

Abstract
Action potential duration (APD) alternans (APD-ALT), defined as beat-to-beat oscillations in APD, has been proposed as an important clinical marker for chronic atrial fibrillation (cAF) risk when it occurs at pacing rates of 120–200 beats/min. Although the ionic mechanisms for occurrence of APD-ALT in human cAF at these clinically relevant rates have been investigated, little is known about the effects of myofilament protein kinetics on APD-ALT. Therefore, we used computer simulations of single cell function to explore whether remodeling in myofilament protein kinetics in human cAF alters the occurrence of APD-ALT and to uncover how these mechanisms are affected by sarcomere length and the degree of cAF-induced myofilament remodeling. Mechanistically based, bidirectionally coupled electromechanical models of human right and left atrial myocytes were constructed, incorporating both ionic and myofilament remodeling associated with cAF. By comparing results from our electromechanical model with those from the uncoupled ionic model, we found that intracellular Ca2+ concentration buffering of troponin C has a dampening effect on the magnitude of APD-ALT (APD-ANM) at slower rates (150 beats/min) due to the cooperativity between strongly bound cross-bridges and Ca2+-troponin C binding affinity. We also discovered that cAF-induced enhanced thin filament activation enhanced APD-ANM at these clinically relevant heart rates (150 beats/min). In addition, longer sarcomere lengths increased APD-ANM, suggesting that atrial stretch is an important modulator of APD-ALT. Together, these findings demonstrate that myofilament kinetics mechanisms play an important role in altering APD-ALT in human cAF.
NEW & NOTEWORTHY Using a single cell simulation approach, we explored how myofilament protein kinetics alter the formation of alternans in action potential duration (APD) in human myocytes with chronic atrial fibrillation remodeling. We discovered that enhanced thin filament activation and longer sarcomere lengths increased the magnitude of APD alternans at clinically important pacing rates of 120–200 beats/min. Furthermore, we found that altered intracellular Ca2+ concentration buffering of troponin C has a dampening effect on the magnitude of APD alternans.
Keywords: action potential duration alternans, atrial fibrillation, computer modeling, mechanoelectric feedback
INTRODUCTION
Atrial fibrillation (AF) is the most common type of human cardiac arrhythmia, which, due to its association with increased morbidity and mortality and its rising prevalence, has become a global health care concern (1, 8, 26). Unfortunately, present therapies do not directly target the underlying mechanisms of AF (13, 16), which is due in part to an incomplete understanding of atrial arrhythmogenesis. Therefore, to enhance prevention and treatment of AF, our understanding of its underlying disease mechanisms needs to be expanded.
Action potential duration (APD) alternans (APD-ALT) is defined as beat-to-beat oscillations in APD. In human chronic AF (cAF), APD-ALT has been found to occur at heart rates of 120–200 beats/min (31). Consequently, APD-ALT has been proposed as an important clinical marker for AF risk since its onset is at slower rates in patients with cAF than in healthy subjects and since it always precedes AF initiation (31). In addition, an increased propensity for APD-ALT in cAF has been shown to increase arrhythmia complexity and persistence (6). These findings indicate that APD-ALT occurring at slower rates (120–200 beats/min) may be an important therapeutic target in patients with AF.
APD-ALT at these clinically relevant slower rates has been found to be driven by Ca2+ transient alternans (CA-ALT) (5). Specifically, altered ryanodine receptor (RyR) kinetics and downregulation of L-type Ca2+ current have been identified as the components of the cAF-associated ionic remodeling that drive APD-ALT formation at the clinically relevant slow heart rates (5). Although the ionic mechanisms of APD-ALT formation in human cAF have been elucidated, little is known about the effects of myofilament protein kinetics on APD-ALT, particularly at the clinically relevant slower rates. Understanding whether myofilament protein kinetics modulate APD-ALT is important because it has been previously shown to alter Ca2+-driven voltage alternans in failing human ventricular myocytes (45). Myofilament remodeling has been documented in myocytes from patients with cAF, including alterations in sarcomeric protein expression and phosphorylation, active and passive tension, myofilament Ca2+ sensitivity, and cross-bridge (XB) cycling rates (2, 10, 11). Finally, mechanical stretch, common in cAF (40), has been shown to increase the susceptibility of atrial myocytes to APD-ALT and enhance their spatial discordance (42). Therefore, our goal in the present study was to use the capability of mechanistic computer simulations to investigate whether remodeling in myofilament protein kinetics in human cAF alters APD-ALT at the clinically relevant slow heart rates and how the established relationships are affected by sarcomere length (SL) and the degree of cAF-induced myofilament remodeling.
METHODS
Human atrial electromechanical myocyte model.
To achieve the goals of the present study, a human atrial electromechanical myocyte model was developed that combined the human atrial ionic model by Chang et al. (5), as shown in Fig. 1A, and the human myofilament kinetics model by Zile and Trayanova (44, 45), as shown in Fig. 1B. This human atrial ionic model was chosen due to its ability to generate APD-ALT at slower clinically relevant pacing rates (5, 6), consistent with findings in human cAF (32). We modeled both right atrial (RA) and left atrial (LA) myocytes, as done by Chang et al. (5).
Fig. 1.
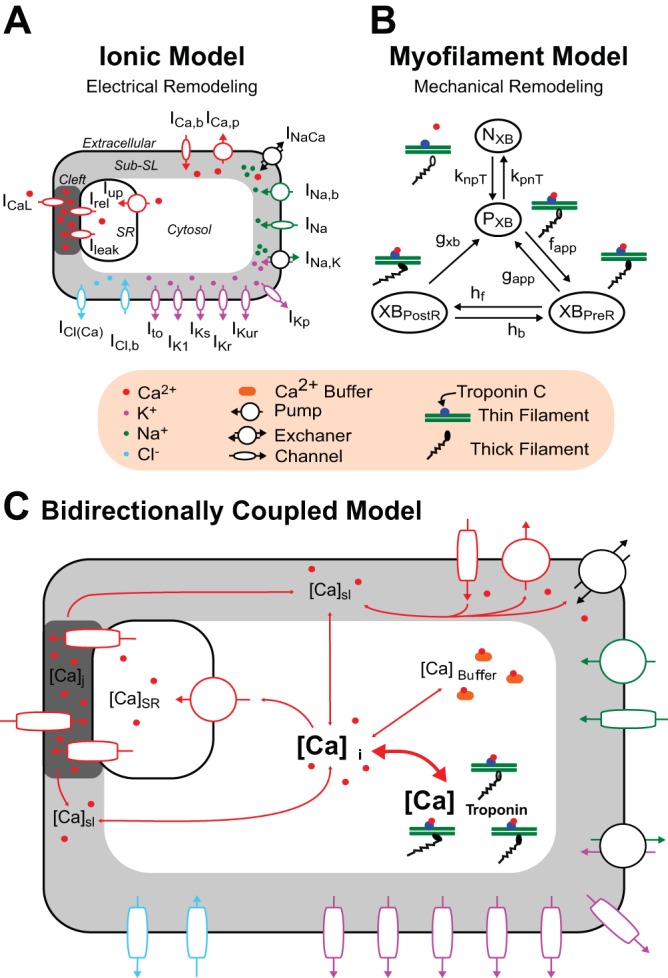
Bidirectionally coupled electromechanical model of the atrial myocyte. The human atrial ionic model by Chang et al. (5) (A) and the human myofilament model by Zile and Trayanova (44, 45) modified to match human atrial force data (B) were bidirectionally coupled (C) by having thin filament activation depend on free intracellular Ca2+ concentration ([Ca2+]i) and by incorporating mechanoelectric feedback via [Ca2+]i buffering of troponin C [thick double-headed red arrow linking [Ca2+]i and total Ca2+ bound to troponin C ([Ca2+]troponin)]. NXB and PXB are thin filament states where cross-bridge (XB) formation is inhibited (NXB) and where weakly bound XB formation is possible (PXB).
