

Abstract
This article reviews our current understanding and modern treatment of multiple sclerosis (MS). MS is a disabling condition resulting in devastating social and economic impacts. As MS can affect any part of the central nervous system, the presentation is often diverse; however, there are key features that can be useful in the clinic. We comment on the diagnostic criteria and review the main subtypes of MS, including clinically isolated syndrome, relapsing remitting MS, secondary progressive MS and primary progressive MS. Although the underlying aetiology of MS is still not known, we summarise those with most evidence of association. Finally, we aim to present treatment strategies for managing the acute relapse, disease-modifying therapies and MS symptoms. This review highlights that progressive MS is an area where there is currently a paucity of available disease-modifying treatments and this will be a major focus for future development.
Keywords: Diagnostic criteria, epidemiology, multiple sclerosis, progressive multiple sclerosis, treatments
What is multiple sclerosis?
Multiple sclerosis (MS) is an acquired disabling neurological disease of young adults, affecting approximately 2.3 million people worldwide. It is most prevalent in North America (140 cases per 100,000) and Europe (108 cases per 100,000); the prevalence is lowest in sub-Saharan Africa (2.1 cases per 100,000) and East Asia (2.2 cases per 100,000).1 Overall, there are approximately 120,000 people with MS in the UK.2
MS is an inflammatory disease of the central nervous system (CNS), which causes a heterogeneous array of symptoms and signs because of differential involvement of motor, sensory, visual and autonomic systems. Optic neuritis (inflammation of the optic nerve), Uhthoff's phenomenon (transient fluctuation or worsening of MS symptoms with a rise in body temperature) and Lhermitte's phenomenon (an abnormal electric-shock like sensation down the spine or limbs on neck flexion) are characteristic of MS.3–5 MS relapses occur because of focal areas of demyelination evolving over 24 hours and persisting for days or weeks before generally, though not exclusively, improving.4,6
Magnetic resonance imaging (MRI) is a key supplementary clinical tool in terms of diagnosis and therapeutic monitoring, with the dominant marker being areas of T2 signal change, which may also enhance with gadolinium, if recent activity. The 2010 revisions to the McDonald diagnostic criteria advise that there could be additional evidence of dissemination of MS lesions in time and in space, shown by MRI evidence.7–9 Many other MRI indices exist, eg T1 black holes, or are being developed, eg atrophy measures of reduction in brain volume, although these are not yet incorporated fully into standard clinical practice.3,10 The diagnosis of MS can be further reinforced by paraclinical data in the form of increased latency of evoked potentials, eg visual evoked potentials, and lumbar puncture with the assessment of unmatched oligoclonal immunoglobulin IgG bands in the cerebrospinal fluid (CSF) that are seen in up to 90% of MS patients.3,5,11 Other disease processes must be considered and actively excluded. A phenocopy that has more recently been established as separate from MS is neuromyelitis optica (NMO) spectrum disorder due in part to the aquaporin-4 antibody and subsequent complement activation.4,11,12
Subtypes of MS
There are different phenotypes of MS; here we discuss the most common (Fig 1).7 Clinically isolated syndrome (CIS) is the initial presentation in 80% of MS cases. CIS encompasses an acute clinical attack affecting one or more CNS sites and can convert to relapsing remitting MS (RRMS). The rate of conversion from CIS to RRMS at 20 years is 21% of patients with a normal MRI scan at baseline versus 82%, if there is one or more clinically silent white matter lesions on MRI.9,13 It has been shown that the McDonald 2010 criteria and increased use of MRI are revolutionising the diagnoses of clinically definite MS for the CIS group.14 With RRMS there is usually good recovery from each clinical episode or relapse.3,8 These early stages of MS with underlying demyelination are thought to have an inflammatory pathological substrate with migration of autoreactive lymphocytes across the blood-brain barrier. This triggers a cascade of inflammation with T- and B-cell clonal expansion, and microglia activation and oxidative damage, mitochondrial injury and energy failure, leading to the development of the characteristic plaque.15,16 Female:Male sex ratio of MS with relapse-onset is 2:1 and age of onset is around 30 years.1,2
Fig 1.

