

Abstract
The gut-liver axis connects the liver with the intestine via bile acid metabolism. Bile acid dysregulation leads to intestinal dysbiosis, that allows enterogenous pathogenic bacteria, including Gram-negative bacteria and their products lipopolysaccharide (LPS), into the liver via the portal vein, triggering inflammation in the liver. The inflammasome serves as an intracellular pattern recognition receptor that detects pathogens or danger signals and mediates innate immunity in the liver or gut. Specifically, the NACHT, LRR and PYD domains-containing protein (NLRP)6 inflammasome maintains intestinal microbial balance, by promoting interleukin (IL)-18-dependent antimicrobial peptide synthesis and mucus secretion from goblet cells. The NLRP3 inflammasome, in contrast, primarily induces IL-1β and aggravates inflammatory liver injury. Furthermore, the NLRP3 inflammasome affects the epithelial integrity of cholangiocytes by inducing the production of pro-inflammatory cytokines. In addition, bile acids, including deoxycholic acid and chenodeoxycholic acid, are able to activate the NLRP3 inflammasome in macrophages; however, bile acids have the potential to exert the opposite role by interacting with the membrane-bound Takeda G-protein receptor 5 or by activating nuclear farnesoid-X receptor. Therefore, further investigation of the molecular mechanisms underlying the inflammasome, involved in the gut-liver axis, may provide important insights into the identification of a potential therapeutic target for the treatment of liver and gut diseases. The present review discusses the roles of the inflammasome in the gut-liver axis, and the emerging associations between the inflammasome and the intestinal microbiota or the bile acids in the gut-liver axis.
Keywords: bile acid, gut-liver axis, inflammation, intestinal microbiota, NLRP3 inflammasome, NLRP6 inflammasome
1. Introduction
The liver and intestine are connected by the gut-liver axis (1). The liver is an endocrine gland that secretes bile acids (BAs) into the intestine to maintain the stability of the intestinal flora (2,3). Gut-liver axis dysfunction is characterized by metabolic disorders of Bas (1). Excessive BAs in the liver induce hepatocyte death and aggravate inflammatory injury (4–7). Decreased BAs in the gut, in contrast, lead to intestinal dysbiosis that impairs intestinal barrier function, inducing bacterial translocation to allow pathogens, including Bacteroidetes (Gram-negative bacteria) and their products, lipopolysaccharide (LPS), into the liver, aggravating hepatic inflammation (8,9). Previous studies demonstrated the significance of the gut-liver axis (1,10,11). Retrospective analyses of a large cohort of clinical samples demonstrated that patients with inflammatory bowel disease had a higher incidence of primary sclerosing cholangitis (12,13). These previous studies suggested that bacterial translocation and bacteremia in the portal vein aggravated primary sclerosing cholangitis. Similarly, loss of intestinal epithelial stemness contributed to bile duct ligation-induced cholestatic liver injury (14). Furthermore, postnatal development of intestinal microbiota, including Proteobacteria, was identified as an important susceptibility factor for biliary atresia in mice (15).
The inflammasome is a large multiprotein complex that recognizes diverse microbial-, stress- and danger-associated signals, and subsequently triggers the maturation of pro-inflammatory cytokines, including interleukin (IL)-1β and IL-18, promoting innate immunity (16). IL-1β, activated by the inflammasome, is involved in liver inflammation (17), whereas, IL-18 has been demonstrated to be involved in modulating the gut microbiota (18). Previous studies demonstrated that the intestinal epithelial NACHT, LRR and PYD domains-containing protein (NLRP)6 inflammasome maintains the intestinal barrier and the intestinal microbial balance (19,20), whereas, the NLRP3 inflammasome serves as an intracellular pattern recognition receptor (PRR) that mediates innate immunity and aggravates inflammatory liver injury (21–23). Furthermore, inflammasome inhibitors, including the caspase inhibitor IDN-6556, have exhibited protective effects in liver injury (24).
In the gut-liver axis, metabolic disorders of BAs caused the imbalance of intestinal microflora and hepatic inflammatory injury (25), and the inflammasome has been demonstrated to be associated with gut barrier integrity, microbial composition and liver injury (18,26). Furthermore, BAs serve as danger signals that may have a direct or indirect effect on the intracellular inflammasome (21,27–29). Therefore, understanding the role of the inflammasome in the gut-liver axis may provide insights into effective treatments for liver and gut diseases. The present review discusses the roles of the inflammasome in the gut-liver axis and the associations between the inflammasome and the intestinal microbiota or the BAs.
2. Inflammasomes are intracellular PRRs
The inflammasome, whose term was initially proposed in 2002 (30), is comprised of multiple proteins present in the cytoplasm and serves as a PRR to recognize various inflammatory stimulations (Table I), including exogenous pathogens [pathogen-associated molecular patterns (PAMPs)], and endogenous signals from damaged or dying cells [damage-associated molecular patterns (DAMPs)], thereby recruiting and regulating the production of inflammatory cytokines (16). In addition, the inflammasome is a key component of innate immunity and serves a role in the regulation of inflammation under various injury conditions. In previous studies, it was demonstrated that the inflammasome is involved in a number of diseases, including liver fibrosis, primary sclerosing cholangitis, cholestasis and biliary obstruction (21,23,31,32).
Table I.
Inflammasome activators.
| A, Whole pathogens | |
|---|---|
| Activators | Associated diseases |
| Bacteria | Infection |
| fungus | Infection |
| Virus | Infection |
| Parasite | Infection |
| B, PAMPs | |
| Activators | Associated diseases |
| LPS | Infection |
| Bacterial pore-forming toxins | Infection |
| Hemozoin | Malaria |
| C, Environmental insults | |
| Activators | Associated diseases |
| Silica | Silicosis |
| Asbestos | Asbestosis |
| Ultraviolet light | Sunburn |
| D, DAMPs | |
| Activators | Associated diseases |
| ATP | Necrosis |
| Glucose | Metabolic syndrome |
| Uric acid | Gout |
| Amyloid β | Alzheimer's disease |
| Cytochrome C | Apoptosis |
| ROS | Allergy |
| Heat-shock protein | Japanese encephalitis virus infection |
| Defensins | Tuberculosis infection |
| HMGB1 | Glaucoma |
| Fatty acids | Obesity/Type 2 diabetes |
| Hyaluronic acid | Airway hyperresponsiveness |
| Mitochondrial DNA | Autophagy and apoptosis |
| Cytoplasmic DNA | Pyroptosis |
| S100 proteins | Rheumatoid arthritis/Crohn's disease |
| Albumin | Renal tubular injury |
ATP, adenosine 5′-triphosphate; DAMPs, damage-associated molecular patterns; HMGB1, High-mobility group box 1; LPS, lipopolysaccharide; PAMPs, pathogen-associated molecular patterns; ROS, reactive oxygen species.
The inflammasome is comprised of a sensor protein, an adaptor protein and caspase-1 (16). The sensor protein family includes NOD-like receptors (NLRs), including NLRP1, NLRP3, NLR family CARD domain-containing protein 4, NLRP6 and the PYHIN family protein interferon-inducible protein AIM2 (33). The NLRP3 inflammasome is the best characterized inflammasome and consists of NLRP3, the apoptosis-associated speck-like protein containing a CARD (ASC) adaptor and pro-caspase-1 (Fig. 1) (34). Furthermore, NLRP3 contains C-terminal leucine-rich repeats (LRRs), a central nucleotide-binding and oligomerization (NACHT) domain, and an N-terminal pyrin domain (PYD) (35). In the presence of PAMPs and DAMPs, NLRP3 oligomerizes and combines with ASC to assemble the inflammasome, recruiting and activating caspase-1, that cleaves the precursors of inflammatory cytokines, in order to produce and release the mature forms of IL-1β and IL-18 (16).
