

ABSTRACT
Gout is the most prevalent inflammatory arthritis and affects 2.5% of the general population in the UK. It is also the only arthritis that has the potential to be cured with safe, inexpensive and well tolerated urate-lowering treatments, which reduce serum uric acid by either inhibiting xanthine oxidase – eg allopurinol, febuxostat – or by increasing the renal excretion of uric acid. Of these, xanthine oxidase inhibitors are used first line and are effective in ‘curing’ gout in the vast majority of patients. Gout can be diagnosed on clinical grounds in those with typical podagra. However, in those with involvement of other joints, joint aspiration is recommended to demonstrate monosodium urate crystals and exclude other causes of acute arthritis, such as septic arthritis. However, a clinical diagnosis of gout can be made if joint aspiration is not feasible. This review summarises the current understanding of the pathophysiology, clinical presentation, investigations and treatment of gout.
KEYWORDS: colchicine, gout, hyperuricemia, tophi, urate-lowering treatment
Key points
Gout is associated with metabolic syndrome and patients should be screened for obesity, diabetes mellitus, hypertension, hyperlipidaemia and cardiovascular diseases
A definite diagnosis of gout requires joint aspiration and polarised light microscopy of aspirated fluid
Serum uric acid may be normal during an acute attack of gout
Acute gout can be managed by rest, ice packs and oral NSAIDs, low-dose colchicine or corticosteroids. If feasible, especially in a hospital setting, aspiration and injection of corticosteroid is recommended as the fastest acting option for acute gout
Indications for urate-lowering treatment (ULT) are more than one acute attack of gout in 12 months (in uncomplicated gout), or less frequent attacks in the presence of tophi, renal impairment, uric acid nephrolithiasis or a need to continue on diuretics for heart failure. However, ULT should be discussed and considered with patients even around the time of first diagnosis
ULT dose should be up-titrated gradually aiming for a serum uric acid (SUA) ≤300 μmol/L. Once the SUA is below this level, it should be checked annually
Introduction
Gout is the commonest inflammatory arthritis with a prevalence of 2.5% in the UK.1 Although it has the potential to be cured, its treatment remains suboptimal, with 30–40% of eligible patients receiving urate-lowering treatment (ULT) generally at suboptimal doses and with adherence <40% at 12 months.1 This review summarises the current understanding of aetio-pathogenesis, clinical features and management of gout.
Pathogenesis
Hyperuricaemia and monosodium urate crystal deposition
Gout results from sustained hyperuricaemia (serum uric acid (SUA) ≥360 μmol/L), which leads to intra- and/or peri-articular monosodium urate (MSU) crystal deposition. Uric acid (UA) is the end product of the breakdown of dietary and endogenous purines. SUA ranges 120–180 μmol/L in most animals because they possess uricase, which breaks down UA to soluble allantoin.2 SUA is higher in primates (eg humans, great apes) because of missense mutations in the uricase gene, occurring 10–20 million years ago, and may have resulted in a survival advantage from its anti-hypotensive and anti-oxidant effects.2
Hyperuricaemia arises from over-production (from increased breakdown of exogenous or endogenous purines) or renal under-excretion of UA. Renal under-excretion accounts for 70% of urate load.3 Recently, several transporters have been identified in the proximal renal tubules, collecting ducts and also in the gastrointestinal tract; this has advanced the understanding of the pathogenesis of hyperuricaemia (Fig 1).
Fig 1.

Urate transport in the kidney. A – mechanism of reabsorption of uric acid; B – mechanism of uric acid secretion. Differences in the activity of these urate transporters result in hyperuricaemia.
