 The following is an abridged abstract from the diary of Ira Smith, Taft’s ‘chief of mails’ at the White House:
The President dieted, all right, but not when he could escape supervision. I remember once when I accompanied him on a journey to Ohio. When we got on the train, leaving the doctor and Mrs. Taft behind, the President began to perk up. He also apparently began to think about food, although it was then ten o’clock in the evening … the President appeared at the door of his sitting room … I took one look and knew what was on his mind.
‘Anybody seen the conductor?’ he asked.
The conductor came a-running.
‘The dining car…’ Mr. Taft began shyly. ‘Could we get a snack?’
The conductor looked surprised. ‘Why, Mr. President, there isn’t any dining car on this train.’
The President’s sun-tanned face turned pink, with perhaps a few splashes of purple …
‘Norton!’ he called in a cold voice. ‘Mr. Norton!’[Norton was his private secretary] …‘there is no diner on this train.’
Norton … reminded Mr. Taft that they had had dinner at the White House, and assured him that they would not go without breakfast. He recalled that the President’s doctor had warned him about eating between meals. The President brushed him aside, turning back to the conductor.
‘What’s the next stop, dammit?’ he asked. ‘The next stop where there’s a diner?’
The conductor believed that would be Harrisburg. Mr. Taft glared at Norton and addressed the conductor:
‘I am President of the United States, and I want a diner attached to this train at Harrisburg. I want it well stocked with food, including filet mignon …’ He silenced the secretary’s protests with a roar. ‘What’s the use of being President,’ he demanded, ‘if you can’t have a train with a diner on it?’
The appetite for food differs greatly between people in its intensity and ‘greed’ is not confined to the feckless. Obesity is not a new disease; nor is it particular to a social underclass.
Do these findings have any direct relevance for patient care or public policy?
As a result of the genetic science undertaken over the past 20 years, we can now give definitive diagnoses to many children who previously would have been labelled as having ‘simple obesity’, with all the pejorative implications regarding improper parental care that the term has traditionally carried. Some of these disorders are not uncommon. Melanocortin 4 receptor deficiency occurs in about 1 per 1,000 people,63 making it commoner than other high-profile genetic diseases such as Duchenne muscular dystrophy or cystic fibrosis. The discovery that a child with severe obesity actually harbours a pathogenic mutation in a gene known to cause obesity is increasingly occurring in scenarios when there has been a previous or ongoing threat to take a child from the care of their parents.54 In the rare case of leptin deficiency, leptin therapy is life-changing.59 Unfortunately, because of the dose–response curve of leptin, this hormone, while hugely effective in children with very low concentrations, is not efficacious (at least alone) in producing weight loss when leptin concentrations are in the normal or high range.64
Gut-derived peptides are a major area of pharmaceutical activity. A long-acting form of GLP1 has been licensed as an obesity therapeutic.65 It seems likely that much of the salutary effect of gastric bypass surgery on weight loss is mediated not, as is commonly and mistakenly thought, through restriction or malabsorption, but through the profound effects on the secretion of these postprandial hormones that occur secondary to alterations in the anatomy of the gut.66 Forms of surgery that attempt to limit food intake by simply restrictive measures (such as gastric banding) have little or no impact on such hormonal milieu and poorly sustained efficacy. Much effort is ongoing to identify the precise nature of the hormonal perturbances that are mediating this effect and with the hope that multi-hormonal therapies will, in time, be able to replace bariatric surgery.
The fact that obesity-prone people are biologically predisposed to overeat provides ammunition to those who encourage public policies to reduce the factors within the environment that encourage excessive caloric ingestion. The road from scientific knowledge of the causes of obesity to the implementation of such knowledge in public health policy remains a twisting and precarious one, a road which some powerful interest groups are keen to strew with obstacles and diversions.
The widespread availability of a relatively inexpensive medicine that would be both safe and effective in promoting and maintaining weight loss is a holy grail. This will be challenging, as it is likely that such a drug or drug combination would probably have to be, at least in part, targeted at the central nervous system. The brain tends to use identical chemical signals to subserve one function in one anatomical area and a different function in another. This was the problem that caused the withdrawal of rimonabant, an anti-cannabinoid drug that powerfully suppressed appetite and promoted weight loss, but which unfortunately had anti-hedonic properties that promoted suicidal behaviour in some recipients.67
Should we give up? I don’t think so. I believe that using hypertension as an analogy is helpful and can put our current issues with development of anti-obesity drugs into a historical perspective. For the first half of the 20th century, hypertension was a major public health problem in many countries, with many deaths from renal and cardiac failure and malignant hypertension being a common medical emergency.68 There were no effective drugs, and drastic surgical approaches such as cervical sympathectomy needed to be undertaken in severe cases. Some useful drugs such as trimethaphan, reserpine and methyldopa emerged, but all had common and severe side effects. Gradually a better understanding of hypertension as a disorder of renal sodium retention began to emerge and drugs that acted in those processes began to develop. Now we have at least four classes of antihypertensive drugs, all of which are now generic and cheap and, when used in combination, there is almost no one whose blood pressure cannot be satisfactorily controlled. These advances in clinical pharmacology have been accompanied by public health efforts focused on primary prevention through limitation of salt intake. Using such a multidisciplinary approach, remarkable advances have been made in the prevention and treatment of blood pressure.68 Obesity has some striking similarities to hypertension, both conditions simply representing terms for the upper ends of a continuously distributed trait at which people begin to exhibit increased morbidity and mortality. Compared with our mature understanding of the control of blood pressure, the fundamental science of the control of human body weight is still in its infancy. It would seem premature to abandon hopes for effective pharmacotherapy for obesity when there is still so much to learn and to exploit for the benefit of human health.
The consequences of obesity
Increasing adiposity is significantly associated with a number of causes of morbidity and mortality. Of these some, such as osteoarthritis of the knee, obstructive sleep apnoea and reflux oesophagitis, are contributed to by the mechanical and gravitational effects of an excessive weight and volume of adipose tissue. The links of obesity with uterine, breast and bowel cancer are statistically robust, but as yet the mechanisms remain unclear.69 One very common disorder where the link with obesity is particularly strong is type 2 diabetes. Indeed, much of the public health concern surrounding obesity relates to its impact on the incidence of type 2 diabetes with all its disabling and expensive complications.70 It is not necessarily intuitively obvious why carrying around excess triglycerides in adipose tissue leads to a disturbance of carbohydrate metabolism characterised by uncontrolled elevation of plasma glucose levels.
