

Abstract
Chronic lymphocytic leukemia (CLL) is a malignant clonal proliferative disorder of B cells. Inhibition of cell apoptosis and cell cycle arrest are the main pathological causes of this disease, but its molecular mechanism requires further investigation. The purpose of the present study was to identify biomarkers for the early diagnosis and treatment of CLL, and to explore the molecular mechanisms of CLL progression. A total of 488 differentially expressed genes (DEGs) and 32 differentially expressed microRNAs (miRNAs; DEMs) for CLL were identified by analyzing the gene chips GSE22529, GSE39411 and GSE62137. Functional and pathway enrichment analyses of DEGs demonstrated that DEGs were mainly involved in transcriptional dysregulation and multiple signaling pathways, such as the nuclear factor-κB and mitogen-activated protein kinase signaling pathways. In addition, Cytoscape software was used to visualize the protein-protein interactions of these DEGs in order to identify hub genes, which could be used as biomarkers for the early diagnosis and treatment of CLL. Cytoscape software was also used to analyze the association between the predicted target mRNAs of DEMs and DEGs and increase knowledge about the miRNA-mRNA regulatory network associated with the progression of CLL. Taken together, the present study provided a bioinformatics basis for advancing our understanding of the pathogenesis of CLL by identifying differentially expressed hub genes, miRNA-mRNA target pairs and molecular pathways. In addition, hub genes may be used as novel biomarkers for the diagnosis of CLL and to guide the selection of CLL drug combinations.
Keywords: chronic lymphocytic leukemia, bioinformatics analysis, protein-protein interaction network, hub genes, microRNA-mRNA network
Introduction
Chronic lymphocytic leukemia (CLL) is a clonal proliferative tumor of mature B lymphocytes characterized by the accumulation of lymphocytes in the peripheral blood, bone marrow, lymph nodes and spleen (1). It is the most common type of leukemia in western populations, causing ~5,000 mortalities in the United States of America annually (2). Unfortunately, the exact molecular mechanism underlying the development of CLL remains unclear, which limits early diagnosis and timely treatment. Therefore, exploring the molecular mechanism of CLL progression and its biomarkers to improve the likelihood of early diagnosis and treatment have become the focus in CLL research. Increasing evidence suggests that numerous genes, miRNAs and cellular pathways are involved in the occurrence and progression of CLL (3,4).
In recent years, the rapid development of computer-based technologies, such as molecular dynamics simulation, has gradually revealed novel molecular mechanisms of the disease (5,6). Through the combination of Gene Expression Omnibus (GEO) chip, The Cancer Genome Atlas data mining (cancergenome.nih.gov/) and computer-based analysis techniques, a series of highly specific and sensitive markers have been reported (7–9). Gene chips are widely used as a genetic testing tool as they can be used to detect rapid gene expression of a sample at a certain time point and are particularly suitable for the screening of differentially expressed genes (10). MicroRNAs (miRNAs/miRs) are endogenous non-coding small RNAs which have several important regulatory roles in cells (11). Computer-based analysis techniques have demonstrated a mutation of a miRNA binding site that is associated with cancer and revealed its mechanism of gene regulation (12).
In the present study, two original gene microarray datasets (GSE22529 and GSE39411) and one original miRNA microarray dataset (GSE622137) were downloaded from the National Center for Biotechnology Information-Gene Expression Omnibus database (NCBI-GEO). The raw gene microarray datasets were initially processed online using the Gene-Cloud of Biotechnology Information (GCBI) software to screen for differentially expressed genes (DEGs) in CLL and normal samples. In addition, the Gene Ontology (GO) Consortium and the Kyoto Encyclopedia of Genes and Genomes (KEGG), in conjunction with the Database for Annotation, Visualization, and Integrated Discovery (DAVID) software program were used to analyze the pathway enrichment of DEGs. The STRING online database and Cytoscape software were then used to develop the protein-protein interaction (PPI) network of the DEGs and for modular analysis to identify CLL central genes. In addition, GEO2R analysis of the miRNA dataset was employed to obtain the differentially expressed miRNAs (DEMs). Cytoscape software was used to analyze the association between the predicted target mRNAs of DEMs and DEGs, which helped to define the regulatory network of miRNA-mRNA in relation to the development of CLL. Our findings suggest that data mining and integration may be useful methods for predicting and understanding the mechanisms of CLL disease development and progression. Ultimately, the early diagnosis and personalized treatment of CLL may be facilitated by identifying new biomarkers (3).
Materials and methods
Microarray data
Data of gene chips GSE22529 (13), GSE39411 (14) and GSE62137 (15) were obtained from the GEO database (www.ncbi.nlm.nih.gov/geo). GSE22529 was based on Agilent GPL96 (Agilent Technologies, Inc., Santa Clara, CA, USA; [HG-U133A] Affymetrix Human Genome U133A Array, Affymetrix; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and GPL97 [(HG-U133B) Affymetrix Human Genome U133B Array, Affymetrix; Thermo Fisher Scientific, Inc.)]. To reduce the offset, based on the GCBI online laboratory (www.gcbi.com.cn/gclib/html/index), the GPL96 annotation data set as the research object, was screened. The GSE22529 dataset included 52 samples, containing 41 CLL samples and 11 normal samples. The GSE39411 data were based on GPL570 [(HG-U133_Plus_2) Affymetrix Human Genome U133 Plus 2.0 Array; Affymetrix, Thermo Fisher Scientific, Inc.], consisting of 41 CLL samples and 11 normal samples. The GSE62137 data were based on GPL14767 [(Agilent-021827 Human miRNA Microarray G4470C)], consisting of 15 CLL samples and 3 normal samples.
Identification of DEGs and DEMs
To identify DEGs in the CLL and normal samples, a web-based online tool, GCBI, was utilized. GCBI is an interactive online tool that allows users to compare two or more groups of samples in a GEO series and analyzes the majority of GEO series with gene symbols. DEGs were screened according to P<0.05 and |log fold change (FC)|>1.5. DEMs were obtained by analyzing miRNA expression profiles using the GEO2R tool (https://www.ncbi.nlm.nih.gov/geo/geo2r/) and screening according to the |log FC|>2 and P<0.05. In addition, DEG heat maps from datasets GSE22529 and GSE39411, were created using Morpheus online software (software.broadinstitute.org/morpheus).
GO and pathway enrichment analysis of DEGs
DAVID version 6.8 (david.abcc.ncifcrf.gov/), an open source platform, was utilized to determine the association among target molecules (16). To gain insight into the biological functions of these misaligned genes, GO and (KEGG) pathway enrichment analyses were performed on the DEGs based on DAVID, and P<0.05 was set as the cut-off criterion. The molecular function, biological processes, cell component and KEGG pathways of these DEGs and target genes were investigated using DAVID.
Protein-protein interaction (PPI) network and module analysis
As a search tool for the retrieval of interacting genes, STRING version 10.5 (string-db.org) was used to assess PPI information (17). To further examine the potential association between these DEGs, STRING was used to characterize the PPI network, and a confidence score >0.4 was set as the cut-off criterion. Cytoscape_v3.5.1 software was used to visualize the resulting PPI network (18). The Molecular Complex Detection (MCODE version 1.4.2) application (http://apps.cytoscape.org/apps/mcode) was used to select significant modules from the PPI network in Cytoscape with MCODE scores >5 and number of nodes >10 as the standard. Furthermore, a topological analysis of the PPI network was performed and the key genes were screened for further biological analysis of the node degree of these DEGs.