Relapse onset multiple sclerosis leads to the progressive accumulation of disability with neurodegeneration after 10–15 years, with less focal inflammation as highlighted by fewer relapses and MRI T2 lesions or gadolinium-enhancing lesions. MRI = magnetic resonance imaging
Over time, there is accumulation of disability and incomplete recovery from each relapse. 10–15 years after the diagnosis of RRMS, up to 80% of people develop secondary progressive MS (SPMS).1,3,7,17 There is further axonal injury and atrophy in both white and grey matter, with a likely underlying neurodegenerative pathogenesis, but less inflammation.16,18
These subtypes of MS should be cross-defined further as active or non-active. If active, there should be occurrence of a clinical relapse or new T2 or gadolinium-enhancing lesions on MRI over at least 1 year. Disease progression is the term used when patients have worsening disability over time compared with those who are worse because of a relapse.7,19
10–15% of patients have progressive disability from the outset, usually due to spinal cord disease. This is defined as primary progressive MS (PPMS).1,3,20 The age of onset is 40 years, roughly a decade older than for relapse onset MS, and the sex ratio is 1:1 in most studies, although there may be a slight male preponderance.1,20 Due to spinal cord dysfunction, patients often have a progressive spastic paraparesis. The pathological basis appears to be more diffuse brain axonal loss and further microglial activation with atrophy. There is anterograde and retrograde neurodegeneration with oxidative damage and energy failure. Therefore, SPMS and PPMS are likely to share a common pathological substrate.15,16
The underlying aetiology of MS is still not known, but is thought to be related to an interplay of genetic susceptibility and environmental factors. Several factors have been investigated, including putative viruses using molecular mimicry, low vitamin D, distance from the equator in early childhood, diet, smoking and toxins.1,3 Meta-analyses suggest that the strongest evidence of association is related to Epstein-Barr virus biomarker positivity, infectious mononucleosis and smoking.21 In terms of genetic susceptibility, the results of large genome-wide association studies (GWAS) have demonstrated the strongest susceptibility signal maps to HLA-DRB1 in the class II region of the major histocompatibility complex (MHC, 6p21.3) and accounts for 10% of the genetic variance underlying risk.22,23
As MS affects a young adult population, the disease has a huge socioeconomic impact. Studies have shown that the costs of lost employment and productivity far outweigh the costs for health and social care in the UK. It is estimated that the service costs of progressive MS in the UK are £3 billion per annum.24,25
Treatments
Treatments for MS can be divided into three categories:
acute relapse management
disease-modifying treatments (DMTs)
symptomatic treatments.
Acute relapse management
The initial difficulty with managing a relapse comes from defining whether an episode is a true relapse or an exacerbation or fluctuation due to an existing demyelinating lesion.6 Either way, the priority is to exclude and treat any concomitant infection (eg urinary tract infections), which can cause such perturbations. If there is uncertainty, MRI with gadolinium may be supportive and may show new enhancing lesions up to 6 weeks after the onset of a relapse. If the relapse is of moderate functional severity or worse, then high-dose methylprednisolone therapy should be considered, at a dose of 500–1,000 mg per day for 3–5 days as per local guidelines. The recent COPOUSEP trial (NCT00984984, ClinicalTrials.gov) demonstrated non-inferiority of oral versus intravenous methylprednisolone, provided the dose is high enough (1 g/day for 3 days of either).26 While not disease-modifying, corticosteroids tend to shorten the duration of the relapse. Plasma exchange is occasionally used, as an adjunctive therapy or alone, if the relapse is rapidly progressive or severe. Therapy interventions, such as physiotherapy, should also be used early to enhance recovery.4
Disease-modifying treatment
A revolution in the treatment of MS has occurred over the last 20 years, and particularly the last 5 years with the advent of more potent disease-modifying treatments (DMTs) – these are illustrated in Fig 2 and Table 1.11,27–30 The ultimate aim of DMT is the integration of clinical (no MS relapses or disability progression) and MRI (no new T2 lesions or atrophy) parameters, condensed as ‘no evidence of disease activity’ (NEDA). Disease-modifying therapy in the UK is prescribed and generally monitored by neurologists with a special interest in MS, as per the National Institute for Health and Care Excellence (NICE) guidelines.6 In the early 1990s, there were only two licensed disease-modifying therapies available in the UK: β-interferon and glatiramer acetate. Considerable developments have led to a total of 11 licensed formulations in the UK, with occasional case-specific use of mitoxantrone.3,4,6,11
Fig 2.