The first clues as to how these two disorders might be linked came shortly after insulin could be measured in the plasma. Bierman et al71 showed that, in non-diabetic people, there was a strong positive correlation between adiposity and fasting insulin, ie for any given level of blood glucose, obese people needed to make more insulin to maintain normal blood glucose. Many subsequent studies, including those using more gold-standard measures of insulin action such as the euglycaemic hyperinsulinaemic clamp, have confirmed the association with obesity and an even stronger association with central rather than peripheral fat distribution.72 Reaven was among the first to report that, even among non-diabetic people, insulin resistance was often accompanied by a constellation of cardiovascular risk factors, including high triglycerides, low high-density lipoprotein (HDL) cholesterol and high blood pressure, a cluster that has subsequently become known as the ‘metabolic syndrome’.73 It is widely accepted that the insulin-resistant state, which drives a chronic increase of insulin secretory activity from the pancreatic beta cell, will – at least in susceptible individuals – lead to the eventual ‘exhaustion’ of islet function and to the hyperglycaemia that is the diagnostic feature of diabetes.74
Thus, obesity is clearly associated with a state of ‘resistance’ to the glucose-lowering effects of insulin. How might that happen? To understand this, one has to have some understanding of how insulin normally controls carbohydrate metabolism.
How does insulin normally exert its effect on carbohydrate metabolism?
It must be remembered that the principal evolutionary purpose of having mechanisms for the tight control of circulating levels of glucose is largely related to the fact that the brain has an absolute requirement for glucose and that even a short period of significant hypoglycaemia is extremely deleterious to brain function. Thus, the liver is set up as a glucose-producing factory that, even under conditions of extreme and prolonged starvation, can continue to provide the brain with glucose (and, as starvation progresses, ketones) at the expense of depleting other nutritional resources of the body.75 The system that controls blood glucose at its lower levels and prevents hypoglycaemia relies on a balance between a range of glucose-raising hormones and the rapid suppression of insulin, a hormone that has a circulating half-life of only around 4.5 minutes.76 At the other end of the normal blood glucose excursion, insulin levels rise in response to increasing levels of blood glucose in a manner that, in healthy individuals, prevents the blood glucose level ever exceeding the renal threshold for glucose resorption; this situation would represent a loss of calories that, in evolutionary terms, would likely have been deleterious. If sustained hyperglycaemia is allowed to persist, certainly if it does so for a matter of years, this also leads to widespread damage to the endothelium of small blood vessels that characterises diabetic retinopathy, neuropathy and nephropathy. Thus, understanding how insulin acts to control carbohydrate metabolism is critical to the understanding of diabetes.
Insulin, a peptide hormone produced only by the beta cells of the pancreatic islet, signals through a widely expressed dimeric transmembrane receptor tyrosine kinase. This acts through adaptor molecules to activate phosphatidylinositol 3-kinase, which generates a lipid signal, phosphatidylinositol 3-phosphate (PIP3), within the plasma membrane. That particular lipid avidly binds to a region of the serine/threonine kinase AKT2, recruiting it to the plasma membrane, where it is activated. Depending on the precise cell type, this initiates a series of events that alter the cellular handling of nutrients.77 In liver cells, AKT2 activation triggers a catalogue of changes in post-translational modification of enzymes and in gene expression, which serve to switch off glucose production (gluconeogenesis) in the liver and promote glycogen storage in several tissues. In skeletal and cardiac muscle (and in adipocytes, which, however, in total take up a much smaller proportion of circulating glucose than does muscle), insulin’s stimulation of AKT2 has an additional impact. It stimulates the rapid translocation to the plasma membrane of vesicles containing the glucose transporter GLUT4, thus promoting rapid uptake of glucose into the tissues expressing this transporter.78 Tissues such as brain and haematopoeitic organs use large amounts of glucose but, in those sites, the uptake of glucose is not dependent on insulin.
The earliest defect discernible in individuals who are insulin resistant and ‘pre-diabetic’ appears to be an impaired ability of insulin to recruit GLUT4-containing vesicles to the plasma membrane in muscle.79 Only later in the disease is it possible to discern that insulin’s suppression of glucose production by the liver is also impaired. It is important to point out that a pure state of insulin resistance is, alone, seldom enough to produce the sustained hyperglycaemia of type 2 diabetes.80 The healthy pancreatic islet has a remarkable ability to upregulate its secretion of insulin to compensate for even profound degrees of insulin resistance. However, it can rarely do so indefinitely and indeed, if it is itself genetically vulnerable, can only do so for short periods of time.
How does over-nutrition lead to defects in insulin’s control of carbohydrate metabolism?
The inflammatory hypothesis
One particular hypothesis currently appears to be in the ascendant. It derives from the uncontroversial and highly replicated observation that the adipose tissue of obese people contains an excess of cells primarily involved in immunity and/or host defence, and shows more evidence of activation of those cells than does adipose tissue from lean people.81
Supporting the importance of these observations is the undoubted finding that infusion of a range of cytokines into animals can make them insulin resistant; an even more compelling set of observations shows that the link between obesity and insulin resistance can be interfered with by genetic manipulations, which secretively disable a range of signalling pathways specifically within macrophages and other cells of the immune/host defence system.82 However, the evidence that this work translates to humans is, at present, rather less convincing. Indeed, to date there is little published evidence that the use of non-specific83 or specific84 anti-inflammatory agents has any sustained, significant impact on human insulin sensitivity.
Numerous genetic variants influencing the expression of cytokine molecules or their receptors exist in the human genome and many of these significantly and reproducibly influence the risk of a range of diseases that are known to have an inflammatory or autoimmune basis.85 While there is extensive overlap between a wide range of common inflammatory diseases in terms of common genetic variants and the cells and tissues in which such variants are thought likely to act, there is little overlap or shared features of inflammatory disease with metabolic disease85 or indeed with adiposity.86 The ongoing absence of convincing human pharmacological or genetic evidence to support the relevance of the inflammation hypothesis in humans suggests that we must continue to look elsewhere for alternative mechanistic explanations for the link between human over-nutrition and impaired insulin action.
An alternative hypothesis: insulin resistance occurs when the adipose depot is no longer able to safely store all excess energy, and other tissues are subjected to macronutrient overload
We know that exposing either liver or skeletal muscle cells, in vitro or in vivo, to an acute overload of nutrients, particularly in the form of free fatty acid, consistently impairs insulin’s ability to influence glucose metabolism,87,88 including impairing GLUT4 translocation in skeletal muscle, the earliest discernible abnormality of insulin action in people on their way to developing type 2 diabetes.79 Obesity is, by definition, a state of overprovision of calories in relation to energy expenditure. But we have professional cells, the adipocytes, designed to detect any such excess of ingested nutrients and lay them down for a rainy day. What if this storage depot did not function properly, either through being inadequate in size or defective in function? The excess ingested nutrients would have to be dealt with somewhere. This is not just a thought experiment; this is what happens in a range of human conditions called lipodystrophies.