Figure 1.
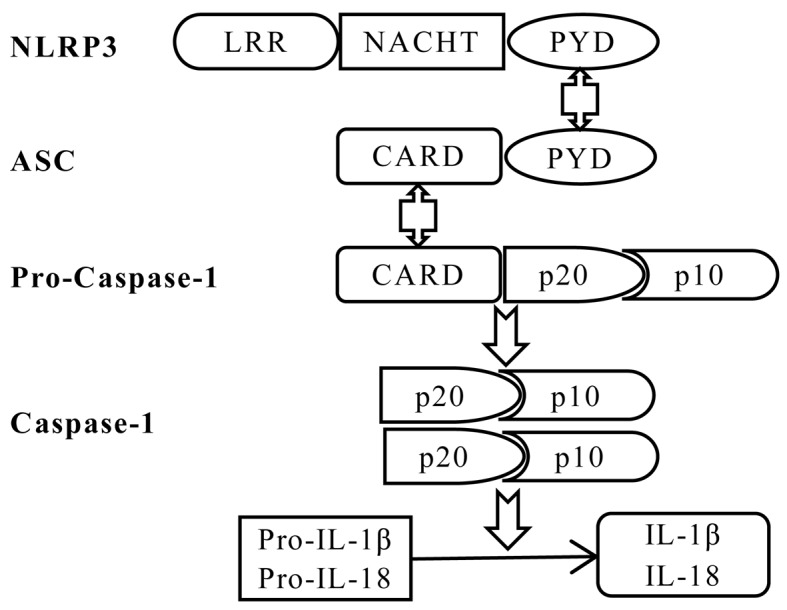
Schematic diagram of NLRP3 inflammasome assembly. The NLRP3 inflammasome is assembled by NLRP3, the ASC adaptor and pro-caspase-1. Upon NLRP3 activation, NLRP3 interacts with ASC via PYDs, and the CARD domain of ASC recruits the CARD of pro-caspase-1, leading to autocleavage of the inactive CARD domain from pro-caspase-1. This cleavage allows the formation of the active caspase-1 p10/p20 tetramer, which cleaves cytokine precursors to produce and release mature IL-1β and IL-18. IL, interleukin; CARD, caspase recruitment domain; LRR, leucine-rich repeat; NACHT, nucleotide-binding and oligomerization domain; PYD, pyrin domain; NLRP3, NACHT, LRR and PYD domains-containing protein 3; ASC, apoptotic speck-like protein containing a CARD.
In total, two signals lead to the activation of the NLRP3 inflammasome (Fig. 2). In the first signal, PAMPs or DAMPs interact with toll-like receptors (TLRs) on the cell surface, leading to nuclear factor-κB (NF-κB)-dependent transcription and translation of pro-IL-1β and pro-IL-18 (36). In addition, NF-κB activates NLRP3 gene transcription by binding to its promoter, which is the limiting step for activation of the NLRP3 inflammasome (37). The second signal involves three concomitant molecular mechanisms. In the first, extracellular adenosine 5′-triphosphate (ATP) induces P2X purinoceptor 7-dependent pore formation on the cell membrane and promotes an intracellular K+ efflux (38), resulting in the translocation of large-pored pannexin-1 channels into the membrane (39). Subsequently, PAMPs or DAMPs enter the cell and activate the NLRP3 inflammasome. Furthermore, phagocytosis of large molecules, including crystalized cholesterol and uric acid, induces lysosomal disruption, resulting in the release of its components and activation of the NLRP3 inflammasome (40). Additionally, thioredoxin-interacting proteins dissociate from thioredoxin and bind to the NLRP3 inflammasome to trigger its activation under the effect of reactive oxygen species (ROS) derived from mitochondria (41). Notably, the aforementioned mechanisms occur simultaneously. Furthermore, the second signal leads to the cleavage and release of pro-IL-1β and pro-IL-18 into IL-1β and IL-18, respectively (16). Therefore, activation of the NLRP3 inflammasome is associated with the recognition of pathogens involved in innate immunity, and senses an imbalance in cell homeostasis, including K+ efflux, Ca2+ signaling, mitochondrial dysfunction and lysosomal rupture.
Figure 2.

Mechanisms involved in NLRP3 inflammasome activation. Two signals are required for the activation of the NLRP3 inflammasome. For signal 1, PAMPs and DAMPs combine with TLRs on the cell membrane to activate NF-κB-dependent transcription and translation of NLRP3, pro-IL-1β and pro-IL-18. For signal 2, three mechanisms have been described. In the first, ATP interacts with P2X7, leading to intracellular K+-depletion and opening of a large-pored pannexin-1 channel, through which PAMPs and DAMPs enter the cell and activate the NLRP3 inflammasome. Furthermore, endocytosis of large molecules, including crystals, results in lysosomal disruption, leading to the release of its components and activation of the NLRP3 inflammasome. Additionally, mitochondria-derived ROS detach TXNIP from thioredoxin and enable activation of the NLRP3 inflammasome. The second signal results in caspase-1 activation, and cleavage of pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18. ASC, apoptosis-associated speck-like protein containing a CARD; DAMPs, danger-associated molecular patterns; NF-κB, nuclear factor-κB; P2X7, P2X purinoceptor 7; PAMPs, pathogen-associated molecular patterns; ROS, reactive oxygen species; TLR, Toll-like receptor; TXNIP, thioredoxin-interacting protein; IL, interleukin; NLRP3, NACHT, LRR and PYD domains-containing protein 3; ATP, adenosine 5′-triphosphate.
3. Inflammasome and intestinal homeostasis
Intestinal homeostasis requires an intact intestinal barrier and a balanced intestinal flora. In the following sections, different roles of the NLRP6 and NLRP3 inflammasome in the two components of intestinal homeostasis are discussed.
Intestinal epithelial NLRP6 inflammasome maintains the intestinal barrier and microbial balance
In humans and mice, NLRP6 is highly expressed in epithelial cells of the small intestine, colon and goblet cells (19,42,43), and is co-expressed with ASC and caspase-1 in the intestinal epithelium (20). The NLRP6 inflammasome mediates the interaction between the intestinal immune system and gut microbes, and its activation mechanisms are summarized in Fig. 3. In infancy, the colonization of gut microbes, including Proteobacteria, Firmicutes and Actinobacteria (44), led to the upregulation of the NLRP6 inflammasome and its downstream cytokines (20,45). Therefore, gut commensal microbes represent one of the first signals to induce NLRP6 inflammasome-dependent antimicrobial responses. Furthermore, microbial metabolites regulate the NLRP6 inflammasome metabolism via the second signal; microbiota-associated metabolite taurine promotes NLRP6 signaling and IL-18 synthesis, whereas, spermine and histamine inhibit NLRP6 inflammasome activity (20). Notably, the protozoan Tritrichomonas musculis was described to induce epithelial IL-18 secretion through ASC inflammasome activation (46). In addition to microbial activation mechanisms, the stress-induced corticotrophin-releasing hormone inhibited the expression of intestinal NLRP6 inflammasome in rats deprived of water (47), whereas, the nuclear transcription factor peroxisome proliferator-activated receptor-γ activated the NLRP6 inflammasome by binding to the NLRP6 promoter (45).
Figure 3.

