

ABSTRACT
The idiopathic inflammatory myopathies are a group of conditions characterised by inflammation of muscles (myositis) and other body systems. The diagnosis can be challenging because of the many potential clinical features and extra-muscular manifestations, which may be seemingly unrelated. An accurate diagnosis requires up-to-date understanding of the clinical manifestations, different clinical subtypes and appropriate interpretation of investigations, including newly described serological subtypes.
This review will detail the approach to the diagnosis of an idiopathic inflammatory myopathy, based on up-to-date knowledge. The recently updated classification criteria and treatment options will also be described.
KEYWORDS: Dermatomyositis, idiopathic inflammatory myopathies, inclusion body myositis, myositis, polymyositis
Introduction
The idiopathic inflammatory myopathies (IIMs) are a group of autoimmune conditions characterised by inflammation of muscle (myositis) and other organ systems, resulting in widespread organ dysfunction, increased morbidity and early mortality. Knowledge of the clinical manifestations of the IIMs and our ability to treat them have dramatically progressed since publication of the Bohan and Peter criteria in 1975.1,2 This review aims to outline the clinical features of the IIMs, current classification criteria, the association of autoantibodies to clinical features and current recommended treatment.
Epidemiology
The IIMs are relatively rare conditions with an overall reported incidence of 11/million person-years (10 for men and 13 for women) and a prevalence of 14/100,000.3
The peak age range of IIM onset is 45–60 years for adults and 5–15 years for children. Apart from inclusion body myositis (IBM), all IIM subtypes occur more commonly in women (ratio 2:1); IBM occurs twice as commonly in men as women.4
Subtypes
Traditionally, the IIMs have been divided into subtypes according to their clinical features. Recent advances have identified that more clinically distinct subtypes exist than were originally recognised.
Dermatomyositis and polymyositis
Dermatomyositis (DM) and polymyositis (PM) are both characterised by myositis (muscle inflammation) and systemic involvement.5,6 Myositis usually involves the proximal musculature of the limbs leading to weakness and fatigue, especially on tasks such as standing from a seated position or reaching up for highly placed objects.
The systemic manifestations (ie involvement of organs other than skeletal muscle) are important aspects of each disease. Possible systemic features include dysphagia,7 dysphonia, interstitial lung disease (ILD),8 subcutaneous calcification,9 dilated cardiomyopathy, arrhythmias, atrioventricular defects and constitutional features, such as fatigue, fever and weight loss. DM is particularly associated with an increased risk of cancer development (cancer-associated myositis).
Myositis can occur without any other systemic manifestations. This so-called ‘lone PM’ is a rare occurrence and can pose a diagnostic challenge in differentiating this from other causes of muscle weakness.
DM can be distinguished from PM by its typical cutaneous features, which include Gottron’s papules (Fig 1), Gottron’s sign, heliotrope rash (Fig 2), V-sign rash, mechanic’s hands (Fig 3), shawl sign rash and erythroderma.10,11 Gottron’s papules are red, scaly papules that occur over the dorsal aspect of the metacarpophalangeal, proximal and distal interphalangeal joints, whereas Gottron’s sign is the same red, scaly papular rash occurring elsewhere on the body. These cutaneous lesions typically worsen with sun exposure.
Fig 1.
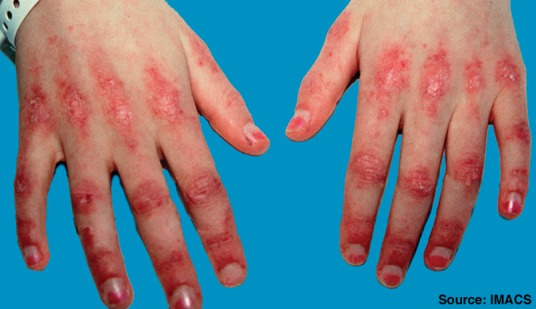
Gottron’s papules over metacarpophalangeal and interphalangeal joints in a patient with dermatomyositis. Reproduced with permission from Dugan et al.11
Fig 2.