A lipodystrophy is a condition characterised by an inherent inability to make fat cells or to synthesise triglycerides within fat cells.89 It can be caused by genetic defects impairing adipocyte differentiation or triglyceride synthesis, or by destruction of fat cells and their precursors by autoimmune mechanisms. It is increasingly recognised, at least in a partial form, as a consequence of total body irradiation for certain childhood cancers,90 and of chronic antiviral therapy for HIV infection.91 What characterises all generalised forms of lipodystrophy, whatever their causes, is the early development of insulin resistance, dyslipidaemia and fatty liver reminiscent of an aggressive version of more typical ‘metabolic syndrome’. Indeed, there is no feature of the metabolic syndrome, including increased risks of atherosclerosis and severe fatty liver disease, that is not reliably present in patients with lipodystrophy. Of course, in cases where no adipose tissue is present, the cause of the insulin resistance cannot be ascribed to inflammation in an adipose depot that simply isn’t there.
A few years ago, in work led by my colleague David Savage with colleagues in Paris, we described a new form of human lipodystrophy, which, although rare, I believe to be particularly illuminating (Fig 3). Affected individuals had diminished, but not absent, fat cell mass and were afflicted with every feature of the metabolic syndrome from an early age. They carried one of two different heterozygous frameshift mutations within the perilipin 1 gene that resulted in retention and continued expression of the molecule’s normal N-terminal portion, but a structural disruption of its C-terminal region.92,93 Perilipin 1 is a protein expressed only in white adipocytes, where it coats the large unilocular triglyceride droplet unique to that cell type. At the location, it serves a crucial regulatory role. When we are in a fed state, we have no need to access the energy stored in our adipocyte triglyceride droplet and, in these conditions, perilipin 1 acts as its guardian. It binds a protein called CGI58 tightly to its C-terminus. When allowed to roam free, CGI58 zips around the surface of the droplet, binding tightly to the enzyme adipose triglyceride lipase (ATGL), which initiates the cascade of triglyceride breakdown. The N-terminus of perilipin 1 plays an important role in continuing the lipolytic cascade by attracting hormone-sensitive lipase (HSL), which hydrolyses diglycerides, to the surface of the droplet. Thus, in the fasting state, signalling cascades in the fat cell result in phosphorylation of the C-terminus of perilipin 1, freeing CGI58 to bring ATGL to the droplet to initiate the process of triglyceride breakdown and also phosphorylating the N-terminus to attract HSL, which performs the second step. In the patients with one mutant copy of perilipin 1, there is always a component of CGI58 which is free to continually bring ATGL to the droplet and drive continuing fat droplet breakdown, even during the fed state. Remarkably, this subtle disturbance affecting only the white fat cell and, within that cell, only impacting on the control of lipolysis on the surface of the lipid droplet, is sufficient to result in every feature of the metabolic syndrome in patients carrying this form of defective allele. Although very rare, this syndrome provides a compelling model whereby a relatively subtle disturbance of the lipid storage function of the fat cell, which is itself not a major quantitatively important site for glucose disposal in the whole organism, is sufficient, alone, to cause severe insulin resistance and all its associated pathologies in humans.
Fig 3.

Coordination of the control of triglyceride breakdown on the surface of the adipocyte fat droplet in the fasting and fed state in a healthy individual (A) and in a patient with a mutation disrupting the C-terminus of perilipin 1 (B). In the fasting state, perilipin 1 (P1), a protein that sits on the surface of the triglyceride droplet of the fat cell, is phosphorylated near its C-terminus. This prevents it from binding CGI58 and allows the latter to bind and activate adipocyte triglyceride lipase (ATGL), the enzyme that initiates triglyceride breakdown. The diglycerides formed by this reaction are broken down by hormone-sensitive lipase (HSL), the active phosphorylated form of which binds to perilipin 1 when the latter is phosphorylated at a residue close to its N-terminus. Monoglyceride lipase (MGL) is constitutively active. Thus, in the fasting state, the fat cell efficiently releases abundant free fatty acid into the systemic circulation for use by other organs. In the fed state, perilipin 1 is dephosphorylated and binds tightly to CGI58, preventing it from interacting with and activating ATGL; HSL does not interact with dephosphorylated perilipin 1 and thus the process of adipocyte lipolysis is efficiently shut down in the fed state while triglyceride synthesis is activated, with free fatty acids derived from triglycerides delivered to the fat tissue in lipoproteins generated in the gut and the liver, with lesser amounts coming from re-esterification and from de novo lipogenesis in fat. Patients with mutations that disrupt the C-terminal phosphorylation site of a single copy of their perilipin 1 protein have a syndrome characterised by all the features of the metabolic syndrome in the face of a diminished, but not absent, fat cell mass. This occurs because the mutant C-terminal tail of perilipin 1 is unable to bind CGI58 so, even in the fed state, ATGL remains at the lipid droplet surface and active; thus, free fatty acids are generated from the adipocyte irrespective of the nutritional state. That disruption of lipid droplet triglyceride regulation is sufficient, alone, to result in every feature of the metabolic syndrome. ATGL = adipocyte triglyceride lipase; C = C-terminus of perilipin 1; DG = diglyceride; FA = fatty acid; FFA = free fatty acid; HSL = hormone-sensitive lipase; LPL = lipoprotein lipase; MG = monoglyceride; MGL = monoglyceride lipase; N = N-terminus of perilipin 1; P1 = perilipin 1; TG = triglyceride
Might such mechanisms be relevant in common forms of insulin resistance? It has certainly been suggested by several investigators that impaired adipose storage capacity and lipid overflow may have broad importance;94–96 however, it has been challenging to obtain any reliable estimates of the extent to which it might be responsible for insulin resistance in the general human population. We have recently worked with colleagues in the MRC Epidemiology Unit within our institute to test this hypothesis more robustly.97 We first examined genetic and phenotypic data from more than 150,000 people to identify genetic variants that were associated with a ‘triad’ of elevated BMI-adjusted fasting insulin, higher serum triglycerides and low serum HDL cholesterol, and identified 53 SNPs that were replicably associated with these measures (as well as with type 2 diabetes and cardiovascular disease in independent analyses).