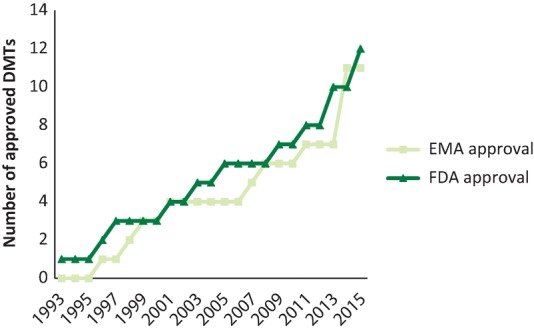
The number of multiple sclerosis disease-modifying therapies (DMTs) that have European Medicines Agency (EMA) or US Food and Drug Administration (FDA) approval. DMTs approved in 2016: β-interferon-1a (Avonex), β-interferon-1b (betaferon), glatiramer acetate (Copaxone), mitoxantrone (Novantrone)*, β-interferon-1a (Rebif), natalizumab (Tysabri), teriflunomide (Aubagio), alemtuzumab (Lemtrada), fingolimod (Gilenya), dimethyl fumarate (Tecfidera), interferon beta 1b (Extavia), β-interferon-1a (Plegridy). *Mitoxantrone (Novantrone) has FDA approval only.
Table 1.
UK-licensed disease-modifying treatments (DMTs) for relapsing remitting MS11,27–30
| Treatment | Mode and frequency | Side effects | Indication | Notes |
|---|---|---|---|---|
| Interferon beta 1a (Avonex) | Intramuscular injection 30 μg once per week | >1:100: flu-like, injection site reactions, headache, lymphopaenia, insomnia, diarrhoea, nausea and vomiting, depression, hair loss, liver function derangement, thyroid disease | First-line: active RRMS with ≥2 relapses, or 1 relapse and new MRI lesions as per McDonald 2010 criteria in ≤2 years EDSS<6.5 (able to walk 20 m) | Fridge storage Caution if depression/suicidal ideation |
| Pegylated interferon beta 1a (Plegridy) | Subcutaneous injectionStart with a dose of 63 μg on day 1, followed by 94 μg on day 15, then 125 μg on day 29 and once every 2 weeks thereafter | Caution re: neutralising antibodies with interferon beta 1b in up to 46% of patients. Monitor antibody titres | CIS and at high risk of developing MS with MRI T2 lesions | Fridge storage Keep out of sunlight |
| Interferon beta 1a (Rebif) | Subcutaneous injection Start at 22 μg three times weekly and escalate over 4 weeks to 44 μg three times weekly. | SPMS with ≥2 relapses in ≤2 years and EDSS progression ≤2 points over 2 years | Fridge storage | |
| Interferon beta 1b (Betaferon) | Subcutaneous injection Alternate days Escalate to 250 μg over 3–6 weeks |
|||
| Interferon beta 1b (Extavia) | ||||
| Glatiramer acetate (Copaxone) | Subcutaneous injection 20 mg once daily or 40 mg three times per week. | >1:100: injection site reactions including lipoatrophy, headache, depression, anxiety and nausea. Also IPIR with flushing, palpitations, dyspnoea and chest pain lasting 15–30 minutes | First-line: active RRMS with ≥2 relapses, or 1 relapse and new MRI lesions, as per McDonald 2010 criteria, in ≤2 years. Able to walk more than 100 m without aids CIS and at high risk of developing MS with MRI T2 lesions SPMS with ≥2 relapses in ≤2 years and EDSS progression ≤2 points over 2 years |
If IPIR lasts longer than 30 minutes then patients should seek urgent medical attention |
| Teriflunamide (Aubagio) | Oral 7 or 14 mg once daily | >1:100: liver function derangement, diarrhoea, nausea, hair thinning and loss | First-line: active RRMS with ≥2 relapses, or 1 relapse and new MRI lesions in ≤2 years | |
| Dimethyl fumarate (Tecfidera) | Oral Initially 120 mg twice daily, then increase after 7 days to 240 mg twice daily |
>1:100: flushing, gastrointestinal upset, lymphopaenia, rash, ketonuria, proteinuria, liver function derangement Very rare cases of PML reported |
First-line: active RRMS with ≥2 relapses, or 1 relapse and new MRI lesions in ≤2 years | Taking oral doses with food may reduce the incidence of flushing and gastrointestinal effects |
| Fingolimod (Gilenya) | Oral 0.5 mg once daily | >1:100: cough, headache, back pain, diarrhoea, infections, liver function derangement, lymphopaenia. <1:100: macular oedema Very rare cases of PML reported |