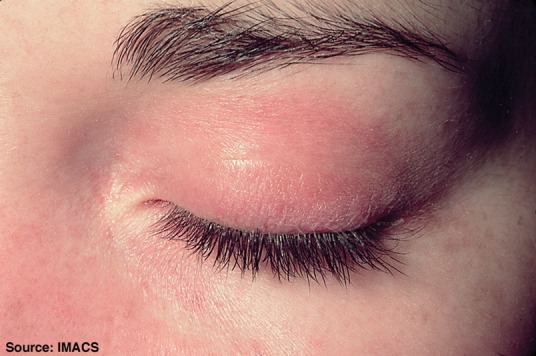
Heliotrope rash over the left eyelid with associated periorbital oedema. Reproduced with permission from Dugan et al.11
Fig 3.
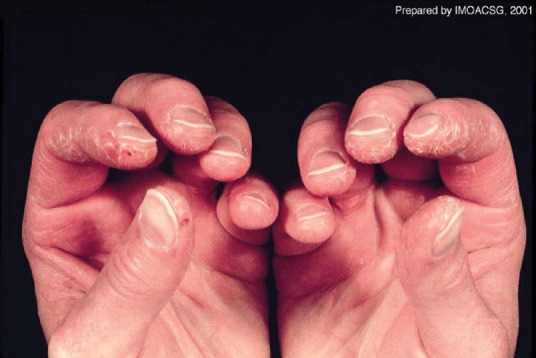
Mechanic’s hands: scaling, cracking and fissuring with bleeding along the lateral and palmar aspects of the fingers. Reproduced with permission from Dugan et al.11
Juvenile DM
Juvenile DM has similar clinical features to adult DM and is characterised by myositis, cutaneous features and systemic involvement in patients under the age of 18 years.12 Features particular to juvenile DM include cutaneous ulcerations, calcinosis cutis and vasculopathy.
Amyopathic DM
DM can exist without myositis, this uncommon subtype is termed clinically amyopathic DM.13 Apart from muscle involvement, the clinical features of clinically amyopathic DM are the same as typical DM in terms of risk of ILD, cutaneous manifestations and malignancy. Hypomyopathic disease may be seen in DM patients with magnetic resonance or muscle biopsy evidence of myositis without weakness. Confusingly, patients may also present with histological features of DM without an overt rash (dermatomyositis sine dermatitis).
Antisynthetase syndrome
The antisynthetase syndrome is a particularly severe IIM subtype associated with myositis, ILD and inflammatory symmetrical polyarthritis of the small joints of the hands and feet.14 Fever, Raynaud’s phenomenon and mechanic’s hands are also characteristic of the antisynthetase syndrome. Antisynthetase syndrome is defined by the presence of an antisynthetase autoantibody and ≥1 of the following:
myositis
ILD
Raynaud’s phenomenon
Mechanic’s hands
arthritis
fever.
Immune-mediated necrotising myopathy
Immune-mediated necrotising myopathy (IMNM), a rare but severe IIM subtype, is characterised by muscle necrosis and regeneration resulting in proximal muscle weakness.15 In contrast to the previously discussed IIMs, inflammatory infiltrates on muscle biopsy are not a predominant feature. IMNM can be idiopathic, may follow viral infections or be associated with statin use, malignancy or connective tissue diseases (CTDs), particularly scleroderma. The statin-associated form of IMNM is associated with autoantibodies directed against HMGCR (3-hydroxy-3-methyl-glutaryl-coenzyme A reductase) and characteristically does not improve despite withdrawal of the statin. IMNM is also associated with presence of the anti-signal recognition particle autoantibody. In the cancer-associated cases, treatment of malignancy (if possible) can lead to resolution of the myopathy.
Inclusion body myositis
IBM is a condition characterised by both inflammatory and degenerative changes within the muscle.16 IBM occurs in patients aged over 50 years. The muscle weakness pattern is notable for both proximal and distal involvement with asymmetry being a common feature. The pattern of muscle weakness particular to IBM is early involvement of the finger flexors, ankle dorsiflexors and knee extensors. Head drop and camptocormia may manifest due to axial muscle involvement. Dysphagia occurs in over half of all patients with IBM.17 The condition follows a slowly progressive course and is unresponsive to immunotherapy. High degrees of disability are seen, especially due to falls and weakness of grip.