Remarkably, when studied in independent cohorts in whom regional fat distribution had been measured by DEXA, the strongest association of the combined 53 SNP genetic risk score was not with an increase in central or visceral fat as might have been expected, but with a marked reduction in femoral and gluteal fat. The importance of adipose tissue genes in mediating this phenotype was supported by the 53 loci containing genes highly expressed in adipose tissue and substantial overlap with adipose tissue-active enhancer elements, as well as by functional analysis of adipocytes in which several of the novel genes had been disrupted. In summary, a hypothesis-free analysis looking for genetic variants that predispose to ‘metabolic syndrome’ revealed a set of variants that are strongly implicated as affecting adipose tissue and that impair its ability to differentiate, and expand. It would seem reasonable to conclude that a ‘forme fruste’ of lipodystrophy is a major mechanism leading to insulin resistance in contemporary societies where over-nutrition is widespread. In simple terms, those who are able to greatly expand their adipose mass in the face of chronic over-nutrition can avoid insulin resistance and metabolic syndrome, while those who have continued imbalance of energy input over expenditure but are unable to safely store this in adipose tissue are the people who will suffer the most severe metabolic derangements (Fig 4).
Fig 4.

The ‘soggy bathroom carpet’ model of over-nutrition-related metabolic disease. A – In health, chronic energy intake (the flow through the tap) is matched by energy expenditure (the flow through the open plughole) such that there is a moderate but healthy amount of excess energy stored (the water rises, but does not overflow the bathtub). B – In an individual with marked obesity that has led to diabetes, the energy inflow chronically exceeds the energy outflow such that the amount of stored energy exceeds the capacity to safely store it and excess nutrients flow elsewhere, causing damage (the soggy carpet). C – In individuals with a limited ability to expand adipose tissue storage capacity (a smaller bathtub), even a small excess of energy intake over expenditure will lead to energy being misdirected (overflowing the bathtub) and to adverse metabolic outcomes including fatty liver, insulin resistance and diabetes.
Orthogonal evidence supporting the ‘nutritional’ rather than the ‘inflammatory’ hypothesis has come from recent elegant (and heroic) studies from Boden and colleagues, who clearly showed how marked insulin resistance could be induced by just a few days of severe caloric overload at a stage when there was no evidence of increased inflammation, or indeed endoplasmic reticulum (ER) stress, in fat or muscle biopsies.98
It is somewhat ironic that it was actually our laboratory that made the first published observations showing that a macrophage product was more highly expressed in fat from obese than from lean humans.99 We didn’t make much of this at the time, as we thought it likely to reflect a higher rate of cell death in overloaded adipose tissue, a supposition that was subsequently elegantly confirmed by Cinti et al.100
The primacy of pancreatic beta cell dysfunction in determining the onset of hyperglycaemia
Recent large-scale, genome-wide studies comparing patients with type 2 diabetes and controls have identified multiple associated loci,101 the majority of which are implicated in insulin secretory capacity rather than insulin sensitivity.102 This is consistent with the fact that the vast majority of single gene disorders causing diabetes do so by impairing the function of the pancreatic beta cell.103 It seems clear that, despite the fact that over-nutrition leads to insulin resistance and this is likely to be playing a key role in driving the epidemic of type 2 diabetes, it is the intrinsic health of the islet that ultimately determines whether a person remains hyperinsulinaemic but does not develop hyperglycaemia or whether type 2 diabetes ensues. Of course insulin-resistant, hyperinsulinaemic individuals may have all the other features of the metabolic syndrome, including higher risk of macrovascular disease, but will not develop the microvascular complications of retinopathy, nephropathy and neuropathy unless hyperglycaemia occurs. I have gained some personal satisfaction from these recent genetic discoveries, as work I had undertaken at the beginning of my scientific career, in the lab of the late Professor Robert Turner in Oxford, demonstrated that quantitative104 and qualitative105 defects of insulin secretion were detectable in first-degree relatives of patients with type 2 diabetes long before they developed diabetes.
Do these findings have any direct relevance for patient care or public policy?
Caloric restriction is a keystone of type 2 diabetes therapy. Its efficacy has been known since the Allen diets106 at the beginning of the last century and is rediscovered every few decades or so.107,108 The model whereby individuals develop lipotoxicity in organs such as liver and muscle (and possibly pancreas) when they reach the limits of safe energy storage in adipose tissue provides a useful explanation for the rapid improvement in glycaemic control occurring long before any significant loss of weight or indeed cellular composition of adipose tissue, and also why different individuals develop insulin resistance at different levels of adiposity. The key challenge is the fact that caloric restriction is very hard for most people to maintain in the long term, as the drive to eat is enhanced by changes in hormonal milieu that tend to drive the weight back towards its starting point.109 Bariatric surgery is so remarkably effective in diabetes because it provides an ongoing and sustained signal to reduce food intake and therefore maintain a healthy energy balance.66
The idea of ‘safe fat’ may seem paradoxical, given the epidemiological relationship between BMI and insulin resistance, but there is a large amount of variation around that distribution and many obese people, particularly those with large gluteofemoral subcutaneous depots, remain remarkably healthy. Of note, thiazolidinedione drugs, which are among the most potent insulin sensitisers, appear to sensitise to a substantial extent by promoting adipocyte differentiation and increasing ‘safe’ triglyceride storage in adipose tissue.110 Conversely, it would appear that pursuing anti-inflammatory strategies may not be a highly effective way of interrupting the link between obesity and insulin resistance.
There are many populations worldwide with high risks of type 2 diabetes, even though they have generally lower levels of adiposity than those seen in the west. It will be fascinating to find out whether people from those groups have an intrinsic limitation in the generation of new fat cells and whether that relates to genetics or to early life events. Indeed, if the early nutritional environment did indeed somehow ‘programme’ the stem cells from which adipocytes are derived and limit the number of subcutaneous adipocytes that could be recruited to cope with later over-nutrition, this could be an important target for early pre and postnatal nutritional interventions. It is tempting to reflect, as others have done previously,111 that the particular susceptibility of ethnic groups such as south Asians to develop insulin resistance and diabetes might have a related mechanism.
Conclusions
Much of what we have learned about the control of human body weight and insulin sensitivity and secretion has come, not from the study of patients with common complex forms of disease, but from the identification of rare individuals with precise single gene defects where one can unequivocally trace the causal chain of pathogenesis. Even in conditions as common as obesity and type 2 diabetes, insights into disease mechanisms can come from the careful characterisation of such human ‘experiments of nature’. This has been a recurrent theme of Harveian Orations, including those from Archibald Garrod (1924) The debt of science to medicine,112 David Weatherall (1992) The role of nature and nurture in common diseases: Garrod’s legacy113 and Keith Peters (2004) Exceptional matters.114 The power and reach of this approach will probably increase as comprehensive genomic interrogation of individual unusual patients becomes more routine.
Endocrine and metabolic disorders are quintessentially disorders of communication across the whole organism and cannot be understood solely by the study of isolated cells. The function and nature of leptin (or, for that matter, insulin) would never have been discovered in a culture dish. We need to recognise how important studies undertaken in animals have been to this and other fields. While we should study humans wherever possible, we will be unable, for example, to understand issues related to the sites of action of hormone action without using rodent models in which we can alter gene expression and function in a tissue-specific and/or temporally controlled fashion. We must study animals sparingly and compassionately, but the advance of science and medicine will, for the foreseeable future, remain highly dependent on such studies.