The Zile et al. (44, 45) myofilament kinetics model, an extension of the Rice et al. (35) model to the human myocyte from the original rabbit model, describes the activation of the thin filament by intracellular Ca2+ binding to the regulatory site on troponin C as well as thin filament binding to thick filament XBs using a Markov model. Because the Zile et al. (44, 45) myofilament model was developed to match human ventricular myocyte force data, here we made additional adjustments to match human atrial force data, as described below.
The ionic and myofilament models were bidirectionally coupled by having thin filament activation depend on the amount of free Ca2+ in the cytosol [intracellular Ca2+ concentration ([Ca2+]i)] and by incorporating mechanoelectric feedback on Ca2+ dynamics. Specifically, the [Ca2+]i transient derived from the ionic model was used to drive thin filament activation in the myofilament model. Furthermore, similarly to our previously developed approach (44, 45), we incorporated mechanoelectric feedback via [Ca2+]i buffering of troponin C (CaBTn). Coupling the ionic and myofilament models was necessary here because such coupling has been shown to be crucial for accurately reproducing contractile experimental data in myocyte simulations (18).
Using this bidirectionally coupled electromechanical model, we adjusted myofilament parameters [previously tuned in Zile et al. (45) to reproduce human ventricular force data] to match normal human atrial force data. To do this, we solved a minimization problem similar to the approach by de Oliveira et al. (9), where the myofilament parameters (par) were adjusted to minimize the error between simulated and experimental values of total twitch time (TT), time to peak tension (TPT), and time to 50% relaxation of force (Trel50%) using the following equation:
| (1) |
where TTexp, TPTexp, and Trel50%,exp are the values (shown in Table 1) of each atrial force characteristic averaged across three sets of experimental data (3, 15, 27). In these experiments and also in our simulations, human RA myocytes (with a SL of 2.4 µm and at a temperature of 37°F) were isometrically paced at 1 Hz [1,000-ms pacing cycle length (CL)] until steady state was reached. The variables TTsim(par), TPTsim(par), and Trel50%,sim(par) were computed from our simulations with human RA myocytes, each run with a unique set of 11 adjusted myofilament parameters (par), as listed in the following notation: par = (knp, kpn, gapp, fapp, hf, hb, gxb, perm50, kon, koffH, and koffL), which are discussed below.
Table 1.
Atrial force characteristics
| Experimental Values | TT | TPT | Trel50% |
|---|---|---|---|
| Bisping et al. (3) | NA | 89 ± 10 | NA |
| Goetzenich et al. (15) | NA | 95 ± 17 | 89 ± 14 |
| Maier et al. (27) | 300 ± 24 | 91 ± 11 | 85 ± 7 |
| Average | 300 | 92 | 87 |
| Range | 276–324 | 78–112 | 75–103 |
| Simulation | 301 | 77 | 91 |
Values are means ± SE (in ms). TT, total twitch time, TPT, time to peak tension; Trel50%, time to 50% relaxation of force; NA, not available. Force characteristics of human right atrial (RA) myocytes were from experiments (3, 15, 27) and from simulations with our healthy human RA myocyte model using the myofilament parameter values parmin shown in Table 2. Force characteristics were averaged from the available experimental data (3, 15, 27) and appear in Eq. 1 as TTexp, TPTexp, and Trel50%,exp, respectively.
For each unique set of par, the myofilament parameters above were each assigned a value between 25% and 400% (in increments of 25%) of their values from our earlier electromechanical model of the normal human ventricular myocyte (45). The set of adjusted myofilament parameters that resulted in the smallest error (0.2093) between simulation results and experimental data (the set to be referred to as parmin), as calculated from Eq. 1, was used in this study. In parmin, the majority of myofilament parameters ended up not being altered, with the exception of knp, kpn, and gapp, which were changed to 25%, 150%, and 200%, respectively, of their original values in the normal ventricular myocyte model. These adjusted myofilament parameter values are shown in Table 2. The force characteristic values [TTsim(parmin), TPTsim(parmin), and Trel50%,sim(parmin)] from simulations with this normal human RA myocyte model are shown in Table 1 for comparison with experimental values. The comparison of force versus time traces between our simulations and experimental data is shown in Fig. 2. Because no experimental human LA force data could be found, we also used parmin for our model of the myofilament kinetics of the healthy human LA myocyte.
Table 2.
Myofilament model parameter values
| Myofilament Parameter | Normal | cAF | Effect of cAF Remodeling on APD-ANM |
|---|---|---|---|
| kon | 47.5 1/µMs | Normal × 100% to normal × 300% (25% increments) | Increases |
| kn_p* | 152.5 s−1 | Normal × 100% to normal × 300% (25% increments) | None |
| gapp* | 186 s−1 | Normal × 100% to normal × 300% (25% increments) | None |
| hb | 34 s−1 | Normal × 100% to normal × 300% (25% increments) | None |
| perm50 | 0.5 (unitless) | Normal × 100% to normal × 20% (10% increments) | Increases |
| koffL | 250 s−1 | Normal × 100% to normal × 20% (10% increments) | None |
| koffH | 25 s−1 | Normal × 100% to normal × 20% (10% increments) | Increases |
| kp_n* | 24 s−1 | Normal × 100% to normal × 20% (10% increments) | None |
| fapp | 4,800 s−1 | Normal × 100% to normal × 20% (10% increments) | None |
| hf | 10 s−1 | Normal × 100% to normal × 20% (10% increments) | None |
| gxb | 30 s−1 | Normal × 100% to normal × 20% (10% increments) | None |
cAF, chronic atrial fibrillation; APD-ANM, action potential duration alternans-normalized magnitude. Normal and cAF values of myofilament model parameters in the human left and right atrial myocyte models are shown. Myofilament parameters in parmin are indicated by an asterisk in the Myofilament Parameter column. The effect of cAF remodeling on APD-ANM column presents the effects of cAF remodeling on the normalized magnitude of APD-alterans (APD-ANM).
Fig. 2.