Target mRNA prediction of DEMs and construction of miRNA-mRNA regulatory networks
The target mRNAs of DEMs were predicted using TargetScan (www.targetscan.org/), miRDB (www.mirdb.org/miRDB/) (19), and miRTarBase version 7 (mirtarbase.mbc.nctu.edu.tw/) (20) online analysis tools. To further improve the reliability of bioinformatics analysis, overlapping target genes between DEGs and predicted target mRNAs were identified using a Venn diagram. The miRNA-mRNA networks were visualized using the Cytoscape platform software.
Results
Identification of DEGs and DEMs
NCBI-GEO is a free database containing microarray/gene profiles and next-generation sequences. Data for gene expression in CLL patients and normal samples was obtained from GSE22529 and GSE39411 along with miRNA expression data from GSE62137. The original gene data files were uploaded to the GCBI online laboratory to screen for the DEGs in CLL and normal samples. Using P<0.05 and |log FC|>1.5 as cut-off criteria, 781 and 1,607 DEGs were extracted from the expression profile datasets GSE22529 and GSE39411, respectively. Following integrated bioinformatics analysis, a total of 488 consistently expressed genes were identified from the two profile datasets, including 204 upregulated and 284 downregulated genes in the CLL samples compared with the normal population. Using a novel web-based tool, Morpheus, heat maps (illustrating the top 100 upregulated and top 100 downregulated genes) of DEGs were constructed from the GSE22529 and GSE39411 datasets (Fig. 1A and B). In addition, the miRNA profile dataset, GSE62137, was analyzed to screen for DEMs in CLL using the GEO2R tool. According to the cut-off criteria (P<0.05 and |logFC|>2.0), a total of 34 differentially expressed miRNAs were identified from this microarray dataset, including 29 upregulated and 5 downregulated miRNAs (Table I). Among them, miR-144 and miR-181a were the most downregulated miRNAs in the GSE62137 dataset, whereas miR-582 was the most upregulated. The 488 consistently expressed genes and 34 differentially expressed miRNAs were used for further functional analysis.
Figure 1.

Heat map of Gene Expression Omnibus series. Top 100 upregulated and 100 downregulated genes from (A) GSE22529 and (B) GSE39411 datasets. Red corresponds to gene upregulation and blue to gene downregulation.
Table I.
Main dysregulated miRNAs in chronic lymphocytic leukemia.
| miRNA | LogFC | P-value |
|---|---|---|
| hsa-miR-582-5p | 8.23 | 2.78×10−21 |
| hsa-miR-451 | −6.25 | 1.91×10−5 |
| hsa-miR-548c-3p | 3.65 | 9.27×10−5 |
| hsa-miR-181c-3p | 3.22 | 1.03×10−4 |
| hsa-miR-181a | −8.66 | 1.12×10−4 |
| hsa-miR-95 | 4.34 | 1.16×10−4 |
| hsa-miR-132 | 6.46 | 2.84×10−4 |
| hsa-miR-144 | −8.52 | 3.25×10−4 |
| hsa-miR-486-5p | −5.57 | 3.88×10−4 |
| hsa-miR-885-3p | 3.23 | 5.86×10−3 |
| hsa-miR-199a-3p | 5.76 | 7.00×10−3 |
| hsa-miR-126 | 6.45 | 9.25×10−3 |
| hsa-miR-145 | 2.59 | 1.98×10−2 |
| hsa-miR-136 | 2.78 | 1.98×10−2 |
| hsa-miR-326 | 2.02 | 1.98×10−2 |
| hsa-miR-346 | 2.03 | 1.98×10−2 |
| hsa-miR-376c | 2.96 | 1.98×10−2 |
| hsa-miR-139-5p | 2.14 | 1.98×10−2 |
| hsa-miR-410 | 2.33 | 1.98×10−2 |
| hsa-miR-339-5p | 2.05 | 1.98×10−2 |
| hsa-miR-584 | 2.44 | 1.98×10−2 |
| hsa-miR-1 | 2.24 | 1.98×10−2 |
| hsa-miR-495 | 2.11 | 1.98×10−2 |
| hsa-miR-376a | 2.89 | 1.98×10−2 |
| hsa-miR-377 | 2.76 | 1.98×10−2 |
| hsa-miR-26b-3p | 2.1 | 1.98×10−2 |
| hsa-miR-181c | 2.09 | 1.98×10−2 |
| hsa-miR-381 | 2.37 | 1.98×10−2 |
| hsa-miR-296-3p | 2.16 | 1.98×10−2 |
| hsa-miR-409-3p | 2.36 | 1.98×10−2 |
| hsa-miR-199a-5p | 3.42 | 1.98×10−2 |
| hsa-miR-126-5p | 3.11 | 1.98×10−2 |
| hsa-miR-664a-5p | −5.32 | 1.98×10−2 |
| hsa-miR-501-5p | 4.2 | 1.98×10−2 |
FC, fold-change; miR, microRNA.
GO function and KEGG pathway enrichment analysis of the DEGs
To gain a deeper understanding of the selected DEGs, GO and KEGG pathway analyses were performed using DAVID. GO analysis demonstrated that upregulated and downregulated DEGs were particularly abundant in the ‘molecular function’, ‘biological processes’ and ‘cell component classification’ (Table II). As demonstrated in this Table, in the ‘molecular function group’, upregulated genes were mainly enriched in ‘protein binding’, ‘protein kinase binding’ and ‘non-membrane spanning protein tyrosine kinase activity’, while the downregulated DEGs were enriched in various combinations, such as ‘antigen binding’, ‘immunoglobulin receptor binding’ and ‘cell adhesion molecule binding’. In the ‘biological processes’ group, upregulated DEGs were mainly associated with ‘intracellular signaling transduction’, the ‘transmembrane receptor protein tyrosine kinase signaling pathway’ and ‘positive regulation of granulocyte differentiation’, and downregulated DEGs were significantly enriched in ‘immune response’ and the ‘B cell receptor signaling pathway’. In addition, GO cell component analysis demonstrated that the upregulated DEGs were enriched in ‘cytosol and plasma membrane’, and downregulated DEGs were enriched in ‘cell surface’, ‘extracellular exosome’ and ‘cell-cell adherent junction’. These results demonstrated that most DEGs are significantly enriched in ‘binding’, ‘adhesion’ and ‘cell cycle’.
Table II.