Mechanisms involved in NLRP6 inflammasome activation. Intestinal microbiota initiate two signals for the activation of the NLRP6 inflammasome. In the first signal, the commensal microbiota serve as a TLR ligand and promotes the transcription of NLRP6 and pro-IL-18. For the second signal, microbial metabolites, including taurine, promote the multiprotein complex assembly to activate the NLRP6 inflammasome. In particular, commensal protozoans promote epithelial IL-18 secretion via activation of the ASC inflammasome. In addition to the microbial roles, CRH inhibits the transcription of NLRP6 inflammasome components, whereas, the nuclear transcription factor PPAR-γ activates NLRP6 by binding to its promoter region. Arrows indicate ‘promotion’, whereas, the symbol ‘┴’ indicates ‘inhibition’. CRH, corticotrophin-releasing hormone; PPAR-γ, peroxisome proliferator-activated receptor-γ; NLRP6, NACHT, LRR and PYD domains-containing protein 6; IL-18, interleukin 18; ASC, apoptosis-associated speck-like protein containing a CARD; TLR, Toll-like receptor.
Accumulating evidence suggested that the NLRP6 inflammasome regulates host-gut commensal microbiota interaction via two mechanisms. In the first mechanism, the NLRP6 inflammasome promotes the secretion of mucus by goblet cells (19). NLRP6−/− mice with defective goblet cell exocytosis were vulnerable to colonization by Citrobacter rodentium, and bacteria penetrated in the epithelial crypts deeper, compared with wild-type mice (19). One possible mechanism for this phenotype may involve intestinal epithelium autophagy dysfunction. A recent study demonstrated that sentinel goblet cells secrete mucus when activated by the NLRP6 inflammasome in a calcium-dependent manner (48). In the second mechanism, the NLRP6 inflammasome maintains the balance of the intestinal flora by promoting intestinal epithelial cells to synthesize antimicrobial peptides, including angiogenin-4, intelectin-1 and resistin-like molecule β (20). In this process, gut microbiota initially activate the NLRP6 inflammasome and induce intestinal epithelial synthesis of IL-18, which promotes the synthesis of antimicrobial peptides in an autocrine or in an IL-22-dependent manner (49). In addition, the NLRP6 inflammasome is involved in preventing enterovirus infection by binding to viral RNA, promoting the expression of interferon-stimulated genes (50).
The microbial metabolites/NLRP6 inflammasome/IL-18/antimicrobial peptide axis serves a critical role in the regulation of the intestinal inflammatory response (20). Due to the presence of the aforementioned metabolic axis, NLRP6−/− mice were unable to synthesize the protective antimicrobial peptides (42,51), resulting in intestinal flora disorder, characterized by the increase of Prevotellaceae species and members of the TM7 phylum, and the decrease of bacteria belonging to the genus Lactobacillus, phylum Firmicutes (42). Notably, the phenotype of exacerbated colitis in NLRP6−/− mice may be transferred to wild-type mice (20). The reason for this phenomenon involves the transfer of dysbiotic microbiota from inflammasome-deficient mice to wild-type mice. Although wild-type mice have an intact NLRP6 inflammasome, dysbiotic microbiota-produced metabolites, including spermine and histamine have the ability to inhibit NLRP6 inflammasome-associated signals and suppress colonic IL-18 expression levels, thereby reducing antimicrobial peptide synthesis, leading to the development of dysbiosis and aggravated colitis in wild-type mice. Nevertheless, antibiotic treatment in addition to exogenous administration of IL-18 abrogated all the aforementioned effects (20). Collectively, these data demonstrated that inflammasome-deficient mice possess a communicable dysbiotic microbiota that enhances susceptibility to colitis. Furthermore, NLRP6-deficient mice had increased susceptibility to colitis-associated colon tumorigenesis, which may be caused by microbiota-induced chemokine (C-C motif) ligand 5 (CCL5)-driven inflammation. The subsequent inability to resolve inflammation and repair damaged epithelium, promotes epithelial cell proliferation, leading to the formation of cancer (52–54).
A previous study suggested that the NLRP6 inflammasome has an indirect effect on the liver by regulating gut microbiota, and mice lacking the NLRP6 inflammasome presented dysbiotic microbiota consisting of Porphyromonadaceae and Prevotellaceae. Furthermore, these intestinal bacteria or their derived products, including LPS and double-stranded bacterial DNA, triggered activation of TLR4 and TLR9 in the portal circulation, and enhanced tumor necrosis factor (TNF)-α expression in the liver, aggravating the progression of non-alcoholic fatty hepatitis in mice (18). Collectively, the NLRP6 inflammasome serves a key role in maintaining the intestinal flora and preventing metabolic liver injury.
Intestinal NLRP3 inflammasome is involved in mucosal defense
The NLRP3 inflammasome has been extensively studied in myeloid cells; however, the presence of NLRP3 in the gut epithelium is controversial. Previous studies demonstrated that the NLRP3 gene is highly expressed in intestinal bone marrow-derived cells, including macrophages and monocytes (55–57). Therefore, in the majority of the previous studies, the role of the NLRP3 inflammasome in intestinal diseases has been investigated by examining its role in myeloid cells. In colonic epithelial cells, NLRP3 expression level is low or approximately undetectable (57,58). However, it was recently observed that the NLRP3 protein was expressed in human intestinal epithelial cells (59).
The NLRP3 inflammasome exacerbates intestinal inflammation. In a rat colitis model, increased expression levels of the NLRP3 inflammasome were observed (60). Intestinal pathogens activated the NLRP3 inflammasome and induced IL-β production in monocytes, aggravating intestinal inflammation (55). In addition, titanium dioxide-containing nanoparticles exacerbated dextran sodium sulfate (DSS)-induced colitis by activating the NLRP3 inflammasome in intestinal epithelial cells and macrophages (59). Furthermore, Escherichia coli isolated from patients with inflammatory bowel disease activated the NLRP3 inflammasome, exacerbating the inflammatory response (61). Conversely, in NLRP3−/− mice, colonic inflammation in a model of inflammatory bowel disease was attenuated (62).
The NLRP3 inflammasome exerts a protective effect on intestinal inflammation by maintaining the intestinal mucosa. Allen et al (57) demonstrated that the NLRP3 inflammasome served a protective role in DSS-induced colitis, and that NLRP3−/− mice exhibited more severe colitis. These observations may be explained by the following mechanisms. Hirota et al (63) demonstrated that the expression levels of IL-1β-induced anti-inflammatory cytokine IL-10 and transforming growth factor β1 (TGF-β1) were decreased in NLRP3−/− mice. Additionally, peritoneal macrophages isolated from NLRP3−/− mice did not exhibit a response to bacterial muramyldipeptide and neutrophils exhibited decreased chemotaxis and increased apoptosis. Furthermore, NLRP3−/− mice exhibited decreased expression levels of β-defensin and antimicrobial peptides. Conversely, the level of pathogenic microorganisms, including Enterobacteriaceae and Mycobacterium was increased. Additionally, NLRP3−/− mice exhibited decreased expression levels of IL-18, resulting in the loss of intestinal epithelial integrity, systemic leukocyte infiltration, and increased chemokine levels, aggravating DSS-induced colitis (64).
The controversial role of the NLRP3 inflammasome in intestinal inflammation may be attributed to one or more of the following factors. Previous studies on NLRP3 inflammasome function primarily relied on transgenic animal models (62–64), in which breeding, environmental factors or different models of inflammation may affect the course of the disease. Furthermore, the role of the NLRP3 inflammasome in intestinal inflammation is predominantly based on the cell types affected. As mentioned above, in case the NLRP3 inflammasome is activated in intestinal epithelial cells, the synthesis of defensin is promoted by inducing IL-18 to maintain the intestinal microbial balance, thereby exerting a protective effect (64). However, once the intestinal epithelial barrier is destroyed, micro-organisms and antigens may access the intestinal lamina propria. At this level, PAMPs may be recognized by macrophages and by dendritic cells via PRRs. Subsequently, the NLRP3 inflammasome in myeloid cells is activated, inducing IL-1β, thereby aggravating inflammation (55).