Association with cancer
The association between the IIMs and increased risk of cancer has been well reported.18–23 The risk of cancer in the IIMs is between 2 and 7 times higher than the general population. The risk is highest in DM (2–7 times higher) compared to PM (2 times higher). The risk of malignancy is highest in the year prior to and the year after IIM onset. DM confers a particularly higher risk of lung, ovarian and breast cancer, whereas PM is more associated with non-Hodgkin lymphomas, lung and bladder cancer.
Current cancer screening recommendations for a patient newly diagnosed with PM or DM comprise comprehensive history and examination, including rectal, breast, pelvic and testicular examination. Prostate-specific antigen measurement should be performed in men. The increased risk of ovarian cancer makes ovarian imaging via ultrasound prudent. The presence of breast cancer should be investigated via mammogram. The clinical utility of further imaging via computerised tomography scan of the thorax, abdomen and pelvis is not yet clarified; however, this would be recommended to identify solid tumours that may otherwise remain undetected. As the risk of cancer remains high up to 3 years after IIM onset, it may be appropriate to repeat screening investigations; however, studies have not yet revealed the utility of this.
Typical presentation of IIMs
Diagnosis of an IIM can be straightforward if a patient presents with typical muscle weakness, clear multisystem involvement and a consistent serological profile. However, many patients in clinical practice present with non-specific clinical features, such as fever, fatigue, myalgia and arthralgia.
Acute or subacute symmetrical muscle weakness of the proximal arm and leg muscles is typical of the IIMs. Muscle weakness may cause a patient to complain about difficulty in combing their hair or progressive difficulty in rising from a chair. Weakness of other muscle groups may cause dysphonia or dysphagia.
As previously described, a diagnosis of DM can be suspected in the presence of muscle weakness and typical DM cutaneous features.
A patient may first present with an extra-muscular manifestation of an IIM, such as ILD or Raynaud’s phenomenon, therefore the IIMs should be included as a secondary cause when investigating for an underlying process.
Approach to diagnosis
A careful history should be taken to identify important information, such as family history of muscle disorders, current or past use of myopathic drugs (Table 1), excess alcohol intake and presence of dysphagia or dysphonia.
Table 1.
Causes of proximal muscle weakness other than idiopathic inflammatory myopathies
| Inherited myopathies | Muscular dystrophies: Becker/Duchenne, facioscapulohumeral, limb girdle |
| Congenital myopathies: nemaline, centronuclear, central core | |
| Myotonic disease: dystrophica myotonica (1 & 2), myotonia congenita | |
| Neurologic | Neuropathic conditions: spinal muscular atrophies, amyotrophic lateral sclerosis, Guillain-Barré syndrome, chronic inflammatory demyelinating polyneuropathy |
| Neuromuscular junction disorders: Eaton-Lambert syndrome, myasthenia gravis | |
| Metabolic | Glycogen storage diseases: acid maltase deficiency (Pompe), McArdle disease |
| Hypokalaemia | |
| Vitamin D deficiency | |
| Mitochondrial cytopathies | |
| Nutritional: vitamin D deficiency, malabsorption | |
| Other: uraemia, hepatic failure, alcoholism, acute intermittent porphyria, diabetic plexopathy | |
| Endocrine myopathies | Hyper/hypothyroidism, acromegaly, Cushing’s syndrome, Addison’s disease, hyper/hypocalcaemia |
| Drug-induced myopathies | Lipid lowering agents eg statins, and clofibrate; D-penicillamine, chloroquine, amiodarone, vincristine, zidovudine |
| Infections | Acute viral: influenza, hepatitis B, echovirus, rickettsia, coxsackie virus, rubella, vaccine-associated |
| Bacterial pyomyositis: staphylococcus, streptococcus, Clostridium perfringens, leprosy | |
| Parasites: toxoplasma, trichinella, schistosoma, cysticercus | |
| Myositis/CTD overlap | Myositis – overlapping with systemic sclerosis, mixed CTD, Sjögren’s syndrome, systemic lupus erythematosus and rheumatoid arthritis |
| Miscellaneous | Periodic paralyses, carcinomatous neuromyopathy, acute rhabdomyolysis, myositis ossificans, microembolisation by atheroma or carcinoma, sarcoidosis with myopathy |
CTD = connective tissue disease
Adapted from Oddis CV. Rheum Dis Clin North Am 2002;28:979–1001.