Gene ontology analysis of differentially expressed genes associated with chronic lymphocytic leukemiaa.
| Category | Term | Count | P-value |
|---|---|---|---|
| Upregulated | |||
| GOTERM_BP_DIRECT | GO:0035556~intracellular signal transduction | 16 | 2.64×10−5 |
| GO:0007169~transmembrane receptor protein tyrosine kinase signaling pathway | 7 | 5.49×10−4 | |
| GO:0008283~cell proliferation | 12 | 1.89×10−3 | |
| GO:0030854~positive regulation of granulocyte differentiation | 3 | 2.27×10−3 | |
| GO:1901385~regulation of voltage-gated calcium channel activity | 3 | 2.27×10−3 | |
| GOTERM_MF | GO:0005515~protein binding | 122 | 6.86×10−6 |
| _DIRECT | GO:0019901~protein kinase binding | 14 | 1.72×10−4 |
| GO:0005524~ATP binding | 31 | 4.04×10−4 | |
| GO:0004715~non-membrane spanning protein tyrosine kinase activity | 5 | 1.35×10−3 | |
| GO:0042169~SH2 domain binding | 4 | 3.39×10−3 | |
| GOTERM_CC | GO:0005886~plasma membrane | 65 | 1.81×10−4 |
| _DIRECT | GO:0005829~cytosol | 53 | 7.60×10−4 |
| GO:0009897~external side of plasma membrane | 9 | 1.63×10−3 | |
| GO:0045121~membrane raft | 8 | 5.46×10−3 | |
| GO:0030175~filopodium | 5 | 6.11×10−3 | |
| Downregulated | GO:0006955~immune | 31 | 1.58×10−12 |
| GOTERM_BP | response | ||
| _DIRECT | GO:0050776~regulation of immune response | 21 | 2.84×10−12 |
| GO:0006958~complement activation, classical pathway | 14 | 2.97×10−9 | |
| GO:0006911~phagocytosis, engulfment | 8 | 7.61×10−7 | |
| GO:0050853~B cell receptor signaling pathway | 9 | 1.36×10−6 | |
| GOTERM_MF | GO:0003823~antigen binding | 15 | 3.62×10−10 |
| _DIRECT | GO:0042803~protein homodimerization activity | 29 | 4.07×10−6 |
| GO:0034987~immunoglobulin receptor binding | 5 | 5.43×10−4 | |
| GO:0004872~receptor activity | 11 | 1.51×10−3 | |
| GO:0005515~protein binding | 154 | 2.01×10−3 | |
| GOTERM_CC | GO:0070062~extracellular exosome | 97 | 8.36×10−18 |
| _DIRECT | GO:0005886~plasma membrane | 113 | 1.27×10−13 |
| GO:0009986~cell surface | 24 | 3.43×10−6 | |
| GO:0072562~blood microparticle | 12 | 1.19×10−5 | |
| GO:0005887~integral component of plasma membrane | 37 | 5.02×10−4 |
If there were more than five terms in this category, the first five terms were only selected based on the P-value. ‘Count’ corresponds to the number of enriched genes in each term. BP, biological processes; CC, component classification; MF, molecular function.
The results of the enrichment of DEG function and signaling pathways (Table III) demonstrated that the downregulated genes were mainly associated with the complex biological behavior of CLL. Using P<0.05 as a cut-off criterion, 17 KEGG pathways were over-represented in downregulated DEGs, including ‘hematopoietic cell lineage’, ‘transcriptional dysregulation in cancer’ and ‘pathways in cancer’, while the four KEGG pathways involved in the upregulated DEGs were enriched in multiple signaling pathways, the ‘cyclic adenosine monophosphate (cAMP) signaling pathway’ and the ‘mitogen-activated protein kinase (MAPK) signaling pathway’. These most significantly enriched GO terms and KEGG pathways demonstrated the interactions of DEGs at the functional level.
Table III.
Kyoto Encyclopedia of Genes and Genomes pathway analysis of differentially expressed genes associated with chronic lymphocytic leukemiaa.
| Category | Term | Count | P-value | Genes |
|---|---|---|---|---|
| Upregulated DEGs | hsa04261:Adrenergic signaling in cardiomyocytes | 7 | 9.44×10−3 | ADRB2, ATP2B4, BCL2, PPP2R5C, CREB3L2, CACNB2, RAPGEF3 |
| hsa00561:Glycerolipid metabolism | 4 | 3.52×10−2 | DGKA, LPL, AGPAT5, ALDH3A2 | |
| hsa04024:cAMP signaling pathway | 7 | 3.63×10−2 | ADRB2, ATP2B4, PDE4A, RRAS2, CREB3L2, RAPGEF3, MYL9 | |
| hsa04010:MAPK signaling pathway | 7 | 3.89×10−2 | PTPN7, MAP3K5, RASGRF1, RRAS2, CACNB2, MYC, RASA1, CDC25B | |
| Downregulated DEGs | hsa05144:Malaria | 8 | 3.32×10−5 | KLRB1, GYPC, CR1, KLRC4-KLRK1, ITGB2, HBA1, HBB, TGFB |
| hsa04640:Hematopoietic cell lineage | 9 | 1.90×10−4 | CR1, CR2, CD44, CD1C, CD22, ITGA4, IL7R, ITGAM, CD1D | |
| hsa05202:Transcriptional dysregulation in cancer | 12 | 3.22×10−4 | JUP, CD86, CEBPB, ID2, REL, LMO2, BCL2A1, AFF1, ZBTB16, MYC, ITGAM, KLF3 | |
| hsa05140:Leishmaniasis | 8 | 3.64×10−4 | FOS, CR1, NFKBIA, ITGB2, ITGA4, ITGAM, TGFB1, IFNGR1 | |
| hsa05323:Rheumatoid arthritis | 7 | 6.30×10−3 | FOS, CD86, CCL3, ITGB2, IL15, LTB, TGFB1 |
If there were more than five terms in this category, the first five terms were only selected based on the P-value. ‘Count’ corresponds to the number of enriched genes in each term.
PPI network construction and module selection
Using the STRING online database and Cytoscape software, a total of 354 DEGs were filtered into the complex DEG PPI network, containing 354 nodes and 1,392 edges (Fig. 2); 134 of the 488 DEGs did not fall into the DEG PPI network. Among the 354 nodes, 12 differentially expressed genes with a high degree of node were screened from the PPI network, and the most significant 12 node degree genes were MYC, interleukin (IL)8, B-cell lymphoma 2 (BCL2), integrin subunit alpha M (ITGAM), transforming growth factor (TGF)B1, cluster of differentiation (CD)44, LCK, FYN, Janus kinase (JAK)2, FOS, CD86 and z chain of T cell receptor associated protein kinase (ZAP)-70 (Fig. 3). According to their degree of importance, 2 important modules from the PPI network complex were selected for further analysis based on Cytoscape MCODE. Functional and pathway enrichment analysis of the genes in these two modules were performed using DAVID. The results revealed that Module 1 consisted of 14 nodes and 82 edges (Fig. 4A and B), which were mainly associated with cytokine-cytokine receptor interaction, the JAK/signal transducer and activator of transcription (Jak-STAT) signaling pathway, and that Module 2 consisted of 28 nodes and 101 edges (Fig. 4C and D), which were enriched in the B cell receptor signaling pathway and the chemokine signaling pathway. The results of the module analysis revealed that eight of the 12 DEGs with a high degree were clustered in Module 1, and the other four genes were in Module 2. In order to further understand the interaction of these 12 key genes, the PPI network of these genes was constructed by STRING (Fig. 5). These results suggested that these central genes are closely associated with CLL and interact to promote the development of disease, which may suggest novel therapeutic approaches against CLL.
Figure 2.

Protein-protein interaction network of differentially expressed genes in chronic lymphocytic leukemia.
Figure 3.