Second-line: highly active RRMS only with relapses despite treatment with beta interferon for at least 1 year or relapses on glatiramer acetate or tecfidera RRMS on natalizumab at high risk of PML (2014 post-NICE approval) First-line: across UK, ABN guidance suggests fingolimod can be used if highly active RRMS with 2 or more disabling relapses in 1 year and new MRI lesions |
Note: bradycardia and atrioventricular conduction slowing can occur while taking the first dose of fingolimod, therefore the first dose is taken under medical supervision and cardiac monitoring for 6 hours |
| Natalizumab (Tysabri) | Intravenous infusion 300 mg once every 4 weeks | >1:100: headache, dizziness, pruritic rash, infections Clear risk gradient of PML. |
First-line: RES RRMS with ≥2 relapses ≤1 year and no previous DMT and either ≥1 gadolinium-enhancing MRI lesion(s) or ≥9 T2-hyperintense MRI brain lesions, if MRI available Second-line: RES RRMS with ≥1 relapses ≤1 year and previous beta interferon not meeting stopping criteria and either ≥1 gadolinium-enhancing MRI lesion or ≥9 T2-hyperintense MRI brain lesions, if MRI available |
|
| Alemtuzumab (Lemtrada) | Intravenous infusion Initially, 12 mg daily for 5 consecutive days to a total dose of 60 mg Second course: 12 months post initial dose, 12 mg daily for 3 consecutive days to a total dose of 36 mg |
>1:100: acute cytokine release syndrome (headaches, rash, fever, nausea, diarrhoea, hypotension), infections including herpes virus, endocrinopathy (especially thryoid) | First-line: active RRMS with ≥2 relapses in ≤2 years, or 1 relapse and new MRI lesions Second-line: highly active RRMS on DMT and relapse within the last year and new MRI lesions |
Pre-medications: oral or intravenous corticosteroid, oral antihistamine and analgesic to prevent cytokine release syndrome Oral prophylaxis with aciclovir for herpes infection and continued for 1 month |
NHS England has taken on the commissioning of drugs since 2013 and advises that patients must be under the care of a designated MS centre that is registered to take part in the national risk sharing scheme involving the three beta interferon products and glatiramer acetate. The Association of British Neurologists (ABN) advises an annual review while on DMTs that will need to be conducted by the MS specialist neurologist who is also best placed to determine whether MRI scanning is required.
CIS = clinically isolated syndrome; DMT = disease-modifying treatment; EDSS = Expanded Disability Status Scale; IPIR = immediate post-injection reaction; MRI = magnetic resonance imaging; MS = multiple sclerosis; NICE = National Institute for Health and Care Excellence; PML = progressive multifocal leucoencephalopathy; RES = rapidly evolving severe; SPMS = secondary progressive multiple sclerosis; RRMS = relapsing remitting multiple sclerosis.
RRMS treatment
In the UK, DMTs are generally used according to NICE criteria, which are dependent on variables that include the number of clinical relapses, MRI radiological activity and disability level (Table 1).6 However, given the heterogeneity of the condition, the choice remains an individual one alongside clinical guidance to allow consideration of benefit versus risk, monitoring requirements and administration route (eg oral versus injectable). Early data via the UK risk-sharing scheme has shown the benefit of DMT in possibly reducing the accrual of disability31 and it is hoped and expected that more effective DMTs will reduce the burden further.
There are two main schools of thought as to how DMTs should be used:
Induction – where high efficacy drugs are used earlier in disease to possibly prevent disability accrual, despite their significant side effect profile.