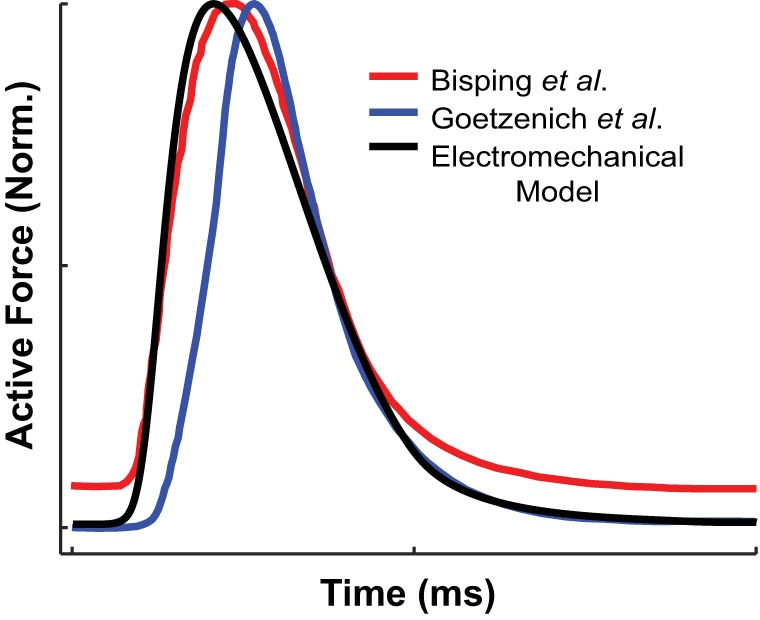
Normalized active force plotted over time for simulations with our normal human right atrial (RA) electromechanical myocyte model (black line) isometrically paced at a cycle length (CL) of 400 ms, sarcomere length (SL) of 1.9 µm, and temperate of 37°F. Normalized active force traces were reproduced from experimental data by Bisping et al. (3) (red line) and Goetzenich et al. (15) (blue line).
Incorporating cAF-induced remodeling.
Ionic remodeling in cAF resulting in the development of APD-ALT at clinically relevant slow rates as well as arrhythmogenesis was incorporated in LA and RA myocytes, as previously described (5, 6). Specifically, Chang et al. (5) decreased gNa by 10%, decreased gCaL by 50%, increased IbarNCX by 40%, increased RyR Ca2+-dependent activation rate (koCa) threefold, decreased RyR Ca2+-dependent inactivation rate (kiCa) by 50%, increased sarcoplasmic reticulum (SR) Ca2+ leak by 25%, and increased gKs and gK1 each twofold compared with normal in both LA and RA myocytes. Chang et al. (5) also added a late component to the Na+ current (INaL) in LA and RA myocytes. Finally, Chang et al. (5) decreased gKur by 45% and 55% and gto by 45% and 80% compared with normal in LA and RA myocytes, respectively. Although one study (42) found that mechanical stretch decreased the expression of SERCA2 in a HL-1 atrial myocyte monolayer, no evidence of this has been found in human AF atrial myocytes and thus no changes to SERCA uptake current were made in the present study.
Myofilament remodeling representing abnormal force generation and myofilament Ca2+ sensitivity found in cAF was incorporated next. Active tension generation and relaxation rates have been shown to be slower and maximum active and passive force generation to be decreased in cAF myofibrils, suggesting that XB cycling rates are depressed (2, 10). However, the exact mechanisms and level of change have not been described. In addition, myofilament Ca2+ sensitivity in human cAF myofibrils, compared with normal, was found to be either significantly increased (2) or unchanged (10). Furthermore, research has found alterations in sarcomeric protein expression and phosphorylation levels (2, 4, 10, 11), suggesting that myofilament Ca2+ sensitivity is increased (2, 4).
To incorporate decreased force generation rates and enhanced Ca2+ sensitivity in our atrial myofilament model, we increased thin filament activation and reduced XB cycling rates. We increased thin filament activation by increasing two parameters (kon and kn_p) and decreasing four parameters (perm50, koffL, koffH, and kp_n). The parameters knpT and kpnT are nonlinear transition rates that are functions of these six parameters and represent Ca2+-based activation of the thin filament, which is shown in Fig. 1 as the transition of the thin filament from the NXB state (XB formation is inhibited) to the PXB state (weakly bound XB formation is possible). The parameter perm50 is the half-activation constant for the shift of a thin filament regulatory unit (RU) from NXB to PXB, koffH (koffL) is the rate constant for Ca2+ unbinding from the high (low) affinity binding site of troponin C, and kon is the rate constant for Ca2+ binding to troponin C. The parameters kn_p and kp_n are constant scaling factors of the knpT and kpnT transition rates. We reduced XB cycling rates by increasing two parameters (gapp and hb) and decreasing another three parameters (fapp, hf, and gxb). The rates fapp and gapp regulate the transition of the thin filament from the PXB state to the strongly bound XB state, where the myosin head has not yet rotated and thus not induced strain in the neck region (XBPreR). The parameters hf and hb are transition rates between the XBPreR and XBPostR states (thin filament is strongly bound to a XB, which has a rotated myosin head and has induced distortion). The parameter gxb represents the ATP-consuming transition rate from XBPostR to PXB. Because cAF-induced changes in mechanical properties (described above) do not directly translate to changes in myofilament model parameters, and due to the progressive nature of remodeling in cAF (32), we explored cAF-induced remodeling of these myofilament model parameters within the range of 100% to 300% of their control values (increments of 25%) for kon, kn_p, gapp, and hb and within the range of 100% to 20% of their control values (increments of 10%) for perm50, koffL, koffH, kp_n, fapp, hf, and gxb (Table 2).
Alternans protocol.
To induce APD-ALT in our RA and LA cAF atrial myocyte models, we used a pacing protocol similar to that used in a clinical study by Narayan et al. (32) and in alternans computational studies (5, 6). We first isometrically paced the myocyte model at a CL of 750 ms (80 beats/min) for 1,000 beats to ensure that steady state was reached. We then paced it for 74 beats at each successive CL, starting at 500 ms and descending, in 50-ms increments, to 300 ms. Because we are inherently changing the equilibrium of the simulation each time we change the pacing rate, the model is not at steady state immediately after the pacing rate is changed. This result is consistent with that found by clinical (32) and modeling studies (5, 6). However, a practical steady state is reached within the first 20 beats of pacing at each rate from 500 to 300 ms. Isometric pacing was used, consistent with previous studies investigating the effects of mechanoelectric feedback on cellular electrophysiology (21, 25). The analysis of simulation results focused particularly on assessing alternans levels from beats paced at a CL of 400 ms (150 beats/min), since Narayan et al. (32) documented that that was the average alternans onset CL in patients with cAF.
To determine how the severity of cAF-induced myofilament remodeling alters APD-ALT, we incorporated different amounts of cAF-induced myofilament remodeling in our simulations. One of the eleven myofilament parameters (perm50, koffL, koffH, kp_n, fapp, hf, gxb, kon, kn_p, gapp, and hb) was assigned a noncontrol value (as described in Incorporating cAF-induced remodeling) for a given simulation. To determine how SL modifies the effects of CaBTn on APD-ALT formation, we ran each of these simulations for a SL value between 1.7 and 2.3 µm (in increments of 0.1 µm). These SLs were chosen because they represent the SL range that occurs during a normal cardiac cycle (41). Note that the 11 myofilament parameters can be modified independently of SL.