A comprehensive examination should be carried out to identify the multitude of signs that can manifest. In particular, careful examination for DM-associated cutaneous changes and manual muscle testing should be carried out. The strength of the following muscle groups as a minimum should be quantified: deltoid, biceps, wrist extensors, quadriceps, ankle dorsiflexors, neck flexors, gluteus medius and gluteus maximus.
Recommended baseline investigations include concentration of muscle enzymes, such as creatinine kinase (CK), alanine transaminase, aspartate aminotransferase, aldolase and lactate dehydrogenase. The concentration of CK has diagnostic and monitoring utility. Although a raised CK is sensitive for a diagnosis of an IIM, there are many other causes of a raised CK (Table 2). Patients with a myositis-spectrum disorder but no muscle disease may present with a normal CK. Less frequently, and sometimes in association with cancer-associated myositis, IIM patients with associated myopathic involvement may also not mount a raised CK response.
Table 2.
Causes of elevated creatinine kinase other than active idiopathic inflammatory myopathy
Muscle trauma
|
Diseases affecting muscle
|
Drug/toxin-induced myopathy
|
Drug-induced myositis
|
Endocrine and metabolic abnormalities
|
Elevation of CK–BB
|
Elevation without disease
|
CK = creatinine kinase; CNS = central nervous system; EMG = electromyography; GI = gastrointestinal
Adapted from Targoff IN. Rheum Dis Clin North Am 2002;28:859–90.
Pathological examination of a muscle biopsy will allow confirmation of muscle inflammation, identify IIM subtype and distinguish non-IIM forms of myopathy.
Testing for myositis specific autoantibodies (MSAs) and myositis-associated autoantibodies (MAAs) can further identify clinical subtype, inform the requirement for further investigations and predict treatment response.
Magnetic resonance imaging of muscle is being increasingly performed. Imaging protocols that include the fat-suppressed short tau inversion recovery sequence are particularly helpful for identifying muscle oedema, which indicates active myositis although is not specific. T1-weighted images may demonstrate fatty replacement of muscle and scar tissue, which indicates irreversible muscle damage.
In myopathies, electromyography testing typically shows low voltage polyphasic potentials. Fibrillation is only present in the acute phase of myositis. Unfortunately, there are no myositis-specific electromyography changes and interpretation can be variable due to operator dependency.
As ILD is a feature of the IIMs, particularly the antisynthetase syndrome, pulmonary function testing and high resolution computerised tomography scanning of the lungs should be carried out. Cardiac failure due to cardiac involvement should be investigated via echocardiogram and electrocardiogram.
Differential diagnosis
Due to the variety of clinical features, age ranges of presentation and systemic features, a differential diagnosis should be considered before a diagnosis of IIM is arrived at, especially in patients without diagnostic serology. Indeed, it is not uncommon for an alternative diagnosis to become apparent months or years after a diagnosis of IIM has been made. The differential diagnoses that should be considered are listed in Table 1.
Autoantibody profiles
Although identifying each patient’s IIM as one of the previously discussed clinical subtypes can inform prognosis, response to treatment and risk of cancer, the marked heterogeneity within each subtype limits this approach. The recent identification of a large number of certain MSAs and MAAs and the observation that clinically distinct IIM syndromes are associated with each allows for much greater investigation and treatment stratification. MSAs are highly specific and therefore their presence strongly indicates an underlying IIM. MSAs usually exist exclusively; however, a small minority of patients may be positive for more than one. Table 3 displays each MSA, the corresponding prevalence and associated clinical features.24
Table 3.