Escalation – starting with a less efficacious, but potentially safer, therapy and then moving up the treatment ladder if there is treatment failure.
Clinical and MRI monitoring are paramount to measuring response, but longitudinal data regarding induction versus escalation regimens are currently lacking. Additionally, in considering the DMTs, one note of caution is that there have been only a few direct head-to-head comparison trials, and therefore the inter-study results have to be interpreted with some caution.8,11
In the UK, DMTs are divided into those of moderate efficacy with a reduction in annualised relapse rate (ARR) of 30–50% (category 1: β-interferons, glatiramer acetate, teriflunamide, dimethyl fumarate and fingolimod) and those of higher efficacy with a reduction in ARR of more than 50% (category 2: alemtuzumab and natalizumab).11 Full details are given in Table 1.
The most serious side effect occurred unexpectedly about a decade ago with natalizumab – a humanised monoclonal antibody against the cell adhesion molecule a4-integrin – where activation of the John Cunningham virus (JCV) resulted in progressive multifocal leukoencephalopathy (PML). JCV is present in about 50% of the population. The risk-dynamics in developing PML have now been well defined and include natalizumab treatment for more than 2 years and prior chemotherapy or immunosuppression, which causes a risk of developing PML of 1 in 200 or higher. Close MRI and JCV status monitoring is required while taking natalizumab.24 PML has also been recently observed, much more rarely, with the oral agents fingolimod and dimethylfumarate.4,11
Switching between agents depends on clinical and radiological criteria. Rules are being developed, based on relapse rate and new T2 lesions or new gadolinium enhancing lesions, eg the Modified Rio Criteria for β-interferon.11 For all licensed DMTs, the advice is to start treatment before the advent of significant disability (as early as possible) and certainly when patients are still ambulant. Stopping a DMT completely can be a difficult decision in the context of progressive disability where there are no licensed DMTs currently, but this needs to be balanced against the risks and the likelihood of effectiveness. Natural history studies suggest an increased risk of relapse in the postpartum period and therefore DMTs should be restarted as early as possible, depending on individual preference.4,11
CIS treatment
In trials, DMTs have been shown to delay clinically definite MS in patients with CIS by approximately 1 year. β-interferons and glatiramer acetate are currently the only licensed therapies for CIS in the UK. These may be prescribed in those with CIS within 12 months of the episode, if MRI evidence establishes the diagnosis of MS according to the 2010 McDonald criteria or predicts a high likelihood of recurrent episodes.4,11 Additionally, there may be improvement in cognitive outcome measures if injectable therapy is started before clinically definite MS, as highlighted by the BENEFIT study (NCT00544037, ClinicalTrials.gov). It is noted that 30–50% of those with CIS have cognitive impairment.32
Progressive MS treatment
Unfortunately, there are no licensed DMTs for progressive MS.23 However, there is a caveat for those with SPMS with two or more relapses within 2 years and slow progression of less than 2 Expanded Disability Status Score (EDSS) point progression in terms of prescribing β-interferon-1b or glatiramer acetate (Table 1). In rapidly progressing SPMS patients, intravenous mitoxantrone can be considered, but this requires close monitoring for cardiac side effects and acute myelogenous leukaemia. Intravenous methylprednisolone pulsed therapy can be considered in SPMS, but again long-term data are lacking.4,11
There are a large number of phase III trials using agents such azathioprine, ciclosporin, β-interferon, dirucotide (myelin basic protein), immunoglobulins and cladribine, as well as other immunomodulators, in progressive MS that have been reported over the last 25 years. Unfortunately, the results have essentially been negative.10 This was reinforced by the negative INFORMS trial of fingolimod in PPMS.33 However, the counter to this may be the recently announced groundbreaking results from the ORATORIO trial (NCT01194570, ClinicalTrials.gov) of ocrelizumab – an anti-CD20 depleting molecule – in PPMS and the EXPAND trial (NCT01665144, ClinicalTrials.gov) of siponimod – a selective sphingosine-1-phosphate receptor modulator – in SPMS that delayed the confirmed progression of MS by about 20–25% over 2–3 years34,35