Additional information regarding the simulation protocol, parameter values, and initial ion concentrations may be found at https://gitlab.com/MZile/AJP_2017.git and in Table 3.
Table 3.
Ion concentrations
| Ion | Initial Concentration |
|---|---|
| Extracellular | |
| K+ | 5.4 mM |
| Na+ | 140 mM |
| Ca2+ | 1.8 mM |
| Intracellular | |
| K+ | 120 mM |
| Na+ | 9.71 mM |
| Ca2+ | 0.224 µM |
| Sarcoplasmic reticulum | |
| Ca2+ | 0.509 mM |
Shown are selected initial ion concentrations for the train of beats isometrically paced at a cycle length of 750 ms for all models.
Analysis of alternans.
APD was defined as the time from maximal upstroke velocity to 90% repolarization of transmembrane voltage (Vm) from peak amplitude (17, 24). APD alternans-normalized magnitude (APD-ANM) was calculated (5, 6, 32) as the mean magnitude of change in APD over the last 10 pairs of beats (11 total beats) at a given pacing CL divided by the mean APD over the final 10 beats, using the equation below:
| (2) |
[Ca2+]i and the total Ca2+ bound to troponin C ([Ca2+]troponin; CA-ALT and CaT-ALT respectively) were quantified in the same manner as APD-ALT, with the change in each variable defined as the difference in amplitude between beats. Alternans-normalized magnitude (ANM) of >5% was the threshold for ANM to be considered significant (5, 6, 32).
RESULTS
Effect of bidirectionally coupling a myofilament model to the ionic model: CaBTn alters normal membrane kinetics.
Coupling our model of the normal myocyte bidirectionally (i.e., representing the effect of CaBTn on the ionic component of the model) altered the membrane kinetics in the ionic component of our electrotechnical model compared with the uncoupled ionic model of the normal myocyte. The altered membrane kinetics were solely due to the effects of bidirectional coupling since the numerical values of the ionic model parameters in the uncoupled model were identical to those in the bidirectionally coupled model. This is shown in Fig. 3 for the action potential, [Ca2+]i, and [Ca2+]troponin. APD at 90% repolarization (APD90) and [Ca2+]i transient behavior are slightly different in our electromechanical RA model compared with the RA ionic model (APD90: 300 vs. 284 ms, [Ca2+]i,diastolic: 0.1997 vs. 0.2066 µM, and [Ca2+]i transient amplitude: 0.3631 vs. 0.2570 µM). Importantly, APD90, [Ca2+]i,diastolic, and [Ca2+]i transient amplitude derived from our electromechanical model are in better agreement with presently available experimental human RA data (range: 255–344 ms, 0.1197 µM, and 0.3449 µM, respectively) (7, 43) than the uncoupled ionic model. Additionally, the amplitude of the [Ca2+]troponin transient is greater in the electromechanical model compared with the uncoupled ionic model due to the difference in how the models represent troponin C buffering of free cytosolic Ca2+. The uncoupled ionic model uses an equation that does not incorporate cooperativity mechanisms, such as the ability of strongly bound XBs to increase the binding affinity of Ca2+ to troponin C on nearby RUs. In the electromechanical model, when the amplitude of the [Ca2+]i transient increases, Ca2+ binds to troponin C, increasing the percentage of RUs in the strongly bound states (XBPreR and XBPostR states). The presence of strongly bound XBs then enhances the Ca2+-troponin C binding affinity, propelling more Ca2+ to bind to troponin C. Because the binding affinity of Ca2+ to troponin C is higher in the electromechanical model compared with the uncoupled ionic model, the amplitude of the [Ca2+]troponin transient is accordingly greater in the electromechanical model.
Fig. 3.

A–C: transmembrane voltage (Vm; A), free intracellular Ca2+ concentration ([Ca2+]i; B), and total Ca2+ bound to troponin C ([Ca2+]troponin; C) plotted over time for our electromechanical model (blue) and the uncoupled ionic model of the normal right atrial (RA) myocyte (red), with both isometrically paced to steady state at a cycle length (CL) of 1,000 ms and sarcomere length (SL) of 2.4 µm. D: normalized active force plotted only over time for our electromechanical model.
Effect of coupling a myofilament model to the cAF ionic model: presence of CaBTn diminishes APD-ANM.
Because CaBTn changed the [Ca2+]i transient in our bidirectionally coupled electromechanical models compared with the uncoupled ionic models of normal RA and LA myocytes (5, 6), we investigated whether CaBTn could also modulate alternans in our cAF RA and LA electromechanical myocyte models. Incorporating only cAF-induced ionic remodeling (without myofilament remodeling; i.e., the myofilament model was that of a normal RA or LA myocyte) in our RA and LA electromechanical myocyte models resulted in subthreshold alternans (ANM < 5%) in APD, [Ca2+]i, and [Ca2+]troponin for the last 62 beats of the 74-beat train. An example is shown by the blue traces in Fig. 4, A–C, where Vm, [Ca2+]i, and [Ca2+]troponin are plotted for the RA myocyte as functions of time for the final two beats. APD-ANM, CA-ALT-normalized magnitude (CA-ANM), and CaT-ALT-normalized magnitude (CaT-ANM) values are 0.03%, 0.02%, and 0.04% respectively. Although ANM was below threshold for the final 62 beats, there were significant albeit progressively diminishing alternans for the first 12 beats of the 74-beat train (not shown). This is in contrast to results from the uncoupled ionic model, which demonstrated that ANM was maintained above threshold for the duration of the 74 beats. An example is shown by the red traces in Fig. 4, A–C, for Vm, [Ca2+]i, and [Ca2+]troponin (ANM of 16%, 75%, and 99%, respectively). The dampening effect of CaBTn on alternans is owed to the presence of key cooperativity mechanisms only in the electromechanical model, specifically the ability of strongly bound XBs to increase the binding affinity of Ca2+ to troponin C on nearby RUs. Therefore, Ca2+-troponin C affinity is enhanced when the amplitude of the [Ca2+]i transient is greater, resulting in more cytosolic Ca2+ ions binding to troponin C during a long compared with short beat. Because Ca2+ is removed from the cytosol when it binds to troponin C, as the beat number progresses within the train of 74 beats, there is a progressive dampening of CA-ANM and thus APD-ALM.
Fig. 4.

A–C: transmembrane voltage (Vm; A), free intracellular Ca2+ concentration ([Ca2+]i; B), and total Ca2+ bound to troponin C ([Ca2+]troponin; C) plotted over time for simulations with our electromechanical right atrial (RA) myocyte model with only chronic atrial fibrillation (cAF)-induced ionic remodeling incorporated (blue lines) and the uncoupled cAF RA ionic model (red lines). The final two beats (of the 74 total beats) isometrically paced at a cycle length (CL) of 400 ms and sarcomere length (SL) of 1.9 µm are shown for each model. D: normalized active force plotted only over time for our electromechanical model.
Increased cAF-induced myofilament remodeling predominantly increases APD-ANM.