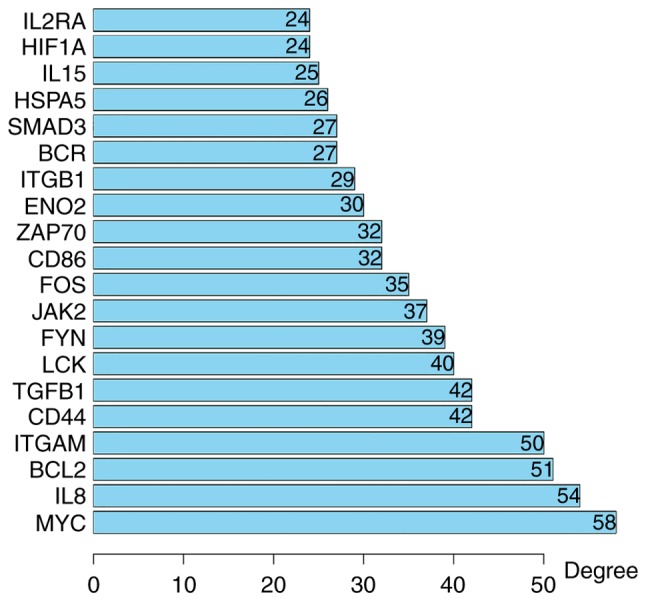
Top 20 differential hub genes in chronic lymphocytic leukemia.
Figure 4.

Top 2 modules from the protein-protein interaction network analysis. (A) Module 1 and (B) the top five most significantly enriched pathways based on the P-value. (C) Module 2 and (D) the top five most significantly enriched pathways based on the P-value.
Figure 5.
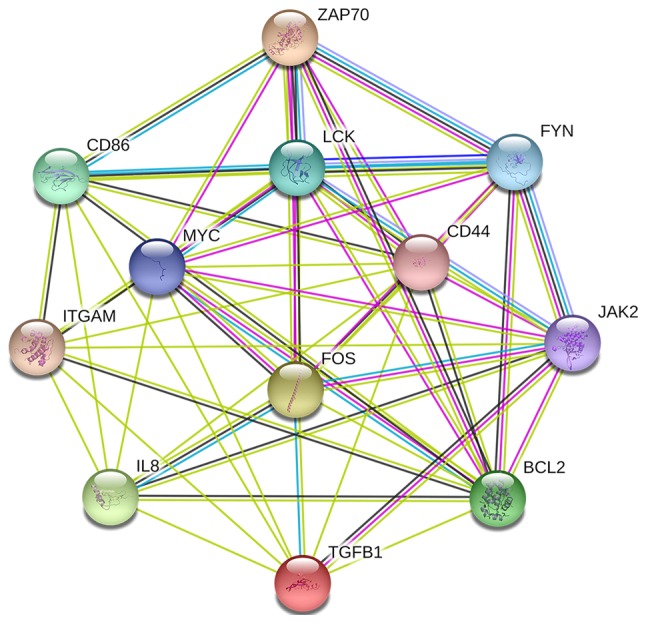
Protein-protein interaction network of the top 12 hub genes in chronic lymphocytic leukemia. Colored lines indicate the type of interaction evidence.
Construction of miRNA-mRNA regulatory network in the pathogenesis of CLL
The target mRNAs of DEMs were predicted using the TargetScan, miRDB and miRTarBase online analysis tools. A total of 621 target mRNAs were predicted, and 32 target mRNAs associated with CLL were screened further by analyzing the association between them and the corresponding DEGs. In the present study, miR-582 was the most significantly upregulated miRNA and was predicted to target the ETS2 and ArfGAP with RhoGAP domain, ankyrin repeat and PH domain (ARAP)2 genes. MiR-144 and miR-181a were the most significantly downregulated miRNAs. MiR-144 was predicted to target the titin (TTN) gene, and miR-188a was predicted to target the zinc finger (ZNF)266, NOTCH2, FK506 binding protein (FKBP)1A, DNA damage inducible transcript (DDIT)4, oxysterol binding protein like (OSBPL)3 and heat shock protein 90 β family member (HSP90B)1 genes. The results also demonstrated that miR-181c and miR-145 targeted six different differentially expressed genes, while miR-584, miR-132 and miR-548c-3p targeted 3 different genes. In addition, DDIT4 could be regulated by hsa-miR-181a, miR-181c, miR-199a-3p and miR-495, while ZNF266, influenza virus NS1A binding protein (IVNS1ABP), OSBPL3, HSP90B1, HSPA5 and NOTCH2 could be regulated by two different miRNAs. A total of 32 target mRNAs corresponded to 17 differentially expressed miRNAs. To better understand the pathogenesis of CLL, miRNA-mRNA regulatory networks (Fig. 6) were constructed and analyzed, and a total of 42 miRNA-mRNA pairs were identified, including 16 positively associated target pairs and 26 negatively associated target pairs (Table IV).
Figure 6.

miRNA-mRNA regulatory network of chronic lymphocytic leukemia. Purple corresponds to the differentially expressed miRNAs and green to the differentially expressed genes (mRNAs) screened. miR, microRNA.
Table IV.
A total of 42 miRNA-mRNA pairs that identified, including 16 positively associated target pairs and 26 negatively associated target pairs.
| Category | miRNA | mRNA |
|---|---|---|
| Positively | hsa-mir-145 | PDGFD |
| associated | hsa-mir-145 | PHACTR2 |
| target pairs | hsa-mir-548c-3p | OGT |
| hsa-mir-548c-3p | IGF2BP3 | |
| hsa-mir-181a | NOTCH2 | |
| hsa-mir-377 | STARD7 | |
| hsa-mir-381 | WEE1 | |
| hsa-mir-145 | SERINC5 | |
| hsa-mir-132 | ASF1A | |
| hsa-mir-377 | RALGPS1 | |
| hsa-mir-132 | RASA1 | |
| hsa-miR-181c-3p | ZNF266 | |
| hsa-mir-181a | FKBP1A | |
| hsa-mir-181a | DDIT4 | |
| hsa-mir-181a | OSBPL3 | |
| hsa-mir-181a | HSP90B1 | |
| Negatively | hsa-mir-145 | IVNS1ABP |
| associated | hsa-mir-501-5p | IVNS1ABP |
| IVNS1ABP | hsa-mir-410 | LDHA |
| target pairs | hsa-mir-181a | ZNF266 |
| hsa-miR-181c-3p | NOTCH2 | |
| hsa-mir-199a-5p | HIF1A | |
| hsa-mir-495-3p | MARCKS | |
| hsa-miR-181c-3p | FKBP1A | |
| hsa-miR-181c-3p | DDIT4 | |
| hsa-mir-199a-3p | DDIT4 | |
| hsa-mir-495 | DDIT4 | |
| hsa-miR-181c-3p | OSBPL3 | |
| hsa-miR-181c-3p | HSP90B1 | |
| hsa-mir-339-5p | BLCAP | |
| hsa-mir-132 | ARHGAP3 | |
| hsa-mir-199a-5p | HSPA5 | |
| hsa-mir-495 | HSPA5 | |
| hsa-mir-582-5p | ETS2 | |
| hsa-mir-548c-3p | PIK3C2B | |
| hsa-mir-381 | TES | |
| hsa-mir-139-5p | FOS | |
| hsa-mir-145 | SMAD3 | |
| hsa-mir-145 | NEDD9 | |
| hsa-mir-582-5p | ARAP2 | |
| hsa-mir-144 | TTN | |
| hsa-mir-346 | PTGS1 |
miRNA, microRNA.