Returning to the patient with whom I began this lecture, what can we offer patient M? What she clearly needs is a procedure that permanently reduces her drive to eat and renders her persistently and substantially leaner than her body ‘wants to be’. Only that way will we ‘unload’ the nutrient excess that is overwhelming her limited capability to lay down safe fat stores. The beneficial effects of bariatric surgery on diabetes are often dramatic and are not even restricted to those who are formally obese.
There are increasing reports of lipodystrophic patients whose severe insulin resistance and poorly controlled diabetes are effectively cured as a result of surgery.115 This is not that surprising if one considers the mechanisms of lipid overflow and lipotoxicity associated with limited safe fat storage capacity, discussed in detail earlier in this lecture. Unfortunately for M, the UK lags far behind most advanced countries in the provision of ‘metabolic surgery’116 and she will probably face many barriers to accessing this highly successful treatment.117 The National Institute for Health and Care Excellence (NICE) has recently published a quality standard for the clinical assessment and management of obesity,118 which includes the explicit recognition of the important role that bariatric surgery plays. It is to be hoped that this will lead to a marked change in behaviour of health commissioners and providers, so that this highly cost-effective form of care can be delivered to patients such as M.
I believe that there are grounds for optimism, that we can reduce the human suffering, disability and early death resulting from the medical consequences of over-nutrition, much as we have done to reduce the adverse outcomes resulting from the sustained elevation of blood pressure. Interventions that usefully render our environment less obesogenic will require sustained lobbying until the necessary courageous political decisions are made. The burgeoning basic science of energy balance and metabolism will uncover novel therapeutic targets and pathways with exciting opportunities for improved drugs and devices, which may be combined with better subclassification of the ‘obesities’ and more effective targeted therapeutics. However, progress is threatened by the fact that, despite the global importance of obesity as a threat to public health, expenditure on research in metabolic disorders is only a small fraction of that spent on, for example, cancer or heart disease. Where obesity and hypertension differ most strikingly is in the degree to which they are stigmatised. A person’s high blood pressure is not obvious to others and few people attribute hypertension to the fault of the individual. A person’s severe obesity is all too obvious to casual observers, many of whom are prone to attributing the condition solely to personal failing. This commonly held view has contributed to widespread social and economic discrimination against people who are obese119,120 and to our reluctance to provide effective therapies, even when they are available and cost-effective. As the science of human obesity continues to advance, such views will increasingly be seen as illogical and unsupportable.
Conflicts of interest
The author has no conflicts of interest to declare.
References
- 1.Keynes G. The life of William Harvey. Oxford:: Clarendon Press; 1966. [Google Scholar]
- 2.International Diabetes Federation IDF diabetes atlas. 7th. Brussels, Belgium:: International Diabetes Federation; 2015. [PubMed] [Google Scholar]
- 3.Bray GA. Obesity: historical development of scientific and cultural ideas. Int J Obes. 1990;14:909–26. [PubMed] [Google Scholar]
- 4.Flegal KM. Carroll MD. Kuczmarski RJ, et al. Overweight and obesity in the United States: prevalence and trends, 1960–1994. Int J Obes Relat Metab Disord. 1998;22:39–47. doi: 10.1038/sj.ijo.0800541. [DOI] [PubMed] [Google Scholar]
- 5.Drewnowski A. Darmon N. The economics of obesity: dietary energy density and energy cost. Am J Clin Nutr. 2005;82:265S–73S. doi: 10.1093/ajcn/82.1.265S. [DOI] [PubMed] [Google Scholar]
- 6.Department for Environment, Food and Rural Affairs, National Statistics Family food 2014. London:: DEFRA; 2015. [Google Scholar]
- 7.Young LR. Nestle M. Expanding portion sizes in the US marketplace: implications for nutrition counseling. J Am Diet Assoc. 2003;103:231–40. doi: 10.1053/jada.2003.50027. [DOI] [PubMed] [Google Scholar]
- 8.Burgoine T. Forouhi NG. Griffin SJ. Wareham NJ. Monsivais P. Associations between exposure to takeaway food outlets, takeaway food consumption, and body weight in Cambridgeshire, UK: population based, cross sectional study. BMJ. 2014;348:g1464. doi: 10.1136/bmj.g1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Monsivais P. McLain J. Drewnowski A. The rising disparity in the price of healthful foods: 2004–2008. Food Policy. 2010;35:514–20. doi: 10.1016/j.foodpol.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wareham NJ. van Sluijs EMF. Ekelund U. Physical activity and obesity prevention: a review of the current evidence. Proc Nutr Soc. 2005;64:229–47. doi: 10.1079/pns2005423. [DOI] [PubMed] [Google Scholar]
- 11.Johnson F. Mavrogianni A. Ucci M. Vidal-Puig A. Wardle J. Could increased time spent in a thermal comfort zone contribute to population increases in obesity? Obes Rev. 2011;12:543–51. doi: 10.1111/j.1467-789X.2010.00851.x. [DOI] [PubMed] [Google Scholar]
- 12.Ekelund U. Ong K. Linné Y, et al. Upward weight percentile crossing in infancy and early childhood independently predicts fat mass in young adults: the Stockholm Weight Development Study (SWEDES) Am J Clin Nutr. 2006;83:324–30. doi: 10.1093/ajcn/83.2.324. [DOI] [PubMed] [Google Scholar]
- 13.Sonnenburg JL. Bäckhed F. Diet–microbiota interactions as moderators of human metabolism. Nature. 2016;535:56–64. doi: 10.1038/nature18846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llewellyn C. Wardle J. Behavioral susceptibility to obesity: Gene-environment interplay in the development of weight. Physiol Behav. 2015;152:494–501. doi: 10.1016/j.physbeh.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 15.Stunkard AJ. Harris JR. Pedersen NL. McClearn GE. The body-mass index of twins who have been reared apart. N Engl J Med. 1990;322:1483–7. doi: 10.1056/NEJM199005243222102. [DOI] [PubMed] [Google Scholar]
- 16.Yang J. Bakshi A. Zhu Z, et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat Genet. 2015;47:1114–20. doi: 10.1038/ng.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marteau TM. Hollands GJ. Fletcher PC. Changing human behavior to prevent disease: the importance of targeting automatic processes. Science. 2012;337:1492–5. doi: 10.1126/science.1226918. [DOI] [PubMed] [Google Scholar]