Similar to NLRP6, the NLRP3 inflammasome may aggravate liver damage by affecting intestinal flora in the gut-liver axis. Pierantonelli et al (26) demonstrated that NLRP3−/− mice fed a Western diet, provided with water containing fructose, exhibited impaired intestinal antimicrobial peptide synthesis. This event led to an increased permeability of the intestinal mucosal barrier, and induced dysbiosis in the form of an increased Firmicutes/Bacteroidetes ratio and increase in Proteobacteria, causing bacterial translocation, leading to the increased expression of the LPS receptor TLR4 and double-stranded bacterial DNA receptor TLR9 in the liver. Antibiotic treatment decreased bacterial translocation and alleviated liver inflammation.
4. Inflammasome increases liver injury by inducing inflammation
Apart from the aforementioned roles of the inflammasomes in the gut, the present review focuses on the roles of inflammasomes in the liver. The inflammasome is expressed in liver parenchymal cells and immune cells (65). In hepatocytes, inflammasome activation induces hepatocyte death via pyroptosis and aggravates non-alcoholic steatohepatitis (16). In immune cells, however, gut-derived PAMPs activate the intracellular inflammasome via PRRs, resulting in increased synthesis of IL-1β and IL-18, with the former being a crucial pro-inflammatory cytokine in liver inflammation (17). A previous study demonstrated that IL-1β promoted the synthesis of monocyte chemoattractant protein 1 (MCP-1) and TNF-α (66), thereby potentiating TNF-α cytotoxicity in hepatocytes, and activating hepatic stellate cells to promote liver fibrosis (67). In addition, DAMPs that are released from dead hepatocytes may activate inflammatory responses in immune cells and hepatic stellate cells (68). Therefore, the inflammasome serves a key role in the hepatic inflammatory network. Previous studies investigating the pathogenesis of liver disease primarily focused on NLRP3 and AIM2 inflammasomes (65,69). In contrast, the NLRP6 inflammasome does not have a direct effect on the liver; however, it was demonstrated to indirectly influence liver disease via the gut-liver axis (18).
5. Inflammasome regulates the barrier of biliary epithelium
Similar to the protective roles exerted by the inflammasome in maintaining the intestinal barrier, the inflammasome regulates the integrity of the biliary epithelium barrier. The biliary epithelial barrier serves a significant role in BA metabolism (70). Once a barrier, including tight junctions, is disrupted, BAs may leak into the portal tracts, thereby inducing the recruitment of leukocytes around the bile duct, activating the pro-inflammatory cytokines TNF-α, IL-1β and fibrosis factor TGF-β1, leading to periductal inflammation and fibrosis (70). Subsequent to fibrosis, bile duct epithelial cells may separate from the peribiliary plexus, and induce autophagy and cell death in the bile duct epithelium, leading to obstructive jaundice (71). A recent study has suggested that the inflammasome is involved in the regulation of biliary epithelial barrier function. Maroni et al (72) demonstrated that NLRP3 inflammasome activation affected epithelial barrier function in vitro and in vivo. In patients with primary sclerosing cholangitis and mouse models, an increased expression of the NLRP3 inflammasome was observed, that stimulated IL-18 expression in bile duct epithelial cells. No significant alterations in IL-1β were identified. In contrast to the protective role in the intestine, NLRP3 inflammasome activation in wild-type mice decreased the expression of E-cadherin and Zonulin-1, and increased the permeability of cholangiocytes. Furthermore, in NLRP3−/− mice, only minor alterations were observed in the aforementioned cell adhesion markers and cholangiocyte permeability (72). Collectively, these findings suggested that the NLRP3 inflammasome is expressed in reactive cholangiocytes and that activation of the NLRP3 inflammasome affected epithelial integrity of cholangiocytes by inducing pro-inflammatory cytokine production.
6. BA metabolism and its effect on the inflammasome in the gut-liver axis
In the gut-liver axis, BAs serve a principal role. BAs are synthesized from cholesterol in hepatocytes by cholesterol 7α-hydroxylase (CYP7A1). Subsequently, primary BAs cholic acid (CA) and chenodeoxycholic acid (CDCA) conjugate with glycocholic acid (GCA) and glycochendeoxycholic acid or taurine (taurocholate and taurodeoxycholic acid) to generate conjugated primary Bas (73). BAs are secreted from the hepatocytes into bile via the canalicular bile salt export pump (66,67), subsequently reabsorbed in the terminal ileum by apical sodium-dependent bile salt transporter, and returned to the liver via the portal vein (74,75). Finally, 95% of BAs are taken up by the basolateral transport systems into hepatocytes. The remaining primary BAs in the intestine are dehydroxylated to secondary BAs, including deoxycholic acid (DCA) and lithocholic acid (LCA), by bacteria (76). Therefore, factors affecting the metabolism of BAs may lead to gut-liver axis dysfunction (77).
BAs serve as endocrine signaling molecules by binding to the nuclear farnesoid-X receptor (FXR) and membrane Takeda G-protein receptor 5 (TGR5) in the gut-liver axis (78–80). FXR is highly expressed in enterocytes and hepatocytes, whereas, TGR5 is not present in parenchymal cells; however, is present in enteroendocrine cells, Kupffer cells, sinusoidal endothelial cells, stellate cells and cholangiocytes (81,82). Among the BAs, CDCA is the strongest FXR agonist (CDCA > DCA > CA > LCA; in order of decreasing potency) (79), whereas, LCA is the most potent in activating TGR5 (LCA > DCA > CDCA > CA) (83). Accumulating evidence has suggested that BAs are involved in a series of pathological processes in the gut-liver axis by activating FXR. For example, CA and DCA inhibited bacterial overgrowth and had a positive effect on mucosal injury in ileum caused by bile duct ligation (84), whereas, mice lacking FXR had impaired gut barrier integrity (85). BAs modulated gut microbial composition and mice fed with CA exhibited a marked decrease in the Bacteroidetes/Firmicutes ratio (2,86). In addition, FXR is a negative regulator of NF-κB-mediated hepatic inflammation and inhibited liver fibrosis, promoting regeneration (87–89). Therefore, FXR may represent a novel therapeutic target for a broad range of gut and liver diseases.
At physiological levels, BAs are unable to activate the NLRP3 inflammasome. However, intrahepatic BAs may reach a critical concentration in cholestasis, causing DCA and CDCA to function as DAMPs to induce a strong activation of the macrophage NLRP3 inflammasome in a dose- and time-dependent manner via signal 1 and signal 2 (Fig. 4) (21,28). The activation of the NLRP3 inflammasome may result in cholestatic liver injury and liver fibrosis, and its activation mechanisms may be involved in calcium influx, ROS production, K+ efflux and intracellular ATP release (21,28). In addition to the aforementioned direct roles, BAs are able to indirectly regulate the NLRP3 inflammasome via their receptors (Fig. 4). Guo et al (27) demonstrated that specific BAs, including CA, GCA, CDCA, DCA, ursodeoxycholic acid, LCA and taurolithocholic acid interacted with the macrophage membrane receptor TGR5 and induced cyclic adenosine 3′,5′-monophosphate-dependent protein kinase A (PKA) activation. PKA kinase in turn phosphorylated NLRP3, thereby preventing NLRP3 inflammasome activation, thus exerting anti-inflammatory effects. In addition, Hao et al (28) identified another mechanism via which the BA-activated nuclear receptor FXR negatively regulated the NLRP3 inflammasome. FXR interacted with NLRP3 and caspase-1, thereby preventing assembly of the NLRP3 inflammasome. However, the expression of FXR in cholestasis was downregulated in the liver, leading to the extensive activation of the NLRP3 inflammasome in macrophages, aggravating cholestatic liver injury. Furthermore, Xie et al (29) confirmed that the FXR activator GW4064 is able to inhibit NLRP3 inflammasome activation, mitigating liver inflammation. The BA receptors FXR and TGR5 not only inhibited intrahepatic BA synthesis and reduced cholestatic liver injury (90,91); however, additionally negatively regulated the NLRP3 inflammasome, exerting anti-inflammatory effects. Accordingly, using novel ligands of FXR and TGR5 may represent a potential therapeutic strategy for the treatment of cholestasis.