Myositis specific autoantibodies, prevalence and associated clinical features
| Antibody | Prevalence | Clinical features | |
|---|---|---|---|
| Antisynthetase antibodies (Jo-1, Zo, OJ, PL12, KS, PL7, EJ, Ha) | Jo-1 9–24% in adult DM/PM | Myositis, ILD, non-erosive arthritis, mechanic’s hands, Raynaud’s, fever | |
| 75% of antisynthetase antibodies are Jo-1 | |||
| Non-Jo-1 <5% in adult DM/PM | |||
| 2–4% in JDM | |||
| MDA5 | 10–48% in Asian adults with DM | CADM | |
| 0–13% in Caucasian adults with DM | Rapidly progressive ILD, alopecia, skin ulceration, palmar papules, panniculitis | ||
| 7–38% in JDM | |||
| SAE1 | Exclusive to DM | CADM, dysphagia preceding muscle weakness | |
| Mi2 | 9–24% adult IIM | Marked cutaneous DM, generally favourable response to treatment | |
| 4–10% JDM | |||
| NXP2 | 1–17% adult PM/DM | Calcinosis in JDM | |
| 23–25% JDM | |||
| TIF1 | 13–31% adult PM/DM | Strong association with cancer – up to 69% | |
| 22–29% JDM | |||
| SRP | 5% Caucasian adults PM/DM | Severe necrotising myopathy, dysphagia | |
| 8–13% Asian/African adult PM/DM | Lower risk of ILD | ||
| <2% JDM | |||
| HMGCR | 6% adult PM/DM | Necrotising myopathy | |
| <1% JDM | Statin use | ||
| cN1A | 33–34% IBM | IBM – moderate specificity | |
| 36% Sjogren’s syndrome | |||
| 20% SLE | |||
| 0–5% adult DM/PM |
CADM = clinically amyopathic dermatomyositis; DM = dermatomyositis; HMGCR = 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase; IBM = inclusion body myositis; IIM = idiopathic inflammatory myopathies; ILD = interstitial lung disease; JDM = juvenile dermatomyositis; MDA5 = melanoma differentiation-associated protein 5; PM = polymyositis; SAE1 = SUMO-activating enzyme subunit 1; SLE = systemic lupus erythematosus; SRP = signal recognition particle; TIF1 = transcriptional intermediary factor 1
Adapted from Betteridge and McHugh.24
Myositis-associated autoantibodies
An IIM can exist alongside CTDs and these are referred to as ‘overlap’ cases. Particular MAAs occur in overlap cases and have diagnostic utility. Although they occur more frequently than the MSAs, they are also found in other CTDs, such as systemic lupus erythematosus and scleroderma, thus reducing their diagnostic specificity.
The anti-PM/Scl autoantibody occurs most commonly in patients who have PM with overlapping sclerodermatous features. It is mutually exclusive to other MSAs and MAAs and is associated with similar clinical features to the antisynthetase syndrome – increased risk of ILD, mechanic’s hands and Raynaud’s phenomenon.25
Coexisting IIM and mixed CTD is associated with positivity for anti-U1-snRNP autoantibodies, which confers good response to steroid treatment and lower prevalence of myositis.26
Patients positive for the anti-Ku autoantibody are more likely to suffer Raynaud’s phenomenon, ILD, arthralgia and myositis. Anti-Ku autoantibody positive patients require higher doses of corticosteroids to induce myositis remission and the ILD has been shown to be particularly steroid resistant.27
The anti-SSA and SSB autoantibodies are also associated with IIM/CTD overlap cases. A 14–25% frequency of anti-SSA autoantibodies has been reported in adult PM/DM overlap cases, compared with 9–19% in IIM-non-overlap cases. Anti-SSA autoantibodies may coexist with anti-Jo1 positivity, and are also observed in IBM. The anti-SSB autoantibodies exist in 4–12% of overlap patients and only 2–7% of IIM-non-overlap cases. 28
Classification criteria
In 1975, Bohan and Peter published their classification criteria for DM and PM (Table 4).1,2 These criteria encompassed clinical features, muscle biopsy evidence of myositis, elevation of muscle-associated enzymes, electromyographic features and the characteristic skin rashes of DM. Importantly, these criteria were devised before IBM and IMNM were recognised as distinct clinical entities. Separate criteria for these conditions and antisynthetase syndrome are now available.29
Table 4.