Discussion
As an incurable B-cell malignancy, CLL is the most common leukemia in the western hemisphere (21). Recently, methods such as chemical compounds, stem cell transplantation and monoclonal antibody treatment have demonstrated high remission rates in the treatment of CLL (22,23); however, these treatments have a weak effect on the overall survival of CLL (24). Therefore, there is an urgent need to identify sensitive and specific biomarkers of CLL and to find novel therapeutic targets.
Recently, the rapid development of microarray technology has provided a research platform for further investigation of disease progression and identification of tumor biomarkers, which may facilitate the identification of targets for diagnosis, therapeutics, and prognosis of tumors (25,26). In the present study, genes whose expression was significantly different between CLL and normal samples were identified, and a series of bioinformatics analyses were conducted. A total of 42 miRNA-mRNA pairs, including 17 differential miRNAs and 32 differential mRNAs were identified, and a miRNA-mRNA network analysis was performed to explore the pathogenesis of CLL.
GO and KEGG analysis demonstrated that the DEGs enrichment pathway reflected the complex biological behavior of CLL, including ‘transcriptional dysregulation in cancer’ and ‘hematopoietic cell lineage’. Upregulated DEGs were associated with multiple signaling pathways, such as cAMP and MAPK. MAPK signaling pathway is fundamental for the maintenance of basic cellular processes including proliferation, differentiation, migration and survival (27). A previous study demonstrated that dysregulation of the MAPK signaling pathway is a common mechanism in the pathogenesis of CLL (28,29). These results indicate that the development and progression of cancer cells are consistent with the close association between cell cycle/cell proliferation regulation and the dysfunction of cancer cells.
In addition, a study reported that nodes with a high degree of connectivity serve an important role in maintaining the entire PPI network and are indispensable (30–32). The 12 hub genes screened from the PPI network are the 12 most central nodes in the network and are considered to be the key genes associated with the development of CLL. Based on the significant association between the node structure and its functional importance in the PPI network (32), the 12 hub genes could be used to gain further insight into therapeutic studies of CLL at the molecular level.
As a gene involved in the induction of apoptosis, MYC was identified as one of the mutant genes that demonstrated the highest degree of connectivity in the present study. It is known that MYC serves an oncogenic role in many cancer types, including prostate and bladder cancers (33,34), and serves an important role in leukemia and lymphomas (35,36). In the present study, MYC was differentially expressed and was enriched in the transcriptional dysregulation of cancer and chronic myelogenous leukemia. BCL2 is the first discovered inhibitor of apoptosis and serves a key role in promoting cell survival. Many studies have demonstrated that BCL2 is one of the most important oncogenes involved in cancer (37–39), inducing the development of lymphoma, together with c-MYC overexpression, in particular (40). In the present study, MYC and BCL2 were identified as the top-ranking regulated candidates among the differentially expressed genes in CLL, as previously demonstrated (41,42).
A persistent inflammatory response is the result of excessive production of inflammatory mediators, leading to inflammatory diseases and cancer (43). A study demonstrated that several CLL-associated cytokines, such as IL8, are associated with disease stage, and the correlation of IL-8 expression levels has been described as one of the prognostic factors for CLL (44). It was also demonstrated that IL8 is differentially expressed in CLL and can serve as a biomarker, further demonstrating that the present results have a certain degree of accuracy. CD44, another member of the adhesion factor family, participates in the process of cell proliferation, differentiation, adhesion and migration (45). Studies have demonstrated that anti-CD44 antibodies can interfere with the formation of B lymphocytes and that the downregulation of CD44 leading to the arrest of cell cycle at the G0/G1 phase, resulting in the decrease of proliferation of K562 cells (46–48). In addition, the expression of CD86 in many types of cancerous tissues was significantly lower than that in paracancerous tissues and normal tissues (49). As a member of the Janus family of tyrosine kinases, JAK2 is a key component of chemokine signaling of B lymphocytes in CLL (50). Previous studies demonstrated that inhibition of JAK2 activity in leukemia cells can promote oncogene silencing (51). The mutation of JAK2 activates the JAK-STAT signaling pathway and is involved in the development of myeloproliferative neoplasms (52), suggesting that upregulation of the JAK pathway may be a key factor in the progression of CLL. In addition, a series of studies demonstrated that dysregulated expression of TGFB1, ITGAM, LCK and FYN are involved in the occurrence of cancer and play an important role in tumor progression (53–56).
As a tyrosine protein kinase, ZAP-70 is involved in the signal transduction of T cells (57). Under normal conditions, B cells have a very low expression of ZAP-70, but the expression of ZAP-70 in B cells can be used as a specific and sensitive indicator of B cell diseases, particularly CLL (58). In the present study, the expression of ZAP-70 was upregulated, which is consistent with previous studies (57,59). Gene transcription is regulated by the transcription factor complex, which is widely involved in cell proliferation, differentiation and regulation of transformation (60). As a nuclear proto-oncogene, FOS encoded protein is an important transcription factor, which can induce the transcription and protein expression of its downstream gene that is involved in the regulation of cell proliferation and apoptosis (61). The abnormal expression of FOS can induce cell transformation and tumor formation (62). In the present study, the dysregulation of FOS expression was observed in CLL patients, which may be involved in abnormal gene transcription in CLL.
Module analysis of the 12 central genes from the PPI network revealed that 8 genes belonged to module 1, and 4 belonged to module 2. Functional and pathway enrichment analysis of genes in module 1 indicated that multiple pathways contribute to the development of CLL, including the NF-κB signaling pathway, which is involved in immune and inflammatory responses, and the phosphoinositide 3-kinase (PI3K)-protein kinase B (Akt) signaling pathway, which is related to chronic myelocytic leukemia resistance and cell proliferation regulation (63). In module 2, the genes were mainly involved in the B cell receptor signaling pathway and the Toll-like receptor signaling pathway. The results suggest that dysregulation of these molecular pathways in CLL may serve a role in its pathogenesis and may eventually serve as additional biomarkers to facilitate early diagnosis and treatment.
The aforementioned analyses demonstrated that these genes have a variety of biological functions in tumorigenesis and progression. A previous study demonstrated the presence of a regulatory control network between miRNAs and mRNAs (64), in which miRNAs could silence mRNA translation through sequence-specific targeting; therefore, it was reasonable to investigate the pathogenesis and treatment of tumors by studying the specific miRNA-mRNA co-regulation effect. In the present study, 34 DEMs were identified, including 29 upregulated and 5 downregulated miRNAs in CLL. Among them, miR-582 was the most significantly upregulated miRNA in the GSE62137 dataset, whereas miR-144 and miR-181a were the most significantly downregulated. A total of 42 miRNA-mRNA pairs were identified, including 17 differential miRNAs and 32 differential mRNAs and comprising 16 positively correlated and 26 negatively correlated target pairs.
miR-144 is expressed at low levels in various tumors, such as hepatocellular carcinoma (65), B-cell lymphoma (66) and chronic lymphocytic leukemia (67). In the present study, TTN was predicted to be the target of miR-144, which was upregulated in CLL according to the GSE22529 and GSE39411 datasets. TTN may be a good predictor of disease progression, survival, and treatment response in metastatic prostate cancer (68). Other studies have demonstrated that miR-582 was highly expressed in a variety of tumors, such as colorectal cancer (69) and non-small cell lung carcinoma (70), and that the upregulation of miR-582 contributed to an increase in the proliferation of prostate cancer cells (71) and to the reduction of human bladder cancer proliferation and invasion by suppressing the expression of target genes (72). In the present study, miR-582 was significantly overexpressed in CLL cells compared to normal samples and that it targeted the downregulated ETS2 and ARAP2 genes. There are few reports regarding the role of ETS2 and ARAP2 in cancer. Some studies indicated that the expression of ETS2 might be associated with the progression of CML (73). Further study is needed to determine the role of ETS2 and ARAP2 in the development of CLL. The results of a previous study confirmed that miR-181a was significantly downregulated in CLL (74). NOTCH2, one of the target genes of miR-181a, is involved in the regulation of cellular processes including differentiation, proliferation and apoptosis (75), and dysregulation of NOTCH2 signaling is involved in aberrant CD23 expression in B-cell CLL (76).