Hyperuricaemia is central to the development of gout. It reduces urate solubility and promotes MSU crystal nucleation and growth.4 The risk of gout increases exponentially with increasing SUA; nevertheless, only a minority of people with hyperuricaemia develop gout. For example, in the normative aging study, the 1-year incidence of gout in men was <0.1%, 0.4%, 0.8%, 4.3% and 7.0% for SUA <416 μmol/L, 416–475 μmol/L, 476–534 μmol/L, 535–594 μmol/L, ≥595 μmol/L, respectively.5
MSU crystal-induced inflammation
Once MSU crystals are released, they are phagocytosed by resident macrophages and other mononuclear cells and induce inflammation by activating the NALP-3 inflammasome, which results in cleavage of pro-IL-1β and secretion of IL-1β.6,7 NALP-3 inflammasome activation is central to MSU crystal-induced inflammation, as macrophages deficient in the NALP-3 inflammasome do not mount an inflammatory response to MSU crystals.6 Once activated, monocytes and macrophages release chemokines (eg CXCL-1, IL-8, C-GSF) that promote neutrophil chemotaxis, survival and proliferation.6 Other chemokines, such as CCL-2, recruit additional macrophages and monocytes,6 resulting in secretion of TNF-α (promoting cellular activation, endothelial adhesion and phagocytosis) and further IL-1β (promoting endothelial adhesion, endogenous pyrogen) causing a self-propagating intense inflammatory response in which neutrophils release lysozymes on phagocytosing MSU crystals. Components of the innate immune system, eg toll-like receptors and complement pathways, are also activated by MSU crystals.6
Epidemiology
The prevalence of gout has increased dramatically in the UK.1 It is more common in men and increases with age, being uncommon in men younger than 40 years.1 It is also rare in premenopausal women because of the uricosuric effect of oestrogen, progesterone and lower insulin resistance; consequently, the gender difference seen in prevalence reduces after the age of 60 years.1,8
Other risk factors for gout include ethnicity (eg Afro-Caribbean, South Pacific islanders); obesity; excess consumption of beer or spirits, red meat, seafood, fructose-sweetened beverages and fruit juices; medications (particularly diuretics); and hypertension, renal impairment and insulin resistance.8 There is evidence that moderate consumption of purine-rich vegetables, fresh fruits, soya and wine do not increase the risk of gout.8 The increased risk from excessive consumption of beer and spirits relates to their high purine content.8 Other factors such as excessive breakdown of ATP and lactic acidosis after alcohol consumption – which generate a UA load and reduce UA excretion, respectively – also contribute.8 Fructose is the only carbohydrate that increases UA levels by increasing ATP degradation and insulin resistance, which further reduces renal urate excretion.8
Several dietary items reduce the risk of gout. For example, regular daily consumption of skimmed milk or at least alternate day consumption of yogurt, coffee (including decaffeinated coffee ≥4 cups/day) and vitamin C (0.5 gm/day) reduces SUA and risk of gout.8 Cherry consumption reduces the risk of recurrent gout attacks and also lowers SUA modestly.8
Clinical features
Acute gout
Acute gout attacks are characterised by typical features of acute crystal synovitis, such as rapid onset (symptoms peaking within 12–24 hours of onset), excruciating joint pain, exquisite tenderness to touch, erythema and articular/periarticular swelling.9,10 The attack usually resolves in 1–2 weeks. The sites commonly affected include the first metatarsophalangeal joint (most frequently affected), knees, ankles, midfoot and, less commonly, wrists, elbows and bursae (eg the olecranon and pre-patellar bursae). Lower limb and mono-articular involvement are common early in the course of the disease, and oligo- and polyarticular presentations occur infrequently. Acute attacks can be precipitated by infection, injury, dehydration, excess alcohol or purine intake, and initiation of ULT.11 Slowly uptitrated ULT, as recommended by several international guidelines, carries a lower risk of triggering acute attacks of gout than starting at a moderate to high dose of ULT.12–15
Chronic tophaceous gout
Chronic tophaceous gout usually develops after many years of recurrent acute attacks; however, it can occasionally develop more quickly, over a few years, with relatively few attacks. Gouty tophi are nodular masses of MSU crystals and inflammatory tissues, and appear as white to yellow firm subcutaneous deposits with often a heterogenous consistency, generally on the finger tips, feet and in the olecranon and pre-patellar bursae. Tophaceous gout presents with chronic joint pain, stiffness and tenderness, with superimposed episodes of acute gout.
Transplant-associated gout
Immunosuppressed solid organ transplant recipients on low-dose prednisolone and calcineurin inhibitors, such as ciclosporine, can present with rapidly progressive tophaceous gout. Unlike patients with primary gout, where tophi typically take over 10 years to develop, transplant recipients can develop tophi within 3–5 years.15
Comorbidities
Obesity, insulin resistance, hypertension, hyperlipidaemia, renal impairment, ischaemic heart disease and congestive cardiac failure are common in people with gout and their presence should be sought specifically and treated appropriately.15
Investigations
While the definite diagnosis of gout requires joint aspiration and polarised light microscopy of the synovial fluid, a confident clinical diagnosis of gout can be made in those with typical features of crystal-induced inflammation affecting the first metatarsophalangeal joint (‘podagra’) without recourse to joint aspiration.15 Septic arthritis is the main differential diagnosis, but others include acute calcium pyrophosphate crystal arthritis (formerly ‘pseudogout’) and reactive arthritis. Gouty tophi can sometimes be confused with rheumatoid nodules although these are usually homogenous, not white in colour and occur in patients with rheumatoid factor or anti-cyclic citrullinated peptide antibodies.