Figure 4.
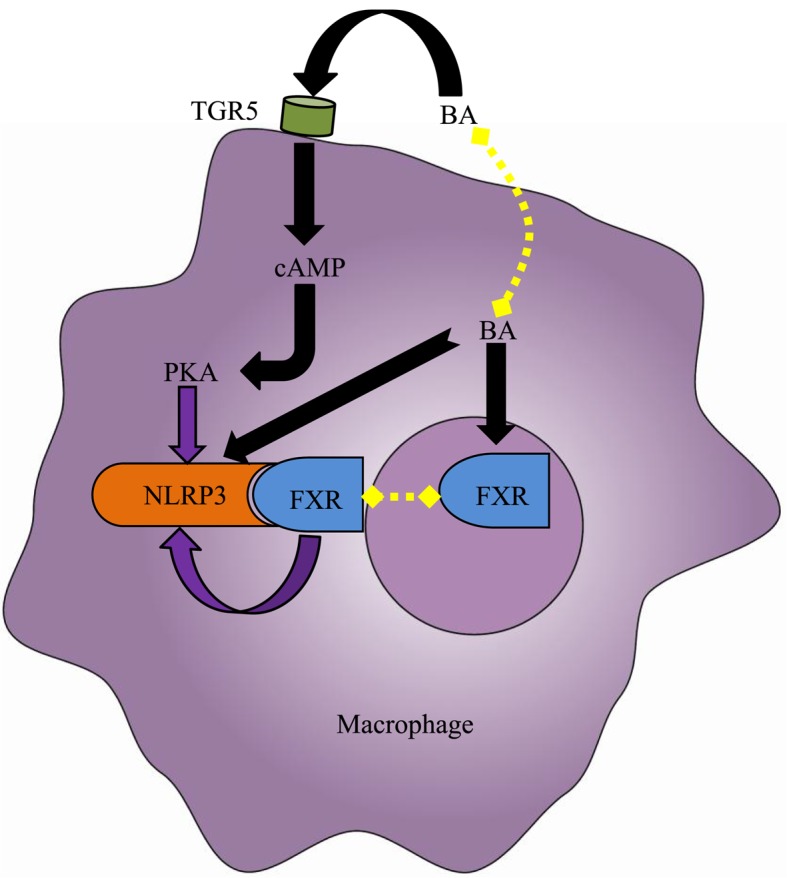
Effect of BA on the NLRP3 inflammasome in the macrophage. Elevated intracellular BAs, including deoxycholic acid and chenodeoxycholic acid, directly activate the NLRP3 inflammasome in macrophages. However, the BA nuclear receptor FXR interacts with NLRP3 to prevent the assembly of NLRP3 inflammasome components, thereby repressing its activation. Furthermore, the BA membrane receptor TGR5 may negatively regulate NLRP3 inflammasome activation by TGR5-cAMP-PKA axis-dependent NLRP3 phosphorylation and ubiquitination. However, due to the limited expression of FXR and TGR5 under cholestasis conditions, the aforementioned protective mechanisms fail to counteract the cytotoxic effects of BAs. Black arrows indicate ‘promotion’, whereas, purple arrow indicates ‘inhibition’. BA, bile acid; cAMP, cylic adenosine monophosphate; FXR, farnesoid-X receptor; PKA, protein kinase A; TGR5, Takeda G-protein receptor 5; NLRP3, NACHT, LRR, and PYD domains-containing protein 3.
7. Inflammasome connects gut and liver
Similar to BAs, the inflammasome has been demonstrated to connect the gut and liver. According to De Minicis et al (8), intestinal NLRP3 inflammasome expression was downregulated in a bile duct ligation mouse model. Collectively, the roles of the inflammasome in the gut-liver axis are presented in Fig. 5. In gut-liver dysfunction, decreased BAs in the intestine lead to dysbiosis characterized by increased Gram-negative bacteria, whose metabolites, including spermine and histamine may inhibit the expression of the NLRP6 inflammasome in the intestinal epithelium. Additionally, dysbiosis leads to a decreased synthesis of IL-18, intestinal antimicrobial peptides, and mucus secretion by goblet cells, increasing intestinal permeability. Increased permeability leads to intestinal flora disruption and bacterial translocation, and eventually results in gut-derived PAMPs and DAMPs to enter the liver via the portal vein. In the liver, PAMPs and DAMPs interact with TLRs to activate the intracellular NLRP3 inflammasome that leads to the synthesis of IL-1β in macrophages or Kupffer cells (17). It has been previously reported that IL-1β exacerbates liver injury in the following ways. IL-1β aggravates inflammation by recruiting inflammatory cells. Additionally, in combination with TNF-α, IL-1β induces pyroptosis of hepatocytes. Furthermore, IL-1β activates hepatic stellate cells and promotes fibrosis (67).
Figure 5.

Roles of the inflammasome in the gut-liver axis. Decreased intestinal BAs induce intestinal flora disorders, increase intestinal permeability, and impair intestinal barrier function. The NLRP6 inflammasome in the intestinal epithelium physiologically induces IL-18 synthesis, and promotes the production of antimicrobial peptides and mucus secretion by goblet cells, which eventually inhibits intestinal barrier disruption and maintains intestinal microbial balance. However, in gut-liver dysfunction, protective effects of the NLRP6 inflammasome may be inhibited, resulting in bacterial translocation, and the transfer of PAMPs and DAMPs to the liver via the portal vein. In the liver, accumulative PAMPs and DAMPs act on TLRs on the cell membrane to activate the intracellular inflammasome. Furthermore, in immune cells, NLRP3 and AIM2 inflammasome activation induces the synthesis of IL-1β, which primarily mediates inflammation in the liver, and increases the expression of MCP-1 and TNF-α. In hepatocytes, MCP-1 further aggravates hepatocyte steatosis. Furthermore, IL-1β sensitizes hepatocytes to TNF-mediated cellular toxicity and induces pyroptosis of hepatocytes in combination with TNF-α. In hepatic stellate cells, NLRP3 inflammasome activation upregulates the expression level of TGF-β1 and promotes hepatic fibrosis. Arrows indicate ‘promotion’, whereas, the symbol ‘┴’ indicates ‘inhibition’. BA, bile acid; DAMPs, danger-associated molecular patterns; MCP-1, monocyte chemoattractant protein 1; PAMPs, pathogen-associated molecular patterns; TLR, Toll-like receptor; IL, interleukin; NLRP6, NACHT, LRR and PYD domains-containing protein 6; AIM, interferon-inducible protein AIM2; TNF, tumor necrosis factor; TGF-β1, transforming growth factor β1.
8. Conclusions and future perspectives
In conclusion, the inflammasome is associated with the gut-liver axis by affecting the intestinal mucosal barrier and microbial composition, and by modulating liver inflammation. However, the majority of the previous studies focused on the role of the inflammasome in either the gut or liver, and ignored its complex role in the gut-liver axis. Furthermore, the inflammasome is assembled by self-oligomerizing scaffold proteins and involves multiple NLR or non-NLR families, including the NLRP3, NLRP6 and the AIM2 inflammasome. As hypothesized, various inflammasomes serve different roles in the same disease. Although the inflammasome may be the same, its roles in maintaining the integrity of the intestinal mucosa may differ. In addition, the NLRP3 inflammasome was downregulated in the gut; however, upregulated in the liver in the same disease (8). Therefore, to understand the exact mechanisms of inflammasome in the gut-liver axis, it is crucial to identify the role of the inflammasome in the liver and gut, and investigate the interactions among multiple types of inflammasomes in the same disease.