Measuring disability in progressive MS is difficult and classical measures such as the EDSS, an ordinal scale, have been widely used to date, although it is imperfect and focuses on motor disability. There is an urgent need to develop interim markers for phase II studies.10 One such parameter is MRI atrophy, for example in the MS-STAT trial (NCT00647348, ClinicalTrials.gov) – using high-dose simvastatin as a neuroprotective agent, the reduction in annualised brain atrophy rate versus placebo was about 40%.36 This technique and others, such as optical coherence tomography (OCT) and spinal cord atrophy, are starting to be harnessed in more modern trial platforms, eg the phase II multiple sclerosis secondary-progressive multi-arm randomisation trial (MS-SMART; NCT01910259, ClinicalTrials.gov) where three putative neuroprotective agents (amiloride, fluoxetine and riluzole) are being compared with placebo.37
Symptomatic treatments
Patients with MS experience a large array of significant and disabling symptoms, including fatigue, cognitive impairment, bladder dysfunction, pain and spasticity. Not all symptoms have effective therapies and the James Lind Alliance Priority Setting Partnership, a patient-care-clinician initiative, along with the UK MS Society, has highlighted that one of the top 10 priorities is to find ‘which treatments are effective to improve cognition in people with MS’ given that this has devastating socioeconomic effects in this young population. Currently available modalities are pharmaceutical and non-pharmaceutical and we have given an approach to symptom management in Table 2. The key is to remove co-existent underlying causes, eg infection or anaemia; deal with provoking factors such as poor sleep; and use the available treatments logically and persistently, in series, until relief is obtained.4,6
Table 2.
Suggested symptomatic treatments in MS
| Drug* | Interventional | Multi-disciplinary input | |
|---|---|---|---|
| Fatigue | Amantadine | Occupational therapy and physiotherapy: fatigue management assessment and exercise programme | |
| Cognition/low mood | Depression: eg citalopram duloxetine | Neuropsychology service, cognitive behavioural therapy, occupational therapy | |
| Spasticity | Baclofen, gabapentin, tizanidine, clonazepam, dantrolene | Intrathecal baclofen, botulinum toxin | Physiotherapy |
| Bladder | Frequency/urgency: oxybutynin, solifenacin, tolterodine, mirabegron
Nocturia: desmopressin/DDAVP spray |
Residual bladder volume >100 ml: intermittent self-catheterisation or permanent catheter; intravesicular botulinum toxin, | Uro-neurology |
| Sexual dysfunction | Sildenafil, tadalafil, alprostadil, yohimbine | Uro-neurology | |
| Constipation | Fibre/fluid, bulking agents, osmotic stimulant laxatives, suppositories, transanal irrigation | ||
| Faecal incontinence | Codeine, loperamide | Biofeedback, neuro-gastroenterology | |
| Pain | Amitriptyline, pregabalin, gabapentin, lamotrigine | ||
| Ataxia/tremor | Propranolol, clonazepam, levetiracetam, isoniazid (with pyridoxine), carbamazepine, ondansetron | Botulinum toxin, thalamotomy | Physiotherapy, occupational therapy, audiovestibular therapy |
| Oscillopsia | Gabapentin, memantine, levetiracetam, clonazepam, baclofen | Neuro-ophthalmology | |
| *These may be unlicensed indications | |||
Conclusions
A sea-change has occurred in the treatment of relapsing MS, with a large variety of available DMTs that need to be closely tailored to the individual person with MS in terms of efficacy and safety. It is a battle half-won though, for it is progression in MS that has to be conquered and where the global effort now turns.23
Conflicts of interest
The authors have no conflicts of interest to declare.
Acknowledgements
JC is supported by the National Institute for Health Research (NIHR), University College London Hospitals (UCLH)/UCL Biomedical Research Centre (BRC).