- 18.Adolph EF. Urges to eat and drink in rats. Am J Physiol. 1947;151:110–25. doi: 10.1152/ajplegacy.1947.151.1.110. [DOI] [PubMed] [Google Scholar]
- 19.Kennedy GC. The role of depot fat in the hypothalamic control of food intake in the rat. Proc R Soc Lond B Biol Sci. 1953;140:578–92. doi: 10.1098/rspb.1953.0009. [DOI] [PubMed] [Google Scholar]
- 20.Hetherington AW. Ranson SW. Hypothalamic lesions and adiposity in the rat. Anat Rec. 1940;78:149–72. [Google Scholar]
- 21.Anand BK. Brobeck JR. Hypothalamic control of food intake in rats and cats. Yale J Biol Med. 1951;24:123–40. [PMC free article] [PubMed] [Google Scholar]
- 22.Hervey GR. The effects of lesions in the hypothalamus in parabiotic rats. J Physiol. 1959;145:336–52. doi: 10.1113/jphysiol.1959.sp006145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ingalls AM. Dickie MM. Snell GD. Obese, a new mutation in the house mouse. J Hered. 1950;41:317–8. doi: 10.1093/oxfordjournals.jhered.a106073. [DOI] [PubMed] [Google Scholar]
- 24.Hummel KP. Dickie MM. Coleman DL. Diabetes, a new mutation in the mouse. Science. 1966;153:1127–8. doi: 10.1126/science.153.3740.1127. [DOI] [PubMed] [Google Scholar]
- 25.Coleman DL. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia. 1973;9:294–8. doi: 10.1007/BF01221857. [DOI] [PubMed] [Google Scholar]
- 26.Coleman DL. Hummel KP. Effects of parabiosis of normal with genetically diabetic mice. Am J Physiol. 1969;217:1298–304. doi: 10.1152/ajplegacy.1969.217.5.1298. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y. Proenca R. Maffei M, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–32. doi: 10.1038/372425a0. 1995, 374-479, Erratum in. [DOI] [PubMed] [Google Scholar]
- 28.Halaas JL. Gajiwala KS. Maffei M, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–6. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 29.Ahima RS. Prabakaran D. Mantzoros C, et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–2. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- 30.Satoh N. Ogawa Y. Katsuura G, et al. The arcuate nucleus as a primary site of satiety effect of leptin in rats. Neurosci Lett. 1997;224:149–52. doi: 10.1016/S0304-3940(97)00163-8. [DOI] [PubMed] [Google Scholar]
- 31.Cheung CC. Clifton DK. Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology. 1997;138:4489–92. doi: 10.1210/endo.138.10.5570. [DOI] [PubMed] [Google Scholar]
- 32.Boston BA. Blaydon KM. Varnerin J. Cone RD. Independent and additive effects of central POMC and leptin pathways on murine obesity. Science. 1997;278:1641–4. doi: 10.1126/science.278.5343.1641. [DOI] [PubMed] [Google Scholar]
- 33.Stephens TW. Basinski M. Bristow PK, et al. The role of neuropeptide Y in the antiobesity action of the obese gene product. Nature. 1995;377:530–2. doi: 10.1038/377530a0. [DOI] [PubMed] [Google Scholar]
- 34.Ollmann MM. Wilson BD. Yang YK, et al. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–8. doi: 10.1126/science.278.5335.135. 1998, 281-1615, Erratum in. [DOI] [PubMed] [Google Scholar]
- 35.Luquet S. Perez FA. Hnasko TS. Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–5. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- 36.Branco T. Tozer A. Magnus CJ, et al. Near-perfect synaptic integration by Nav1.7 in hypothalamic neurons regulates body weight. Cell. 2016;165:1749–61. doi: 10.1016/j.cell.2016.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.West DB. Greenwood MR. Marshall KA. Woods SC. Lithium chloride, cholecystokinin and meal patterns: evidence that cholecystokinin suppresses meal size in rats without causing malaise. Appetite. 1987;8:221–7. doi: 10.1016/0195-6663(87)90021-3. [DOI] [PubMed] [Google Scholar]
- 38.Spreckley E. Murphy KG. The L-cell in nutritional sensing and the regulation of appetite. Front Nutr. 2015;2:23. doi: 10.3389/fnut.2015.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parker HE. Gribble FM. Reimann F. The role of gut endocrine cells in control of metabolism and appetite. Exp Physiol. 2014;99:1116–20. doi: 10.1113/expphysiol.2014.079764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Müller TD. Nogueiras R. Andermann ML, et al. Ghrelin. Mol Metab. 2015;4:437–60. doi: 10.1016/j.molmet.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Talukdar S. Owen BM. Song P, et al. FGF21 regulates sweet and alcohol preference. Cell Metab. 2016;23:344–9. doi: 10.1016/j.cmet.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Rahilly S. Gray H. Humphreys PJ, et al. Impaired processing of prohormones associated with abnormalities of glucose homeostasis and adrenal function. N Engl J Med. 1995;333:1386–90. doi: 10.1056/NEJM199511233332104. [DOI] [PubMed] [Google Scholar]
- 43.Jackson RS. Creemers JW. Ohagi S, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303–6. doi: 10.1038/ng0797-303. [DOI] [PubMed] [Google Scholar]
- 44.Montague CT. Farooqi IS. Whitehead JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–8. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 45.Leibel RL. And finally, genes for human obesity. Nat Genet. 1997;16:218–20. doi: 10.1038/ng0797-218. [DOI] [PubMed] [Google Scholar]
- 46.Yeo GS. Farooqi IS. Aminian S, et al. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20:111–2. doi: 10.1038/2404. [DOI] [PubMed] [Google Scholar]
- 47.Vaisse C. Clement K. Guy-Grand B, et al. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998;20:113–4. doi: 10.1038/2407. [DOI] [PubMed] [Google Scholar]
- 48.Yeo GS. Connie Hung CC. Rochford J, et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004;7:1187–9. doi: 10.1038/nn1336. [DOI] [PubMed] [Google Scholar]
- 49.Gray J. Yeo GS. Cox JJ, et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. 2006;55:3366–71. doi: 10.2337/db06-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krude H. Biebermann H. Luck W, et al. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. 1998;19:155–7. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- 51.Clément K. Vaisse C. Lahlou N, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- 52.Jr Holder JL. Butte NF. Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000;9:101–8. doi: 10.1093/hmg/9.1.101. [DOI] [PubMed] [Google Scholar]
- 53.Pearce LR. Atanassova N. Banton MC, et al. KSR2 mutations are associated with obesity, insulin resistance, and impaired cellular fuel oxidation. Cell. 2013;155:765–77. doi: 10.1016/j.cell.2013.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Rahilly S. Farooqi IS. The genetics of obesity in humans. In: De Groot LJ, editor; Chrousos G, editor; Dungan K, et al., editors. Endotext. South Dartmouth MA: MDText.com Inc; 2000. [Google Scholar]