Bohan and Peter criteria
| 1 | Symmetrical weakness of limb-girdle muscles and anterior neck flexors | ||
| 2 | Muscle biopsy evidence typical of myositis | ||
| 3 | Elevation of serum skeletal muscle enzymes, particularly CK | ||
| 4 | Typical EMG features of myositis | ||
| 5 | Typical DM rash, including heliotrope and Gottron’s papules | ||
| For the diagnosis of PM | For the diagnosis of DM | ||
| Definite | All of items 1–4 | Definite | Item 5 plus 3 of items 1–4 |
| Probable | 3 of items 1–4 | Probable | Item 5 plus 2 of items 1–4 |
| Possible | 2 of items 1–4 | Possible | Item 5 plus 1 of items 1–4 |
Exclusion criteria: congenital muscular dystrophies, central or peripheral neurological disease, infectious myositis, metabolic/endocrine myopathies and myasthenia gravis
CK = creatinine kinase; DM = dermatomyositis; EMG = electromyogram; PM = polymyositis
Depending on the number of criteria fulfilled, a label of possible, probable or definite DM/PM could be assigned. In an effort to include advances in IIM presentation and identification of MSAs, the International Myositis Assessment and Clinical Studies (IMACS) Group are developing the International Myositis Classification Criteria Project (IMCCP) classification criteria.29
The IMCCP classification criteria will take the form of an individual’s probability of having an IIM. Classification as an IIM using the IMCCP criteria requires a probability higher than 50% and the cut off for inclusion into a clinical trial is a probability higher than 90%.
Treatment
Pharmacological management of the IIMs involves induction of remission followed by maintenance of remission.30 The current accepted treatment regimen is based on observational studies as no randomised controlled trials have been carried out, partly because of the rarity of the IIMs and their clinical heterogeneity. Aside from pharmacological intervention, non-fatiguing resistance exercises and general rehabilitation measures are important treatments in all IIMs, especially IBM.
Induction of remission
Glucocorticoids still form the cornerstone of IIM remission induction. For individuals with moderate disease severity, oral prednisolone at a dose of 0.5–1 mg/kg/day (usually 40–60 mg, maximum 100 mg per day) is recommended. For those with more severe disease, intravenous methylprednisolone (250–500 mg in 2–3 doses on alternate days) can be used initially and then converted to oral prednisolone once disease severity has reduced. The initial dose should be continued until the CK level is lower than twice the upper limit of normal. Following adequate reduction of disease activity and CK level, the daily prednisolone dose can be weaned alongside early introduction of a second line agent.
For those patients in whom the CK does not improve after 4–5 weeks, methylprednisolone can be added, if not already used, or administration of intravenous cyclophosphamide, intravenous immunoglobulin or intravenous rituximab should be considered under specialist advice.
Dermatological manifestations can improve with sunlight avoidance, topical steroids or calcineurin inhibitors (tacrolimus, ciclosporin).
Maintenance of remission
As with other rheumatological diseases, disease modifying anti-rheumatic drugs can be used as steroid-sparing agents to maintain IIM remission and allow the dose reduction or withdrawal of glucocorticoid treatment.
Methotrexate, azathioprine and mycophenolate mofetil are effective maintenance treatments for most patients; however, some will remain resistant and require escalation of treatment. In this situation, combination treatment with ciclosporin and methotrexate or methotrexate/azathioprine with mycophenolate mofetil can be effective. Treatment with tacrolimus, cyclosphosphamide, rituximab, tocilizumab or intravenous immunoglobulin can also be considered under specialist care for more resistant cases. Cyclophosphamide or tacrolimus should be considered where ILD is a predominant feature.