Incorporating both cAF-induced ionic and myofilament remodeling in our human RA and LA electromechanical myocyte models resulted predominantly in APD-ANM increases (Fig. 5). Specifically, bifurcations in APD as functions of myofilament parameters occurred as the level of thin filament remodeling increased after changes in kon, koffH, and perm50 in RA and LA myocyte models (Fig. 5, top). The increase in thin filament activation caused a transition from stable APD to beat-to-beat APD oscillations. In RA and LA models, significant APD-ANM (Fig. 5, bottom) occurred for cAF-induced remodeling of kon > 200% (RA) or kon > 250% (LA), koffH < 40% (RA), or koffH < 30% (LA) and perm50 < 30% or < 60% (both LA and RA) of control values. cAF-induced remodeling of other myofilament parameters that increased thin filament activation (knp, kpn, and koffL) or those that led to a reduced rate of XB cycling (gapp, hb, fapp, hf, and gxb) did not alter APD-ANM (not shown).
Fig. 5.

Bifurcation diagrams for action potential duration (APD; A, C, and E) and sensitivity of APD alternans-normalized magnitude (APD-ANM; B, D, and F) as functions of chronic atrial fibrillation (cAF)-induced myofilament remodeling in simulations with our right atrial (RA; blue lines) and left atrial (LA; green lines) electromechanical models with human chronic atrial fibrillation (cAF)-induced ionic remodeling and a sarcomere length (SL) of 1.9 µm. To incorporate cAF-induced increased thin filament activation, each unique simulation has a cAF-remodeled value of kon (A and B), koffH (C and D), or perm50 (E and F) ranging from normal (100% of control) to diseased (300% of control for kon and 20% of control for koffH and perm50). The pink dashed lines in B, D, and F indicate the division between subthreshold ANM and significant ANM (>5%). AF, atrial fibrillation.
An increase in thin filament activation enhanced the binding affinity of troponin C for Ca2+, thus increasing the fraction of troponin C with bound Ca2+ during both long and short beats. However, as more Ca2+ binds to troponin C, the fraction of troponin C with bound Ca2+ reached saturation (i.e., a value of one) during the long beat. As a result, the fraction of troponin C bound to Ca2+ during the short beat increased relative to that during the long beat, causing a concomitant relative increase in the fraction of strongly bound XBs. Because of the cooperativity between strongly bound XBs and Ca2+-troponin C binding affinity on nearby RUs, this caused a further increase in Ca2+-troponin C affinity. As a result, [Ca]troponin increased, and the peak magnitude of [Ca2+]i decreased during the short beat relative to the long beat, increasing CA-ANM. Therefore, as cAF-induced thin filament activation increased, APD-ANM also became larger. An example of this is shown in Fig. 6, where we compared Vm, [Ca2+]i, the fraction of strongly bound XBs, and [Ca]troponin for simulations with different amounts of cAF-induced remodeling of thin filament activation. When cAF-induced remodeling of thin filament activation was increased from 150% to 225% of control, increases were found in APD-ANM (to 9.4% from 0.03%), CA-ANM (to 43% from 0.02%), CaT-ANM (to 74% from 0.03%), and the maximum percentage of XBs in a strongly bound state (to 25% from 1.8% during the first beat; to 11% from 1.8% during the second beat).
Fig. 6.

Transmembrane voltage (Vm; A), free intracellular Ca2+ concentration ([Ca2+]i; B), percentage of regulatory units with strongly bound cross-bridges (XBs; C), and [Ca2+]troponin (D) plotted over time for our right atrial (RA) electromechanical myocyte models with chronic atrial fibrillation (cAF)-induced ionic remodeling. The final two beats (of the 74 total beats) isometrically paced at a sarcomere length (SL) of 1.9 µm with cAF-induced myofilament remodeling with a value of kon = 150% (blue lines), at a SL of 1.9 µm with kon = 225% (red lines), or at a SL of 2.3 µm with kon = 150% (green lines) are shown. The percentage of regulatory units (RUs) with strongly bound cross-bridges (XBs) were calculated as the sum of RUs in the XBPreR and XBPostR states divided by the total number of RUs.
Subthreshold APD-ANM (Fig. 5, bottom) occurred for RA and LA models with values of perm50 ≤ 30%. In these cases, the fraction of troponin C with bound Ca2+ became close to saturation during both short and long beats. Therefore, the binding affinity of troponin C to Ca2+ was reduced in the short beat relative to that in the long beat, causing the difference in the amount of Ca2+ buffered by troponin C between successive beats to progressively diminish. Because Ca2+ is removed from the cytosol when it binds to troponin C, this leads to progressive dampening of CA-ALT and thus of APD-ALT.
Longer SLs predominantly increase APD-ANM.
Finally, we investigated the effect of SL on APD-ANM, since stretch is common in cAF (40) and has been shown to increase the susceptibility of atrial myocytes to APD-ALT (42). The results of the simulations revealed that longer SLs predominantly increase APD-ANM for both RA and LA electromechanical myocyte models, as shown in Fig. 7 for simulations with cAF-induced myofilament remodeling represented by kon (Fig. 7, left), koffH (Fig. 7, middle), and perm50 (Fig. 7, right). An increase in SL increases the thin filament single overlap region, resulting in greater thin filament activation and enhanced binding affinity of troponin C to Ca2+. As described in Increased cAF-induced myofilament remodeling predominantly increases APD-ANM, when binding affinity is heightened, the fraction of troponin C with bound Ca2+ reaches saturation during only the long beats, resulting in larger APD-ANM. An example of this is shown in Fig. 6 for simulations at two different SLs. When SL was increased from 1.9 to 2.3 µm, increases were found in APD-ANM (to 15% from 0.03%), CA-ANM (to 94% from 0.02%), CaT-ANM (to 124% from 0.03%), and the maximum percentage of XBs in a strongly bound state (to 42% from 1.8% during the first beat; to 16% from 1.8% during the second beat). For simulations with SLs > 2.1 µm and with the greatest cAF-induced myofilament remodeling (kon > 275% for RA only, koffH < 30%, and perm50 < 50% for both RA and LA), the binding affinity of troponin C to Ca2+ was largest. When the binding affinity is greatest, the fraction of troponin C with bound Ca2+ reaches saturation during both the long and short beats, resulting in smaller APD-ANM.
Fig. 7.

Sensitivity of action potential duration alternans-normalized magnitude (APD-ANM) to sarcomere length (SL) in simulations with our right atrial (RA; A, C, and E) and left atrial (LA; B, D, and F) electromechanical myocyte models with chronic atrial fibrillation (cAF)-induced ionic remodeling and varying degrees of cAF-induced myofilament remodeling. cAF-induced myofilament remodeling was incorporated as in Fig. 5. Each white dot represents a unique simulation in which the model was isometrically paced at a specific SL ranging from 1.7 to 2.3 µm in increments of 0.1 µm. Colors represent APD-ANM. Contour lines were drawn at APD-ANM of 5%, 10%, 15%, and 20%.