- 55.Farooqi IS. Keogh JM. Yeo GS, et al. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–95. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 56.Farooqi IS. Matarese G. Lord GM, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krakoff J. Ma L. Kobes S, et al. Lower metabolic rate in individuals heterozygous for either a frameshift or a functional missense MC4R variant. Diabetes. 2008;57:3267–72. doi: 10.2337/db08-0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Farooqi IS. Jebb SA. Langmack G, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341:879–84. doi: 10.1056/NEJM199909163411204. [DOI] [PubMed] [Google Scholar]
- 59.Paz-Filho G. Wong ML. Licinio J. Ten years of leptin replacement therapy. Obes Rev. 2011;12:e315–23. doi: 10.1111/j.1467-789X.2010.00840.x. [DOI] [PubMed] [Google Scholar]
- 60.Farooqi IS. Bullmore E. Keogh J, et al. Leptin regulates striatal regions and human eating behavior. Science. 2007;317:1355. doi: 10.1126/science.1144599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Locke AE. Kahali B. Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Levine DI. Corpulence and correspondence: President William H. Taft and the medical management of obesity. Ann Intern Med. 2013;159:565–570. doi: 10.7326/0003-4819-159-8-201310150-00012. [DOI] [PubMed] [Google Scholar]
- 63.Alharbi KK. Spanakis E. Tan K, et al. Prevalence and functionality of paucimorphic and private MC4R mutations in a large, unselected European British population, scanned by meltMADGE. Hum Mutat. 2007;28:294–302. doi: 10.1002/humu.20404. [DOI] [PubMed] [Google Scholar]
- 64.DePaoli AM. 20 years of leptin: leptin in common obesity and associated disorders of metabolism. J Endocrinol. 2014;223:T71–81. doi: 10.1530/JOE-14-0258. [DOI] [PubMed] [Google Scholar]
- 65.Pi-Sunyer X. Astrup A. Fujioka K, et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373:11–22. doi: 10.1056/NEJMoa1411892. [DOI] [PubMed] [Google Scholar]
- 66.Madsbad S. Dirksen C. Holst JJ. Mechanisms of changes in glucose metabolism and bodyweight after bariatric surgery. Lancet Diabetes Endocrinol. 2014;2:152–64. doi: 10.1016/S2213-8587(13)70218-3. [DOI] [PubMed] [Google Scholar]
- 67.Ward SJ. Raffa RB. Rimonabant redux and strategies to improve the future outlook of CB1 receptor neutral-antagonist/inverse-agonist therapies. Obesity (Silver Spring) 2011;19:1325–34. doi: 10.1038/oby.2011.69. [DOI] [PubMed] [Google Scholar]
- 68.Kotchen TA. Historical trends and milestones in hypertension research: a model of the process of translational research. Hypertension. 2011;58:522–38. doi: 10.1161/HYPERTENSIONAHA.111.177766. [DOI] [PubMed] [Google Scholar]
- 69.Lauby-Secretan B. Scoccianti C. Loomis D, et al. Body fatness and cancer – viewpoint of the IARC working group. N Engl J Med. 2016;375:794–8. doi: 10.1056/NEJMsr1606602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lotta LA. Abbasi A. Sharp SJ, et al. Definitions of metabolic health and risk of future type 2 diabetes in BMI categories: a systematic review and network meta-analysis. Diabetes Care. 2015;38:2177–87. doi: 10.2337/dc15-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bierman EL. Bagdade JD. Porte D., Jr Obesity and diabetes: the odd couple. Am J Clin Nutr. 1968;21:1434–7. doi: 10.1093/ajcn/21.12.1434. [DOI] [PubMed] [Google Scholar]
- 72.DeFronzo RA. Ferrannini E. Insulin resistance: A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14:173–94. doi: 10.2337/diacare.14.3.173. [DOI] [PubMed] [Google Scholar]
- 73.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 74.Bogardus C. Lillioja S. Bennett PH. Pathogenesis of NIDDM in Pima Indians. Diabetes Care. 1991;14:685–90. doi: 10.2337/diacare.14.7.685. [DOI] [PubMed] [Google Scholar]
- 75.Felig P. Marliss E. Owen OE. Cahill GF., Jr Blood glucose and gluconeogenesis in fasting man. Arch Intern Med. 1969;123:293–8. [PubMed] [Google Scholar]
- 76.Matthews DR. Lang DA. Burnett MA. Turner RC. Control of pulsatile insulin secretion in man. Diabetologia. 1983;24:231–7. doi: 10.1007/BF00282705. [DOI] [PubMed] [Google Scholar]
- 77.Whiteman EL. Cho H. Birnbaum MJ. Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab. 2002;13:444–51. doi: 10.1016/s1043-2760(02)00662-8. [DOI] [PubMed] [Google Scholar]
- 78.Larance M. Ramm G. James DE. The GLUT4 code. Mol Endocrinol. 2008;22:226–33. doi: 10.1210/me.2007-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morino K. Petersen KF. Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55(Suppl 2):S9–S15. doi: 10.2337/db06-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Parker VE. Savage DB. O’Rahilly S. Semple RK. Mechanistic insights into insulin resistance in the genetic era. Diabet Med. 2011;28:1476–86. doi: 10.1111/j.1464-5491.2011.03463.x. [DOI] [PubMed] [Google Scholar]
- 81.Weisberg SP. McCann D. Desai M, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gregor MF. Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–45. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 83.Goldfine AB. Conlin PR. Halperin F, et al. A randomised trial of salsalate for insulin resistance and cardiovascular risk factors in persons with abnormal glucose tolerance. Diabetologia. 2013;56:714–23. doi: 10.1007/s00125-012-2819-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lo J. Bernstein LE. Canavan B, et al. Effects of TNF-alpha neutralization on adipocytokines and skeletal muscle adiposity in the metabolic syndrome. Am J Physiol Endocrinol Metab. 2007;293:E102–E109. doi: 10.1152/ajpendo.00089.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Farh KK. Marson A. Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–43. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bulik-Sullivan B. Finucane HK. Anttila V, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–41. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dresner A. Laurent D. Marcucci M, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103:253–9. doi: 10.1172/JCI5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boden G. Chen X. Ruiz J. White JV. Rossetti L. Mechanisms of fatty acid-induced inhibition of glucose uptake. J Clin Invest. 1994;93:2438–46. doi: 10.1172/JCI117252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Robbins AL. Savage DB. The genetics of lipid storage and human lipodystrophies. Trends Mol Med. 2015;21:433–8. doi: 10.1016/j.molmed.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 90.Adachi M. Asakura Y. Muroya K. Goto H. Kigasawa H. Abnormal adipose tissue distribution with unfavorable metabolic profile in five children following hematopoietic stem cell transplantation: a new etiology for acquired partial lipodystrophy. Clin Pediatr Endocrinol. 2013;22:53–64. doi: 10.1297/cpe.22.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Galescu O. Bhangoo A. Ten S. Insulin resistance, lipodystrophy and cardiometabolic syndrome in HIV/AIDS. Rev Endocr Metab Disord. 2013;14:133–40. doi: 10.1007/s11154-013-9247-7. [DOI] [PubMed] [Google Scholar]