As miRNAs can participate in complex biological functions in the body, they can influence a variety of biological processes and pathways through miRNA-mRNA regulatory networks (77,78). Providing that each miRNA potentially regulates a large number of targets, understanding the function of miRNAs in tumorigenesis may provide insight into the key questions that remain in cancer research. The miRNA-mRNA regulatory network highlighted in the present study provides new theoretical guidance for further exploring the mechanism of CLL and provides a novel perspective for understanding the underlying biological processes of CLL and miRNA-targeted therapy.
Taken together, the present data demonstrated that the molecular mechanisms of CLL can be understood and that biomarker prediction can be achieved through data mining and integration. Overall, 488 candidate genes and 32 different miRNAs from multiple datasets were screened by bioinformatics analysis. From the PPI network of DEGs, 12 major altered hub genes and multiple pathways associated with the development of CLL were identified, such as the Jak-STAT signaling pathway, the PI3K-Akt signaling pathway and the MAPK signaling pathway. In addition, the comprehensive bioinformatics analysis of DEGs and the construction of miRNA-mRNA regulatory network provides a bioinformatics approach to further study the pathogenesis of CLL and lay a solid foundation for the personalized treatment of CLL in the future. However, further studies are needed to validate these results.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the National Natural Science Foundation of China (grant no. 81673799) and the National Natural Science Foundation of China Youth Fund (grant no. 81703915).
Availability of data and materials
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.
Authors' contributions
CG, CS and CZ conceived and designed the study. JZ and LL performed data analysis. JW, CL, and HL integrated the data and created the figures. CZ reviewed and revised the article. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352:804–815. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 2.DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, Alteri R, Robbins AS, Jemal A. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014;64:252–271. doi: 10.3322/caac.21235. [DOI] [PubMed] [Google Scholar]
- 3.Fernando TR, Rodriguez-Malave NI, Rao DS. MicroRNAs in B cell development and malignancy. J Hematol Oncol. 2012;5:7. doi: 10.1186/1756-8722-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martini V, Frezzato F, Severin F, Raggi F, Trimarco V, Martinello L, Molfetta R, Visentin A, Facco M, Semenzato G, et al. Abnormal regulation of BCR signalling by c-Cbl in chronic lymphocytic leukaemia. Oncotarget. 2018;9:32219–32231. doi: 10.18632/oncotarget.25951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajendran V. Structural analysis of oncogenic mutation of isocitrate dehydrogenase 1. Mol Biosyst. 2016;12:2276–2287. doi: 10.1039/C6MB00182C. [DOI] [PubMed] [Google Scholar]
- 6.Rajendran V, Gopalakrishnan C, Sethumadhavan R. Pathological role of a point mutation (T315I) in BCR-ABL1 protein-A computational insight. J Cell Biochem. 2018;119:918–925. doi: 10.1002/jcb.26257. [DOI] [PubMed] [Google Scholar]
- 7.Luo P, Yang Q, Cong LL, Wang XF, Li YS, Zhong XM, Xie RT, Jia CY, Yang HQ, Li WP, et al. Identification of miR124a as a novel diagnostic and prognostic biomarker in nonsmall cell lung cancer for chemotherapy. Mol Med Rep. 2017;16:238–246. doi: 10.3892/mmr.2017.6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamaraj B, Gopalakrishnan C, Purohit R. In silico analysis of miRNA-mediated gene regulation in OCA and OA genes. Cell Biochem Biophys. 2014;70:1923–1932. doi: 10.1007/s12013-014-0152-9. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Han X, Wu H, Zhou Y. Bioinformatics analysis of transcription profiling of solid pseudopapillary neoplasm of the pancreas. Mol Med Rep. 2017;16:1635–1642. doi: 10.3892/mmr.2017.6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim L, Lau N, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 12.Bhaumik P, Gopalakrishnan C, Kamaraj B, Purohit R. Single nucleotide polymorphisms in microRNA binding sites: Implications in colorectal cancer. ScientificWorldJournal. 2014;2014:547154. doi: 10.1155/2014/547154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gutierrez A, Jr, Tschumper RC, Wu X, Shanafelt TD, Eckel-Passow J, Huddleston PM III, Slager SL, Kay NE, Jelinek DF. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B-cell lymphocytosis. Blood. 2010;116:2975–2983. doi: 10.1182/blood-2010-02-269878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vallat L, Kemper CA, Jung N, Maumy-Bertrand M, Bertrand F, Meyer N, Pocheville A, Fisher JW III, Gribben JG, Bahram S. Reverse-engineering the genetic circuitry of a cancer cell with predicted intervention in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2013;110:459–464. doi: 10.1073/pnas.1211130110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruiz-Lafuente N, Alcaraz-García MJ, Sebastián-Ruiz S, García-Serna AM, Gómez-Espuch J, Moraleda JM, Minguela A, García-Alonso AM, Parrado A. IL-4 up-regulates MiR-21 and the MiRNAs hosted in the CLCN5 gene in chronic lymphocytic leukemia. PLoS One. 2015;10:e0124936. doi: 10.1371/journal.pone.0124936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 17.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong N, Wang X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43:D146–D152. doi: 10.1093/nar/gku1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou CH, Shrestha S, Yang CD, Chang NW, Lin YL, Liao KW, Huang WC, Sun TH, Tu SJ, Lee WH, et al. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018;46:D296–D302. doi: 10.1093/nar/gkx1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nabhan C, Rosen ST. Chronic lymphocytic leukemia: A clinical review. JAMA. 2014;312:2265–2276. doi: 10.1001/jama.2014.14553. [DOI] [PubMed] [Google Scholar]
- 22.Cang S, Iragavarapu C, Savooji J, Song Y, Liu D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J Hematol Oncol. 2015;8:129. doi: 10.1186/s13045-015-0224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rai KR. Therapeutic potential of new B cell-targeted agents in the treatment of elderly and unfit patients with chronic lymphocytic leukemia. J Hematol Oncol. 2015;8:85. doi: 10.1186/s13045-015-0165-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwok M, Davies N, Agathanggelou A, Smith E, Oldreive C, Petermann E, Stewart G, Brown J, Lau A, Pratt G, et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53 or ATM defective chronic lymphocytic leukemia cells. Blood. 2016;127:582–595. doi: 10.1182/blood-2015-05-644872. [DOI] [PubMed] [Google Scholar]