Conclusions
In conclusion, the IIMs are rare autoimmune CTDs characterised by distinct clinical subtypes and varied multisystem involvement. Identification of MSA/MAAs can inform diagnosis and risk of secondary organ involvement and cancer development. The anticipated IMCCP classification criteria improve on the Bohan and Peter criteria. Treatment constitutes exclusion of other causes of muscle weakness and raised CK, then use of immunosuppression with induction of remission followed by maintenance.
Author contributions
AO prepared the manuscript and collated information. JL provided information on neurological aspects of the manuscript. HC provided senior leadership, review and guidance.
Conflicts of interest
The authors have no conflicts of interests to declare.
References
- 1.Bohan A. Peter JB. Polymyositis and dermatomyositis (first of two parts) N Engl J Med. 1975;292:344–7. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 2.Bohan A. Peter JB. Polymyositis and dermatomyositis (second of two parts) N Engl J Med. 1975;292:403–7. doi: 10.1056/NEJM197502202920807. [DOI] [PubMed] [Google Scholar]
- 3.Svensson J. Arkema EV. Lundberg IE. Holmqvist M. Incidence and prevalence of idiopathic inflammatory myopathies in Sweden: a nationwide population-based study. Rheumatology. 2017;56:802–10. doi: 10.1093/rheumatology/kew503. [DOI] [PubMed] [Google Scholar]
- 4.Molberg Ø. Dobloug C. Epidemiology of sporadic inclusion body myositis. Curr Opin Rheumatol. 2016;28:657–60. doi: 10.1097/BOR.0000000000000327. [DOI] [PubMed] [Google Scholar]
- 5.Callen JP. Dermatomyositis. Lancet. 2000;355:53–7. doi: 10.1016/S0140-6736(99)05157-0. [DOI] [PubMed] [Google Scholar]
- 6.Milisenda JC. Selva-O’Callaghan A. Grau JM. The diagnosis and classification of polymyositis. J Autoimmun. 2014;48–49:118–21. doi: 10.1016/j.jaut.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 7.Cunningham JD. Lowry LD. Head and neck manifestations of dermatomyositis-polymyositis. Otolaryngol Head Neck Surg. 1985;93:673–7. [PubMed] [Google Scholar]
- 8.Marie I. Hachulla E. Chérin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Care Res. 2002;47:614–22. doi: 10.1002/art.10794. [DOI] [PubMed] [Google Scholar]
- 9.Valenzuela A. Chung L. Casciola-Rosen L. Fiorentino D. Identification of clinical features and autoantibodies associated with calcinosis in dermatomyositis. JAMA Dermatol. 2014;150:724–9. doi: 10.1001/jamadermatol.2013.10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaba LC. Fiorentino DF. Skin disease in dermatomyositis. Curr Opin Rheumatol. 2012;24:597–601. doi: 10.1097/BOR.0b013e3283585748. [DOI] [PubMed] [Google Scholar]
- 11.Dugan EM. Huber AM. Miller FW. Rider LG. International Myositis Assessment and Clinical Studies Group Photoessay of the cutaneous manifestations of the idiopathic inflammatory myopathies. Dermatol Online J. 2009;15:1. [PubMed] [Google Scholar]
- 12.Rider LG. Katz JD. Jones OY. Developments in the classification and treatment of the juvenile idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 2013;39:877–904. doi: 10.1016/j.rdc.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Udkoff J. Cohen PR. Amyopathic dermatomyositis: a concise review of clinical manifestations and associated malignancies. Am J Clin Dermatol. 2016;17:509–18. doi: 10.1007/s40257-016-0199-z. [DOI] [PubMed] [Google Scholar]