DISCUSSION
An increased propensity for APD-ALT has been shown to render the human atria vulnerable to ectopy-induced arrhythmia and to increase arrhythmia complexity and its persistence in computational models of human cAF (6). These simulation results are consistent with studies demonstrating that atrial alternans is linked to disease progression in patients with AF (32) and is associated with enhanced AF susceptibility after myocardial infarction in clinical studies (22, 28) and atrial tachycardia in animal models (19, 29). Recent studies have also examined the cellular mechanism underling the occurrence of APD-ALT at clinically relevant slow pacing rates, i.e.,120–200 beats/min (5, 6). Although researchers have examined the ionic mechanisms underlying APD-ALT, the effects of myofilament protein kinetics on APD-ALT have not been elucidated. Here, we investigated whether myofilament protein kinetics mechanisms modulate APD-ALT formation in cAF at clinically relevant pacing rates. By comparing our human electromechanical atrial myocyte model with the human ionic atrial model, we showed that CaBTn has a dampening effect on APD-ANM. These results are consistent with our previous simulation study, where we showed that CaBTn diminished Ca2+ and voltage alternans, driven by heart failure-induced downregulation of SR Ca2+ uptake current, in simulations of failing human ventricular myocytes (45). Because APD-ALT has been advocated as a clinical marker for AF risk and has been suggested as a new therapeutic target in patients with cAF (5, 6, 31), our results propose that targeting myofilament protein kinetics may offer an alternative approach for prevention of arrhythmia in patients with cAF.
We also examined how APD-ALT is affected by cAF-induced myofilament remodeling. Previous studies have suggested that myofilament Ca2+ sensitivity, the amount of Ca2+ necessary to produce half-maximal force, is increased in AF (2, 10), although the degree to which it is increased and whether it promotes arrhythmogenicity in AF have not been elucidated. Therefore, in the present study, we investigated how varying degrees of increased thin filament activation, which enhances Ca2+ sensitivity, altered the formation of APD-ALT. Our results show that increasing thin filament activation and thus Ca2+ sensitivity via CaBTn predominantly enhanced APD-ANM. Because APD-ALT has been identified as a dynamic arrhythmogenic substrate underlying human AF and because APD-ANM is larger in patients with cAF than in patients with paroxysmal AF (31), our observations suggest that myofilament remodeling may occur in patients with cAF but not in patients with paroxysmal AF. This is supported by Belus et al. (2), who found that myofilament Ca2+ sensitivity was increased in human cAF (long-standing persistent AF), whereas Eiras et al. (10) found that there was no significant difference in myocytes from healthy subjects and patients with persistent AF. Therefore, enhanced Ca2+ sensitivity may be responsible for the enhanced arrhythmogenicity found in patients with cAF compared with patients with persistent AF. However, organ-scale simulations will be necessary to verify this hypothesis. Furthermore, our results suggest that therapies decreasing the disease-enhanced thin filament activation could reduce APD-ANM, thus potentially reducing the propensity for arrhythmia in patients with AF.
Acute and chronic stretch are known to cause abnormal atrial remodeling and to increase vulnerability to AF (40). Acute atrial stretch causes transient electrophysiological changes such as conduction slowing and altered atrial refractoriness in animal (33, 36, 37) and human (12, 34) studies. Chronic atrial stretch has been implicated in structural atrial remodeling and persistent conduction slowing in both animal (38) and human (30) studies. Although investigators have examined some of the proposed mechanisms by which atrial stretch can lead to AF (14), such as stretch-activated ion channels (23, 39), few studies have examined the effects of atrial stretch on APD-ALT. One study by Tsai et al. (42) found that mechanical stretch in a HL-1 atrial myocyte monolayer led to increased susceptibility to CA-ALT and APD-ALT due to slower Ca2+ reuptake kinetics via decreased expression of SR adenosine triphosphatase 2. However, only rapid field pacing could induce APD-ALT in this study. This is in contrast to the clinical observations (32) showing that APD-ALT occurs in patients with cAF at slower rates, and thus these finding may be more relevant to patients with paroxysmal AF (20). Therefore, we investigated how stretched sarcomeres affected the formation of APD-ALT at clinically relevant pacing rates in human cAF. Our findings at slower clinically relevant rates of 150 beats/min (400 ms CL), similar to those from Tsai et al. (42) at faster pacing rates of 240–360 beats/min (250– 67 ms CL), showed that simulated myocytes with longer SLs resulted in enhanced APD-ANM due to larger CA-ANM, suggesting that atrial stretch is an important modulator of APD-ALT. Therefore, therapies designed to reduce the mechanical stretch commonly found in cAF may reduce the propensity for arrhythmia in these patients.
This study focused on alternans onset at modest heart rates in patients with cAF and the role of CaBTn in it; the generation of such alternans is a cell-level phenomenon. As previous research has demonstrated, cell-level alternans that originate at slow rates might transition into spatially discordant alternans at high rates, sustaining arrhythmogenesis (6). The role of contraction in this process at such high rates has not been investigated. The cell-level model developed here would be a useful tool in such investigations.
This study has a number of limitations. Other types of mechanoelectric feedback, including stretch-activated channels and xRos signaling, may influence APD-ALT in atrial myocytes but were outside the scope of this study. In addition, the myofilament model empirically rather than physiologically represents enhanced Ca2+-troponin C buffering at longer SLs. Specifically, at longer SLs, the myofilament model increases the effective Ca2+-troponin C binding rate rather than diminishing Ca2+ unbinding rate due to the cooperative activity of active nearby XBs. This limitation is inherent to the original formulation of the myofilament model (35). Also, despite optimizing our model to accurately reproduce human atrial force data, we were unable to validate the state occupancy of the Markov model due to the phenomenological nature of the original Rice et al. (35) model and lack of available experimental data. Finally, because of the phenomenological nature of the original Rice et al. (35) model, we were unable to validate the numerical values of myofilament model parameters in cAF against experimental data.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01-HL1-26802 and PD1-HL-123271 (to N. A. Trayanova) and a Leducg grant.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.A.Z. and N.A.T. conceived and designed research; M.A.Z. performed experiments; M.A.Z. analyzed data; M.A.Z. and N.A.T. interpreted results of experiments; M.A.Z. prepared figures; M.A.Z. drafted manuscript; M.A.Z. and N.A.T. edited and revised manuscript; M.A.Z. and N.A.T. approved final version of manuscript.