- 25.Chen F, Shen C, Wang X, Wang H, Liu Y, Yu C, Lv J, He J, Wen Z. Identification of genes and pathways in nasopharyngeal carcinoma by bioinformatics analysis. Oncotarget. 2017;8:63738–63749. doi: 10.18632/oncotarget.19478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun C, Yuan Q, Wu D, Meng X, Wang B. Identification of core genes and outcome in gastric cancer using bioinformatics analysis. Oncotarget. 2017;8:70271–70280. doi: 10.18632/oncotarget.20082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou YY, Li Y, Jiang WQ, Zhou LF. MAPK/JNK signalling: A potential autophagy regulation pathway. Biosci Rep. 2015;35:e00199. doi: 10.1042/BSR20140141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shukla A, Shukla V, Joshi SS. Regulation of MAPK signaling and implications in chronic lymphocytic leukemia. Leuk Lymphoma. 2018;59:1565–1573. doi: 10.1080/10428194.2017.1370548. [DOI] [PubMed] [Google Scholar]
- 29.Pandzic T, Larsson J, He L, Kundu S, Ban K, Akhtar-Ali M, Hellström AR, Schuh A, Clifford R, Blakemore SJ, et al. Transposon mutagenesis reveals fludarabine resistance mechanisms in chronic lymphocytic leukemia. Clin Cancer Res. 2016;22:6217–6227. doi: 10.1158/1078-0432.CCR-15-2903. [DOI] [PubMed] [Google Scholar]
- 30.He X, Zhang J. Why do hubs tend to be essential in protein networks? PLoS Genet. 2006;2:e88. doi: 10.1371/journal.pgen.0020088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barabási A, Oltvai Z. Network biology: Understanding the cell's functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 32.Hahn M, Kern A. Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks. Mol Biol Evol. 2005;22:803–806. doi: 10.1093/molbev/msi072. [DOI] [PubMed] [Google Scholar]
- 33.Fisher KW, Montironi R, Lopez Beltran A, Moch H, Wang L, Scarpelli M, Williamson SR, Koch MO, Cheng L. Molecular foundations for personalized therapy in prostate cancer. Curr Drug Targets. 2015;16:103–114. doi: 10.2174/1389450115666141229154500. [DOI] [PubMed] [Google Scholar]
- 34.Wu X, Liu D, Tao D, Xiang W, Xiao X, Wang M, Wang L, Luo G, Li Y, Zeng F, Jiang G. BRD4 regulates EZH2 transcription through upregulation of C-MYC and represents a novel therapeutic target in bladder cancer. Mol Cancer Ther. 2016;15:1029–1042. doi: 10.1158/1535-7163.MCT-15-0750. [DOI] [PubMed] [Google Scholar]
- 35.Sánchez-Beato M, Sánchez-Aguilera A, Piris MA. Cell cycle deregulation in B-cell lymphomas. Blood. 2003;101:1220–1235. doi: 10.1182/blood-2002-07-2009. [DOI] [PubMed] [Google Scholar]
- 36.Delgado MD, León J. Myc roles in hematopoiesis and leukemia. Genes Cancer. 2010;1:605–616. doi: 10.1177/1947601910377495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 38.Dawson SJ, Makretsov N, Blows FM, Driver KE, Provenzano E, Le Quesne J, Baglietto L, Severi G, Giles GG, McLean CA, et al. BCL2 in breast cancer: A favourable prognostic marker across molecular subtypes and independent of adjuvant therapy received. Br J Cancer. 2010;103:668–675. doi: 10.1038/sj.bjc.6605736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng H, Wang X, Li T, Chen L. Bcl-2 expression and patient survival in gastric cancer: A systematic review of the literature with meta-analysis. Med Oncol. 2015;32:389. doi: 10.1007/s12032-014-0389-6. [DOI] [PubMed] [Google Scholar]
- 40.Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 41.Landau D, Tausch E, Taylor-Weiner A, Stewart C, Reiter JG, Bahlo J, Kluth S, Bozic I, Lawrence M, Böttcher S, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–530. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robertson L, Plunkett W, McConnell K, Keating M, McDonnell T. Bcl-2 expression in chronic lymphocytic leukemia and its correlation with the induction of apoptosis and clinical outcome. Leukemia. 1996;10:456–459. [PubMed] [Google Scholar]
- 43.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wierda WG, Johnson MM, Do KA, Manshouri T, Dey A, O'Brien S, Giles FJ, Kantarjian H, Thomas D, Faderl S, et al. Plasma interleukin 8 level predicts for survival in chronic lymphocytic leukaemia. Br J Haematol. 2003;120:452–456. doi: 10.1046/j.1365-2141.2003.04118.x. [DOI] [PubMed] [Google Scholar]
- 45.Slevin M, Krupinski J, Gaffney J, Matou S, West D, Delisser H, Savani RC, Kumar S. Hyaluronan-mediated angiogenesis in vascular disease: Uncovering RHAMM and CD44 receptor signaling pathways. Matrix Biol. 2007;26:58–68. doi: 10.1016/j.matbio.2006.08.261. [DOI] [PubMed] [Google Scholar]
- 46.Chang G, Zhang H, Wang J, Zhang Y, Xu H, Wang C, Zhang H, Ma L, Li Q, Pang T. CD44 targets Wnt/β-catenin pathway to mediate the proliferation of K562 cells. Cancer Cell Int. 2013;13:117. doi: 10.1186/1475-2867-13-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krause DS, Lazarides K, von Andrian UH, Van Etten RA. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med. 2006;12:1175–1180. doi: 10.1038/nm1489. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Yang J, Li J, Wang RC, Yuan J, Li Y, Wang SY, Wang C, Hao HL. Effect of arsenic trioxide on K562 cell proliferation and its mechanism. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2017;25:90–93. doi: 10.7534/j.issn.1009-2137.2017.01.015. (In Chinese) [DOI] [PubMed] [Google Scholar]
- 49.Yang WF, WANG FZ. Expression of CD1a, CD80, CD86 in dendritic cell of tumor tissue and regional lymph node in esophageal carcinoma. Ai Zheng. 2004;23:189–192. (In Chinese) [PubMed] [Google Scholar]
- 50.Montresor A, Toffali L, Mirenda M, Rigo A, Vinante F, Laudanna C. JAK2 tyrosine kinase mediates integrin activation induced by CXCL12 in B-cell chronic lymphocytic leukemia. Oncotarget. 2015;6:34245–34257. doi: 10.18632/oncotarget.5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tefferi A, Pardanani A. JAK inhibitors in myeloproliferative neoplasms: Rationale, current data and perspective. Blood Rev. 2011;25:229–237. doi: 10.1016/j.blre.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 52.Zhang HY, Zhai XW. Research advances in the role of JAK2 mutations in acute leukemia. Zhongguo Dang Dai Er Ke Za Zhi. 2015;17:644–649. (In Chinese) [PubMed] [Google Scholar]
- 53.Blahna MT, Hata A. Smad-mediated regulation of microRNA biosynthesis. FEBS Lett. 2012;586:1906–1912. doi: 10.1016/j.febslet.2012.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fabregat I, Moreno-Càceres J, Sànchez A, Dooley S, Dewidar B, Giannelli G, Ten Dijke P. IT-LIVER Consortium: TGF-β signalling and liver disease. FEBS J. 2016;283:2219–2232. doi: 10.1111/febs.13665. [DOI] [PubMed] [Google Scholar]