- 92.Kozusko K. Tsang VH. Bottomley W, et al. Clinical and molecular characterization of a novel PLIN1 frameshift mutation identified in patients with familial partial lipodystrophy. Diabetes. 2015;64:299–310. doi: 10.2337/db14-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gandotra S. Le Dour C. Bottomley W, et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med. 2011;364:740–8. doi: 10.1056/NEJMoa1007487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Danforth E., Jr Failure of adipocyte differentiation causes type II diabetes mellitus? Nat Genet. 2000;26:13. doi: 10.1038/79111. [DOI] [PubMed] [Google Scholar]
- 95.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–76. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Virtue S. Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the metabolic syndrome – an allostatic perspective. Biochim Biophys Acta. 2010;1801:338–49. doi: 10.1016/j.bbalip.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 97.Lotta LA. Gulati P. Day RF, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2016 doi: 10.1038/ng0217-317c. doi:10.1038/ng.3714. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 98.Boden G. Homko C. Barrero CA, et al. Excessive caloric intake acutely causes oxidative stress, GLUT4 carbonylation, and insulin resistance in healthy men. Sci Transl Med. 2015;7:304re7. doi: 10.1126/scitranslmed.aac4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Savage DB. Sewter CP. Klenk ES, et al. Resistin/Fizz3 expression in relation to obesity and peroxisome proliferator-activated receptor-gamma action in humans. Diabetes. 2001;50:2199–202. doi: 10.2337/diabetes.50.10.2199. [DOI] [PubMed] [Google Scholar]
- 100.Cinti S. Mitchell G. Barbatelli G, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–55. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 101.Morris AP. Voight BF. Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–90. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dimas AS. Lagou V. Barker A, et al. Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes. 2014;63:2158–71. doi: 10.2337/db13-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hattersley A. Bruining J. Shield J. Njolstad P. Donaghue K. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2009;10:33–42. doi: 10.1111/j.1399-5448.2009.00571.x. [DOI] [PubMed] [Google Scholar]
- 104.O’Rahilly SP. Rudenski AS. Burnett MA, et al. Beta-cell dysfunction, rather than insulin insensitivity, is the primary defect in familial type 2 diabetes. Lancet. 1986;328:360–4. doi: 10.1016/s0140-6736(86)90052-8. [DOI] [PubMed] [Google Scholar]
- 105.O’Rahilly S. Turner RC. Matthews DR. Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. N Engl J Med. 1988;318:1225–30. doi: 10.1056/NEJM198805123181902. [DOI] [PubMed] [Google Scholar]
- 106.Allen FM. Studies concerning diabetes. JAMA. 1914;63:939–43. [Google Scholar]
- 107.Henry RR. Wiest-Kent TA. Scheaffer L. Kolterman OG. Olefsky JM. Metabolic consequences of very-low-calorie diet therapy in obese non-insulin-dependent diabetic and nondiabetic subjects. Diabetes. 1986;35:155–64. doi: 10.2337/diab.35.2.155. [DOI] [PubMed] [Google Scholar]
- 108.Lim EL. Hollingsworth KG. Aribisala BS, et al. Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia. 2011;54:2506–14. doi: 10.1007/s00125-011-2204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sumithran P. Prendergast LA. Delbridge E, et al. Long-term persistence of hormonal adaptations to weight loss. N Engl J Med. 2011;365:1597–604. doi: 10.1056/NEJMoa1105816. [DOI] [PubMed] [Google Scholar]
- 110.Yang X. Smith U. Adipose tissue distribution and risk of metabolic disease: does thiazolidinedione-induced adipose tissue redistribution provide a clue to the answer? Diabetologia. 2007;50:1127–39. doi: 10.1007/s00125-007-0640-1. [DOI] [PubMed] [Google Scholar]
- 111.Sniderman AD. Bhopal R. Prabhakaran D. Sarrafzadegan N. Tchernof A. Why might South Asians be so susceptible to central obesity and its atherogenic consequences? The adipose tissue overflow hypothesis. Int J Epidemiol. 2007;36:220–5. doi: 10.1093/ije/dyl245. [DOI] [PubMed] [Google Scholar]
- 112.Garrod AE. The Harveian oration on the debt of science to medicine. Br Med J. 1924;2:747–51. doi: 10.1136/bmj.2.3330.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weatherall DJ. The role of nature and nurture in common diseases: Garrod’s legacy; the Harveian oration delivered before the Fellows of the Royal College of Physicians of London on Thursday, 22nd October 1992. London:: Royal College of Physicians of London; 1992. [Google Scholar]
- 114.Peters K. Exceptional matters. Lancet. 2004;364:2142–51. doi: 10.1016/S0140-6736(04)17556-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ciudin A. Baena-Fustegueras JA. Fort JM, et al. Successful treatment for the Dunnigan-type familial partial lipodystrophy with Roux-en-Y gastric bypass. Clin Endocrinol (Oxf) 2011;75:403–4. doi: 10.1111/j.1365-2265.2011.04057.x. [DOI] [PubMed] [Google Scholar]
- 116.Owen-Smith A. Kipping R. Donovan J, et al. A NICE example? Variation in provision of bariatric surgery in England. BMJ. 2013;346:f2453. doi: 10.1136/bmj.f2453. [DOI] [PubMed] [Google Scholar]
- 117.Gulliford MC. Charlton J. Booth HP, et al. Costs and outcomes of increasing access to bariatric surgery for obesity: cohort study and cost-effectiveness analysis using electronic health records. Health Serv Deliv Res. 2016:4. doi: 10.3310/hsdr04170. [DOI] [PubMed] [Google Scholar]
- 118.National Institute of Health and Care Excellence Obesity: clinical management and assessment. NICE Quality standard 127. London:: NICE; 2016. [Google Scholar]
- 119.Puhl RM. Brownell KD. Psychosocial origins of obesity stigma: toward changing a powerful and pervasive bias. Obes Rev. 2003;4:213–27. doi: 10.1046/j.1467-789x.2003.00122.x. [DOI] [PubMed] [Google Scholar]
- 120.Puhl RM. Heuer CA. Obesity stigma: important considerations for public health. Am J Public Health. 2010;100:1019–28. doi: 10.2105/AJPH.2009.159491. [DOI] [PMC free article] [PubMed] [Google Scholar]