REFERENCES
- 1.Ball J, Carrington MJ, McMurray JJ, Stewart S. Atrial fibrillation: profile and burden of an evolving epidemic in the 21st century. Int J Cardiol 167: 1807–1824, 2013. doi: 10.1016/j.ijcard.2012.12.093. [DOI] [PubMed] [Google Scholar]
- 2.Belus A, Piroddi N, Ferrantini C, Tesi C, Cazorla O, Toniolo L, Drost M, Mearini G, Carrier L, Rossi A, Mugelli A, Cerbai E, van der Velden J, Poggesi C. Effects of chronic atrial fibrillation on active and passive force generation in human atrial myofibrils. Circ Res 107: 144–152, 2010. doi: 10.1161/CIRCRESAHA.110.220699. [DOI] [PubMed] [Google Scholar]
- 3.Bisping E, Tenderich G, Barckhausen P, Stumme B, Bruns S, von Lewinski D, Pieske B. Atrial myocardium is the predominant inotropic target of adrenomedullin in the human heart. Am J Physiol Heart Circ Physiol 293: H3001–H3007, 2007. doi: 10.1152/ajpheart.01276.2006. [DOI] [PubMed] [Google Scholar]
- 4.Cazorla O, Szilagyi S, Vignier N, Salazar G, Krämer E, Vassort G, Carrier L, Lacampagne A. Length and protein kinase A modulations of myocytes in cardiac myosin binding protein C-deficient mice. Cardiovasc Res 69: 370–380, 2006. doi: 10.1016/j.cardiores.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Chang KC, Bayer JD, Trayanova NA. Disrupted calcium release as a mechanism for atrial alternans associated with human atrial fibrillation. PLOS Comput Biol 10: e1004011, 2014. doi: 10.1371/journal.pcbi.1004011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang KC, Trayanova NA. Mechanisms of arrhythmogenesis related to calcium-driven alternans in a model of human atrial fibrillation. Sci Rep 6: 36395, 2016. doi: 10.1038/srep36395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christ T, Wettwer E, Voigt N, Hála O, Radicke S, Matschke K, Várro A, Dobrev D, Ravens U. Pathology-specific effects of the IKur/Ito/IK,ACh blocker AVE0118 on ion channels in human chronic atrial fibrillation. Br J Pharmacol 154: 1619–1630, 2008. doi: 10.1038/bjp.2008.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, Gillum RF, Kim YH, McAnulty JH Jr, Zheng ZJ, Forouzanfar MH, Naghavi M, Mensah GA, Ezzati M, Murray CJ. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation 129: 837–847, 2014. doi: 10.1161/CIRCULATIONAHA.113.005119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Oliveira BL, Rocha BM, Barra LP, Toledo EM, Sundnes J, Weber dos Santos R. Effects of deformation on transmural dispersion of repolarization using in silico models of human left ventricular wedge. Int J Numer Methods Biomed Eng 29: 1323–1337, 2013. doi: 10.1002/cnm.2570. [DOI] [PubMed] [Google Scholar]
- 10.Eiras S, Narolska NA, van Loon RB, Boontje NM, Zaremba R, Jimenez CR, Visser FC, Stooker W, van der Velden J, Stienen GJ. Alterations in contractile protein composition and function in human atrial dilatation and atrial fibrillation. J Mol Cell Cardiol 41: 467–477, 2006. doi: 10.1016/j.yjmcc.2006.06.072. [DOI] [PubMed] [Google Scholar]
- 11.El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U, Dobrev D. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation 114: 670–680, 2006. doi: 10.1161/CIRCULATIONAHA.106.636845. [DOI] [PubMed] [Google Scholar]
- 12.Elvan A, Adiyaman A, Beukema RJ, Sie HT, Allessie MA. Electrophysiological effects of acute atrial stretch on persistent atrial fibrillation in patients undergoing open heart surgery. Heart Rhythm 10: 322–330, 2013. doi: 10.1016/j.hrthm.2012.10.041. [DOI] [PubMed] [Google Scholar]
- 13.Fabritz L, Guasch E, Antoniades C, Bardinet I, Benninger G, Betts TR, Brand E, Breithardt G, Bucklar-Suchankova G, Camm AJ, Cartlidge D, Casadei B, Chua WW, Crijns HJ, Deeks J, Hatem S, Hidden-Lucet F, Kääb S, Maniadakis N, Martin S, Mont L, Reinecke H, Sinner MF, Schotten U, Southwood T, Stoll M, Vardas P, Wakili R, West A, Ziegler A, Kirchhof P. Expert consensus document: Defining the major health modifiers causing atrial fibrillation: a roadmap to underpin personalized prevention and treatment. Nat Rev Cardiol 13: 230–237, 2016. doi: 10.1038/nrcardio.2015.194. [DOI] [PubMed] [Google Scholar]
- 14.Franz MR, Bode F. Mechano-electrical feedback underlying arrhythmias: the atrial fibrillation case. Prog Biophys Mol Biol 82: 163–174, 2003. doi: 10.1016/S0079-6107(03)00013-0. [DOI] [PubMed] [Google Scholar]
- 15.Goetzenich A, Schroth SC, Emmig U, Autschbach R, Pieske B, Rossaint R, Christiansen S. Hypothermia exerts negative inotropy in human atrial preparations: in vitro-comparison to rabbit myocardium. J Cardiovasc Surg (Torino) 50: 239–245, 2009. [PubMed] [Google Scholar]
- 16.Hanley CM, Robinson VM, Kowey PR. Status of antiarrhythmic drug development for atrial fibrillation: new drugs and new molecular mechanisms. Circ Arrhythm Electrophysiol 9: e002479, 2016. doi: 10.1161/CIRCEP.115.002479. [DOI] [PubMed] [Google Scholar]
- 17.Hayashi H, Miyauchi Y, Chou CC, Karagueuzian HS, Chen PS, Lin SF. Effects of cytochalasin D on electrical restitution and the dynamics of ventricular fibrillation in isolated rabbit heart. J Cardiovasc Electrophysiol 14: 1077–1084, 2003. doi: 10.1046/j.1540-8167.2003.03234.x. [DOI] [PubMed] [Google Scholar]
- 18.Ji YC, Gray RA, Fenton FH. Implementation of contraction to electrophysiological ventricular myocyte models, and their quantitative characterization via post-extrasystolic potentiation. PLoS One 10: e0135699, 2015. doi: 10.1371/journal.pone.0135699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jousset F, Tenkorang J, Vesin JM, Pascale P, Ruchat P, Rollin AG, Fromer M, Narayan SM, Pruvot E. Kinetics of atrial repolarization alternans in a free-behaving ovine model. J Cardiovasc Electrophysiol 23: 1003–1012, 2012. doi: 10.1111/j.1540-8167.2012.02336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalifa J, Yamazaki M. Repolarization alternans in dilated pulsing atria: a preventable “prelude” to atrial fibrillation? J Am Coll Cardiol 58: 2116–2117, 2011. doi: 10.1016/j.jacc.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 21.Katsnelson LB, Solovyova O, Balakin A, Lookin O, Konovalov P, Protsenko Y, Sulman T, Markhasin VS. Contribution of mechanical factors to arrhythmogenesis in calcium overloaded cardiomyocytes: model predictions and experiments. Prog Biophys Mol Biol 107: 81–89, 2011. doi: 10.1016/j.pbiomolbio.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 22.Kettlewell S, Burton FL, Smith GL, Workman AJ. Chronic myocardial infarction promotes atrial action potential alternans, afterdepolarizations, and fibrillation. Cardiovasc Res 99: 215–224, 2013. doi: 10.1093/cvr/cvt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim D. A mechanosensitive K+ channel in heart cells. Activation by arachidonic acid. J Gen Physiol 100: 1021–1040, 1992. doi: 10.1085/jgp.100.6.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim YH, Garfinkel A, Ikeda T, Wu TJ, Athill CA, Weiss JN, Karagueuzian HS, Chen PS. Spatiotemporal complexity of ventricular fibrillation revealed by tissue mass reduction in isolated swine right ventricle. Further evidence for the quasiperiodic route to chaos hypothesis. J Clin Invest 100: 2486–2500, 1997. doi: 10.1172/JCI119791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohl P, Day K, Noble D. Cellular mechanisms of cardiac mechano-electric feedback in a mathematical model. Can J Cardiol 14: 111–119, 1998. [PubMed] [Google Scholar]