Peripheral blood
Neutrophilia and raised inflammatory markers are common in acute gout. SUA is a negative acute phase reactant and is often reduced during the acute attack, so should be rechecked 1–2 weeks after resolution. Hyperuricaemia itself should not be used to diagnose gout as hyperuricaemia is common in the general population. SUA is greater than 360 μmol/L in 50.4% of men and 16.3% of women.16 Renal function, liver function, glucose, lipid profile and full blood count should be checked to screen for chronic kidney disease (CKD), liver disease, diabetes, dyslipidaemia and myeloproliferative disorders.
Synovial fluid
Aspiration of the affected joint and prompt examination of the aspirated synovial fluid, which is often turbid with low viscosity, under plain and polarised light microscopy can readily confirm negatively-birefringent needle-shaped MSU crystals (Fig 2). Gram stain and culture of aspirated synovial fluid should also be performed to exclude septic arthritis, which can rarely coexist with gout. Synovial fluid leukocyte counts are not routinely performed but may be >10,000/mm3. MSU crystals can also be demonstrated in synovial fluid aspirated between attacks (the intercritical period).
Fig 2.
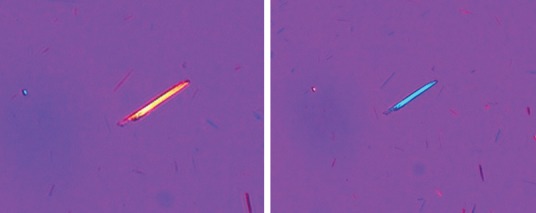
Polarised light microscopy showing needle shaped negatively birefringent (change in colour of the MSU crystal on employing the polariser) monosodium urate crystal. Figure courtesy of the Department of Microbiology, Addenbrookes Hospital, Cambridge, UK.
Urinary uric acid excretion
Urinary uric acid excretion is rarely performed in clinical practice as xanthine oxidase inhibitors (XOIs) are extremely effective in treating gout, and need only be checked in those with premature onset gout, that is disease onset prior to 25 years of age, or if there is a family history of early-onset gout.12–14 If required, a 24-hour urine collection is preferred over a spot uric acid creatinine ratio.
Radiography
Plain radiographic changes of gout (Box 1, Fig 3) take several years to develop and are helpful in making a diagnosis in the later stages of gout. In contrast, ultrasonography is sensitive in detecting MSU crystal deposits. The ultrasonographic findings of gout include double contour sign (MSU crystal deposition on surface of hyaline articular cartilage), intra-articular and intra-bursal tophi, and hyper-echoic aggregates (Fig 4). However, these findings are not 100% specific and may be present in joints with calcium pyrophosphate deposition.17 Dual energy computerised tomography, which can demonstrate MSU crystals, is not widely available and has a limited role in early disease.
Box 1.
Plain radiographic features of gout
| Soft-tissue swelling during acute or tophaceous gout |
| Increased soft tissue density/calcification in tophaceous gout – eccentric |
| Preserved joint space until late disease |
| Cortical erosions – well defined, marginal/intra-articular/away from joint line, with sclerotic margins and overhanging edge |
| Absence of bony proliferation |
Fig 3.

Radiographs showing changes of gout. A – soft-tissue swelling around the index and little finger proximal interphalangeal joints in a patient with tophaceous gout; B – erosion in the first metatarsophalangeal joint with overhanging edges; C – typical punched out erosions in the first metatarsophalangeal joint with sclerosis and preserved bone mineral density (c.f. in rheumatoid arthritis the bones appear osteopaenic).
Fig 4.
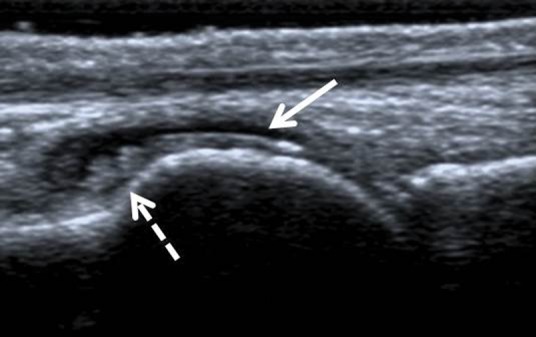
Ultrasound scan of the first metatarsophalangeal joint showing double contour sign (solid white arrow) and tophus (dashed white arrow) in the dorsal synovial recess.