BAs serve as endocrine signaling molecules in the pathogenesis of gut-liver axis. The effect of BAs on the NLRP3 inflammasome is controversial. BAs may directly activate the NLRP3 inflammasome. In contrast, BAs may have the opposite effect by binding to the membrane receptor TGR5 or nuclear receptor FXR. This discrepancy may be explained by condition-specific effects. At physiological levels, BAs were described to inhibit the NLRP3 inflammasome in an indirect way, maintaining the cell homeostasis. However, BAs may exert their cytotoxic effects by directly acting on the NLRP3 inflammasome. Therefore, BA receptors may represent the key molecules for the treatment of liver diseases. For example, FXR agonist obeticholic acid has been approved for the treatment of primary biliary cholangitis, whereas, TGR5 agonists failed to improve cholestatic liver injury in a mouse model of sclerosing cholangitis (92). The underlying mechanism of the dual role of BAs may be associated with the different distribution of these two BA receptors in the liver. Further studies are required to elucidate the interactions among BAs, BA receptors and the inflammasome.
Previous studies have demonstrated the benefits of inflammasome-targeting drugs. For instance, the caspase inhibitor IDN-6556 improved liver injury and liver fibrosis (24). Furthermore, administration of the IL-1 inhibitor anakinra ameliorated liver inflammation, steatosis, hepatocellular damage and fibrosis (17). In conclusion, based on the ongoing mechanistic studies of the inflammasome, novel inflammasome-targeting drugs may provide novel therapeutics for the treatment of gut and liver diseases.
Acknowledgements
The authors thank Mr. Jin-Feng Jin for the technical assistance in figure editing.
Glossary
Abbreviations
- ASC
apoptosis-associated speck-like protein containing a CARD
- ATP
adenosine 5′-triphosphate
- BAs
bile acids
- CA
cholic acid
- CCL5
chemokine (C-C motif) ligand 5
- CDCA
chenodeoxycholic acid
- CYP7A1
cholesterol 7α-hydroxylase
- DAMPs
damage-associated molecular patterns
- DCA
deoxycholic acid
- DSS
dextran sodium sulfate
- FXR
farnesoid-X receptor
- GCA
glycocholic acid
- IL
interleukin
- LCA
lithocholic acid
- LPS
lipopolysaccharide
- LRR
leucine-rich repeat
- MCP-1
monocyte chemoattractant protein 1
- NACHT
nucleotide-binding and oligomerization
- NF-κB
nuclear factor-κB
- NLR
NOD-like receptor
- NLRP
NACHT, LRR and PYD domains-containing protein
- PAMPs
pathogen-associated molecular patterns
- PKA
protein kinase A
- PRR
pattern recognition receptor
- PYD
pyrin domain
- ROS
reactive oxygen species
- TGF-β1
transforming growth factor β1
- TGR5
Takeda G-protein receptor 5
- TLR
Toll-like receptor
- TNF-α
tumor necrosis factor α
Funding
The present study received financial support from The National Natural Science Foundation of China (grant nos. 81770519 and 81771633), The Science Foundation of Shanghai (Shanghai, China; grant nos. 16411952200, 16140902300 and 17411960600), Shanghai Hospital Development Center (Shanghai, China; grant no. SHDC12014106), Shanghai Key Disciplines (Shanghai, China; grant no. 2017ZZ02022), Shanghai Rising-Star Program (Shanghai, China; A type; grant no. 15QA1400800) and The Science Foundation of Shanghai Excellent Youth Scholars (Shanghai, China; grant no. 2017YQ042).
Availability of data and materials
Not applicable.
Authors' contributions
SZ conceived and designed the theme. JW retrieved concerned literatures and wrote the article. RD reviewed and edited the article.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Visschers RG, Luyer MD, Schaap FG, Olde Damink SW, Soeters PB. The gut-liver axis. Curr Opin Clin Nutr Metab Care. 2013;16:576–581. doi: 10.1097/MCO.0b013e32836410a4. [DOI] [PubMed] [Google Scholar]
- 2.Islam KB, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, Ogura Y, Hayashi T, Yokota A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology. 2011;141:1773–1781. doi: 10.1053/j.gastro.2011.07.046. [DOI] [PubMed] [Google Scholar]
- 3.Yokota A, Fukiya S, Islam KB, Ooka T, Ogura Y, Hayashi T, Hagio M, Ishizuka S. Is bile acid a determinant of the gut microbiota on a high-fat diet? Gut Microbes. 2012;3:455–459. doi: 10.4161/gmic.21216. [DOI] [PubMed] [Google Scholar]
- 4.Cai SY, Boyer JL. Studies on the mechanisms of bile acid initiated hepatic inflammation in cholestatic liver injury. Inflamm Cell Signal. 2017;4:e1561. [PMC free article] [PubMed] [Google Scholar]
- 5.Cai SY, Ouyang X, Chen Y, Soroka CJ, Wang J, Mennone A, Wang Y, Mehal WZ, Jain D, Boyer JL. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight. 2017;2:e90780. doi: 10.1172/jci.insight.90780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez MJ, Briz O. Bile-acid-induced cell injury and protection. World J Gastroenterol. 2009;15:1677–1689. doi: 10.3748/wjg.15.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: A novel mechanism of inflammation during obstructive cholestasis. Am J Pathol. 2011;178:175–186. doi: 10.1016/j.ajpath.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Minicis S, Rychlicki C, Agostinelli L, Saccomanno S, Candelaresi C, Trozzi L, Mingarelli E, Facinelli B, Magi G, Palmieri C, et al. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology. 2014;59:1738–1749. doi: 10.1002/hep.26695. [DOI] [PubMed] [Google Scholar]
- 9.Sabino J, Vieira-Silva S, Machiels K, Joossens M, Falony G, Ballet V, Ferrante M, Van Assche G, Van der Merwe S, Vermeire S, Raes J. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut. 2016;65:1681–1689. doi: 10.1136/gutjnl-2015-311004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiest R, Albillos A, Trauner M, Bajaj JS, Jalan R. Intestinal hepatic axis for liver disease. J Hepatol. 2017;67:1084–1103. doi: 10.1016/j.jhep.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, Knight R. The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol. 2018;15:397–411. doi: 10.1038/s41575-018-0031-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Toole A, Alakkari A, Keegan D, Doherty G, Mulcahy H, O'Donoghue D. Primary sclerosing cholangitis and disease distribution in inflammatory bowel disease. Clin Gastroenterol Hepatol. 2012;10:439–441. doi: 10.1016/j.cgh.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 13.Weismüller TJ, Trivedi PJ, Bergquist A, Imam M, Lenzen H, Ponsioen CY, Holm K, Gotthardt D, Färkkilä MA, Marschall HU, et al. Patient age, sex, and inflammatory bowel disease phenotype associate with course of primary sclerosing cholangitis. Gastroenterology. 2017;152:1975–1984. doi: 10.1053/j.gastro.2017.02.038. e1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu R, Li X, Huang Z, Zhao D, Ganesh BS, Lai G, Pandak WM, Hylemon PB, Bajaj JS, Sanyal AJ, Zhou H. C/EBP homologous protein-induced loss of intestinal epithelial stemness contributes to bile duct ligation-induced cholestatic liver injury in mice. Hepatology. 2018;67:1441–1457. doi: 10.1002/hep.29540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jee J, Mourya R, Shivakumar P, Fei L, Wagner M, Bezerra JA. Cxcr2 signaling and the microbiome suppress inflammation, bile duct injury, and the phenotype of experimental biliary atresia. PLoS One. 2017;12:e0182089. doi: 10.1371/journal.pone.0182089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 17.Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, Barrieau M, Min SY, Kurt-Jones EA, Szabo G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;122:3476–3489. doi: 10.1172/JCI60777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, Frankel G, Levy M, Katz MN, Philbrick WM, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156:1045–1059. doi: 10.1016/j.cell.