- 26.Lip GYH, Brechin CM, Lane DA. The global burden of atrial fibrillation and stroke: a systematic review of the epidemiology of atrial fibrillation in regions outside North America and Europe. Chest 142: 1489–1498, 2012. doi: 10.1378/chest.11-2888. [DOI] [PubMed] [Google Scholar]
- 27.Maier LS, Barckhausen P, Weisser J, Aleksic I, Baryalei M, Pieske B. Ca2+ handling in isolated human atrial myocardium. Am J Physiol Heart Circ Physiol 279: H952–H958, 2000. doi: 10.1152/ajpheart.2000.279.3.H952. [DOI] [PubMed] [Google Scholar]
- 28.Miyauchi Y, Zhou S, Okuyama Y, Miyauchi M, Hayashi H, Hamabe A, Fishbein MC, Mandel WJ, Chen LS, Chen PS, Karagueuzian HS. Altered atrial electrical restitution and heterogeneous sympathetic hyperinnervation in hearts with chronic left ventricular myocardial infarction: implications for atrial fibrillation. Circulation 108: 360–366, 2003. doi: 10.1161/01.CIR.0000080327.32573.7C. [DOI] [PubMed] [Google Scholar]
- 29.Monigatti-Tenkorang J, Jousset F, Pascale P, Vesin JM, Ruchat P, Fromer M, Narayan SM, Pruvot E. Intermittent atrial tachycardia promotes repolarization alternans and conduction slowing during rapid rates, and increases susceptibility to atrial fibrillation in a free-behaving sheep model. J Cardiovasc Electrophysiol 25: 418–427, 2014. doi: 10.1111/jce.12353. [DOI] [PubMed] [Google Scholar]
- 30.Morton JB, Sanders P, Vohra JK, Sparks PB, Morgan JG, Spence SJ, Grigg LE, Kalman JM. Effect of chronic right atrial stretch on atrial electrical remodeling in patients with an atrial septal defect. Circulation 107: 1775–1782, 2003. doi: 10.1161/01.CIR.0000058164.68127.F2. [DOI] [PubMed] [Google Scholar]
- 31.Narayan SM, Bode F, Karasik PL, Franz MR. Alternans of atrial action potentials during atrial flutter as a precursor to atrial fibrillation. Circulation 106: 1968–1973, 2002. doi: 10.1161/01.CIR.0000037062.35762.B4. [DOI] [PubMed] [Google Scholar]
- 32.Narayan SM, Franz MR, Clopton P, Pruvot EJ, Krummen DE. Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation 123: 2922–2930, 2011. doi: 10.1161/CIRCULATIONAHA.110.977827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ravelli F, Allessie M. Effects of atrial dilatation on refractory period and vulnerability to atrial fibrillation in the isolated Langendorff-perfused rabbit heart. Circulation 96: 1686–1695, 1997. doi: 10.1161/01.CIR.96.5.1686. [DOI] [PubMed] [Google Scholar]
- 34.Ravelli F, Masè M, del Greco M, Marini M, Disertori M. Acute atrial dilatation slows conduction and increases AF vulnerability in the human atrium. J Cardiovasc Electrophysiol 22: 394–401, 2011. doi: 10.1111/j.1540-8167.2010.01939.x. [DOI] [PubMed] [Google Scholar]
- 35.Rice JJ, Wang F, Bers DM, de Tombe PP. Approximate model of cooperative activation and crossbridge cycling in cardiac muscle using ordinary differential equations. Biophys J 95: 2368–2390, 2008. doi: 10.1529/biophysj.107.119487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sideris DA, Toumanidis ST, Thodorakis M, Kostopoulos K, Tselepatiotis E, Langoura C, Stringli T, Moulopoulos SD. Some observations on the mechanism of pressure related atrial fibrillation. Eur Heart J 15: 1585–1589, 1994. doi: 10.1093/oxfordjournals.eurheartj.a060433. [DOI] [PubMed] [Google Scholar]
- 37.Solti F, Vecsey T, Kékesi V, Juhász-Nagy A. The effect of atrial dilatation on the genesis of atrial arrhythmias. Cardiovasc Res 23: 882–886, 1989. doi: 10.1093/cvr/23.10.882. [DOI] [PubMed] [Google Scholar]
- 38.Takeuchi S, Akita T, Takagishi Y, Watanabe E, Sasano C, Honjo H, Kodama I. Disorganization of gap junction distribution in dilated atria of patients with chronic atrial fibrillation. Circ J 70: 575–582, 2006. doi: 10.1253/circj.70.575. [DOI] [PubMed] [Google Scholar]
- 39.Tavi P, Laine M, Weckström M. Effect of gadolinium on stretch-induced changes in contraction and intracellularly recorded action- and afterpotentials of rat isolated atrium. Br J Pharmacol 118: 407–413, 1996. doi: 10.1111/j.1476-5381.1996.tb15417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thanigaimani S, McLennan E, Linz D, Mahajan R, Agbaedeng TA, Lee G, Kalman JM, Sanders P, Lau DH. Progression and reversibility of stretch induced atrial remodeling: Characterization and clinical implications. Prog Biophys Mol Biol 130: 376–386, 2017. doi: 10.1016/j.pbiomolbio.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 41.Trayanova NA, Rice JJ. Cardiac electromechanical models: from cell to organ. Front Physiol 2: 43, 2011. doi: 10.3389/fphys.2011.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai CT, Chiang FT, Tseng CD, Yu CC, Wang YC, Lai LP, Hwang JJ, Lin JL. Mechanical stretch of atrial myocyte monolayer decreases sarcoplasmic reticulum calcium adenosine triphosphatase expression and increases susceptibility to repolarization alternans. J Am Coll Cardiol 58: 2106–2115, 2011. doi: 10.1016/j.jacc.2011.07.039. [DOI] [PubMed] [Google Scholar]
- 43.Voigt N, Trafford AW, Ravens U, Dobrev D. Abstract 2630: cellular and molecular determinants of altered atrial Ca2+ signaling in patients with chronic atrial fibrillation. Circulation 120: S667–S668, 2009. [Google Scholar]
- 44.Zile MA, Trayanova NA. Myofilament protein dynamics modulate EAD formation in human hypertrophic cardiomyopathy. Prog Biophys Mol Biol 130: 418–428, 2017. doi: 10.1016/j.pbiomolbio.2017.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zile MA, Trayanova NA. Rate-dependent force, intracellular calcium, and action potential voltage alternans are modulated by sarcomere length and heart failure induced-remodeling of thin filament regulation in human heart failure: a myocyte modeling study. Prog Biophys Mol Biol 120: 270–280, 2016. doi: 10.1016/j.pbiomolbio.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]