Treatment
Full patient education about gout and its treatment options, and involvement of the patient in management decisions are the cornerstones of successful treatment.
Acute attack
Management includes rest, application of ice packs, and prompt use of non-steroidal anti-inflammatory drugs (NSAIDs), colchicine or corticosteroids.12–15 These are most effective when started early, eg within the first 12–24 hours of onset. Any fast-acting NSAID at full dose (eg naproxen, diclofenac) with a proton pump inhibitor may be prescribed. Although indomethacin is effective, it is best avoided because of frequent toxicity. Colchicine (0.5 mg two to three times per day) is also effective. These may be required for 1–2 weeks, and in order to prevent rebound attacks the treatment may be continued for 1–2 days after the acute attack has subsided.12–14 Diarrhoea is the commonest side effect of colchicine. The dose of colchicine should be reduced in older patients, in those with CKD (estimated glomerular filtration rate (eGFR)<30 mL/min) and avoided in the presence of interacting drugs – such as macrolides and statins. Statins should be temporarily omitted while the patient is taking colchicine because of increased risk of colchicine toxicity.
Monoarticular gout can be very effectively treated with joint aspiration and intra-articular corticosteroid injection, eg methylprednisolone or triamcinolone. Oral prednisolone at a dose of 20–35 mg/day should be used when NSAIDs and colchicine are contraindicated or poorly tolerated, or for oligo/polyarticular gout. Anti-IL-1 agents, such as anakinra, canakinumab and rilonacept, are effective treatments for acute gout but are not approved by the National Institute for Health and Care Excellence (NICE).
Long-term urate-lowering treatment
Long-term ULT should be explained and discussed with all gout patients at diagnosis. Current guidelines recommend ULT if a patient is getting regular acute attacks (>1 attack/year) or has tophi, renal impairment, transplant-associated gout, nephrolithiasis or needs to continue on diuretics for heart failure.12–15
Although most patients with gout exhibit renal under-excretion rather than overproduction of urate, the efficacy and tolerability of XOIs makes them the mainstay of ULT.12–15 British and European guidelines recommend that ULT should be started 1–2 weeks after the acute attack has subsided as reduction in SUA may prolong the current episode or precipitate another attack. However, the 2012 American guidelines support ULT initiation during an acute attack of gout.12 Patients on ULT who develop an acute attack should continue on it.12–15 ULT should be started at a low dose, and the dose gradually increased with monthly SUA measurement, aiming for a SUA <300 μmol/L.15
As patients starting ULT are at risk of recurrent gout attacks, co-prescription of colchicine (0.5 mg once to twice daily) for a period of 6 months should be considered.15 If colchicine is not tolerated, NSAIDs or COX-2 inhibitors, or low dose prednisolone (eg 7.5 mg/day) may be used after discussion of risks and benefits.15
Xanthine oxidase inhibitors
Allopurinol inhibits synthesis of UA by inhibiting xanthine oxidase. It is metabolised to oxypurinol, an active metabolite that is long acting and renally excreted. Allopurinol should be commenced at an initial dose of 100 mg/day (50 mg/day in presence of CKD stage ≥4), and the dose increased gradually (eg monthly) until a target SUA of <300 μmol/L or the maximum licensed dose is reached. The maximum dose of allopurinol should be reduced in the presence of renal impairment and specialist advice sought. The American College of Rheumatology recommends that the dose of allopurinol may be cautiously increased to over 300 mg/day in patients with renal impairment if it is commenced at a low dose (eg 50 mg/day) and the dose escalated gradually, with careful screening for adverse events.12
Allopurinol may cause adverse events, including allopurinol hypersensitivity syndrome (mortality <25%, incidence <1:1000), which presents as an erythematous desquamating rash, fever, hepatitis, eosinophilia and worsening renal function. It is more common in Koreans and Han Chinese than in Caucasians, and in those with renal failure, especially if commenced on a high dose of the drug (eg 300 mg/day). The treatment of allopurinol hypersensitivity syndrome is supportive. Such patients may be commenced on febuxostat, a newer non-purine XOI, after careful discussion and close monitoring as febuxostat can itself associate with drug reaction with eosinophilia and systemic symptoms (DRESS syndrome). However, there are limited published data to conclude that there is no cross-reactivity between allopurinol and febuxostat.18
Febuxostat is started at 80 mg/day in the UK with the dose increased to 120 mg/day, if required. It is not licensed for use in renal impairment (eGFR<30 mL/min) or in those with ischaemic heart disease. One recent study suggests that febuxostat may be safe in CKD stage 4; however, such use remains unlicensed and should only be used after careful discussion with the patient.19 Allopurinol and febuxostat should not be co-prescribed with azathioprine as inhibition of azathioprine metabolism leads to severe blood dyscrasias. Use of febuxostat in people with ischaemic heart disease or congestive cardiac failure is not approved by NICE.