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, David E, Savidor A, Korem T, Herzig Y, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell. 2015;163:1428–1443. doi: 10.1016/j.cell.2015.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong Z, Zhou J, Zhao S, Tian C, Wang P, Xu C, Chen Y, Cai W, Wu J. Chenodeoxycholic acid activates NLRP3 inflammasome and contributes to cholestatic liver fibrosis. Oncotarget. 2016;7:83951–83963. doi: 10.18632/oncotarget.13796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han J, Bae J, Choi CY, Choi SP, Kang HS, Jo EK, Park J, Lee YS, Moon HS, Park CG, et al. Autophagy induced by AXL receptor tyrosine kinase alleviates acute liver injury via inhibition of NLRP3 inflammasome activation in mice. Autophagy. 2016;12:2326–2343. doi: 10.1080/15548627.2016.1235124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wree A, McGeough MD, Inzaugarat ME, Eguchi A, Schuster S, Johnson CD, Peña CA, Geisler LJ, Papouchado BG, Hoffman HM, Feldstein AE. NLRP3 inflammasome driven liver injury and fibrosis. Roles of IL-17 and TNF. Hepatology. 2017 doi: 10.1002/hep.29523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barreyro FJ, Holod S, Finocchietto PV, Camino AM, Aquino JB, Avagnina A, Carreras MC, Poderoso JJ, Gores GJ. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015;35:953–966. doi: 10.1111/liv.12570. [DOI] [PubMed] [Google Scholar]
- 25.Alaish SM, Smith AD, Timmons J, Greenspon J, Eyvazzadeh D, Murphy E, Shea-Donahue T, Cirimotich S, Mongodin E, Zhao A, et al. Gut microbiota, tight junction protein expression, intestinal resistance, bacterial translocation and mortality following cholestasis depend on the genetic background of the host. Gut Microbes. 2013;4:292–305. doi: 10.4161/gmic.24706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierantonelli I, Rychlicki C, Agostinelli L, Giordano DM, Gaggini M, Fraumene C, Saponaro C, Manghina V, Sartini L, Mingarelli E, et al. Lack of NLRP3-inflammasome leads to gut-liver axis derangement, gut dysbiosis and a worsened phenotype in a mouse model of NAFLD. Sci Rep. 2017;7:12200. doi: 10.1038/s41598-017-11744-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, Zheng M, Zhang X, Xia D, Ke Y, et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity. 2016;45:802–816. doi: 10.1016/j.immuni.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 28.Hao H, Cao L, Jiang C, Che Y, Zhang S, Takahashi S, Wang G, Gonzalez FJ. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis-associated sepsis. Cell Metab. 2017;25:856–867. doi: 10.1016/j.cmet.2017.03.007. e855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie S, Guo C, Chi Z, Huang B, Wu Y, Wang D, Xia D. A rapid administration of GW4064 inhibits the NLRP3 inflammasome activation independent of farnesoid X receptor agonism. FEBS Lett. 2017;591:2836–2847. doi: 10.1002/1873-3468.12782. [DOI] [PubMed] [Google Scholar]
- 30.Martinon F, Burns K, Tschopp J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/S1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 31.Giebeler A, Brandenburg LO, Kaldenbach M, Erschfeld S, Wasmuth H, Wruck C, Trautwein C, Streetz KL. Lack of hepatic c-Met and gp130 expression is associated with an impaired antibacterial response and higher lethality after bile duct ligation. Lab Invest. 2012;92:1726–1737. doi: 10.1038/labinvest.2012.122. [DOI] [PubMed] [Google Scholar]
- 32.Matsushita H, Miyake Y, Takaki A, Yasunaka T, Koike K, Ikeda F, Shiraha H, Nouso K, Yamamoto K. TLR4, TLR9, and NLRP3 in biliary epithelial cells of primary sclerosing cholangitis: Relationship with clinical characteristics. J Gastroenterol Hepatol. 2015;30:600–608. doi: 10.1111/jgh.12711. [DOI] [PubMed] [Google Scholar]
- 33.Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol. 2015;12:387–400. doi: 10.1038/nrgastro.2015.94. [DOI] [PubMed] [Google Scholar]
- 34.Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: An integrated view. Immunol Rev. 2011;243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 35.Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, et al. The NLR gene family: A standard nomenclature. Immunity. 2008;28:285–287. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boaru SG, Borkham-Kamphorst E, Van de Leur E, Lehnen E, Liedtke C, Weiskirchen R. NLRP3 inflammasome expression is driven by NF-κB in cultured hepatocytes. Biochem Biophys Res Commun. 2015;458:700–706. doi: 10.1016/j.bbrc.2015.02.029. [DOI] [PubMed] [Google Scholar]
- 38.Kahlenberg JM, Dubyak GR. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am J Physiol Cell Physiol. 2004;286:C1100–C1108. doi: 10.1152/ajpcell.00494.2003. [DOI] [PubMed] [Google Scholar]
- 39.Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, Vandenabeele P, Núñez G. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. 2007;26:433–443. doi: 10.1016/j.immuni.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 40.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 42.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gremel G, Wanders A, Cedernaes J, Fagerberg L, Hallström B, Edlund K, Sjöstedt E, Uhlén M, Pontén F. The human gastrointestinal tract-specific transcriptome and proteome as defined by RNA sequencing and antibody-based profiling. J Gastroenterol. 2015;50:46–57. doi: 10.1007/s00535-014-0958-7. [DOI] [PubMed] [Google Scholar]
- 44.Del Chierico F, Vernocchi P, Petrucca A, Paci P, Fuentes S, Praticò G, Capuani G, Masotti A, Reddel S, Russo A, et al. Phylogenetic and metabolic tracking of gut microbiota during perinatal development. PLoS One. 2015;10:e0137347. doi: 10.1371/journal.pone.0137347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kempster SL, Belteki G, Forhead AJ, Fowden AL, Catalano RD, Lam BY, McFarlane I, Charnock-Jones DS, Smith GC. Developmental control of the Nlrp6 inflammasome and a substrate, IL-18, in mammalian intestine. Am J Physiol Gastrointest Liver Physiol. 2011;300:G253–G263. doi: 10.1152/ajpgi.00397.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chudnovskiy A, Mortha A, Kana V, Kennard A, Ramirez JD, Rahman A, Remark R, Mogno I, Ng R, Gnjatic S, et al. Host-Protozoan interactions protect from mucosal infections through activation of the inflammasome. Cell. 2016;167:444–456. doi: 10.1016/j.cell.2016.08.076. e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun Y, Zhang M, Chen CC, Gillilland M III, Sun X, El-Zaatari M, Huffnagle GB, Young VB, Zhang J, Hong SC, et al. Stress-induced corticotropin-releasing hormone-mediated NLRP6 inflammasome inhibition and transmissible enteritis in mice. Gastroenterology. 2013;144:1478–1487. doi: 10.1053/j.gastro.2013.02.038. e1471-1487.e1-e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Birchenough GM, Nyström EE, Johansson ME, Hansson GC. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science. 2016;352:1535–1542. doi: 10.1126/science.aaf7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, Hedl M, Zhang W, O'Connor W, Jr, Murphy AJ, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491:259–263. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang P, Zhu S, Yang L, Cui S, Pan W, Jackson R, Zheng Y, Rongvaux A, Sun Q, Yang G, et al. Nlrp6 regulates intestinal antiviral innate immunity. Science. 2015;350:826–830. doi: 10.1126/science.aab3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elinav E, Thaiss CA, Flavell RA. Analysis of microbiota alterations in inflammasome-deficient mice. Methods Mol Biol. 2013;1040:185–194. doi: 10.1007/978-1-62703-523-1_14. [DOI] [PubMed] [Google Scholar]