Note
This article was originally published in the 2016 Clinical Medicine supplement Horizons in Medicine 28. All articles in this supplement are available at www.clinmed.rcpjournal.org/content//16/Suppl_6
References
- 1.Multiple Sclerosis International Federation Atlas of MS mapping multiple sclerosis around the world. London: MSIF, 2013. 2013 [Google Scholar]
- 2.Kingwell E. Marriott JJ. Jetté N, et al. Incidence and prevalence of multiple sclerosis in Europe: a systematic review. BMC Neurol. 2013;13:128. doi: 10.1186/1471-2377-13-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Compston A. Coles A. Multiple sclerosis. Lancet. 2008;372:1502–17. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 4.Stevenson EV. Alexander JS. Yun JW. Becker F. Gonzalez-Toledo E. Chapter 16 – Mechanisms of blood–brain barrier disintegration in the pathophysiology of multiple sclerosis. In: Minagar A, editor. Multiple sclerosis. London:: Academic Press; 2016. pp. 393–413. [Google Scholar]
- 5.Deangelis TM. Miller A. Diagnosis of multiple sclerosis. In: Tselis AC, editor; Booss J, editor. Handbook of clinical neurology. Amsterdam:: Elsevier BV; 2014. pp. 307–12. [DOI] [PubMed] [Google Scholar]
- 6.Perry M. Swain S. Kemmis-Betty S. Cooper P. Multiple sclerosis: summary of NICE guidance. BMJ. 2014;349:g5701. doi: 10.1136/bmj.g5701. [DOI] [PubMed] [Google Scholar]
- 7.Lublin FD. New multiple sclerosis phenotypic classification. Eur Neurol. 2014;72(Suppl 1):1–5. doi: 10.1159/000367614. [DOI] [PubMed] [Google Scholar]
- 8.Lublin FD. Reingold SC. Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83:278–86. doi: 10.1212/WNL.0000000000000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milo R. Miller A. Revised diagnostic criteria of multiple sclerosis. Autoimmun Rev. 2014;13:518–24. doi: 10.1016/j.autrev.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 10.Ontaneda D. Fox RJ. Chataway J. Clinical trials in progressive multiple sclerosis: lessons learned and future perspectives. Lancet Neurol. 2015;14:208–23. doi: 10.1016/S1474-4422(14)70264-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scolding N. Barnes D. Cader S, et al. Association of British Neurologists: revised guidelines for prescribing disease-modifying treatments in multiple sclerosis Pract Neurol. 2015;15:273–9. doi: 10.1136/practneurol-2015-001139. 2015. [DOI] [PubMed] [Google Scholar]
- 12.Miller DH. Weinshenker BG. Filippi M, et al. Differential diagnosis of suspected multiple sclerosis: a consensus approach. Mult Scler. 2008;14:1157–74. doi: 10.1177/1352458508096878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fisniku LK. Brex PA. Altmann DR, et al. Disability and T2 MRI lesions: a 20-year follow-up of patients with relapse onset of multiple sclerosis. Brain. 2008;131:808–17. doi: 10.1093/brain/awm329. [DOI] [PubMed] [Google Scholar]
- 14.Bronwlee WJ. Swanton JK. Altmann DR. Ciccarelli O. Miller DH. Earlier and more frequent diagnosis of multiple sclerosis using the McDonald criteria. J Neurol Neurosurg Psychiatry. 2015;86:584–5. doi: 10.1136/jnnp-2014-308675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lassmann H. Brück W. Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol. 2007;17:210–8. doi: 10.1111/j.1750-3639.2007.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lassmann H. van Horssen J. Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012;8:647–56. doi: 10.1038/nrneurol.2012.168. [DOI] [PubMed] [Google Scholar]
- 17.Eriksson M. Andersen O. Runmarker B. Long-term follow up of patients with clinically isolated syndromes, relapsing-remitting and secondary progressive multiple sclerosis. Mult Scler. 2003;9:260–74. doi: 10.1191/1352458503ms914oa. [DOI] [PubMed] [Google Scholar]
- 18.Mahad DH. Trapp PBD. Lassmann PH. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14:183–93. doi: 10.1016/S1474-4422(14)70256-X. [DOI] [PubMed] [Google Scholar]