- 14.Mahler M. Miller FW. Fritzler MJ. Idiopathic inflammatory myopathies and the anti-synthetase syndrome: a comprehensive review. Autoimmun Rev. 2014;13:367–71. doi: 10.1016/j.autrev.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kassardjian CD. Lennon VA. Alfugham NB. Mahler M. Milone M. Clinical features and treatment outcomes of necrotizing autoimmune myopathy. JAMA Neurol. 2015;72:996–1003. doi: 10.1001/jamaneurol.2015.1207. [DOI] [PubMed] [Google Scholar]
- 16.Greenberg SA. Pathogenesis and therapy of inclusion body myositis. Curr Opin Neurol. 2012;25:630–9. doi: 10.1097/WCO.0b013e328357f211. [DOI] [PubMed] [Google Scholar]
- 17.Badrising UA. Maat-Schieman MLC. Houwelingen JC, et al. Inclusion body myositis. J Neurol. 2005;252:1448–54. doi: 10.1007/s00415-005-0884-y. [DOI] [PubMed] [Google Scholar]
- 18.Qiang JK. Kim WB. Baibergenova A. Alhusayen R. Risk of malignancy in dermatomyositis and polymyositis. J Cutan Med Surg. 2017;21:131–6. doi: 10.1177/1203475416665601. [DOI] [PubMed] [Google Scholar]
- 19.Dobloug GC. Garen T. Brunborg C. Gran JT. Molberg Ø. Survival and cancer risk in an unselected and complete Norwegian idiopathic inflammatory myopathy cohort. Semin Arthritis Rheum. 2015;45:301–8. doi: 10.1016/j.semarthrit.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Olazagasti JM. Baez PJ. Wetter DA. Ernste FC. Cancer risk in dermatomyositis: a meta-analysis of cohort studies. Am J Clin Dermatol. 2015;16:89–98. doi: 10.1007/s40257-015-0120-1. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y-J. Wu C-Y. Huang Y-L, et al. Cancer risks of dermatomyositis and polymyositis: a nationwide cohort study in Taiwan. Arthritis Res Ther. 2010;12:R70. doi: 10.1186/ar2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stockton D. Doherty VR. Brewster DH. Risk of cancer in patients with dermatomyositis or polymyositis, and follow-up implications: a Scottish population-based cohort study. Br J Cancer. 2001;85:41–5. doi: 10.1054/bjoc.2001.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chow WH. Gridley G. Mellemkjaer L, et al. Cancer risk following polymyositis and dermatomyositis: a nationwide cohort study in Denmark. Cancer Causes Control. 1995;6:9–13. doi: 10.1007/BF00051675. [DOI] [PubMed] [Google Scholar]
- 24.Betteridge Z. McHugh N. Myositis-specific autoantibodies: an important tool to support diagnosis of myositis. J Intern Med. 2016;280:8–23. doi: 10.1111/joim.12451. [DOI] [PubMed] [Google Scholar]
- 25.Mahler M. Raijmakers R. Novel aspects of autoantibodies to the PM/Scl complex: clinical, genetic and diagnostic insights. Autoimmun Rev. 2007;6:432–7. doi: 10.1016/j.autrev.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 26.Coppo P. Clauvel JP. Bengoufa D, et al. Inflammatory myositis associated with anti-U1-small nuclear ribonucleoprotein antibodies: a subset of myositis associated with a favourable outcome. Rheumatology. 2002;41:1040–6. doi: 10.1093/rheumatology/41.9.1040. [DOI] [PubMed] [Google Scholar]
- 27.Rigolet A. Musset L. Dubourg O, et al. Inflammatory myopathies with anti-Ku antibodies. Medicine. 2012;91:95–102. doi: 10.1097/MD.0b013e31824d9cec. [DOI] [PubMed] [Google Scholar]
- 28.Defendenti C. Atzeni F. Spina MF, et al. Clinical and laboratory aspects of Ro/SSA-52 autoantibodies. Autoimmun Rev. 2011;10:150–4. doi: 10.1016/j.autrev.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 29.Lundberg IE. Miller FW. Tjärnlund A. Bottai M. Diagnosis and classification of idiopathic inflammatory myopathies. J Intern Med. 2016;280:39–51. doi: 10.1111/joim.12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carstens P-O. Schmidt J. Diagnosis, pathogenesis and treatment of myositis: recent advances. Clin Exp Immunol. 2014;175:349–58. doi: 10.1111/cei.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]