- 52.Chen GY, Liu M, Wang F, Bertin J, Núñez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol. 2011;186:7187–7194. doi: 10.4049/jimmunol.1100412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, Lemoine Y, Hot D, Chamaillard M. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci USA. 2011;108:9601–9606. doi: 10.1073/pnas.1100981108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu B, Elinav E, Huber S, Strowig T, Hao L, Hafemann A, Jin C, Wunderlich C, Wunderlich T, Eisenbarth SC, Flavell RA. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc Natl Acad Sci USA. 2013;110:9862–9867. doi: 10.1073/pnas.1307575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seo SU, Kamada N, Muñoz-Planillo R, Kim YG, Kim D, Koizumi Y, Hasegawa M, Himpsl SD, Browne HP, Lawley TD, et al. Distinct commensals induce interleukin-1β via NLRP3 inflammasome in inflammatory monocytes to promote intestinal inflammation in response to injury. Immunity. 2015;42:744–755. doi: 10.1016/j.immuni.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Filardy AA, He J, Bennink J, Yewdell J, Kelsall BL. Posttranscriptional control of NLRP3 inflammasome activation in colonic macrophages. Mucosal Immunol. 2016;9:850–858. doi: 10.1038/mi.2015.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, Ting JP. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010;207:1045–1056. doi: 10.1084/jem.20100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, Eisenbarth SC, Flavell RA. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci USA. 2010;107:21635–21640. doi: 10.1073/pnas.1016814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ruiz PA, Morón B, Becker HM, Lang S, Atrott K, Spalinger MR, Scharl M, Wojtal KA, Fischbeck-Terhalle A, Frey-Wagner I, et al. Titanium dioxide nanoparticles exacerbate DSS-induced colitis: Role of the NLRP3 inflammasome. Gut. 2017;66:1216–1224. doi: 10.1136/gutjnl-2015-310297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zherebiatiev A, Kamyshnyi A. Expression levels of proinflammatory cytokines and NLRP3 inflammasome in an experimental model of Oxazolone-induced colitis. Iran J Allergy Asthma Immunol. 2016;15:39–45. [PubMed] [Google Scholar]
- 61.De la Fuente M, Franchi L, Araya D, Díaz-Jiménez D, Olivares M, Álvarez-Lobos M, Golenbock D, González MJ, López-Kostner F, Quera R, et al. Escherichia coli isolates from inflammatory bowel diseases patients survive in macrophages and activate NLRP3 inflammasome. Int J Med Microbiol. 2014;304:384–392. doi: 10.1016/j.ijmm.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bauer C, Duewell P, Lehr HA, Endres S, Schnurr M. Protective and aggravating effects of Nlrp3 inflammasome activation in IBD models: Influence of genetic and environmental factors. Dig Dis. 2012;30(Suppl 1):S82–S90. doi: 10.1159/000341681. [DOI] [PubMed] [Google Scholar]
- 63.Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, Lam V, Potentier MS, Ng K, Bawa M, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis. 2011;17:1359–1372. doi: 10.1002/ibd.21478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–391. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol. 2012;57:642–654. doi: 10.1016/j.jhep.2012.03.035. [DOI] [PubMed] [Google Scholar]
- 66.Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: Regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology. 2011;54:2185–2197. doi: 10.1002/hep.24599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139:323–334.e327. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143:1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 69.Csak T, Pillai A, Ganz M, Lippai D, Petrasek J, Park JK, Kodys K, Dolganiuc A, Kurt-Jones EA, Szabo G. Both bone marrow-derived and non-bone marrow-derived cells contribute to AIM2 and NLRP3 inflammasome activation in a MyD88-dependent manner in dietary steatohepatitis. Liver Int. 2014;34:1402–1413. doi: 10.1111/liv.12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rao RK, Samak G. Bile duct epithelial tight junctions and barrier function. Tissue Barriers. 2013;1:e25718. doi: 10.4161/tisb.27772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, Krause R, Lammert F, Langner C, Zatloukal K, et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2004;127:261–274. doi: 10.1053/j.gastro.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 72.Maroni L, Agostinelli L, Saccomanno S, Pinto C, Giordano DM, Rychlicki C, De Minicis S, Trozzi L, Banales JM, Melum E, et al. Nlrp3 activation induces Il-18 synthesis and affects the epithelial barrier function in reactive cholangiocytes. Am J Pathol. 2017;187:366–376. doi: 10.1016/j.ajpath.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 73.Ikegami T, Honda A. Reciprocal interactions between bile acids and gut microbiota in human liver diseases. Hepatol Res. 2018;48:15–27. doi: 10.1111/hepr.13001. [DOI] [PubMed] [Google Scholar]
- 74.Hofmann AF. The enterohepatic circulation of bile acids in mammals: Form and functions. Front Biosci (Landmark Ed) 2009;14:2584–2598. doi: 10.2741/3399. [DOI] [PubMed] [Google Scholar]
- 75.Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res. 2015;56:1085–1099. doi: 10.1194/jlr.R054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 77.Halilbasic E, Claudel T, Trauner M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol. 2013;58:155–168. doi: 10.1016/j.jhep.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 79.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, Lehmann JM. Bile acids: Natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 80.Potthoff MJ, Potts A, He T, Duarte JA, Taussig R, Mangelsdorf DJ, Kliewer SA, Burgess SC. Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am J Physiol Gastrointest Liver Physiol. 2013;304:G371–G380. doi: 10.1152/ajpgi.00400.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11:55–67. doi: 10.1038/nrgastro.2013.151. [DOI] [PubMed] [Google Scholar]
- 82.Reich M, Klindt C, Deutschmann K, Spomer L, Häussinger D, Keitel V. Role of the G protein-coupled bile acid receptor TGR5 in liver damage. Dig Dis. 2017;35:235–240. doi: 10.1159/000450917. [DOI] [PubMed] [Google Scholar]
- 83.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 84.Ding JW, Andersson R, Soltesz V, Willén R, Bengmark S. The role of bile and bile acids in bacterial translocation in obstructive jaundice in rats. Eur Surg Res. 1993;25:11–19. doi: 10.1159/000129252. [DOI] [PubMed] [Google Scholar]
- 85.Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA. 2006;103:3920–3925. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wahlström A, Sayin SI, Marschall HU, Bäckhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016;24:41–50. doi: 10.1016/j.cmet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 87.Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology. 2008;48:1632–1643. doi: 10.1002/hep.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wagner M, Zollner G, Trauner M. Nuclear receptors in liver disease. Hepatology. 2011;53:1023–1034. doi: 10.1002/hep.24148. [DOI] [PubMed] [Google Scholar]
- 89.Zhu C, Fuchs CD, Halilbasic E, Trauner M. Bile acids in regulation of inflammation and immunity: Friend or foe? Clin Exp Rheumatol. 2016;34(4 Suppl 98):S25–S31. [PubMed] [Google Scholar]
- 90.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 91.Péan N, Doignon I, Garcin I, Besnard A, Julien B, Liu B, Branchereau S, Spraul A, Guettier C, Humbert L, et al. The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Hepatology. 2013;58:1451–1460. doi: 10.1002/hep.26463. [DOI] [PubMed] [Google Scholar]
- 92.Baghdasaryan A, Claudel T, Gumhold J, Silbert D, Adorini L, Roda A, Vecchiotti S, Gonzalez FJ, Schoonjans K, Strazzabosco M, et al. Dual farnesoid X receptor/TGR5 agonist INT-767 reduces liver injury in the Mdr2-/- (Abcb4-/-) mouse cholangiopathy model by promoting biliary HCO3 output. Hepatology. 2011;54:1303–1312. doi: 10.1002/hep.24537. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.