- 19.Cross AH. Wingerchuk DM. Weinshenker BG. Active and progressive: a new duality of MS classification. Neurology. 2014;83:206–7. doi: 10.1212/WNL.0000000000000601. [DOI] [PubMed] [Google Scholar]
- 20.Miller DH. Leary SM. Primary-progressive multiple sclerosis. Lancet Neurol. 2007;6:903–12. doi: 10.1016/S1474-4422(07)70243-0. [DOI] [PubMed] [Google Scholar]
- 21.Belbasis L. Bellou V. Evangelou E. Ioannidis JP. Tzoulaki I. Environmental risk factors and multiple sclerosis: an umbrella review of systematic reviews and meta-analyses. Lancet Neurol. 2015;14:263–73. doi: 10.1016/S1474-4422(14)70267-4. [DOI] [PubMed] [Google Scholar]
- 22.Isobe N. Madireddy L. Khankhanian P, et al. An ImmunoChip study of multiple sclerosis risk in African Americans. Brain. 2015;138:1518–30. doi: 10.1093/brain/awv078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sawcer S. Hellenthal G. Pirinen M, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2012;476:214–9. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson AJ. A much-needed focus on progression in multiple sclerosis. Lancet Neurol. 2015;14:133–5. doi: 10.1016/S1474-4422(14)70330-8. [DOI] [PubMed] [Google Scholar]
- 25.McCrone P. Heslin M. Knapp M. Bull P. Thompson A. Multiple sclerosis in the UK: service use, costs, quality of life and disability. Pharmacoeconomics. 2008;26:847–60. doi: 10.2165/00019053-200826100-00005. [DOI] [PubMed] [Google Scholar]
- 26.Le Page E. Veillard D. Laplaud DA, et al. Oral versus intravenous high-dose methylprednisolone for treatment of relapses in patients with multiple sclerosis (COPOUSEP): a randomised, controlled, double-blind, non-inferiority trial. Lancet. 2015;386:974–81. doi: 10.1016/S0140-6736(15)61137-0. [DOI] [PubMed] [Google Scholar]
- 27.Multiple Sclerosis Trust . Letchworth. MS Trust; 2015. Disease modifying drugs: a guide to treatments for relapsing MS. [Google Scholar]
- 28.National Institute for Health and Care Excellence . NICE clinical guideline No 186. London:: NICE; 2014. Multiple sclerosis in adults: management. [PubMed] [Google Scholar]
- 29.NHS England . London:: NHS England; 2014. Clinical commissioning policy: disease modifying therapies for patients with multiple sclerosis (MS). [Google Scholar]
- 30.Brayfield A. London:: Pharmaceutical Press; 2014. Martindale: the complete drug reference, 38th edn. [Google Scholar]
- 31.Palace J. Duddy M. Bregenzer T, et al. Effectiveness and cost-effectiveness of interferon beta and glatiramer acetate in the UK Multiple Sclerosis Risk Sharing Scheme at 6 years: a clinical cohort study with natural history comparator. Lancet Neurol. 2015;14:497–505. doi: 10.1016/S1474-4422(15)00018-6. [DOI] [PubMed] [Google Scholar]
- 32.Edan G. Kappos L. Montalbán X, et al. Long-term impact of interferon beta-1b in patients with CIS: 8-year follow-up of BENEFIT. J Neurol Neurosurg Psychiatry. 2014;85:1183–9. doi: 10.1136/jnnp-2013-306222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lublin F. Miller DH. Freedman MS, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387:1075–84. doi: 10.1016/S0140-6736(15)01314-8. [DOI] [PubMed] [Google Scholar]
- 34.Montalban X. Hemmer B. Rammohan K, et al. Efficacy and safety of ocrelizumab in primary progressive multiple sclerosis - results of the placebo-controlled, double-blind, Phase III ORATORIO study. Neurology. 2016;86(Suppl 16):S49.001. [Google Scholar]
- 35.Kappos L. Bar-Or A. Cree B, et al. Efficacy and Safety of siponimod in secondary progressive multiple sclerosis - results of the placebo controlled, double-blind, Phase III EXPAND study [abstract] ECTRUMS Online Library. 206:147077. [Google Scholar]
- 36.Chataway J. Schuerer N. Alsanousi A, et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet. 2014;383:2213–21. doi: 10.1016/S0140-6736(13)62242-4. [DOI] [PubMed] [Google Scholar]
- 37.MS-SMART Multiple Sclerosis-Secondary Progressive Multi-Arm Randomisation Trial. https://clinicaltrials.gov/ct2/show/NCT01910259. [Accessed 14 November 2016]