- 55.Serafin V, Lissandron V, Buldini B, Bresolin S, Paganin M, Grillo F, Andriano N, Palmi C, Cazzaniga G, Marmiroli S, et al. Phosphoproteomic analysis reveals hyperactivation of mTOR/STAT3 and LCK/Calcineurin axes in pediatric early T-cell precursor ALL. Leukemia. 2017;31:1007–1011. doi: 10.1038/leu.2017.13. [DOI] [PubMed] [Google Scholar]
- 56.Ban K, Gao Y, Amin HM, Howard A, Miller C, Lin Q, Leng X, Munsell M, Bar-Eli M, Arlinghaus RB, Chandra J. BCR-ABL1 mediates up-regulation of Fyn in chronic myelogenous leukemia. Blood. 2008;111:2904–2908. doi: 10.1182/blood-2007-05-091769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen L, Widhopf G, Huynh L, Rassenti L, Rai KR, Weiss A, Kipps TJ. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2002;100:4609–4614. doi: 10.1182/blood-2002-06-1683. [DOI] [PubMed] [Google Scholar]
- 58.Orchard JA, Ibbotson RE, Davis Z, Wiestner A, Rosenwald A, Thomas PW, Hamblin TJ, Staudt LM, Oscier DG. ZAP-70 expression and prognosis in chronic lymphocytic leukaemia. Lancet. 2004;363:105–111. doi: 10.1016/S0140-6736(03)15260-9. [DOI] [PubMed] [Google Scholar]
- 59.Purroy N, Abrisqueta P, Carabia J, Carpio C, Palacio C, Bosch F, Crespo M. Co-culture of primary CLL cells with bone marrow mesenchymal cells, CD40 ligand and CpG ODN promotes proliferation of chemoresistant CLL cells phenotypically comparable to those proliferating in vivo. Oncotarget. 2015;6:7632–7643. doi: 10.18632/oncotarget.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kolla V, Gonzales L, Gonzales J, Wang P, Angampalli S, Feinstein SI, Ballard PL. Thyroid transcription factor in differentiating type II cells: Regulation, isoforms, and target genes. Am J Respir Cell Mol Biol. 2007;36:213–225. doi: 10.1165/rcmb.2006-0207OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Durchdewald M, Angel P, Hess J. The transcription factor Fos: A Janus-type regulator in health and disease. Histol Histopathol. 2009;24:1451–1461. doi: 10.14670/HH-24.1451. [DOI] [PubMed] [Google Scholar]
- 62.Wagstaff SC, Bowler WB, Gallagher JA, Hipskind RA. Extracellular ATP activates multiple signalling pathways and potentiates growth factor-induced c-fos gene expression in MCF-7 breast cancer cells. Carcinogenesis. 2000:2175–2181. doi: 10.1093/carcin/21.12.2175. [DOI] [PubMed] [Google Scholar]
- 63.Zhao L, Shan Y, Liu B, Li Y, Jia L. Functional screen analysis reveals miR-3142 as central regulator in chemoresistance and proliferation through activation of the PTEN-AKT pathway in CML. Cell Death Dis. 2017;8:e2830. doi: 10.1038/cddis.2017.223. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, et al. Mammalian microRNAs: Experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992–1009. doi: 10.1101/gad.1884710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cao T, Li H, Hu Y, Ma D, Cai X. miR-144 suppresses the proliferation and metastasis of hepatocellular carcinoma by targeting E2F3. Tumour Biol. 2014;35:10759–10764. doi: 10.1007/s13277-014-2017-7. [DOI] [PubMed] [Google Scholar]
- 66.Wang H, Wang A, Hu Z, Xu X, Liu Z, Wang Z. A critical role of miR-144 in diffuse large B-cell lymphoma proliferation and invasion. Cancer Immunol Res. 2016;4:337–344. doi: 10.1158/2326-6066.CIR-15-0161. [DOI] [PubMed] [Google Scholar]
- 67.Machová Poláková K, Lopotová T, Klamová H, Burda P, Trněný M, Stopka T, Moravcová J. Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol Cancer. 2011;10:41. doi: 10.1186/1476-4598-10-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tomioka A, Tanaka N, Yoshikawa M, Miyake M, Anai S, Chihara Y, Okajima E, Hirayama A, Hirao Y, Fujimoto K. Nadir PSA level and time to nadir PSA are prognostic factors in patients with metastatic prostate cancer. BMC Urol. 2014;14:33. doi: 10.1186/1471-2490-14-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Song B, Long Y, Liu D, Zhang W, Liu C. MicroRNA-582 promotes tumorigenesis by targeting phosphatase and tensin homologue in colorectal cancer. Int J Mol Med. 2017;40:867–874. doi: 10.3892/ijmm.2017.3059. [DOI] [PubMed] [Google Scholar]
- 70.Fang L, Cai J, Chen B, Wu S, Li R, Xu X, Yang Y, Guan H, Zhu X, Zhang L, et al. Aberrantly expressed miR-582-3p maintains lung cancer stem cell-like traits by activating Wnt/β-catenin signalling. Nat Commun. 2015;6:8640. doi: 10.1038/ncomms9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maeno A, Terada N, Uegaki M, Goto T, Okada Y, Kobayashi T, Kamba T, Ogawa O, Inoue T. Up-regulation of miR-582-5p regulates cellular proliferation of prostate cancer cells under androgen-deprived conditions. Prostate. 2014;74:1604–1612. doi: 10.1002/pros.22877. [DOI] [PubMed] [Google Scholar]
- 72.Uchino K, Takeshita F, Takahashi RU, Kosaka N, Fujiwara K, Naruoka H, Sonoke S, Yano J, Sasaki H, Nozawa S, et al. Therapeutic effects of microRNA-582-5p and −3p on the inhibition of bladder cancer progression. Mol Ther. 2013;21:610–619. doi: 10.1038/mt.2012.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lafage-Pochitaloff M, Courcoul M, Simonetti J, Sainty D, Dastugue N, Tabilio A, Hagemeijer A, Birg F. Expression of the ETS2 and transferrin receptor genes in Philadelphia-positive chronic myeloid leukemia patients with a reciprocal t(3;21) Genes Chromosomes Cancer. 1992;5:1–13. doi: 10.1002/gcc.2870050102. [DOI] [PubMed] [Google Scholar]
- 74.Zhu DX, Miao KR, Fang C, Fan L, Zhu W, Zhu HY, Zhuang Y, Hong M, Liu P, Xu W, Li JY. Aberrant microRNA expression in Chinese patients with chronic lymphocytic leukemia. Leuk Res. 2011;35:730–734. doi: 10.1016/j.leukres.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 75.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: Cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 76.Hubmann R, Schwarzmeier JD, Shehata M, Hilgarth M, Duechler M, Dettke M, Berger R. Notch2 is involved in the overexpression of CD23 in B-cell chronic lymphocytic leukemia. Blood. 2002;99:3742–3747. doi: 10.1182/blood.V99.10.3742. [DOI] [PubMed] [Google Scholar]
- 77.Song HM, Luo Y, Li DF, Wei CK, Hua KY, Song JL, Xu H, Maskey N, Fang L. MicroRNA-96 plays an oncogenic role by targeting FOXO1 and regulating AKT/FOXO1/Bim pathway in papillary thyroid carcinoma cells. Int J Clin Exp Pathol. 2015;8:9889–9900. [PMC free article] [PubMed] [Google Scholar]
- 78.Chruścik A, Lam AK. Clinical pathological impacts of microRNAs in papillary thyroid carcinoma: A crucial review. Exp Mol Pathol. 2015;99:393–398. doi: 10.1016/j.yexmp.2015.08.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.