Uricosuric drugs
Uricosuric drugs have limited availability in the UK. Probenecid and sulfinpyrazone have lower potency than benzbromarone, are less effective in renal impairment (eGFR<30 mL/min) and sulfinpyrazone frequently causes gastrointestinal side effects. Benzbromarone is extremely potent and better tolerated, but is not licensed for use in the UK because of reports of fulminant liver failure in Japanese patients.20 Probenecid and benzbromarone are sourced from Europe and should only be used after documenting informed consent. Benzbromarone is effective in patients with eGFR>20 mmol/L.15 Uricosuric drugs can be combined with XOIs if required.
Fenofibrate and losartan have uricosuric effects and may be used as adjuncts to XOIs when otherwise indicated. Of these, fenofibrate 200–300 mg/day (micronized) reduces SUA by approximately 100 μmol/L, while losartan 50 mg/day reduces SUA by 47 μmol/L with no further reduction at higher doses.21,22
Newer anti-gout treatments
Lesinurad, a new uricosuric drug, has been approved in the EU in combination with an XOI and is currently undergoing NICE appraisal. PEGylated uricase is effective in reducing tophus burden in patients with treatment failure to other ULTs; however, it is not NICE approved and is currently unavailable in the UK.
Dietary and lifestyle adjuncts
Dietary modifications to achieve ideal weight should be attempted, as this reduces insulin resistance and SUA levels. However, there are no randomised controlled trials and effects of dietary modifications on SUA are modest. For instance, vitamin C (≥500 mg/day) reduces SUA by c40 μmol/L with significant heterogeneity of effects.23
Managing comorbidities
Losartan and amlodipine should be used preferentially to treat hypertension and diuretics and beta-blockers should be avoided as they cause hyperuricaemia and associate with gout.24 Hyperlipidaemia should be treated with atorvastatin or fenofibrate.24 The cardiovascular benefit from low-dose aspirin outweighs the insignificant risk of hyperuricaemia from reduced renal urate excretion, and it should be continued if indicated.15 Similarly, diuretics prescribed for heart failure should be continued but bendroflumethiazide prescribed for hypertension should be changed to an alternative antihypertensive.
Conflict of interest
AA has received research grants from Astra Zeneca and Oxford Immunotech. MD has received research grants from Astra Zeneca and Oxford Immunotech, and honoraria for attending ad hoc advisory boards for AstraZeneca, Nordic Biosciences and Roche. ER has no conflicts of interest to declare.
References
- 1.Kuo CF. Grainge MJ. Mallen C. Zhang W. Doherty M. Rising burden of gout in the UK but continuing suboptimal management: a nationwide population study. Ann Rheum Dis. 2015;74:661–7. doi: 10.1136/annrheumdis-2013-204463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pillinger MH. Rosenthal P. Abeles AM. Hyperuricemia and gout: new insights into pathogenesis and treatment. Bull NYU Hosp Jt Dis. 2007;65:215–21. [PubMed] [Google Scholar]
- 3.Choi HK. Mount DB. Reginato AM. Pathogenesis of gout. Ann Intern Med. 2005;143:499–516. doi: 10.7326/0003-4819-143-7-200510040-00009. [DOI] [PubMed] [Google Scholar]
- 4.Chhana A. Lee G. Dalbeth N. Factors influencing the crystallization of monosodium urate: a systematic literature review. BMC Musculoskelet Disord. 2015;16:296. doi: 10.1186/s12891-015-0762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campion EW. Glynn RJ. DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med. 1987;82:421–6. doi: 10.1016/0002-9343(87)90441-4. [DOI] [PubMed] [Google Scholar]
- 6.Busso N. So A. Gout. Mechanisms of inflammation in gout. Arthritis Res Ther. 2010;12:206. doi: 10.1186/ar2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinon F. Petrilli V. Mayor A. Tardivel A. Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 8.MacFarlane LA. Kim SC. Gout: a review of nonmodifiable and modifiable risk factors. Rheum Dis Clin North Am. 2014;40:581–604. doi: 10.1016/j.rdc.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor WJ. Fransen J. Jansen TL, et al. Study for Updated Gout Classification Criteria (SUGAR): identification of features to classify gout. Arthritis Care Res. 2016;68:1894–8. doi: 10.1002/acr.22585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaffo AL. Schumacher HR. Saag KG, et al. Developing a provisional definition of flare in patients with established gout. Arthritis Rheum. 2012;64:1508–17. doi: 10.1002/art.33483. [DOI] [PubMed] [Google Scholar]
- 11.Choi HK. A prescription for lifestyle change in patients with hyperuricemia and gout. Curr Opin Rheumatol. 2010;22:165–72. doi: 10.1097/BOR.0b013e328335ef38. [DOI] [PubMed] [Google Scholar]
- 12.Khanna D. FitzGerald JD. Khanna PP, et al. American College of Rheumatology guidelines for management of gout part i: systematic non-pharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012;64:1431–46. doi: 10.1002/acr.21772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang W. Doherty M. Bardin T, et al. EULAR evidence based recommendations for gout. Part II: management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT) Ann Rheum Dis. 2006;65:1312–24. doi: 10.1136/ard.2006.055269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang W. Doherty M. Pascual E, et al. EULAR evidence based recommendations for gout. Part I: diagnosis. Report of a task force of the Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT) Ann Rheum Dis. 2006;65:1301–11. doi: 10.1136/ard.2006.055251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jordan KM. Cameron JS. Snaith M, et al. British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology. 2007;46:1372–4. doi: 10.1093/rheumatology/kem056a. [DOI] [PubMed] [Google Scholar]
- 16.Zhu Y. Pandya BJ. Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011;63:3136–41. doi: 10.1002/art.30520. [DOI] [PubMed] [Google Scholar]
- 17.Loffler C. Sattler H. Peters L, et al. Distinguishing gouty arthritis from calcium pyrophosphate disease and other arthritides. J Rheumatol. 2015;42:513–20. doi: 10.3899/jrheum.140634. [DOI] [PubMed] [Google Scholar]
- 18.Bardin T. Chalès G. Pascart T, et al. Risk of cutaneous adverse events with febuxostat treatment in patients with skin reaction to allopurinol. A retrospective, hospital-based study of 101 patients with consecutive allopurinol and febuxostat treatment. Joint Bone Spine. 2016;83:314–7. doi: 10.1016/j.jbspin.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 19.Shibagaki Y. Ohno I. Hosoya T. Kimura K. Safety, efficacy and renal effect of febuxostat in patients with moderate-to-severe kidney dysfunction. Hypertens Res. 2014;37:919–25. doi: 10.1038/hr.2014.107. [DOI] [PubMed] [Google Scholar]
- 20.Kydd AS. Seth R. Buchbinder R. Edwards CJ. Bombardier C. Uricosuric medications for chronic gout. Cochrane Database Syst Rev. 2014 doi: 10.1002/14651858.CD010457.pub2. (11):CD010457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Derosa G. Maffioli P. Sahebkar A. Plasma uric acid concentrations are reduced by fenofibrate: a systematic review and meta-analysis of randomized placebo controlled trials. Pharmacol Res. 2015;102:63–70. doi: 10.1016/j.phrs.2015.09.012. [DOI] [PubMed] [Google Scholar]
- 22.Würzner G. Gerster JC. Chiolero A, et al. Comparative effects of losartan and irbesartan on serum uric acid in hypertensive patients with hyperuricaemia and gout. J Hypertens. 2001;19:1855–60. doi: 10.1097/00004872-200110000-00021. [DOI] [PubMed] [Google Scholar]
- 23.Juraschek SP. 3rd Miller ER. Gelber AC. Effect of oral vitamin C supplementation on serum uric acid: a meta-analysis of randomized controlled trials. Arthritis Care Res. 2011;63:1295–306. doi: 10.1002/acr.20519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi HK. Soriano LC. Zhang Y. Rodríguez LAG. Antihypertensive drugs and risk of incident gout among patients with hypertension: population based case-control study. BMJ. 2012;344:d8190. doi: 10.1136/bmj.d8190. [DOI] [PMC free article] [PubMed] [Google Scholar]